文/Sobereva@北京科音
First release: 2023-Nov-7 Last update: 2024-Feb-18
0 前言
對周期性體系,雜化泛函的計算耗時眾所周知遠高于純泛函。著名的第一性原理程序CP2K不光純泛函的計算超級快,對大體系做雜化泛函計算也非常快(盡管還是遠比純泛函貴),遠遠快于Quantum ESPRESSO (QE)、VASP等純粹基于平面波的程序。CP2K結合像樣的基組做雜化泛函的單點計算,在一般雙路服務器上算到上千原子都沒問題。然而,使用CP2K做高效的雜化泛函計算有很多非常關鍵性的要點必須知道,遠遠不是像Gaussian程序那樣光寫個雜化泛函名字就了事的。然而,筆者經常在網上回答CP2K問題時看到很多人完全不具備這些最基本常識,胡用瞎用CP2K做雜化泛函計算,導致耗時巨高、始終出不來結果、任務崩潰、SCF完全不收斂等各種問題。因此筆者覺得很有必要寫個小文將CP2K做雜化泛函計算的最關鍵常識和要點挨個提一下。Multiwfn程序(http://www.shanxitv.org/multiwfn)具有極其便利的創建CP2K輸入文件的功能,在本文的最后會還用Multiwfn產生C60晶體的CP2K雜化泛函計算的輸入文件作為實際例子。
本文的內容至少對CP2K 2024.1,以及2023-Nov-7及以后更新的Multiwfn是適用的。
筆者在北京科音CP2K第一性原理計算培訓班(http://www.keinsci.com/workshop/KFP_content.html)的“能量的計算及相關問題”部分里講雜化泛函計算遠比本文全面、深入得多,并列舉諸多經驗技巧和例子,還詳細介紹HSE06、CAM-B3LYP、wB97X等各種范圍分離形式的雜化泛函以及revDSD-PBEP86-D3(BJ)等各種雙雜化泛函的用法和要點,使用雜化泛函結合k點計算的要點,歡迎想學習更多知識者關注和參加。
1 CP2K雜化泛函計算的特性
---------
2024-Feb-18更新:以下關于k點的說法已經過時。從2024.1版開始支持了雜化泛函計算時考慮k點,并且可以結合ADMM。此時需要用RI計算HF交換部分。
首先要知道CP2K做雜化泛函計算的一個關鍵不足是不支持考慮k點,因此如果被計算的是周期性體系,晶胞必須大到只考慮gamma點就足夠。如果你要計算的體系的晶胞原本比較小,顯然必須先擴胞到足夠大才行。如果不知道擴到多大夠用,嚴格來說需要做你感興趣的屬性的計算結果隨擴胞倍數的收斂性測試。雖然CP2K能夠對含有非常多原子的超胞做得動雜化泛函計算,但耗時會明顯高于QE等程序對原胞在考慮k點的情況下做雜化泛函計算。
最能充分體現CP2K雜化泛函計算效率優勢的情況是算一些較大分子晶體、MOF、COF等一些大晶胞體系,以及做第一性原理動力學等情況。對小晶胞體系如上所述經過擴胞后也能算,只不過不體現CP2K的長處。
CP2K顯然沒法用雜化泛函算能帶,因為不支持k點,這種目的只能用QE等程序代替。必須用CP2K算的話,應當用CP2K支持的純泛函里算帶隙相對比較好的比如HLE17。
---------
由于雜化泛函遠比普通泛函昂貴,尤其是用于幾何優化、NEB、動力學等涉及到結構變化的任務。因此如果你算的問題用純泛函就足以描述得不錯、用雜化泛函不會帶來明顯好處的話,顯然應該用純泛函。記住,非必要甭考慮雜化泛函,純泛函的耗時僅有雜化泛函的一個微不足道的零頭。對幾何優化、振動分析、NEB等高耗時任務用純泛函,算勢壘、反應能的時候用更好的雜化泛函是可以接受且常見的策略。
周期性體系的雜化泛函計算不僅耗時遠高于純泛函計算,對內存要求更是遠遠高于純泛函計算。經常用CP2K做雜化泛函任務的服務器的內存容量絕對是多多益善,選擇服務器的時候要注意這一點。也別天真地試圖拿個人計算機將就著跑CP2K的雜化泛函計算。
CP2K做雜化泛函計算的前提是編譯時必須帶著libint電子積分庫,否則沒法算雜化泛函中HF交換項中涉及的雙電子積分。CP2K的編譯方法見http://www.shanxitv.org/586。
雜化泛函用的贗勢基組和贗勢就用常見的給PBE等純泛函優化的就可以。從CP2K 9.1開始,data目錄下的BASIS_MOLOPT_UZH和POTENTIAL_UZH文件里還分別給出了專門給PBE0優化的贗勢基組和贗勢,如C的DZVP-MOLOPT-PBE0-GTH-q4和GTH-PBE0-q4,原理上來說它們用于雜化泛函計算會更理想一些(但實際未必有什么優勢)。
2 庫侖截斷
雜化泛函計算之所以比純泛函昂貴得多,在于計算雜化泛函的HF交換項時要計算大量的雙電子積分(two-electron integral, ERI),尤其是對于周期性體系來說,不光要計算晶胞內基函數之間的ERI,還要計算中心晶胞與周圍鏡像晶胞基函數之間的ERI。再加上由于只能算gamma點而晶胞必然較大,原子數、基函數通常非常多,無疑要算的ERI是巨量的。
由于周期邊界條件,原理上來說要計算的ERI是無窮的,顯然這會導致計算無法進行。因此實際計算時必須使用庫侖截斷(Coulomb truncation),也就是將ERI中描述電子庫侖相互作用的1/r算符截斷在某個距離(截斷半徑),超過截斷半徑時1/r視為0,相應地ERI就不需要計算了,這個策略使得ERI的計算數目是有限的。如果你計算的是非周期性體系,由于本身ERI數目就是有限的,因此雖然也可以用庫侖截斷,但這就不是必須的了。
雜化泛函的庫侖截斷的半徑設置顯然既影響耗時也影響結果。截斷半徑越小,要計算的ERI越少,耗時就越低,但結果越偏離泛函原本的精確結果。顯然不能為了一味地節約時間而把截斷半徑設得太小,否則結果會很垃圾。6埃通常是耗時和精度的較好權衡,設得明顯更小會令精度有明顯損失,設得更大并不會令精度有明顯提升卻會增加耗時不少。如果對精度要求很高也可以用8埃,設得更大就完全沒必要了。
要注意像6、8埃這種程度的截斷半徑其實還沒有大到令絕對能量充分收斂,但能量在相互求差時,由截斷半徑帶來的系統性誤差能很大程度抵消。可以認為用不同的截斷半徑相當于用不同的理論方法,直接對比能量的情況必須保持截斷半徑的設置相統一!
值得一提的是,不一定截斷半徑小結果就一定差,有可能截斷半徑造成的對ERI忽略的誤差和泛函本身的誤差相抵消導致恰好對某些體系某些問題的計算比用更大截斷半徑時結果還更好。
截斷半徑應當小于晶胞最短尺寸的一半以免導致得到缺乏物理意義的結果。這里所謂的最短尺寸,是(001)晶面間距、(010)晶面間距、(100)晶面間距三者中的那個最小值。也因此為了能夠使用6埃截斷半徑,晶胞最短尺寸應當大于12埃,如果晶胞不夠大則應當擴胞(即便拋開庫侖截斷問題不談,單從只能計算gamma點這點來說,晶胞也不能過小)。
Multiwfn創建的雜化泛函輸入文件中,若周期性設成了NONE,則不使用庫侖截斷,若是周期性,Multiwfn會自動加上庫侖截斷的設置。如果晶胞最短尺寸夠大,Multiwfn會自動把截斷半徑設為6埃,如果不夠大,則會根據當前晶胞最短尺寸自動設成能設的最大半徑。
以下是庫侖截斷的設置方式,加入到&DFT/&XC里面。POTENTIAL_TYPE TRUNCATED代表用庫侖截斷的1/r算符,CUTOFF_RADIUS設置截斷半徑(埃)。T_C_G_DATA t_c_g.dat是默認的可以不寫,這指定的是庫侖截斷用的gamma函數的數據文件t_c_g.dat,在CP2K的data目錄下。
&HF
&INTERACTION_POTENTIAL
POTENTIAL_TYPE TRUNCATED
CUTOFF_RADIUS 6.0
T_C_G_DATA t_c_g.dat
&END INTERACTION_POTENTIAL
&END HF
3 積分屏蔽
積分屏蔽(integral screening)是指盡可能在不明顯犧牲精度的前提下減少要計算的ERI的量,這對于CP2K雜化泛函計算的耗時和內存使用量有至關重要的影響!上一節的庫侖截斷本身就是一種積分屏蔽策略,CP2K里還有另外兩種非常重要的積分屏蔽方法,也是普遍應用在Gaussian等量子化學程序中的:
(1)利用Schwarz不等式做積分屏蔽:利用Schwarz不等式關系,可以根據數目較少的二指標ERI估計出數目甚巨的三、四指標ERI中哪些數值肯定小于閾值從而可以忽略掉,這可以大幅減少ERI要算的數目(原本總共要算的ERI中占比最大的就是四個基函數序號不同的那些ERI)。閾值通過&DFT/&XC/&HF/&SCREENING中EPS_SCHWARZ設置,此值越大,被忽略的ERI數目越多、計算精度越差。默認的1E-10雖然足夠保證精度,但此時太貴,沒必要。通常設為1E-6就可以了,是精度和耗時的較好權衡,想要精度更好點用1E-8。
(2)結合密度矩陣做積分屏蔽:ERI對能量的貢獻取決于它與特定密度矩陣元的乘積。因此哪怕ERI不是很小,但與之相乘的密度矩陣元很小,那么這樣的ERI也完全不需要計算。CP2K定義了特定方法快速估計一個ERI與相應密度矩陣元乘積值的上限,如果小于上述的EPS_SCHWARZ,則此ERI就會忽略掉。這種依賴于密度矩陣的積分屏蔽默認是關閉的,如果開啟則把&DFT/&XC/&HF/&SCREENING里的SCREEN_ON_INITIAL_P設為T。由于這種積分屏蔽能令耗時巨幅降低,因此我強烈建議總是開啟,也因此Multiwfn產生的雜化泛函的輸入文件里都自動把SCREEN_ON_INITIAL_P設成T。
基于密度矩陣做積分屏蔽有一個極其關鍵的要點是初猜的密度矩陣質量不能太差!然而很多人對此一無所知!默認情況下初始的密度矩陣是CP2K自動做初猜產生的,由于它和SCF收斂時的密度矩陣相差很大、質量很糙,因此此時積分屏蔽通常做得極爛,不僅可以忽略的ERI可能沒忽略,還會把一些重要的ERI給忽略掉,由此可能導致KS矩陣質量很爛、SCF極難收斂。而且忽略哪些ERI是在SCF過程一開始決定好的,即便之后隨著SCF迭代密度矩陣質量不斷變好,也不會由此使得積分屏蔽逐步變得合理,這導致即便最后SCF收斂了,得到的能量的精度也可能很爛。因此當SCREEN_ON_INITIAL_P設T時,必須讀取一個基本合理的波函數當初猜。通常是在雜化泛函計算前先用相同的基組做一個便宜的純泛函計算,比如PBE0計算前先用PBE做一個單點計算,得到記錄了PBE收斂的波函數信息的wfn文件,然后PBE0計算時用SCF_GUESS RESTART,并把WFN_RESTART_FILE_NAME指定成那個wfn文件,以讀取其作為初猜。這不僅使得基于密度矩陣的積分屏蔽可以合理進行,還有一個額外的好處是由于初猜波函數質量較好(至少比默認自動產生的好得多),使得雜化泛函計算時達到SCF收斂所需的迭代次數會比較少,這進一步節約了時間(注意即便拋開ERI計算耗時不談,雜化泛函在構造KS矩陣時花的時間也明顯高于純泛函,因為涉及到密度矩陣元與ERI的大量的乘加操作)。
一般的量子化學程序雖然也基于密度矩陣做積分屏蔽,但不是非得像CP2K這樣得讀取一個像樣的初猜波函數,這是因為它們會根據當前最新的密度矩陣判斷這一輪SCF用的ERI哪些是能被忽略的。CP2K的做法和它們不同,主要是因為CP2K傾向于讓用戶做incore形式的SCF而非一般量化程序一般用的direct形式的SCF,見后文。
當SCREEN_ON_INITIAL_P設T時,對于雜化泛函的幾何優化、分子動力學等任務的續算,要注意光有.restart文件還不行,還必須提供上一步的.wfn文件當初猜波函數,否則也是會由于基于自動初猜的密度矩陣做積分屏蔽非常惡心而令任務無法正常跑下去。
如果你嫌麻煩而不想先做純泛函計算產生收斂的波函數再做雜化泛函計算,那就不要寫SCREEN_ON_INITIAL_P T。對于小體系、小基組特別是非周期性的計算,不基于密度矩陣做積分屏蔽時耗時也不高。而對于計算量較大的雜化泛函計算,則強烈建議不要偷這個懶而多花巨量不必要的時間。
4 ADMM
CP2K還支持其開發者獨創的ADMM (Auxiliary Density Matrix Methods)方法減少要算的ERI數目從而進一步顯著加快雜化泛函的計算,其原理見以下北京科音CP2K第一性原理計算培訓班的ppt。

ppt里提到的主基組就是指平時實際用的基組,諸如DZVP-MOLOPT-SR-GTH。對于不同的主基組,有一些專門構造的與之匹配的ADMM輔助基組,比如MOLOPT系列基組適合搭配的是CP2K的data目錄下的BASIS_ADMM_UZH文件里的那些輔助基組,從小到大依次是admm-dz、admm-dzp、admm-tzp、admm-tz2p。admm-dzp是2-zeta結合極化函數的輔助基組,通常算是保底質量。DZVP-MOLOPT-SR-GTH主基組適合搭配admm-dzp,更好的主基組如TZVP-MOLOPT-GTH結合admm-dzp也可以接受,若結合admm-tzp精度會更好些,這都比起不用ADMM能巨幅節約計算時間和內存。輔助基組的大小與主基組越接近,ADMM給雜化泛函HF交換項帶來的誤差越小,但需要計算越多ERI因而越耗時。如果輔助基組和主基組設為相同的,那么ADMM不會造成任何誤差,相應地也不會節約任何耗時(反倒還會增加一些耗時)。
data目錄下的BASIS_ADMM文件中的FIT3、pFIT3等輔助基組都比較過時了,對元素涵蓋也遠不如BASIS_ADMM_UZH里的完整,因此如今不再建議使用(它們出現在不少官方例子文件和文獻中,如今不要效仿)。目前版本的Multiwfn產生利用ADMM的雜化泛函的輸入文件時默認設的是admm-dzp。
注意前述這些輔助基組都是對GTH贗勢基組用的,千萬別做全電子計算時也用它們。要想全電子計算時用ADMM,可以用(aug-)pcseg系列基組作為主基組,在《在線基組和贗勢數據庫一覽》(http://www.shanxitv.org/309)中介紹的BSE基組數據庫中有和它匹配的(aug-)admm輔助基組,諸如pcseg-2適合搭配admm-2。pcseg是一類構造得很好的對DFT計算非常劃算的基組,《談談量子化學中基組的選擇》(http://www.shanxitv.org/336)里簡要提及了,在《北京科音基礎量子化學培訓班》(http://www.keinsci.com/workshop/KBQC_content.html)里講基組的地方做了具體介紹。
用MOLOPT系列基組做周期性體系的雜化泛函計算幾乎是一定要結合ADMM的,否則極難算得動!因為MOLOPT是完全廣義收縮基組,而CP2K利用的計算源高斯函數(PGTF)之間ERI的libint庫又沒有專門考慮廣義收縮的情況,因此造成等效的PGTF數目甚巨,ERI的數目又與PGTF數目的四次方近似成正比,數目顯然多得恐怖。如果你用片段收縮基組,比如def2-SVP、6-31G**、pcseg-1、pob-DZVP-rev2之類去做雜化泛函計算,由于需要考慮的PGTF數目遠比檔次與之相仿佛的DZVP-MOLOPT-SR-GTH要少,因此ADMM方法不是非用不可,但不用的話耗時會顯著高于DZVP-MOLOPT-SR-GTH搭配admm-dzp的情況。
不能有的原子用ADMM加速計算而有的原子不用。因此如果你用混合基組,其中有的原子用的主基組有ADMM輔助基組,有的沒有,你又想用ADMM,那么沒有ADMM輔助基組的那些原子的輔助基組就只能設定成其主基組。雖然這些原子沒法被ADMM技術加速,但至少其它那部分原子能被加速。
前面提到的各種節約耗時的策略是彼此完全兼容的,同時利用可以最大程度節約時間。即周期性體系雜化泛函計算時一般要用庫侖截斷、利用Schwarz不等式做積分屏蔽、結合密度矩陣做積分屏蔽、開啟ADMM。所有這些設定都會對能量產生影響,因此寫文章的時候最好注明,橫向對比時應統一。
啟用ADMM需要在&KIND里加上BASIS_SET AUX_FIT [輔助基組名],同時增加一行BASIS_SET_FILE_NAME [輔助基組文件名]。并且在&DFT里加入以下內容
&AUXILIARY_DENSITY_MATRIX_METHOD
&END AUXILIARY_DENSITY_MATRIX_METHOD
如果用的是對角化而非OT,還需要在以上字段里加入ADMM_PURIFICATION_METHOD NONE要求不做純化。
5 ERI的儲存和內存使用量的控制
SCF有兩種常見形式,incore SCF是指SCF計算一開始將所有要用到的ERI全都算出來并儲存在內存里,之后SCF每一輪在構造KS矩陣時會直接從內存里讀取ERI而不再重新計算。direct SCF是指SCF每一輪都重新計算要用到的ERI,這也叫on-the-fly方式計算ERI。顯然,incore比direct總耗時低得多,但代價是機子的必須內存非常大,因為要存的ERI通常非常多。在CP2K的雜化泛函計算中,如果你給的內存足夠存得下所有ERI,那么只有SCF第一輪的時候由于要計算所有ERI所以耗時很高,而之后SCF每一輪的耗時就很低了。如果給的內存不夠儲存所有ERI,那么能在內存里存多少ERI就存多少,存的那部分在之后每一輪SCF中會直接讀取,存不下的ERI在每一輪SCF中會重新算,相當于介于incore和direct之間的情況。顯然,做大體系雜化泛函計算時應當盡量在大內存機子上做、分配給CP2K盡可能多的內存,以讓盡可能多的ERI能存到內存中、盡可能減少每輪SCF過程中要on-the-fly計算的ERI的量來節約耗時。
常有人問為什么他做雜化泛函計算時老是卡在SCF剛開始的位置不動,這是因為SCF第一輪肯定是要計算所有需要考慮的ERI,耗時顯然往往很高,對大體系、大基組的情況花一、兩個小時都是常事,看上去好像一直停在這里似的。如果你給的內存較少而導致大部分ERI都得on-the-fly方式去算,那么之后SCF每一輪都得花將近這么多時間,整個任務跑完可能得半天甚至一天時間。如果PRINT_LEVEL設的是MEDIUM,在SCF第一輪算完后就會輸出Number of sph. ERI's calculated on the fly和Number of sph. ERI's stored in-core,前者是之后每一輪SCF都需要重新算的ERI數,后者是已存到內存中而之后不需要SCF每一輪重新算的ERI數。顯然前者相對于后者越小,之后每一輪SCF比第一輪時間少得越多。
CP2K用戶通常默認用popt版本,每個MPI進程在計算HF交換項時最大內存使用量由&DFT/&XC/&HF/&MEMORY里的MAX_MEMORY控制,單位為MB,扣除掉儲存零碎數據的占用量外,其余都用來儲存ERI。MAX_MEMORY與MPI并行進程數的乘積必須小于當前機子空余物理內存量,顯然應當盡量給大以盡可能減少on-the-fly方式算的ERI量,但也不要太頂著頭分配,因為進入HF交換項計算模塊之前CP2K還會占一定量的內存,故要有適當余量。在SCF第一輪不斷往內存里寫入ERI、內存占用量不斷增大的過程中,若空余內存已用完時CP2K還在繼續往里存ERI,CP2K就會馬上崩潰,甚至還可能導致計算機暫時失去響應。所以MAX_MEMORY應當設得恰到好處。
如果空余物理內存很有限,為了能讓MAX_MEMORY能設得比較大,可以減小MPI進程數。為了不因此浪費CPU運算能力,此時可以用psmp版,此時每個MPI進程下屬的OpenMP線程都可以共享這個MPI進程儲存的ERI。
6 雜化泛函的定義方式
這一節簡要說一下雜化泛函怎么正確在輸入文件里定義。對于非周期性體系,比如用PBE0,直接在&DFT里寫上以下內容即可。此時不用庫侖截斷而用完整的1/r算符,EPS_SCHWARZ為默認的1E-10、SCREEN_ON_INITIAL_P為默認的F。
&XC
&XC_FUNCTIONAL PBE0
&END XC_FUNCTIONAL
&END XC
顯然對周期性體系絕對不能簡單寫成上面那樣,至少也得自己去指定為使用庫侖截斷。結合前面的講解,周期性體系PBE0計算的泛函定義部分一般寫為下面這樣。PBE0泛函由75%的PBE交換項+25%的HF交換項+100%的PBE相關項構成,所以需要恰當地定義&PBE里面的參數,并在&HF里用FRACTION指定HF成份。
&XC
&XC_FUNCTIONAL
&PBE
SCALE_X 0.75
SCALE_C 1.0
&END PBE
&END XC_FUNCTIONAL
&HF
FRACTION 0.25
&SCREENING
EPS_SCHWARZ 1E-6
SCREEN_ON_INITIAL_P T
&END SCREENING
&INTERACTION_POTENTIAL
POTENTIAL_TYPE TRUNCATED
CUTOFF_RADIUS 6.0
&END INTERACTION_POTENTIAL
&MEMORY
MAX_MEMORY 3000
&END MEMORY
&END HF
&END XC
有的泛函在&XC_FUNCTIONAL里有特定的字段,可以省得自己去定義細節參數,但HF成份依然必須自己定義,否則相當于只做了殘缺的純泛函計算,結果毫無意義。比如HF成分為10%的TPSSh泛函可以寫為
&XC
&XC_FUNCTIONAL
&HYB_MGGA_XC_TPSSH
&END HYB_MGGA_XC_TPSSH
&END XC_FUNCTIONAL
&HF
FRACTION 0.1
...同上
&END HF
&END XC
更多的泛函定義方式可以直接看Multiwfn產生的相應雜化泛函的輸入文件里的寫法,都是嚴格正確的。
7 一個常見警告:The Kohn Sham matrix is not 100% occupied
做雜化泛函計算,特別是用了ADMM時,很容易在SCF開始后看到The Kohn Sham matrix is not 100% occupied警告。具體原因在https://www.cp2k.org/faq:hfx_eps_warning有介紹,這里就不多說了。對于實際計算來說,看到這個警告時,先把&QS里的EPS_PGF_ORB設為比默認明顯更小的1E-12,如果計算正常,即便還有這個警告也可直接無視。EPS_PGF_ORB設得越小,越無法利用重疊矩陣和KS矩陣的稀疏性節約時間,但這對耗時的影響相對于雜化泛函計算的總耗時來說微不足道。目前版本的Multiwfn產生的雜化泛函的輸入文件里自動就把EPS_PGF_ORB設成了1E-12。
對于晶胞較小的情況,本身矩陣稀疏性就差,開發者建議上來就在&QS里設MIN_PAIR_LIST_RADIUS -1從而完全不利用矩陣的稀疏性節約時間,此時絕對不會再出現那個警告,但比起用EPS_PGF_ORB 1E-12時耗時會高一些。
8 簡單例子:C60分子晶體
最后,這里以C60分子晶體的單點任務作為例子演示一個標準的周期性雜化泛函的計算。如果你還不了解Multiwfn創建CP2K輸入文件的功能,建議先閱讀《CP2K第一性原理程序在CentOS中的簡易安裝方法》(http://www.shanxitv.org/586)。
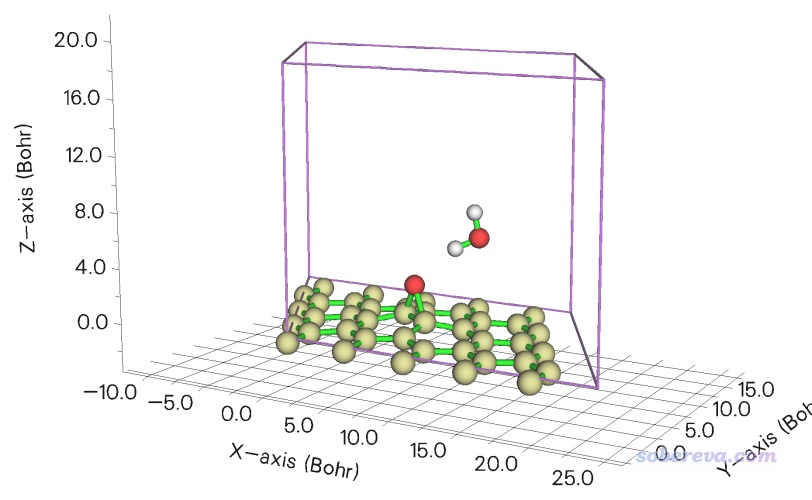
此體系的cif文件和下面涉及的輸入文件都可以在http://www.shanxitv.org/attach/690/file.zip中找到。此體系結構如下所示,一共240個C原子。晶胞邊長是14.07埃,對于分子晶體來說不擴胞的情況下只考慮gamma點可以接受。
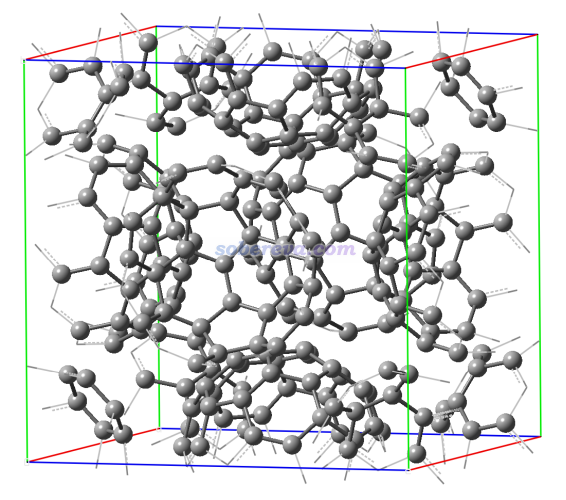
啟動Multiwfn,輸入C60.cif的路徑載入之,然后輸入
cp2k
PBE.inp //要產生的PBE計算的輸入文件名
0 //基于默認設置產生輸入文件
現在當前目錄下就有了PBE.inp,對應PBE/DZVP-MOLOPT-SR-GTH單點任務。然后接著在Multiwfn里輸入
cp2k
PBE0.inp //要產生的PBE0計算的輸入文件名
1 //選擇理論方法
-6 //PBE0結合ADMM
0 //產生輸入文件
由PBE0.inp內容可見,Multiwfn自動指定的計算設置完全合適,對應的是PBE0/DZVP-MOLOPT-SR-GTH結合admm-dzp輔助基組、6埃庫侖截斷、EPS_SCHWARZ 1E-6、SCREEN_ON_INITIAL_P T、開啟OT。
手動把PBE0.inp中的WFN_RESTART_FILE_NAME前頭的#去掉并設為PBE-RESTART.wfn,把SCF_GUESS RESTART開頭的#去掉,從而使得PBE0計算時讀取PBE收斂的波函數當初猜。然后根據你的機子的空余物理內存量和要用的MPI進程數設置合適的MAX_MEMORY。
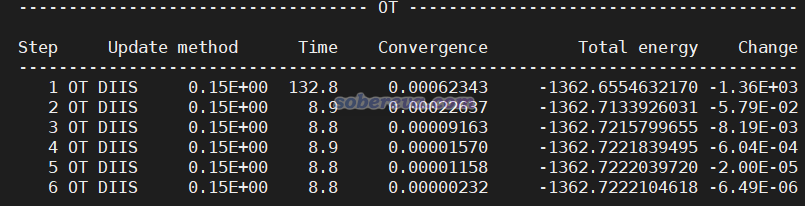
把PBE.inp和PBE0.inp放到同一個目錄下,先運行PBE.inp,算完后當前目錄下就出現了PBE-RESTART.wfn,然后再運行PBE0.inp。在《淘寶上購買的雙路EPYC 7R32 96核服務器的使用感受和雜談》(http://www.shanxitv.org/653)介紹的筆者的2*7R32 512GB雙路服務器上用96核并行、MAX_MEMORY設5000,用CP2K 2023.2 popt版,PBE計算花了8秒,PBE0計算花了178秒。PBE0任務的SCF迭代過程如下所示,可見第一輪耗時較高,由于當前給的內存足夠儲存所有ERI,因而ERI沒有on-the-fly計算的,所以之后每一輪SCF的耗時遠遠低于第一輪。
9 總結
本文把強大的CP2K高效地做雜化泛函計算的各方面要點進行了介紹,以避免初學者對各種關鍵信息毫不知情而憑感覺胡用瞎用。本文的內容也充分體現出用Multiwfn產生CP2K輸入文件非常容易,令研究者從復雜的輸入文件編寫中充分解放,但這絕不意味著CP2K的使用者就可以對關鍵性技術細節毫不知情,否則會各種踩坑、白浪費時間、算出無意義的結果。CP2K用戶必須掌握的知識、要領遠多于大部分其它計算程序。筆者開設的北京科音CP2K第一性原理計算培訓班(http://www.keinsci.com/workshop/KFP_content.html)可以使得學員完整系統透徹掌握全部這些知識。
]]>文/Sobereva@北京科音 2023-Aug-1
0 前言
經常有做計算化學的人在網上問為什么他沒法重復出來文獻里的計算數據。導致重復不出來的原因實在太多了,然而初學者提問此類問題時幾乎總是含糊其辭,提供的有效信息甚少,導致別人根本無從回復,或者需要別人羅列一大堆可能導致差異的原因。我感到每次回復此類問題太費事,于是在這里專門寫個文章說說這事。遇到這種問題的讀者仔細看完此文后,應該能料想出可能是什么原因導致自己和文獻的結果存在差異。就算還搞不明白,至少也應該能明白在網上問別人的時候應當提供什么信息,知道怎么把導致差異的可能原因的范圍盡可能縮小,免得別人打很多字羅列諸多當前不需要考慮的可能性,或者別人嫌你提供的有效信息完全不夠而根本不想回復。
這里首先要強調一點的是,如果你的目的就是為了精確重復文獻數據,那你就要盡最大可能使用和文獻完全相同的方式去做計算,而不要管人家的做法合不合理,哪怕你明知道人家的計算方式不當。下文說的內容都是為了讓你能盡可能準確重復出文獻里的數據,而不是講怎么合理地計算。如果你的實際目的是想獲得相同體系相同研究問題的合理的結果,那你只要確保你自己的計算是合理的就行了,沒必要非得把別人的數據重復出來,說不定人家的計算本來就有問題。如果最終驗證發現別人的計算有硬傷并因此導致結論錯誤,或者憑基本知識就能斷定別人的計算有問題,那么可以發一篇comment文章指正以免以訛傳訛,比如《我對一篇存在大量錯誤的J.Mol.Model.期刊上的18碳環研究文章的comment》(http://www.shanxitv.org/584)里介紹的文章。
1 結構不一樣
自己計算用的結構和文獻不一致,是導致數據無法重現的最常見原因之一。如果你用的結構就是文獻正文或補充材料里給的坐標,那就可以直接排除這個因素了。注意有些文獻的SI里鍵長、笛卡爾坐標單位是Bohr而不是埃,千萬別弄錯了。
如果你計算的基本結構特征都和文獻里不符,也即算的都不是同一個東西,能重現出文獻里的數據就怪了。很多體系有不同的形態,比如酚酞在酸性、中性、堿性環境下主要的存在形態明顯不一樣,氨基酸在真空和水環境下的質子化態明顯不一樣,姜黃素在水和有機溶劑中一種是烯醇式一種是酮式結構,等等。你必須確保你算的和文獻里算的確實是同一種狀態的結構,如果文獻里給了截圖,一定要仔細對照。
做計算要有基本的化學常識,并且要認真仔細,免得搭出來沒意義的結構去做計算,自然和文獻里對不上。比如文獻里給出Lewis結構式時通常是隱去碳上的氫的,如果計算的時候真不畫氫,或者少畫了個別的氫,自然計算毫無意義。再比如,文獻里算一個羧基配位的過渡金屬配合物,如果你在計算時羧基上還留著氫,明顯也是錯的。再比如,文獻里畫了一個帶一個小配體的過渡金屬配合物的Lewis式,若你算的是水溶液中的狀態并忽略了配位水分子,也自然是錯的。再比如,文獻里給出一個聯苯或者碗烯的二維結構式,看上去是在一個平面上,若你去計算其基態時真的擺成平面來優化,得到的也是大概率是錯誤的結構(有破壞平面的虛頻的結構)。還要注意一些結構的構建不要想當然,比如SiO2表面在水中時表面的O主要是以羥基及其去質子化形態存在的,比例和pH有關,如果都當成是烏禿禿的氧就錯了。結構構建的關鍵性信息在文獻里一般都會有具體說明,但也可能一些文獻中在描述上有疏漏。
有些體系在計算的時候結構構建方式不唯一,比如《要善用簇模型,不要盲目用ONIOM算蛋白質-小分子相互作用問題》(http://www.shanxitv.org/597)、《18碳環(cyclo[18]carbon)與石墨烯的相互作用:基于簇模型的研究一例》(http://www.shanxitv.org/615)和《使用量子化學程序基于簇模型計算金屬表面吸附問題》(http://www.shanxitv.org/540)里提到的簇模型,怎么構建簇明顯會影響結果,特別是當簇用得較小的時候。若要重復文獻里的數據,就要用盡可能相同的模型,哪怕其模型很昂貴。
很多分子有不止一種構象(勢能面極小點),柔性大分子甚至有一萬種以上的構象。幾何結構優化一般會優化到與初始結構最接近的勢能面極小點。而不同構象下計算的屬性可能有不小差異,因此你必須確保你用的構象和文獻里一致,也即結構優化使用的初猜結構要盡可能像文獻里給出的。如果文獻沒明確給出三維結構信息,按理說文獻應當用的是能量最低構象(確切來說是自由能最低構象)做的計算,但也有可能作者忽略了構象搜索并得到了不合理的結果。如果你對構象搜索一點概念都沒有,建議看下文了解些常識
《使用molclus程序做團簇構型搜索和分子構象搜索》(http://bbs.keinsci.com/thread-577-1-1.html)
《gentor:掃描方式做分子構象搜索的便捷工具》(http://bbs.keinsci.com/thread-2388-1-1.html)
《將Confab或Frog2與Molclus聯用對有機體系做構象搜索》(http://bbs.keinsci.com/thread-20063-1-1.html)
《使用Molclus結合xtb做的動力學模擬對瑞德西韋(Remdesivir)做構象搜索》(http://bbs.keinsci.com/thread-16255-1-1.html)
也不是所有情況都適合用能量最低構象來計算,比如某化學反應可能是從某個能量相對較高的構象發生的,可以對反應的IRC的反應物一端做幾何優化得到之。
團簇體系的構型搜索和柔性分子的構象搜索同等重要。哪怕水二聚體這么簡單的兩個小分子構成的團簇都都有不同的極小點構型,并且對應的結合能明顯不同。因此計算這種體系也必須確保和文獻用的構型相一致。如果文中沒明確交代,一般默認其用的是能量最低構型結構做的計算,但也可能作者忽略了這個問題,導致誤用了能量不是最低的構型并因此得到了錯誤的結果。如果你對構型搜索沒有概念,建議看此文:《genmer:生成團簇初始構型結合molclus做團簇結構搜索的超便捷工具》(http://bbs.keinsci.com/thread-2369-1-1.html)。
有的時候兩種極小點構型或構象可能結構相差很小。這種情況需要把可視化程序里的視角和文獻里的圖片擺得盡可能一致再去觀察,免得算的其實不是同一個結構。
上面說的是計算極小點結構的情況,自不必說,重復文獻里過渡態的計算結果時也必須嚴格確保和文獻里用的結構一致。
對于研究固體表面的情況,要注意文獻里的結構是怎么來的。表面是從體相直接切出來的未經優化的(unrelaxed),還是經過優化的(relaxed),還是使用的考慮表面重構的(reconstructed),這需要區分清楚。文獻中計算用的是具體哪個晶面也必須要搞清楚。固體表面吸附問題也有類似構象/構型搜索的勢能面存在多種極小點問題,一個小分子可能在表面上不同位點以不同方式吸附,實際算的結構必須和文獻相一致。文獻里算的吸附是物理吸附還是化學吸附也得搞清楚。對于吸附過程有勢壘的情況,初猜結構中你把被吸附物和表面擺得相距很近,一般優化完了的是形成新鍵的化學吸附,如果擺得較遠,一般得到的是弱相互作用維持的物理吸附,二者的結構、相互作用能相差甚大。研究表面吸附還有覆蓋率的問題,要注意文獻中對所用結構的相關描述,研究低覆蓋率和飽和覆蓋率時用的模型明顯不同。
用slab模型研究固體表面還要注意厚度問題。slab太薄的話,得到的功函數、吸附能等性質都不準確。對于二維層狀材料的研究,還要注意層數對計算的性質的影響。這些方面都應當和文獻嚴格一致。對表面類體系優化時還經常牽扯到對一部分原子凍結使之固定為體相的結構,之后振動分析也相應地牽扯凍結,凍結方式必須和文獻一致。
做某些體系的分子動力學模擬也同樣要注意初始結構的差異,否則現象可能和文獻不符。比如有文獻研究表明水中的蛋白質由于疏水效應會自發進入口徑足夠大的碳納米管,如果你一開始把蛋白質放得離碳納米管老遠,由于質量頗大的蛋白質本身整體運動就較慢,再加上碳納米管的隨意旋轉(沒凍結的話),可能文獻里說的那種蛋白質自發進入管內的現象跑挺長時間也出現不了。再比如想模擬結冰過程,哪怕一開始把溫度設得顯著低于冰點,但沒有在水中加入局部的冰的結構的話,跑很長時間也不會看到冰的結構的出現。至于在模擬的時間尺度內一定會出現的現象,初始結構就不重要了。比如水和乙醇的互溶,無論初始結構里把二者混合均勻,還是分成兩相,最后肯定都是互溶的。
做周期性體系的模擬還要注意盒子尺寸與文獻要一致。用第一性原理計算固體時,假設k點數是固定的,盒子尺寸的不同會影響精度。做動力學模擬時,盒子尺寸還間接影響原子運動行為以及模擬得到的屬性。
用Quantum ESPRESSO等平面波程序時,以及CP2K這種將平面波當輔助基函數的程序時,如果算的是某些維度為非周期性體系,還需要注意真空層的厚度,要盡可能和文獻一致。因為真空層可能對結果有不可忽視的影響,比如影響非周期性方向上與相鄰鏡像的作用是否可以忽略。而且比如CP2K里用MT的Psolver時還有非周期性方向上盒子尺寸大于相應方向有電子密度明顯分布的兩倍以上的要求,顯然真空區不能小了。
假設文獻用的是實驗測定的坐標直接進行性質的計算,還要注意實驗來源必須一致。比如文獻或晶體數據庫里對同一種晶體結構可能提供了不同溫度下測定的坐標,會有一定程度的不同(體現熱膨脹)。顯然壓力也直接影響測定的結構。哪怕是相同的測試環境,不同來源的晶體結構也會有差異,特別是氫的位置的確定,無疑這對氫鍵等問題的計算結果會有直接影響。做量子化學/第一性原理研究的人一般都會至少對氫的坐標重新進行優化來refine之,需要注意文獻里是怎么考慮的。蛋白質的高解析度和低解析度測出來的晶體結構里殘基側鏈的位置也可能相差甚大,甚至明顯影響到做酶催化、分子對接之類問題的結果。同樣的屬性,以不同類型實驗測定的結果注定也會有差異,比如氣相電子衍射和單晶衍射測的一個是氣相結構一個是晶體結構,對于柔性分子差異可能很大,這點參考《實驗測定分子結構的方法以及將實驗結構用于量子化學計算需要注意的問題》(http://www.shanxitv.org/569)。
上面列舉了諸多結構層面影響計算結果的可能性,當然不可能列舉得很完備,實際情況多種多樣。總之希望讀者能意識到嚴格保證和文獻里用的結構的一致對于重現數據有多么重要。
2 計算設置不一樣
為了精確重復文獻數據,要盡可能仔細閱讀文章及其補充材料,了解清楚文獻計算細節,使得自己的計算設置能最大程度與文獻相一致。本節里談到的很多可能導致差異的因素在《談談不同量子化學程序計算結果的差異問題》(http://www.shanxitv.org/573)里都有詳細說明,務必注意閱讀。
量子化學和第一性原理計算用的理論方法,以及理論方法中涉及的設定和參數(如范圍分離泛函的w參數、色散校正的經驗參數、CASSCF和多參考方法的活性空間的選擇、TDDFT計算是否用了TDA近似、DFT+U參數、CASPT2計算用的IPEA shift參數、DLPNO方法用的閾值、GW@DFT和DFT-SAPT具體基于什么泛函、sTDA計算用的經驗參數,等等)必須和文獻精確一致。有的理論方法在同一個程序里甚至有兩種定義,比如B3LYP在ORCA里寫成B3LYP還是B3LYP/G是不同的,要搞清楚文獻實際用的哪個(通常是前者)。還要注意理論方法名和關鍵詞的差異,比如里這里提及的:《Gaussian16里用PBE0關鍵詞計算的實際上是PBE0-DH雙雜化泛函》(http://bbs.keinsci.com/thread-13660-1-1.html)。有的文獻的寫法還可能存在含糊性,比如可能文獻就說對某個開殼層體系用了二階微擾理論,但實際上二階微擾理論用于開殼層的實現形式有一堆,比如UMP2、PUMP2、ROMP2、ZAPT2、ROMP、OPT2等等,如果文中給了引用的文獻,要根據文獻判斷具體是哪種。
對計算結果可能有明顯影響的各種條件必須一致,比如:
? 溶劑效應的考慮。如果用的是隱式溶劑模型,那么用的具體是什么方法?描述的是哪種溶劑?用CP2K的SCCS模型時α、β、γ參數是怎么設的?描述混合溶劑的時是怎么確定溶劑參數的?如果用的是顯式溶劑模型,那么溶劑分子排布是怎么考慮的?有沒有同時用隱式溶劑模型?
? 計算熱力學校正量的模型。比如《使用Shermo結合量子化學程序方便地計算分子的各種熱力學數據》(http://www.shanxitv.org/552)里介紹的Shermo程序默認用的以及ORCA用的是Grimme的QRRHO模型,而Gaussian用的是較原始的不適合含低頻體系的RRHO模型
? 計算熱力學量時是否考慮了平動和轉動,顯然算周期性體系時不應當考慮(在Shermo程序中這由settings.ini里的imode決定)
? 計算轉動對熱力學量貢獻時對轉動對稱數的考慮,看Shermo手冊附錄部分了解相關常識
? 計算熱力學校正量、振動分析用的原子質量
? k點數目和分布
? 平面波相關計算的截斷能
? CP2K中,做雜化泛函時用的庫侖截斷半徑、DFT+U計算用的布居計算方法以及考慮的角動量、Poisson solver的選擇
? 對相對論效應的考慮
? 對外場/外勢的考慮
? DFT積分格點精度。參看《密度泛函計算中的格點積分方法》(http://www.shanxitv.org/69)
? 是否用了凍核、怎么凍的
? 雙電子積分的積分屏蔽設置
? 是否用了RI、DLPNO等加速計算方式
? 是否考慮了偶極校正
? QM/MM計算劃分的位置、MM與QM區之間的相互作用的考慮、link原子的設置
等等等等
基組必須和文獻嚴格一致,并注意上述http://www.shanxitv.org/573博文里說的許多細節問題。注意一個基組/贗勢名可能有不止一個指代,比如Stuttgart贗勢,有不同方式考慮相對論效應的版本、不同價電子數和氧化態的版本,Gaussian內置的和Stuttgart官網上的贗勢和贗勢基組定義往往又有所不同。再比如,GAMESS-US用戶寫CCD和CCT關鍵詞分別用的是cc-pV(D+d)Z和cc-pV(T+d)Z,而不是更常見的cc-pVDZ和cc-pVTZ。要弄清楚作者實際用的什么,仔細看文中的描述,并注意作者引的基組原文。
不光一般意義上的基組(叫主基組或者叫軌道基組)要和文獻計算時的一致,輔助基組也應當一致。輔助基組種類很多,比如RIJ用的/J輔助基組,RIJK用的/JK輔助基組,電子相關計算用的/C輔助基組,CP2K里加快雜化泛函計算用的ADMM輔助基組,CP2K里做LRIGPW計算用的輔助基組,等等。同一個主基組能夠或者適合搭配的某種目的的輔助基組可能不止一種,而且有的時候有含糊性和任意性,比如ORCA里用ma-def2-TZVP主基組時,可能有人用autoaux關鍵詞自動產生輔助基組,可能有的人用標準的def2/J輔助基組。
對于基于經典力場的模擬,力場參數顯然必須和文獻嚴格一致。如果參數是從文獻里拷來然后用在自己計算中的,注意單位轉換,以及1-4相互作用規則和LJ參數的混合規則等細節必須和文章相同。文獻給出的LJ參數有的是R0有的是σ需要區分。原子電荷也必須和文獻保持一致,如果原子電荷是作者自己算的,要搞清楚計算原子電荷的方法,并且如果是基于波函數算的,那么波函數是在什么幾何結構、溶劑環境、計算級別下得到的要弄清楚。這點很重要,比如對于RESP電荷,氣相下計算的和水環境下計算的,Hartree-Fock和B3LYP計算的,相差大了去了,參看這些討論:《RESP擬合靜電勢電荷的原理以及在Multiwfn中的計算》(http://www.shanxitv.org/441)、《RESP2原子電荷的思想以及在Multiwfn中的計算》(http://www.shanxitv.org/531)。
做動力學模擬的情況,必須確保模擬相關設置和文獻一致。比如步長、步數、坐標/速度/受力/能量保存頻率、參考溫度和壓力、控溫和控壓方法、熱浴和壓浴的時間常數、可壓縮系數、各項同性還是各向異性控壓、初速度怎么產生的、范德華和靜電作用的計算方式(如簡單截斷、twin-range、Ewald、PME、SPME等,以及其中涉及的截斷半徑,是否用了switching function等處理)、水模型是剛性還是柔性的、是否用了鍵/鍵角約束、用沒用能量/壓力色散校正,等等。
如果你用的計算程序和文獻里不同,或者和文獻里的相同但版本不同,那么由于默認設置不同,以及算法實現的不同,可能對結果帶來很大差異,一定要仔細閱讀《談談不同量子化學程序計算結果的差異問題》(http://www.shanxitv.org/573)。
在量子化學波函數分析領域也有很多細節設置會影響分析結果。這里以非常流行的波函數分析程序Multiwfn(http://www.shanxitv.org/multiwfn)為例:
? 按照《使用Multiwfn做電子密度、ELF、靜電勢、密度差等函數的盆分析》(http://www.shanxitv.org/179)、《使用Multiwfn的定量分子表面分析功能預測反應位點、分析分子間相互作用》(http://www.shanxitv.org/159)、《使用Multiwfn對靜電勢和范德華勢做拓撲分析精確得到極小點位置和數值》(http://www.shanxitv.org/645)等文章介紹的方法進行分析時,格點間距數值越小精度越高,但也越耗時
? 按照《使用Multiwfn繪制態密度(DOS)圖考察電子結構》(http://www.shanxitv.org/482)繪制PDOS和OPDOS圖時,在Multiwfn里選擇的計算軌道成份的方法會直接影響結果
? 按照《使用Multiwfn模擬掃描隧道顯微鏡(STM)圖像》(http://www.shanxitv.org/549)和《使用Multiwfn結合CP2K的波函數模擬周期性體系的隧道掃描顯微鏡(STM)圖像》(http://www.shanxitv.org/671)繪制STM圖時,偏壓的數值和常高模式掃描的平面的選擇都會明顯影響結果
? 計算《衡量芳香性的方法以及在Multiwfn中的計算》(http://www.shanxitv.org/176)和《使用AdNDP方法以及ELF/LOL、多中心鍵級研究多中心鍵》(http://www.shanxitv.org/138)里介紹的多中心鍵級時,基于原本的基函數和基于自然原子軌道(NAO)計算會得到不同的結果
? 計算Hirshfeld原子電荷時需要用到原子自由狀態下的密度。用自行提供的原子波函數文件來計算此密度,還是直接用Multiwfn內置的此密度,會有所不同。
本節提到的這些計算細節是理應充分體現在文章的computational details里或者補充材料里的。但由于一些文獻的作者為了省篇幅,或者用的很多設置和程序默認的一致,或者由于作者計算水平比較外行、意識不到很多細節對計算非常重要,就在文中沒特意說。如果有些要點不知道作者具體怎么考慮的,自己把各種可能性都試試就知道了,文章沒特意說的設置就姑且用程序默認的。
3 計算的電子結構狀態不一樣
凈電荷和自旋多重度的設置必須和文獻里的相同,否則相當于算的是其它電子態,自然算出來的各種性質都不同。比如計算基態的氧氣發生的反應、計算富勒烯的原子化能,如果氧氣和碳原子誤當成了單重態來算,顯然結果是錯的。此外,稍微了解點兩態反應性的概念就會知道,不少體系在不同自旋態下反應機理都明顯不一樣,如果自旋多重度或凈電荷設錯了,自然反應物/產物/中間體/過渡態等無法得到和文獻里一致的結果,而且相差懸殊。
如果文獻沒注明、從文獻中看不出相關信息,那么凈電荷一般為0。如果被研究的體系的自旋多重度不那么顯而易見,或者研究中涉及到了不止一個自旋態,文中一般都會說明自旋多重度。如果確實沒說,那就得查閱文獻或者數據庫,或者自己判斷、測試了。
很容易被忽略的是開殼層單重態或者叫自旋極化單重態。用KS-DFT等方法來計算的話應當做對稱破缺計算,參看《談談片段組合波函數與自旋極化單重態》(http://www.shanxitv.org/82)。比如整體為單重態的反鐵磁性耦合體系、雙自由基體系、共價鍵被拉長的均裂狀態就是最典型的例子。還有一些特殊的體系也是如此,比如18碳環的D18h點群的過渡態結構、乙烯二面角扭轉成90度的結構、六并苯。重復文獻里的這些體系的計算時別誤當成了閉殼層單重態來計算(文中如果也是做了對稱破缺計算,一般都會明確說。如果沒說,沒準文獻也誤當成了閉殼層計算)。
對于第一性原理計算磁性材料時必須謹慎,很多情況下不是光設個整體的自旋多重度就完事的,而必須恰當設置初始磁矩才能收斂到真正要考察的磁性狀態。為了重復文獻(或材料數據庫)中的數據,若它們給了收斂的波函數對應的各個原子的自旋磁矩/自旋布居信息的話,最好根據其來設置初猜波函數里的原子自旋磁矩,以盡最大可能收斂到和文獻相同的波函數狀態。
要注意波函數穩定性問題。即便凈電荷、自旋多重度的設置是正確的,也不代表SCF收斂后得到的波函數就是實際想研究的態、和文獻里算的狀態相一致。如果文獻里研究的是基態,你算的結果和文獻對不上,而且體系又不是簡單有機體系而是電子結構特殊的體系,那么應當做一下波函數穩定性檢測,比如Gaussian里可以用stable關鍵詞,發現不穩定的話可以用stable=opt嘗試搞出穩定波函數。不穩定的波函數可能意味著這是某個激發態,還可能意味著此波函數是沒有任何物理意義的虛假的狀態。
4 計算的東西或方式不一樣
重復不出文獻的數據時還要想清楚到底你算的東西、計算的方式是否和文獻一致、有可比性。下面列舉一些典型的需要注意的情況。
比如通過能量差E(AB)-E(A)-E(B)考察倆分子A和B間相互作用強度,復合物結構肯定是要優化的,而單分子是直接從復合物優化后的結構里直接取出來,還是也做優化,這在不同文中的做法就不一樣了。單分子不優化情況下得到的能量差一般稱為(復合物結構下的)相互作用能,單分子也優化情況下得到的能量差一般稱為結合能,二者之間相差各個單體的變形能之和。對于諸如《透徹認識氫鍵本質、簡單可靠地估計氫鍵強度:一篇2019年JCC上的重要研究文章介紹》(http://www.shanxitv.org/513)里面列舉的那些剛性小分子,相互作用能和結合能差異甚微,但如果考察的是兩個容易變形的分子間的作用強度,比如兩個氨基酸分子的結合,那么相互作用能和結合能就必須明確區分了。J. Chem. Phys., 158, 244106 (2023)還專門討論了什么情況變形能會較大。對于鍵能,也是類似地有不同計算方式。所以研究此類問題的時候一定要搞清楚文獻具體是怎么算的。
再比如做SAPT能量分解計算,會得到很多耦合項,比如交換與色散作用的耦合項,這是歸到色散項里,還是歸到交換互斥項里,不同文章的做法可能不一樣,需要注意文章怎么說的。再比如計算反應的自由能問題,是計算標準反應自由能,還是計算特定濃度下的反應自由能,這是不一樣,相差濃度校正項,要搞清楚文獻實際算的是什么。再比如電子親和能、電離能、激發能,都有垂直和絕熱之分,文獻里計算哪種都有,也必須確保和文獻算的東西對應。
文獻里有時候對熱力學量的標注比較含糊,要分清楚文獻算的是什么溫度下什么量。比如E,文獻里可能指電子能量,也可能指內能,需要結合語境判斷。
模擬ECD譜需要轉子強度,有長度形式和速度形式兩種,諸如Gaussian都會輸出,在Multiwfn里按照《使用Multiwfn繪制紅外、拉曼、UV-Vis、ECD、VCD和ROA光譜圖》(http://www.shanxitv.org/224)里說的繪制ECD時可以選采用哪種,一般推薦用速度形式。如果你用的和文獻不一致,會導致峰型存在一定差異。
基組的CBS外推的方法不唯一,有不同公式,有的情況外推方式里也涉及到參數,參看《談談能量的基組外推》(http://www.shanxitv.org/172)。不光是能量外推,還有比如GW計算的準粒子能量隨基組的外推、幾何結構隨基組的外推等。一些文獻對高級別理論方法結合大基組還用一些近似方式估計,比如一種常見的是CCSD(T)/大基組 = CCSD(T)/小基組 + (MP2/大基組-MP2/小基組),這在《各種后HF方法精度簡單橫測》(http://www.shanxitv.org/378)里專門說了。涉及到這些高精度數據估算時,需注意保證和文獻的做法一致。
還有一些量本來就沒有唯一定義,去重復文獻的數據更是得弄清楚文獻里用什么定義、什么方法算的。比如自由體積,我在《使用Multiwfn計算晶體結構中自由區域的體積、圖形化展現自由區域》(http://www.shanxitv.org/617)里介紹了一種合理的計算方式,但顯然這不是唯一方式。再比如分子半徑,我在《談談分子半徑的計算和分子形狀的描述》(http://www.shanxitv.org/190)就已經列舉了多種計算方式。還有諸如原子電荷,計算方法更是多了去了,如ADCH、RESP、Hirshfeld、NPA等,簡要介紹見《原子電荷計算方法的對比》(http://www.whxb.pku.edu.cn/CN/abstract/abstract27818.shtml)。
為了正確理解文獻具體算的是什么,必須把背景知識搞清楚,典型的就是gap這個概念,有HOMO-LUMO gap、band gap、optical gap、fundamental gap等,參見《正確地認識分子的能隙(gap)、HOMO和LUMO》(http://www.shanxitv.org/543)。如果沒搞清楚概念和定義方式,可能重復不出文獻的數據,或者實際上文獻的計算方式或用詞不當,你卻意識不到這一點。
某些問題的計算中可能涉及到一些實驗值,要搞清楚作者用的什么值。比如用量子化學程序計算含碳物質的生成焓,通過構造熱力學循環可知其中需要利用實驗的(或第一性原理計算的)石墨的升華焓,要搞清楚這個量是從哪來的,具體是多少。再比如, 量子化學計算某分子的標準氧化還原勢,其中利用到標準氫電極的電勢,其數值有不同來源,從4.05到4.44 V都有報道,Phys. Chem. Chem. Phys., 16, 15068 (2014)建議對理論計算的情況用4.28 V。
還有一些研究涉及到一些額外的參數需要注意,典型的就是頻率校正因子,見《談談諧振頻率校正因子》(http://www.shanxitv.org/221)。是否使用頻率校正因子,以及對某一特定級別使用什么目的的、哪個來源的校正因子,都會影響結果。
周期性體系的計算中,軌道能量的絕對數值是沒意義的,不同程序用的能級零點也不一樣,需要關心的只有相對數值,比如帶隙。所以不要問為什么DOS圖的形狀和文獻一致但橫坐標和文獻不一致。這也正是為什么文獻在繪制DOS圖時往往一般把價帶頂或者費米能級位置設為0點,因為這樣才便于公平對比。
還有些量在文獻里用的習俗和你用的可能不同。比如算結合能問題,有少數文章把結合過程的能量降低量作為正值記錄。再比如NBO給出的E2超共軛作用能,程序輸出時把正值作為超共軛作用造成的體系能量降低,但有的文獻報道E2值的時候取的是負值。
如以上列舉的情況可見,算某個東西的時候必須仔細閱讀文獻搞清楚作者是怎么算的、用的什么定義、算的到底是什么。
5 數據的可重現性本身就差
有些數據很容易重現,比如在RHF/6-31G**級別下優化水分子的基態結構,不管用什么主流量化程序、怎么優化,得到的都是相同結果,初始結構很隨意。有些數據本身就是難以精確重現,典型的就是凝聚相體系的分子動力學。由于分子動力學過程有混沌效應,而且實際計算中往往不可避免地引入隨機因素,會導致文獻中的動力學模擬的定量結果幾乎無法嚴格精確重現。關于這點我專門寫過文章,參看《數值誤差對計算化學結果重現性的影響》(http://www.shanxitv.org/88)。更具體來說也看重現文獻里的什么動力學模擬數據。比如小分子的液態性質相對來說是容易重復的。比如重復密度值,文獻里是801.92 g/L,你算出來801.88 g/L,這就可以認為是很好重復出來了。如果你算出來是810.21 g/L,而且模擬流程和設置是合理的,那就是有明顯不可忽視的差異了,已經超過了隨機性的影響了,需要根據前面所說的來考慮導致與文獻存在差異的可能原因。也有的量的重現相對比較困難,比如磷脂膜里插入了一個分子,去計算它的側向擴散系數,這就屬于相對比較難算準的,因為本身分子在磷脂膜這種擁擠的環境里擴散就困難,而且體系里就這么一個分子,沒法通過同類分子的平均來減小統計誤差。就算你自己跑的時間非常長以保證統計精度絕對足夠高,但文獻里給出的數據未必統計精度就夠高。像這種問題,對文獻里的數據的重現精度就別要求太高了。
還有一種情況是動力學模擬的現象在有必然性的同時本身也有一定隨機性,跑出什么現象有運氣成分。比如距離蛋白質的口袋一定距離處放一個小分子配體,在水環境下做動力學模擬,考察2 ns模擬時間內配體能否和蛋白質結合,初速度隨機生成。像這種模擬的結果就很有隨機性,如果跑10次模擬,可能有的模擬剛過幾百ps小分子就進入口袋了,也可能有的模擬開始后小分子恰好往蛋白質相反方向游離,跑到2 ns還沒進入口袋。對于這種問題的研究必須做大量的模擬來得到統計結果,較嚴謹的文獻普遍都會這么考察這種現象明顯有隨機性的問題,你若要重復文獻也必須以相同的方式去模擬。如果模擬的樣本數不夠多的話,也不要指望結果特別相符。比如前述問題,可能文獻里跑10次發現有5次進入口袋,你模擬10次發現有3次進入口袋,這未必是模擬設置和文獻有區別,而純粹是隨機性所致,要以正確的態度來看待這種情況。
順帶一提,量子化學、第一性原理做靜態的計算的可重復性比起分子動力學模擬要好得多得多,但也不排除極個別情況前者可能受到隨機性的明顯影響。比如對勢能面很復雜的大分子進行優化,特別是QM/MM計算時可能牽扯到幾千原子的情況的優化,有可能由于微小的隨機性因素(如使用了并行計算所帶來的)導致兩次優化分別收斂到了勢能面上可能相距挺遠的兩個極小點,且有肉眼可察覺的差異。
6 對相符程度要求過高
有人其實可能已經合理重復出了文獻中的數據,但由于他對重復的精度要求過高,導致誤以為沒重復出來。
頭腦里要有收斂限的概念。比如文獻里優化的結構二面角是123.64度,你優化出來的是123.69,這就可以認為是重現出來了,因為這點差異通常都已經小于計算程序的幾何優化的默認收斂限了,差異基本來自初始結構的任意性。再比如,文獻里DFT計算出的反應能是-8.12 kJ/mol,你算出來的是-8.18 kJ/mol,也完全可以接受了。如果本來你用的計算程序和文獻里都不同,在盡可能令設置和文獻相同的情況下,你算出來比如是-8.32 kJ/mol也可以算重復出了文獻,各種雞毛蒜皮不值得一提的因素足矣帶來這種程度的差異。但如果你算出來是比如-9.8 kJ/mol,那就明顯不是不值得一提的差異了。再比如TDDFT計算電子激發能、DFT計算HOMO能級,和文獻里相差0.02 eV,這都可以算一致,但如果相差到0.1 eV,這就不算一致了,畢竟TDDFT算激發能的精度都在0.3 eV左右(泛函選擇得當的前提下),當差異達到接近方法本身的誤差的數量級時明顯就算顯著差異了。
重復文獻數據的時候用的收斂限應當足夠嚴,起碼令收斂限顯著小于你期望的對文獻數據的重現精度。雖然文獻作者用的收斂限未必夠嚴,因此光是你把收斂限設嚴未必有意義,但至少先做自己這里能做的事。
7 自己的計算存在硬傷
重復不出文獻數據有可能是自己計算存在硬傷導致的。初學者缺乏常識、經驗和敏銳性,計算很容易存在硬傷,例如:
? 關鍵詞沒寫對或設置不當,導致數據沒意義,比如:Gaussian里想要用PBE0泛函但寫的關鍵詞是PBE0(此時做的是PBE0-DH泛函計算)、用贗勢基組時只定義了基組部分而沒定義贗勢、自定義基組時漏定義了某些原子或元素、Gaussian里用IOp自定義泛函時用opt freq關鍵詞(IOp沒法自動傳遞到freq這一步導致振動分析不是在與優化相同級別勢能面的極小點計算的)、Gaussian里用后HF方法算偶極矩時沒寫density結果得到的是HF級別的、計算UV-Vis光譜時算的態數太少導致光譜不夠完整、CP2K計算時用雜化泛函并且基于密度矩陣做積分屏蔽時沒讀取純泛函波函數當初猜(《解決CP2K中SCF不收斂的方法》http://www.shanxitv.org/665文中提到了)、CP2K算磁性體系時沒寫UKS結果當成了閉殼層算、Gaussian里按照特定方向加電場計算時不知道要寫恰當用nosymm導致電場加的方向和期望的不符(nosymm用處見《談談Gaussian中的對稱性與nosymm關鍵詞的使用》http://www.shanxitv.org/297)、用sobtop(http://www.shanxitv.org/soft/Sobtop)直接產生拓撲文件后沒有給里面添加原子電荷、用sobtop產生拓撲文件時誤把可旋轉二面角以諧振勢對待、用Multiwfn做空穴-電子分析時沒按照《使用Multiwfn做空穴-電子分析全面考察電子激發特征》(http://www.shanxitv.org/434)說的在Gaussian輸入文件里寫IOp(9/40=4),等等
? 缺乏基本知識瞎算,比如:計算小晶胞體系不知道考慮k點、算極化率時不知道要帶彌散函數、B3LYP算色散主導的弱相互作用卻不加色散校正、CP2K里用MOLOPT基組計算需要CUTOFF很高的Na的時候用的CUTOFF不夠、界面體系用了各向同性控壓而不知道應該用半各向同性控壓、算氣-液界面體系在垂直于界面方向開了控壓結果導致真空區消失、用Multiwfn基于CP2K產生的molden文件分析前沒在里面添加盒子信息(參見《詳談使用CP2K產生給Multiwfn用的molden格式的波函數文件》http://www.shanxitv.org/651)、做Mulliken布居分析用的基組帶了不該帶的彌散函數、算溶劑環境下溶質自由能的時候忘了考慮標準態濃度轉換問題(參看《談談隱式溶劑模型下溶解自由能和體系自由能的計算》http://www.shanxitv.org/327)、試圖用后HF方法計算分子軌道(卻渾然不知程序雖然給出了結果但實際上是HF部分產生的)、用背景電荷表現環境分子的勢場用的是對靜電勢重現性極爛的QEq電荷(相關討論看《基于背景電荷計算分子在晶體環境中的吸收光譜http://www.shanxitv.org/579),等等
? 計算流程/方式不對,比如:算高精度能量和性質之前結構沒經過幾何優化、直接把IRC兩端的結構當做反應物和產物的準確結構用于計算能量和屬性數據、振動分析和IRC計算之前結構沒有在嚴格相同級別下優化過、對動力學軌跡計算某個分子的RMSD衡量內部結構變化的時候不知道先要消除整體平動和轉動、算電子發射光譜之前沒優化激發態、模擬拉曼光譜時沒按照《使用Multiwfn計算特定方向的UV-Vis吸收光譜》(http://www.shanxitv.org/648)里說的將拉曼活性轉化成拉曼強度
? 數據讀取錯誤。比如Gaussian里做了雙雜化泛函計算,結果讀成了其中SCF Done后面的中間量(怎么正確讀看《談談該從Gaussian輸出文件中的什么地方讀電子能量》http://www.shanxitv.org/488)。再比如做幾何優化,Gaussian會對初始和最終結構都做布居分析,本來想讀最終結構下的,結果誤讀成第一次輸出的。再比如做開殼層體系的NBO分析時,NBO會對總密度矩陣、alpha密度矩陣和beta密度矩陣的分析結果依次輸出,如果讀錯地方就糟糕了
? 對計算數據質量缺乏把控。如果數據質量太糟糕,自然可能無法重現文獻數據。各個部分的計算都要保證數據質量。比如算穩定狀態必須確保波函數穩定、確保無虛頻。雖然做波函數穩定性測試和振動分析會增加耗時,但必要時必須做,否則可能得到錯誤的結果而無法重復文獻。不要輕易使用比如Gaussian里opt=loose這種關鍵詞放寬收斂限,否則很可能收斂限太松導致某些情況對文獻數據重復性太差,尤其是此時做振動分析得到的頻率的準確度很可能很糟糕。用取消SCF收斂情況檢查的IOp(5/13=1)這種危險關鍵詞的時候必須謹慎檢查最終SCF收斂情況,千萬別最后收斂精度很爛卻直接使用其數據。再比如對于通過分子動力學計算物質平衡狀態屬性時,必須保證采樣充分使統計誤差足夠小。再比如考察某種磷脂膜體系的特征時,需要模擬時間足夠長以使得體系充分達到平衡,本身這類體系達到平衡所需的時間就偏長,動輒需要100 ns以上
? 沒恰當考慮對稱性問題。對于有對稱性的體系,在Gaussian等支持對稱性的量子化學程序里,要盡可能利用對稱性,這樣計算又好又快,內行的研究者的文章里對于能利用對稱性的情況幾乎都會利用。不利用對稱性甚至可能還會得到和文獻不符的結論,比如文獻報道某個體系是平面的,但你一開始擺的結構是彎曲的,由于優化的收斂不可能無窮精確,最后你得到的結構可能輕微偏離平面,與文獻結論不一致。如果體系本身沒有那么高對稱性,你一開始當成高對稱性結構來計算,最后可能錯誤地得到了高對稱性結構,也和文獻不符(這種情況只要做一下振動分析看有沒有虛頻便知)。
? 其它低級錯誤:單位轉換因子沒用對或者用的不準確(比如百毒搜出來的大量轉換因子都是錯的)、該做構型/構象搜索的情況沒做或者做得不當、該考慮構型/構象權重平均的情況沒考慮、優化表面吸附問題時把被吸附分子放得太遠導致還沒優化到吸附狀態就滿足了收斂判據、處理數據用的Excel表格或者腳本存在問題、科學計數法記錄的數據沒讀對(如漏掉了指數部分),等等
8 文獻自身有問題
千萬不要只懷疑自己而不懷疑文獻,盡信文獻不如無文獻,很多文獻里(包括IF很高的期刊的文章里)的數據是有錯誤的,甚至是非常明顯的錯誤,上一節提到的各種犯錯的例子也都出現在很多文獻里。比如《我對一篇存在大量錯誤的J.Mol.Model.期刊上的18碳環研究文章的comment》(http://www.shanxitv.org/584)里我就指出一篇文章里從頭到尾的一大堆錯誤,諸如作者計算C18的時候大概率由于用的是非平面的初始結構,導致優化出的C18不是精確平面的,然后他們發現C18的極小點結構竟然有ECD信號,明顯這是虛假的結果。
還有些文獻里對數據的解釋、標注是錯的,是他們對數據理解有問題。比如直接把HOMO、LUMO能量的負值說成是氧化、還原勢是很多涉及電化學的文章里常見的事,顯然你如果以正確的方式來算,也即計算溶液下的電極反應的自由能變,結果肯定和文獻對不上。再比如有不少文獻用的費米能級是錯的,對有gap的體系直接就把價帶頂當做費米能級,這明顯不符合正確的確定有限溫度下費米能級的方式(正確定義參看wiki相應條目和Multiwfn手冊3.300.9節)。
還有些文獻里的計算方式從描述上就知道是錯的。比如有的文獻寫G=Eele+ZPE-TS,有本科化學知識的人都知道這明擺著不對,H(T)和U(0)之間的差異上哪去了?就算是作為近似不考慮這部分、假設這部分在求差的時候能極大抵消,至少也應該把等號寫成約等號。這種明顯不合理的數據就別去重復了,非要重復的話那就暫時性降智故意用錯誤的公式。
很多文獻作者也對數據很不負責任。比如之前我在IF很高的文獻里看到補充材料中的Gaussian輸入文件里居然全帶著IOp(5/13=1),這文獻的數據的可靠性沒法令人不放心(雖然作者可能檢查了最終收斂情況,但誰也不敢說作者確實這么做了)。現在有很多無良代算機構,沒一丁點搞理論計算的人的基本操守,為了能給出令做實驗的人滿意的與實驗相符的計算數據來交差,極有可能在算不出期望的數據的情況下捏造假數據,這樣的數據更別指望能被別人重復出來了。甚至有的代算方連計算都不計算,直接憑空胡編亂造數據,建議讀者看看《談談我對計算化學代算的看法》(http://www.shanxitv.org/505)里提及的情況。
有很多文獻對計算的關鍵要點、直接影響數據可重復性的因素交代得非常粗糙、簡略、語焉不詳,甚至根本不提,這無疑給讀者帶來了很多誤解、給別人重復計算造成了障礙。重復這些文章的數據只能是考慮各種可能情況去試了。另外,還有可能文章里用的參數、設置本來就不慎寫錯了,作者寫的程序本身存在bug等等。
對于已經恰當考慮了前面說的所有因素,竭盡全力去重復文獻,但還是重復不出來(且確實是有不可忽視的差異而不是在復現精度上過度較真),如果沒有絕對必要去重復文獻,那就別去重復了,極有可能文獻的數據就是有問題的,或者沒交代關鍵性計算細節。這種情況只要你能保證自己的計算是合理的就夠了。但如果有特殊原因就是非要重復文獻不可,顯然該做的不是在思想家公社QQ群或者計算化學公社論壇(bbs.keinsci.com)等公開場所提問,因為找誰都不如直接找作者,世界上還有誰比作者更清楚數據是怎么來的的人么?世界上還有誰比作者更有回復你的義務?發郵件給通訊作者,向作者索要相應數據的輸入輸出文件,或者向作者提供你復現文獻計算的輸入輸出文件問作者為什么沒重復出來即可。當然很有可能一直沒收到作者的回復,可能性有這些:
(1)作者看漏了你的郵件,或者郵件被誤歸入垃圾郵件,或者正忙著什么事沒來及回復你結果后來又忘了。可以過一個禮拜(且可以換個郵箱)再發一次試試。如果通訊作者有多個,也可以給另外的人發
(2)措辭不當,不夠禮貌和誠懇,而作者脾氣又大或者又比較忙,又覺得回復你也沒什么好處,就不回了。可以過一陣重新措辭再發一次。為了讓作者有動力花時間給你回復,最好表示你會在未來的文章中會引用他的文獻。
(3)文獻中的數據是作者找別人代算的,代算方又是非常不負責任的,相關文件也不提供、計算細節也不交代清楚,甚至代算之后就再也聯系不上了,因而作者也無能為力。
(4)做計算的是學生,學生畢業后找不到人了,導師又不清楚計算細節、沒原始文件。
(5)文獻中的計算本身就有嚴重錯誤或是假數據,可能收到你的郵件后作者才意識到,又或者可能之前就已經明知道用的是假數據,總之都不希望你知道他的數據存在問題,免得面子上掛不住,甚至怕文章最后被撤稿。
文/Sobereva@北京科音
First release: 2022-Mar-10 Last update: 2022-Mar-18
1 前言
電子密度差(electron density difference, EDD)是分析成鍵、電子轉移非常常用的手段。波函數分析程序Multiwfn(http://www.shanxitv.org/multiwfn)在繪制和分析密度差方面非常靈活、方便、強大,筆者之前有不少文章與此相關,見《使用Multiwfn作電子密度差圖》(http://www.shanxitv.org/113)、《使用Multiwfn考察分子軌道、NBO等軌道對密度差、福井函數的貢獻》(http://www.shanxitv.org/502)、《使用Multiwfn計算激發態之間的密度差》(http://www.shanxitv.org/429)、《使用Multiwfn做電子密度、ELF、靜電勢、密度差等函數的盆分析》(http://www.shanxitv.org/179)等。之前筆者寫的密度差有關的文章主要是對于孤立體系寫的,本文的目的之一是介紹怎么將非常流行、免費強大的CP2K第一性原理程序與Multiwfn相結合,對周期性體系繪制密度差圖。在此順帶特別強調,electron density difference的中文是密度差,“差分密度”是對密度差的嚴重不當稱呼,絕對不要用這個詞!
本文另一個目的是介紹怎么通過Multiwfn繪制平面平均密度差(plane-averaged EDD)曲線,它是密度差的衍生。密度差是個三維空間函數,經常通過繪制平面圖、等值面圖方式圖形化考察。雖然這兩種圖很直觀,但平面圖會受到截面選取的影響,而等值面圖會受到等值面數值(isovalue)選取的影響,因此都有一定任意性,也不容易定量討論。對于電子轉移有比較明確方向的情況,可以繪制平面平均密度差曲線,由此可以清楚地考察垂直于選取的方向上每個截面上的密度差積分值,在定量討論、對比上比較便利。例如,固體表面上吸附一個小分子,吸附導致垂直于表面方向有明顯的電子轉移,就可以在垂直于表面的方向上繪制平面平均密度差曲線來準確、細致地考察不同截面處的電子凈增、減情況。還有一種圖叫做電荷位移曲線(charge displacement curve),它相當于平面平均密度差曲線的積分曲線,對于定量討論電荷轉移量情況很有幫助,本文也會介紹怎么通過Multiwfn繪制這種圖。
本文使用的例子是NaCl板吸附一個CO分子,NaCl和CO將被定義為兩個片段,通過密度差考察吸附導致的電子轉移情況。本文文件包里的opt目錄下的NaCl_CO-1.restart是CP2K在PBE-D3(BJ)/DZVP-MOLOPT-SR-GTH級別下對這個體系做優化產生的restart文件,我們將基于這個結構繪制密度差。為簡化問題,在優化的時候整個NaCl板的坐標被凍結為了晶體坐標的狀態,因此不涉及表面重構。
下文涉及到的相關文件可以在http://www.shanxitv.org/attach/638/file.rar下載。cub文件由于體積太大,文件包里就不提供了。
本文的計算使用CP2K 9.1,安裝方法見《CP2K第一性原理程序在CentOS中的簡易安裝方法》(http://www.shanxitv.org/586)。VMD使用的是1.9.3版,可在http://www.ks.uiuc.edu/Research/vmd/免費下載。讀者務必使用Multiwfn最新版,如果不知道是不是最新的就現在立刻去主頁http://www.shanxitv.org/multiwfn下載一個。如果對Multiwfn不了解,看《Multiwfn FAQ》(http://www.shanxitv.org/452)和《Multiwfn入門tips》(http://www.shanxitv.org/167)。不了解cub文件的話可以看看《Gaussian型cube文件簡介及讀、寫方法和簡單應用》(http://www.shanxitv.org/125)。
2 計算密度差格點數據
這一節我們要得到整個體系、NaCl板和CO的各自的電子密度的cub文件,然后由Multiwfn求差。
我們先創建一個計算整個體系的電子密度cub文件的CP2K輸入文件。啟動Multiwfn,然后輸入
NaCl_CO-1.restart //在本文文件包里
cp2k //創建CP2K輸入文件。在《使用Multiwfn非常便利地創建CP2K程序的輸入文件》(http://www.shanxitv.org/587)一文里有詳述
NaCl_CO.inp //產生的輸入文件的路徑
現在進入了創建CP2K輸入文件的界面。為了之后繪制的平面平均密度差曲線比較好觀看,最好把當前體系挪到盒子中央附近。先看一眼當前的結構是什么樣,輸入
-11 //進入幾何操作界面。此界面的詳細介紹見《Multiwfn中非常實用的幾何操作和坐標變換功能介紹》(http://www.shanxitv.org/610)
0 //觀看當前結構
現在出現了圖形窗口。點擊菜單上的Other settings - Toggle showing cell frame把盒子邊框顯示出來,再選Other settings - Toggle showing all boundary atoms把邊界原子顯示出來。現在看到的圖如下
可見當前整個體系在盒子底部,NaCl有三層,由于最下層的原子正好在Z=0的位置,因此這些邊界原子在盒子頂端也顯示了出來。點擊右上角的RETURN按鈕返回。為了把體系挪到盒子中央去,輸入
24 //平移體系使得選中的部分在晶胞中居中
1-110 //當前體系里所有原子。也可以直接按回車選中整個體系
0 //再次觀看體系
可見此時體系確實在盒子中央了
關閉圖形窗口,接著輸入
-10 //返回CP2K輸入文件創建界面
-7 //設置周期性
XY //XY方向二維周期性
-3 //要求產生cube文件
1 //電子密度
0 //產生CP2K輸入文件
現在當前目錄下就有了NaCl_CO.inp。讀者可根據要求修改里面的CUTOFF等設置,本例不那么講究,就用默認的。運行之,算完后當前目錄下就有了NaCl_CO-ELECTRON_DENSITY-1_0.cube,將之改名為NaCl_CO.cub。
將NaCl_CO.inp復制為NaCl.inp,用文本編輯器打開,把其中PROJECT NaCl_CO改為PROJECT NaCl,把&COORD里面C和O原子刪掉。用CP2K運行NaCl.inp,將得到的cub文件改名為NaCl.cub。
將NaCl_CO.inp復制為CO.inp,用文本編輯器打開,把PROJECT NaCl_CO改為PROJECT CO,把&COORD里面Na和Cl原子刪掉。用CP2K運行CO.inp,將得到的cub文件改名為CO.cub。
現在用Multiwfn對三個cub文件求差。啟動Multiwfn,然后輸入
NaCl_CO.cub //輸入此文件實際路徑
13 //處理格點數據
11 //格點數據運算
4 //減去另一個格點數據
NaCl.cub //輸入此文件實際路徑
11 //格點數據運算
4 //減去另一個格點數據
CO.cub //輸入此文件實際路徑
現在內存里的格點數據就已經是密度差格點數據了。想觀看一下等值面的話,可以進入選項-2 Visualize isosurface of present grid data。由于物理吸附對應的密度差的數量級往往很小,所以要用比較小的等值面數值才能清楚看到。在圖形窗口右側把等值面數值設為0.0005后,看到的圖像如下(點save picture按鈕保存的圖像比直接在圖形窗口里看到的更好)
綠色和藍色等值面分別代表格點數據數值為正和為負的部分。當前密度差是按照ρ(NaCl...CO)-ρ(NaCl)-ρ(CO)算的,顯然綠色體現吸附后電子密度增加的部分,藍色體現電子密度減少的部分。點擊右上角RETURN關閉圖形窗口。
若想得到密度差的cub文件,就選0 Export present grid data to Gaussian-type cube file (.cub),輸入EDD.cub,然后內存里的格點數據就被導出為了當前目錄下的EDD.cub。
使用《在VMD里將cube文件瞬間繪制成效果極佳的等值面圖的方法》(http://www.shanxitv.org/483)里提供的我寫的腳本,在VMD程序里只需要輸入一行命令cub EDD 0.0004,就可以把EDD.cub的0.0004等值面顯示出來(如果沒顯示出來,一個字一個字看http://www.shanxitv.org/483這篇文章)。在文本窗口里輸入pbc box把盒子顯示出來,再改成正交視角,經過Tachyon渲染看到的就是下面的圖。圖中藍色小球是Na,棕色是Cl,等值面的藍色和綠色和前面Multiwfn里的定義相同。
從此圖可以明顯看出,CO的碳結合到Na上后,C和O上的電子密度整體大幅減少,很大程度往Na的方向上轉移了。這也容易理解,當前體系里Na帶顯著的正電荷,CO結合上去,特別是C還帶著豐富的孤對電子,必然其電子會被往Na那邊顯著地極化。
再來看看把等值面數值設得更小的情況。在VMD文本窗口里輸入cubiso 0.0001把等值面數值調為0.0001,稍微調整視圖后看到的圖如下
可見此時的密度差圖展現了比之前更多的信息。粉色虛線圈住的地方電子密度有一定降低,由于僅在等值面數值很小的時候才能顯現出來,所以這部分降低量很少(想通過積分得到確切值的話需要用Multiwfn的盆分析功能,前面http://www.shanxitv.org/179文中介紹過)。這部分電子密度降低是因為CO向Na這邊轉移了電子,由于交換互斥作用,使得原本這部分的電子被排斥開,向更遠處轉移。
3 繪制密度差局部積分曲線和平面平均密度差曲線
這一節繪制密度差局部積分曲線和平面平均密度差曲線,首先了解一下定義。某個三維空間函數的局部積分曲線(local integral curve)可以對任意方向繪制,如果對比如Z方向繪制,則表達式為
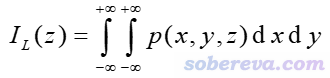
其中p是被考察的函數。如果p取為密度差的話,那么這個曲線就是前述的密度差局部積分曲線了。
還有個概念叫積分曲線(integral curve),就是對局部積分曲線再進一步積分。例如對Z方向如下所示
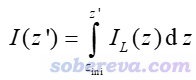
其中積分的起點z_ini由自己決定。如果被考察的函數是密度差,則這個積分曲線對應的就是前述的電荷位移曲線。
平面平均曲線(plane-averaged curve)定義如下,其中A_XY是格點數據對應的盒子在XY方向的面積。這種曲線體現的是被考察的函數在特定截面上的平均值。
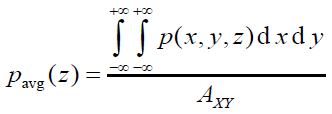
在Multiwfn里可以基于格點數據對任意函數繪制上述三種曲線,需要先把相應的格點數據存到內存里。比如可以通過主功能5格點數據運算功能算出來后自動存到內存里,也可以啟動時從cub等格點數據文件里直接載入,等等。
下面繪制密度差局部積分曲線。如果你之前已經關了Multiwfn,就再啟動Multiwfn,載入密度差格點數據文件EDD.cub,然后進入主功能13,再進入子功能18,這是用來繪制(局部)積分曲線和平面平均曲線的功能。如果你按照上一節操作之后還沒關Multiwfn,則內存里的格點數據正是密度差格點數據,因此直接進入子功能18就行了。
在子功能18里,Multiwfn先問你對哪個方向繪制,我們想垂直于NaCl界面繪制,因此輸入Z,因為NaCl界面平行于XY方向。之后Multiwfn問你繪制范圍的下限和上限位置是什么,此例想對整個Z范圍進行繪制,因此按照屏幕上的提示直接輸入a。之后會看到一個菜單,通過相應選項可以直接繪制積分曲線、局部積分曲線、平面平均曲線,可以保存它們的圖像,還可以把曲線數據導出為文本文件便于用Origin、gnuplot、qtgrace等第三方程序繪圖。
現在選擇屏幕上的選項2 Plot graph of local integral curve,由于當前內存里的格點數據是密度差,所以下圖就是密度差局部積分曲線
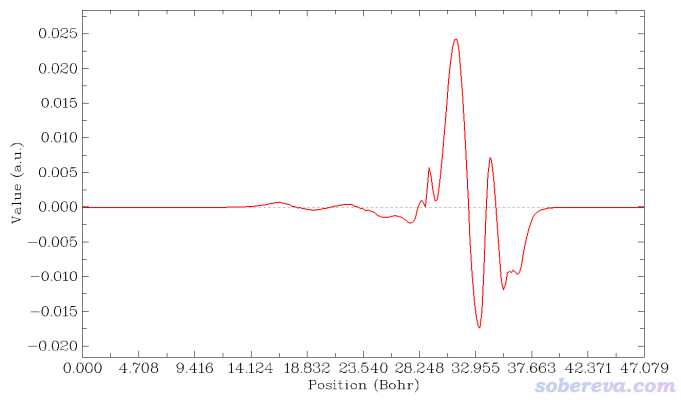
讀者如果想讓等值面圖和密度差局部積分曲線便于準確對照,可以用屏幕上相應選項讓Multiwfn導出局部積分曲線數據為當前目錄下的locintcurve.txt(每一列的含義看導出時屏幕上的提示),然后放到Origin等程序里作圖。之后在VMD里用正交視角,把盒子邊框顯示出來,把保存的等值面圖和局部積分曲線圖放到一起,把前者的白色背景設為透明色或者摳掉,再調節位置和大小讓兩張圖邊框位置正好對上,如下所示。不會用Photoshop的話用版本不很老的PowerPoint也可以完成
再改一下透明度,裁切一下邊緣去掉盒子邊框,最終得到下面的圖,效果很好。由這個圖可以比較準確地捕捉各個截面上電子的凈增減,可見在C與表層的NaCl相接的地方電子密度由于相互作用而增加明顯(但絕對的數量級并不算大)。
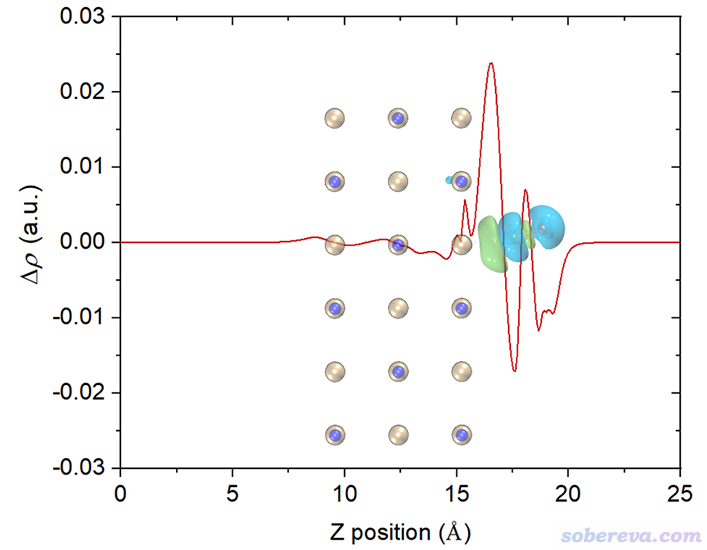
如果想要繪制平面平均密度差曲線,前面選2 Plot graph of local integral curve的地方改成選3 Plot graph of plane-averaged curve即可,其它不變。
4 繪制電荷位移曲線
在Multiwfn當前界面里選1 Plot graph of integral curve,由于當前內存里的格點數據是密度差,因此得到的積分曲線的就是電荷位移曲線了:
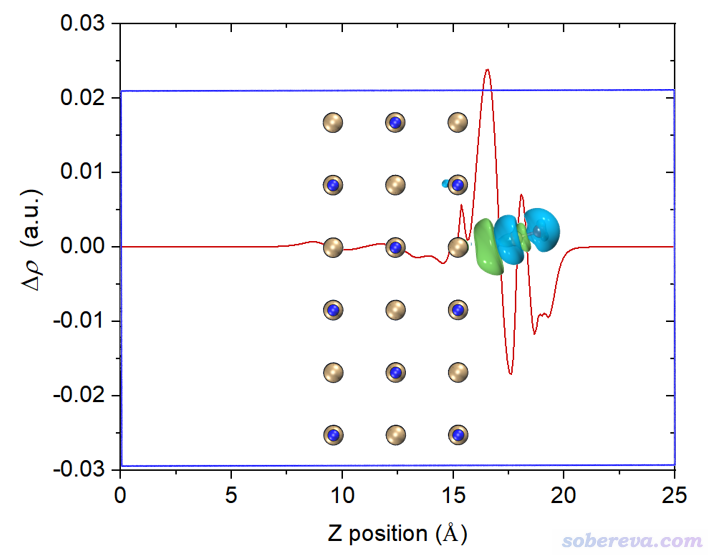
上圖橫軸左端對應于NaCl...CO體系Z=0的位置,橫軸右端對應于NaCl...CO體系Z=25埃(盒子Z尺寸)的位置。這個圖是對上一節的密度差局部積分曲線從左向右積分產生的,從肉眼可以清楚看出來對應關系。曲線最右邊數值為0,體現了密度差全空間積分為0,也即CO與NaCl結合沒有導致總電子數有凈變化。
為了便于觀看,將電荷位移曲線和結構圖+等值面圖疊加起來。我也把曲線最高的峰的橫、縱坐標位置標注上了。電荷位移曲線縱坐標是無量綱的,因為對應的是電子數。
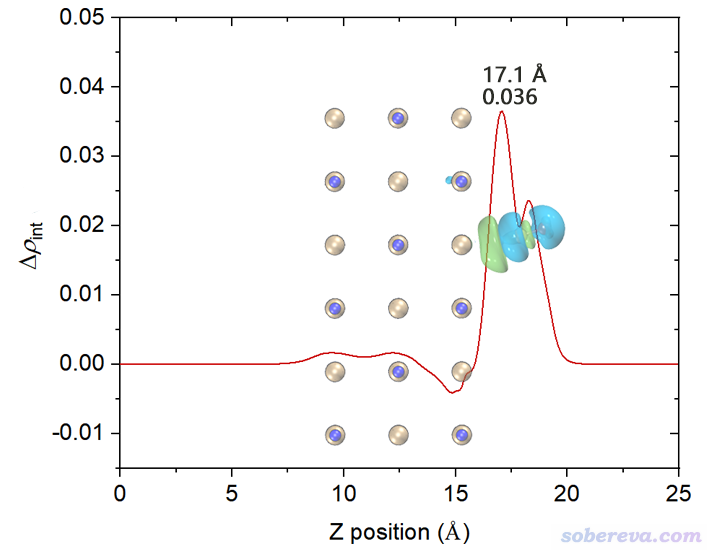
這張圖體現出,在Z<17.1埃的區域里,電子數凈增加了0.036。如果姑且把這個位置作為CO和NaCl的分界,那么說成是CO向NaCl凈轉移了0.036個電子是可以的。對于其它吸附體系,未必在表面和被吸附物之間恰好有一個峰,讀者可以自己決定哪個Z值可作為兩部分的分界面(這有一定任意性),從而從電荷位移曲線上讀出電子凈轉移量。考察電荷位移曲線顯然不是唯一的判斷電荷轉移量的方法,更普適、更常用的是計算片段電荷,即某個片段里所有原子電荷的加和(結果明顯受原子電荷計算方法的選取所影響。原子電荷計算方法有很多,比如基于CP2K的波函數Multiwfn可以計算CM5、Hirshfeld、Mulliken等電荷)。
5 對其它函數計算(局部)積分曲線
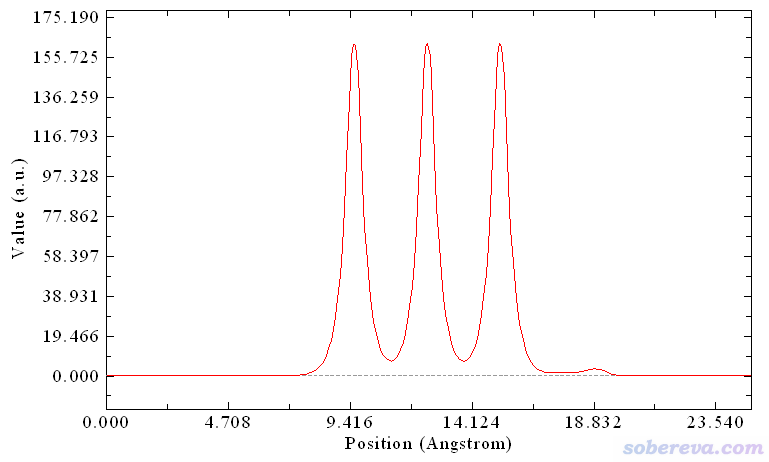
上面介紹的Multiwfn計算(局部)積分曲線和平面平均曲線對任何三維空間函數都適用,這里順帶再舉個例子。比如想考察NaCl...CO這個體系不同Z位置的電子密度分布,就啟動Multiwfn,依次輸入
NaCl_CO.cub
13 //處理格點數據
18 //繪制(局部)積分曲線
Z //順著Z方向繪制
a //整個Z范圍
-1 //將橫坐標單位切換為埃
2 //繪制局部積分曲線
由于載入的NaCl_CO.cub里記錄的是電子密度,所以此時看到的下面的局部積分曲線體現了對應不同Z值的XY截面上的電子量。中間三個很高的峰正對應于NaCl板的每一層,18.8埃附近的小峰來自于CO的電子。
可見,不管什么函數,Multiwfn都能畫(局部)積分曲線或平面平均曲線,只要提供記錄相應函數的格點數據文件就可以。諸如自旋密度也同樣可以繪制這些曲線,比如你有個反鐵磁性體系,位于不同層上的原子自旋磁矩情況各有不同,就可以對自旋密度繪制局部積分或平面平均曲線,清楚地考察電子自旋分布情況。對于靜電勢也可以繪制平面平均曲線,特別是對于界面體系,在垂直于界面方向的這種圖是計算功函數的關鍵。用戶只要給Multiwfn提供靜電勢的格點數據文件即可,比如流行的CP2K就可以直接產生,即在Multiwfn產生CP2K輸入文件的界面里選擇導出cube文件的內容的時候選4 Hartree potential (negative of ESP)。
在http://bbs.keinsci.com/thread-29906-1-1.html一貼里有VASP用戶通過本文介紹的功能基于VASP計算的CHGCAR和LOCPOT文件,用Multiwfn分別考察了平面平均電子密度曲線和平面平均靜電勢曲線,和文獻里的結果相當一致,讀者可以看看。
6 總結&其它
本文介紹了(局部)積分曲線、平面平均密度差曲線、電荷位移曲線這些概念,并詳解介紹了如何將CP2K第一性原理程序與強大的Multiwfn波函數分析程序相結合繪制密度差圖以及這些曲線。從本文的例子可見,密度差等值面圖能很直觀地展現三維空間中各處電子密度的凈增減,而對于恰當的情況(電子轉移和極化以單一方向為主),若將之與平面平均密度差曲線、電荷位移曲線相結合,則能考察得更清楚、更便于定量討論,還便于準確地橫向對比。例如一個固體表面吸附不同分子,想考察電子轉移特征的差異,就可以把不同情況的平面平均密度差曲線同時繪制在一張圖上,差異一目了然。
本文的做法絕不僅限于CP2K用戶使用。記錄電子密度的cub文件用任何其它程序產生也都可以,cub是化學領域最常見的記錄格點數據的格式,因此被支持得很廣泛。Multiwfn支持的格點數據文件格式有很多,還包括dx、grd、vti、VASP的CHGCAR/CHG/ELFCAR/LOCPOT。對于VASP用戶,想得到密度差,可以先用Multiwfn載入CHGCAR,進主功能13用子功能0轉成cub文件,對整體和各個片段都這么做得到各自的電子密度cub文件后,再按上文方式進行操作。
Multiwfn也可以基于波函數文件直接用主功能5計算密度差格點數據。如果你是Gaussian、ORCA等主流量子化學程序用戶,一般都建議先計算出各個狀態的波函數文件(支持的格式和產生方法見《詳談Multiwfn支持的輸入文件類型、產生方法以及相互轉換》http://www.shanxitv.org/379),之后用主功能5的自定義運算功能可輕松得到密度差格點數據(參考《使用Multiwfn作電子密度差圖》http://www.shanxitv.org/113里使用主功能5的過程),然后直接就能去主功能13里的子功能18里繪制密度差局部積分曲線、平面平均密度差和電荷位移曲線。對于CP2K也可以如此,先對涉及的各個狀態產生記錄波函數信息的molden文件,按照《使用Multiwfn結合CP2K通過NCI和IGM方法圖形化考察固體和表面的弱相互作用》(http://www.shanxitv.org/588)里的說明自行把晶胞信息作為[Cell]字段寫進去,然后用主功能5做自定義運算就可以獲得密度差格點數據。
在《全面探究18碳環獨特的分子間相互作用與pi-pi堆積特征》(http://www.shanxitv.org/572)和《一篇文章深入揭示外電場對18碳環的超強調控作用》(http://www.shanxitv.org/570)介紹的我關于18碳環體系的研究中都使用了本文提到的功能繪制了密度差的局部積分曲線,由其中的例子可見這對于考察分子間相互作用和外電場對電子分布的影響特別有好處,非常直觀。
本文的做法是非常普適的,絕不僅限于展現片段間相互作用的密度差。對于電子激發導致激發態相對于基態電子密度分布改變的密度差、外加電場導致電子分布變化的密度差、體系電離或附著電子導致的密度變化的密度差等等,都可以按本文的方法繪制相應的(局部)積分曲線來研究。
使用本文的做法通過Multiwfn產生相關數據若用于發表,記得按照程序啟動時的提示恰當引用Multiwfn。
]]>文/Sobereva@北京科音
First release: 2022-Feb-28 Last update: 2023-Feb-10
0 前言
波函數分析程序Multiwfn(http://www.shanxitv.org/multiwfn)有十分豐富的電子激發分析功能,已被量子化學研究者廣為使用,在《Multiwfn支持的電子激發分析方法一覽》(http://www.shanxitv.org/437)有全面的功能介紹。而且Multiwfn還有靈活強大的繪制電子光譜的功能,見《使用Multiwfn繪制紅外、拉曼、UV-Vis、ECD、VCD和ROA光譜圖》(http://www.shanxitv.org/224)。以往都是Gaussian、GAMESS-US、ORCA等量子化學程序用戶在研究電子激發相關問題時使用Multiwfn的這些功能,而近期,Multiwfn的一些相關功能也已支持了CP2K第一性原理程序。免費、高效的CP2K算周期性大體系的TDDFT相當快。CP2K用戶使用TDDFT計算分子或周期性體系的激發態后,可以非常方便地使用Multiwfn繪制UV-Vis譜和分析電子激發特征。而且Multiwfn具有非常方便的創建CP2K的TDDFT輸入文件的功能,上手計算十分容易。本文的目的是通過一個典型的分子晶體例子,令讀者了解使用Multiwfn和CP2K通過TDDFT研究周期性體系電子激發問題的基本做法,不少關鍵性的細節也一起說明,以便讀者能舉一反三,解決自己研究中的問題。
讀者務必使用2023-Feb-10及以后更新的Multiwfn版本,否則和本文介紹的情況會有所不同。Multiwfn可以在官網http://www.shanxitv.org/multiwfn免費下載,不了解Multiwfn者建議看《Multiwfn FAQ》(http://www.shanxitv.org/452)和《Multiwfn入門tips》(http://www.shanxitv.org/167),使用Multiwfn發表文章別忘了按照程序啟動時的提示進行恰當引用。本文用的CP2K是9.1版,安裝方法見《CP2K第一性原理程序在CentOS中的簡易安裝方法》(http://www.shanxitv.org/586)。如果讀者對TDDFT計算完全缺乏了解,建議閱讀《Gaussian中用TDDFT計算激發態和吸收、熒光、磷光光譜的方法》(http://www.shanxitv.org/314)和《亂談激發態的計算方法》(http://www.shanxitv.org/265)了解相關基本知識。
本文涉及的相關文件都可以在http://www.shanxitv.org/attach/634/file.rar里得到。文件約20MB,如果下載慢請換個時段下。
本文對CP2K的電子激發計算部分僅僅做非常簡要的描述,在筆者講授的北京科音CP2K第一性原理計算培訓班有專門的一節非常詳細講授CP2K做電子激發計算(也包括X光吸收),介紹見http://www.keinsci.com/workshop/KFP_content.html,非常歡迎了解和參加。
1 體系特征和一些計算細節
本文的例子是一個Donor-π-Acceptor型分子4-(N,N-dimethyl-amino)-4′-(2,3,5,6-tetrafluorostyryl)-stilbene構成的晶體,如下所示,晶胞里有4個分子,共192個原子。我們將要繪制它的UV-Vis譜,并對其中對應強吸收峰的電子激發分析其本質。此體系X光衍射得到的晶體結構是本文文件包里的bulk.cif。
這個體系實際上是ACS Omega, 3, 10481 (2018)一文中理論研究的體系,文中深入研究了這個分子晶體的J聚集導致吸收峰相對于單體分子紅移的本質。作者用的是Quantum ESPRESSO做的TDDFT計算,而本例我們將用CP2K做TDDFT。
此晶體的原胞的三個邊長分別是7.59、5.87、41.51埃。由于前兩個晶胞尺寸較短,而CP2K的TDDFT計算時不能考慮k點,所以如果想更準確計算吸收光譜的話建議將體系復制成2*3*1的超胞。這是常見做法,J. Chem. Theory Comput., 17, 5214 (2021)在TDDFT計算分子晶體光譜時還特意對超胞尺寸做了結果的收斂性測試。搞成超胞自然會導致耗時劇增,由于本文只是示例目的,為了節約耗時,就只用原胞來算了。ACS Omega那篇文章里也是直接用的原胞來計算和分析。
由于X光衍射得到的晶體結構中的氫的位置一般都是不準的,《實驗測定分子結構的方法以及將實驗結構用于量子化學計算需要注意的問題》(http://www.shanxitv.org/569)里也說過這個問題,因此電子激發計算前應當至少先對氫做優化,不過本文暫且忽略這個步驟,而自己實際研究中應當注意這個問題。
CP2K的TDDFT只支持TDA近似形式,也可以叫TDA-DFT,計算出的振子強度不如嚴格的TDDFT準確,但激發能的準確度并沒打折扣。
ACS Omega原文里是基于PBE泛函做的TDDFT,我們也用這個泛函來做。眾所周知,雖然純泛函在第一性原理程序里計算比雜化泛函快得多得多,但有明顯低估激發能的傾向(尤其是對于電荷轉移激發),故讀者實際研究時如果對激發能的準確性有要求,屆時別忘了用PBE0等適合的雜化泛函來算,這在CP2K中也是支持的,而且能算不小的體系。
CP2K 9.1 CP2K做TDDFT只支持GPW,即必須用贗勢基組而不能用全電子基組(后記:從2022.1開始支持了GAPW的TDDFT,可以用全電子基組了)。對于TDDFT目的用TZVP-GTH贗勢基組就不錯,精度足夠,而且也不很貴。再好一點還可以用TZV2P-GTH。用CP2K里常用的DZVP-MOLOPT-SR-GTH贗勢基組算TDDFT時還能比TZVP-GTH更便宜一些,但是由于此基組收縮度太高,導致之后用Multiwfn做很多分析的時候在計算格點數據時會非常昂貴,所以本文用TZVP-GTH。
2 使用CP2K做TDDFT計算
用Multiwfn創建CP2K輸入文件非常方便,在《使用Multiwfn非常便利地創建CP2K程序的輸入文件》
(http://www.shanxitv.org/587)里有詳細說明。
啟動Multiwfn,然后輸入
bulk.cif //在本文文件包里提供了
cp2k //產生CP2K輸入文件
bulk_TDDFT.inp //將要生成的CP2K輸入文件的文件名
15 //開啟激發態計算
y //讓計算任務順帶輸出記錄分子軌道的molden文件
30 //把最低30個空軌道算出來并存到molden文件里
16 //設置激發態計算數目
50 //算最低50個激發態
2 //選擇基組
-3 //TZVP-GTH
0 //生成輸入文件
現在當前目錄下就有了bulk_TDDFT.inp。輸入文件默認的CUTOFF等設置對于當前計算來說都是適合的,所以不用做額外改動。直接用CP2K運行此文件,輸出文件是本文文件包里的bulk_TDDFT.out。任務同時產生了molden文件bulk_TDDFT-MOS-1_0.molden。
如《詳談使用CP2K產生給Multiwfn用的molden格式的波函數文件》(http://www.shanxitv.org/651)一文所述,CP2K直接產生的molden文件里不含晶胞信息,沒法用于Multiwfn做分析,因此必須手動把晶胞信息填進去,做法在此文也明確說了。現在把以下內容插入到molden文件的第二行,本文文件包里的molden文件是我已經改好的。這部分內容大家直接從CP2K輸入文件里的&CELL部分復制過去即可。
[Cell]
7.59400000 0.00000000 0.00000000
0.00000000 5.87400000 0.00000000
-3.11381184 0.00000000 41.39304623
下面說一些和當前的TDDFT計算有關的細節。
輸入文件里的&PROPERTIES - &TDDFPT是TDDFT計算的相關設置,含義在Multiwfn自動產生的注釋里都寫明了。其中的MIN_AMPLITUDE 0.01代表組態系數的絕對值大于0.01的才會輸出,這個數設得越小,之后Multiwfn做空穴-電子分析、躍遷密度分析、NTO分析等會越精確。要求較高的話建議改小到0.001,但由于輸出的組態會更多,會令輸出文件信息量更大,而且在Multiwfn的激發態分析過程中耗時也會高一些。如果只需要大概看個分析結果,用Multiwfn默認設的0.01就夠了,至少結果是定性正確的。
之所以此任務要求產生記錄了分子軌道的molden文件,是因為之后使用Multiwfn做許多電子激發分析都需要利用到分子軌道信息。這里只要求記錄最低30個空軌道而非所有空軌道,并非是為了節約耗時(相對于激發態計算過程,算出所有空軌道并記錄也不會增加多少耗時),而是因為記錄所有空軌道的話molden文件可能非常大,對大體系甚至可能達到幾GB,又占硬盤拷貝又慢。TDDFT計算中,每個激發態波函數都是由一大批組態函數線性組合來描述的,不同組態函數形式上對應于不同形式的軌道躍遷,每個組態函數對激發態波函數都各有各的貢獻量,參看《電子激發任務中軌道躍遷貢獻的計算》(http://www.shanxitv.org/230)。我們當前算出來的激發態的組態函數的系數在CP2K輸出文件里可以直接看到,如下所示。對于能量比較低的一批激發態,如果在電子激發分析時只考慮貢獻不可完全忽略的組態函數(如組態系數絕對值大于0.01的),則這些組態函數只可能涉及到占據軌道向比較低階空軌道的躍遷。對本文的例子,算最低30個空軌道基本足夠了,如果想絕對穩妥的話建議算50個空軌道。一般來說,對越高階激發態做空穴-電子分析,建議算越多的空軌道,因為越高階激發態中涉及越高空軌道的組態函數越可能無法被忽略。后文還會再專門提到怎么準確檢驗算的空軌道數目是否確實夠。
-------------------------------------------------------------------------------
- Excitation analysis -
-------------------------------------------------------------------------------
State Occupied Virtual Excitation
number orbital orbital amplitude
-------------------------------------------------------------------------------
1 1.64779 eV
296 297 -0.902183
294 298 0.306244
295 299 0.275238
293 300 -0.124557
296 302 -0.013313
294 304 0.013288
293 303 0.013231
295 301 -0.012515
...略
50 2.97063 eV
295 307 -0.629538
294 306 -0.596348
293 305 -0.350078
296 308 -0.348347
296 310 -0.036322
294 309 0.019915
294 303 -0.016826
293 304 -0.016232
296 301 0.015761
295 302 0.014885
284 297 0.013140
283 298 0.012456
算的激發態數目越多,計算耗時越高。當前的例子只計算了50個激發態,對于獲得較完整的UV-Vis光譜來說并不夠。從輸出文件里以下部分可見,第50激發態的激發能是2.97 eV,折合417 nm,連可見光區都沒覆蓋完整。但考慮到純泛函算的激發能整體明顯偏低,如果用諸如PBE0算,可能最高能達到比如3.6 eV (344 nm)左右,屆時可見光部分就都覆蓋了。如果要把近紫外區也都算出來,需要算的態數建議翻倍。
State Excitation Transition dipole (a.u.) Oscillator
number energy (eV) x y z strength (a.u.)
------------------------------------------------------------------------
TDDFPT| 1 1.64779 -3.4173E-08 4.5488E-09 -1.0672E-08 5.25777E-17
TDDFPT| 2 1.66084 -1.4486E-08 -8.1846E-08 9.6347E-09 2.84886E-16
TDDFPT| 3 1.66146 8.5244E-08 3.6451E-02 5.0929E-08 5.40824E-05
...略
TDDFPT| 49 2.96978 2.8362E-01 -9.3226E-07 2.7092E-01 1.11931E-02
TDDFPT| 50 2.97063 -6.2827E-06 -1.6851E-01 -3.5284E-05 2.06651E-03
從以上信息中也可以看到振子強度、躍遷偶極矩這些信息,和Gaussian等量子化學程序做TDDFT給出的信息形式非常類似。
3 繪制電子光譜
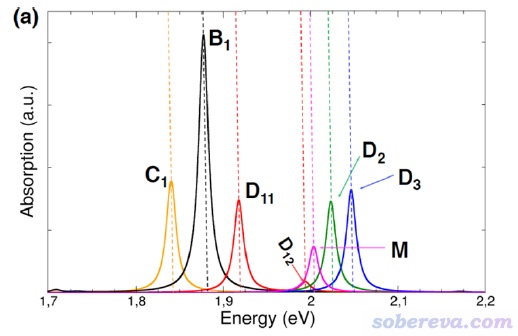
用Multiwfn繪制電子光譜功能的全面介紹、各種細節以及相關的基本原理請讀者看《使用Multiwfn繪制紅外、拉曼、UV-Vis、ECD、VCD和ROA光譜圖》(http://www.shanxitv.org/224),這里僅畫個很簡單的圖。多數UV-Vis譜橫坐標用的是nm,但在前述的ACS Omega文章里研究這個晶體的光譜時橫坐標用的是eV,如下所示。其中黑線是和我們計算模型一致的bulk狀態的吸收曲線,我們試圖重現一下。圖中M、D、C分別是作者自行基于晶體結構構造的分子單體、二聚體、鏈狀排列時的吸收曲線,這不是我們本例關心的。
啟動Multiwfn然后載入bulk_TDDFT.out,之后輸入
11 //繪制光譜
3 //UV-Vis
10 //修改橫坐標單位
1 //eV
8 //設置峰展寬用的半高全寬(FWHM)
0.07 eV //和ACS Omega文章里用的一致。順帶一提,Multiwfn默認用的FWHM比這個大得多,一般適合溶液的吸收光譜。算晶體的話可以設小一些
16 //設置顯示光譜極大極小點的標簽
1 //設置標簽狀態
1 //把曲線的極大點數值顯示出來
3 //設置標簽上的小數位數
2 //兩位小數
0 //返回
0 //作圖
現在看到的圖如下所示(注:筆者為了光譜曲線更平滑,此處先選擇-4把導出格式設為了pdf,然后選了1導出pdf文件,下圖實際上是pdf的截圖,這比直接在屏幕上顯示的光滑得多,且可以無損縮放)。讀者也可以自行再用Multiwfn界面上的選項修改橫、縱坐標范圍和標簽間隔,使得坐標軸的標簽更整齊。
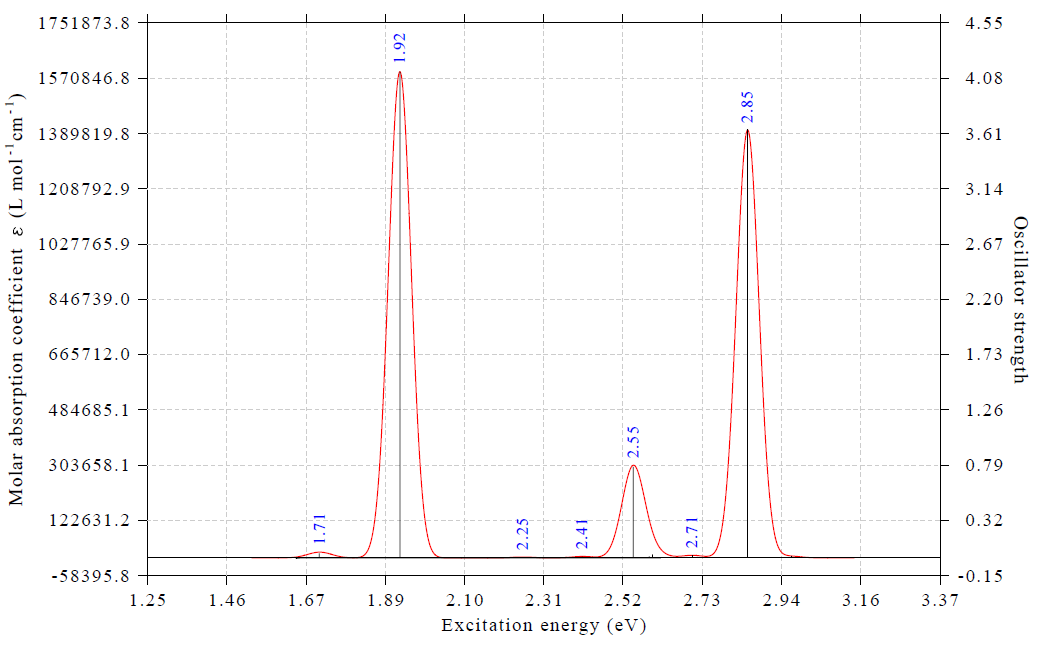
由于我們算了50個激發態,比文獻里算的更多,因此上面的光譜差不多把3 eV激發能以內的電子激發對應的吸收都算出來了。上圖中紅色曲線是吸收光譜,對應左邊坐標軸,藍色標簽標注的是吸收峰的位置。由圖可見,在1.71 eV處有個很弱的吸收峰(振子強度0.04),而在1.92 eV處的吸收巨強,振子強度超過4(具體是4.13)。這和ACS Omega文章中的譜圖的特征非常吻合。那篇文章補充材料中的表S1里,正好在1.71 eV處也有個激發,振子強度僅為0.08,而在1.887 eV處有個振子強度7.96的賊強的激發。我們算的電子激發能和文獻里相符極好,但振子強度比文獻里的小了將近一倍,這主要是由于CP2K用了TDA近似所致。不過由于不同的峰的相對高度受TDA的影響遠小于絕對高度受其影響,而我們一般只關心曲線形狀,所以基于CP2K的TDDFT算的光譜是可以用的。
想搞清楚各個吸收峰對應哪個電子激發很簡單。在Multiwfn當前的繪制光譜的界面里選-1,就能把激發態序號、不同單位的激發能以及振子強度都直接顯示出來,如下所示。上面的譜圖里黑色豎線橫坐標位置是激發能,豎線高度對應右邊坐標軸,是振子強度,和以下信息一對照便知各個峰對應什么激發態。顯然,1.71 eV的極弱吸收對應的是第8激發態(S8),1.92 eV的極強吸收對應的是第13激發態(S13)。接下來我們要著重對S13這個重要的電子激發做一些分析,看看它對應的本質為何。
Index Excit.energy(eV nm 1000 cm^-1) Oscil.str.
1 1.64779 752.42719 13.29032 0.00000
2 1.66084 746.51502 13.39558 0.00000
3 1.66146 746.23644 13.40058 0.00005
4 1.67536 740.04512 13.51269 0.00471
5 1.70466 727.32510 13.74901 0.00153
6 1.70527 727.06492 13.75393 0.00000
7 1.70540 727.00950 13.75498 0.00000
8 1.70974 725.16406 13.78998 0.03991
9 1.73479 714.69284 13.99203 0.00786
10 1.74660 709.86030 14.08728 0.00000
11 1.75022 708.39209 14.11648 0.00025
12 1.75461 706.61970 14.15189 0.00000
13 1.92496 644.08715 15.52585 4.13484
14 2.15741 574.69002 17.40069 0.00000
...略
4 考察主要的分子軌道躍遷
特別常見的分析電子激發的方式是看激發中哪些分子軌道的躍遷對激發有主要貢獻,然后去看軌道圖形考察。計算貢獻的方法在《電子激發任務中軌道躍遷貢獻的計算》(http://www.shanxitv.org/230)中專門說了,并且如《使用Multiwfn便利地查看所有激發態中的主要軌道躍遷貢獻》(http://www.shanxitv.org/529)所述,利用Multiwfn還可以極其便利地一鍵得到所有電子激發中的各種軌道躍遷貢獻,這里就對當前體系用一下。
啟動Multiwfn,載入bulk_TDDFT.out,之后輸入
18 //電子激發分析
15 //顯示所有激發態中的分子軌道躍遷情況
馬上看到以下內容
# 1 1.6478 eV 752.43 nm f= 0.00000 Spin multiplicity= 1:
H -> L 81.4%, H-2 -> L+1 9.4%, H-1 -> L+2 7.6%
# 2 1.6608 eV 746.52 nm f= 0.00000 Spin multiplicity= 1:
H -> L+1 75.2%, H-2 -> L 21.1%
...略
# 12 1.7546 eV 706.62 nm f= 0.00000 Spin multiplicity= 1:
H-3 -> L+3 61.6%, H-1 -> L+2 21.2%, H-2 -> L+1 14.5%
# 13 1.9250 eV 644.09 nm f= 4.13484 Spin multiplicity= 1:
H-2 -> L+2 30.3%, H -> L+3 25.6%, H-3 -> L 20.2%, H-1 -> L+1 17.6%
...略
可見基態S0到S13的激發同時由多個軌道躍遷所明顯貢獻,HOMO-2 -> LUMO+2貢獻了30.3%、HOMO -> LUMO+3貢獻了25.6%、HOMO-3 -> LUMO貢獻了20.2%、HOMO-1 -> LUMO+1貢獻了17.6%。像這種情況就極度不適合通過觀看分子軌道來分析,因為得同時看一大堆軌道,極其麻煩。這種情況最理想的分析方法就是用Multiwfn獨家的空穴-電子(hole-electron)分析,對于任何激發都能轉化為“空穴”到“電子”的躍遷,分析時只需要看這兩個函數的分布即可,在下一節我們將做這種分析。
由以上信息可見S0到S1的激發倒是有主導性的分子軌道躍遷,即HOMO -> LUMO貢獻達到81.4 %,此時看這一對軌道的特征就能大致判斷電子激發情況。這里不妨就看一下HOMO和LUMO的分布是什么樣的。關于Multiwfn顯示分子軌道在《使用Multiwfn觀看分子軌道》(http://www.shanxitv.org/269)有詳細介紹。
啟動Multiwfn,載入bulk_TDDFT-MOS-1_0.molden。注意屏幕上顯示了如下的晶胞信息:
Cell angles: Alpha= 90.0000 Beta= 94.3020 Gamma= 90.0000 degree
可見當前晶胞不是正交的,這種情況下Multiwfn在計算格點數據后直接顯示的等值面不完全正確,需要用Multiwfn導出格點數據后通過VMD或VESTA來顯示等值面。不過,由于此例的Beta角偏離90度也不太大,因此直接用Multiwfn觀看等值面也勉強可以。
進入主功能0,此時文本窗口顯示了HOMO-LUMO gap等軌道相關信息。在圖形界面里點擊右側Show Labels關閉原子標簽、點擊Show axis關閉坐標軸。選擇菜單欄Other settings - Toggle showing cell frame關閉盒子邊框。選擇菜單欄Isosur. quality - Good quality。然后在界面右下角的文本框里輸入h然后按回車,代表顯示HOMO。之后把右下角的Isovalue(等值面數值)改成一個合適的值0.026。此時看到的HOMO等值面圖如下圖左側所示。然后在右下角列表里點擊297切換到LUMO軌道,圖像如下圖右側所示。
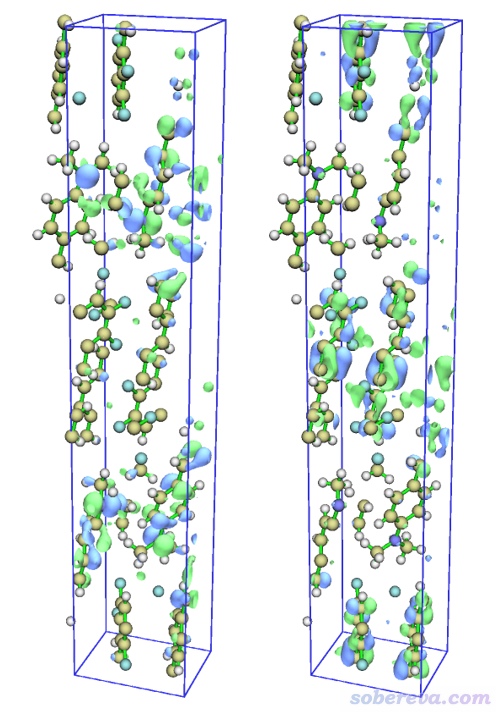
由對比可見,HOMO和LUMO主體分布于不同分子上,因此S0-S1是分子間電荷轉移激發。關于電子激發類型介紹看《圖解電子激發的分類》(http://www.shanxitv.org/284)。
下面我們讓Multiwfn導出HOMO和LUMO的格點數據,從而之后能放到專門的可視化程序里觀看得到更好的效果。點當前圖形窗口右上角的RETURN按鈕返回主菜單,然后輸入
200 //主功能200
3 //計算并導出軌道波函數的格點文件
h //HOMO
9 //根據晶胞設置格點數據的盒子原點、邊長和格點間距
[回車] //用(0,0,0)作為原點
[回車] //格點數據的盒子的三個矢量和當前晶胞相同
[回車] //用默認的0.25 Bohr格點間距(為了降低耗時也可以設大一些,如0.4)
然后當前目錄下就出現了h.cub。將之拖入VESTA,直接就看到了下圖,效果不錯。對LUMO也這么干。
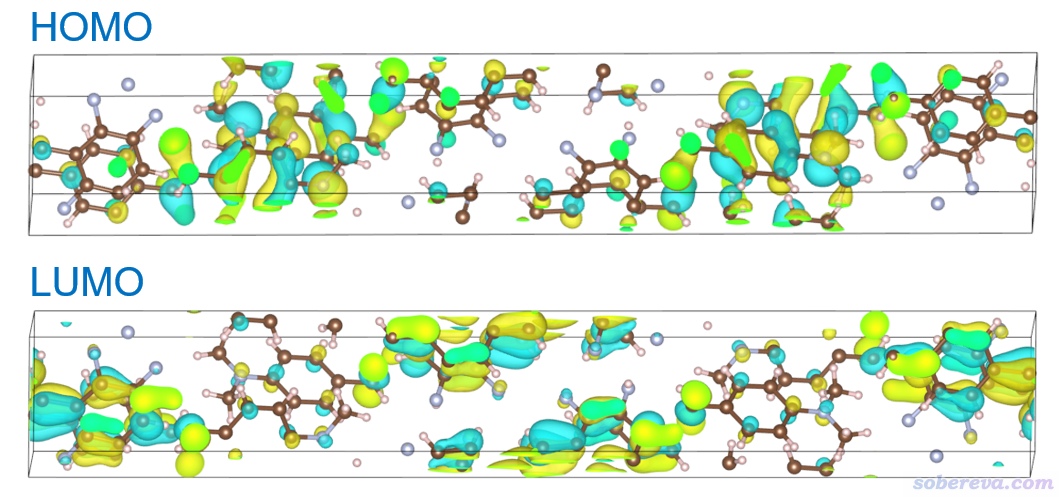
5 空穴-電子分析
下面做空穴-電子分析。此方法的原理、細節、大量用于分子體系的實例在《使用Multiwfn做空穴-電子分析全面考察電子激發特征》(http://www.shanxitv.org/434)都給出了,沒讀過的話務必一讀,也請按照此文所述恰當引用相關文章。
啟動Multiwfn,載入bulk_TDDFT-MOS-1_0.molden,然后輸入
18 //電子激發分析
1 //空穴-電子分析
bulk_TDDFT.out //CP2K輸出文件
13 //分析第13激發態(S13),也就是那個吸收極強的激發態
注意此時屏幕上會看到提示Involved highest unoccupied orbital: 324 (HOMO+ 28),即這個激發態涉及到的組態函數里(即由于系數絕對值>0.001因而被輸出到CP2K輸出文件里的)最高牽扯到的空軌道是第28個空軌道。前面說了,我們當前的TDDFT計算讓CP2K把最低30個空軌道信息寫入了molden文件,比28更大,所以記錄的空軌道數是夠多的。如果記錄的數目小于這里的28,則對這個激發態做的空穴-電子分析可能不準確。每次做空穴-電子分析的時候建議留意一下這里的信息,如果發現存入molden文件里的空軌道數不夠多,應該把CP2K輸入文件里ADDED_MOS后面的值改大,然后重算一次單點任務得到新的molden文件,基于它和之前的TDDFT任務的輸出文件做空穴-電子分析。
接著輸入
1 //可視化空穴、電子、躍遷密度等函數
9 //根據晶胞設置格點數據的盒子原點、邊長和格點間距
[回車] //用(0,0,0)作為原點
[回車] //格點數據的盒子的三個矢量和當前晶胞相同
[回車] //用默認的0.25 Bohr格點間距(為了降低耗時也可以設大一些,如0.4)
在筆者的8核i7-10870H筆記本上兩分鐘算完,然后出現了后處理菜單。雖然用此處的選項在Multiwfn里也能直接顯示空穴、電子等函數的等值面,但由于盒子不是正交的,所以還是先導出格點數據再用VMD、VESTA等程序來顯示。在空穴-電子分析框架里有很多定量指數和函數可以考察電子激發特征,這一節我們就考察空穴、電子以及密度差(charge density difference,當前語境下對應于電子減去空穴),因此在后處理菜單中依次選擇選項10、11、15導出它們的格點數據分別成為hole.cub、electron.cub、CDD.cub。它們在本文的文件包里也提供了。
把這三個cub文件分別拖到VESTA里顯示。等值面數值用0.001(Properties里選Isosurfaces標簽頁,點擊當前僅有的項,把Isosurface level設為0.001),在Objects - Volumetric Data里取消選擇Show Sections,即不顯示截面,然后得到的圖如下所示。
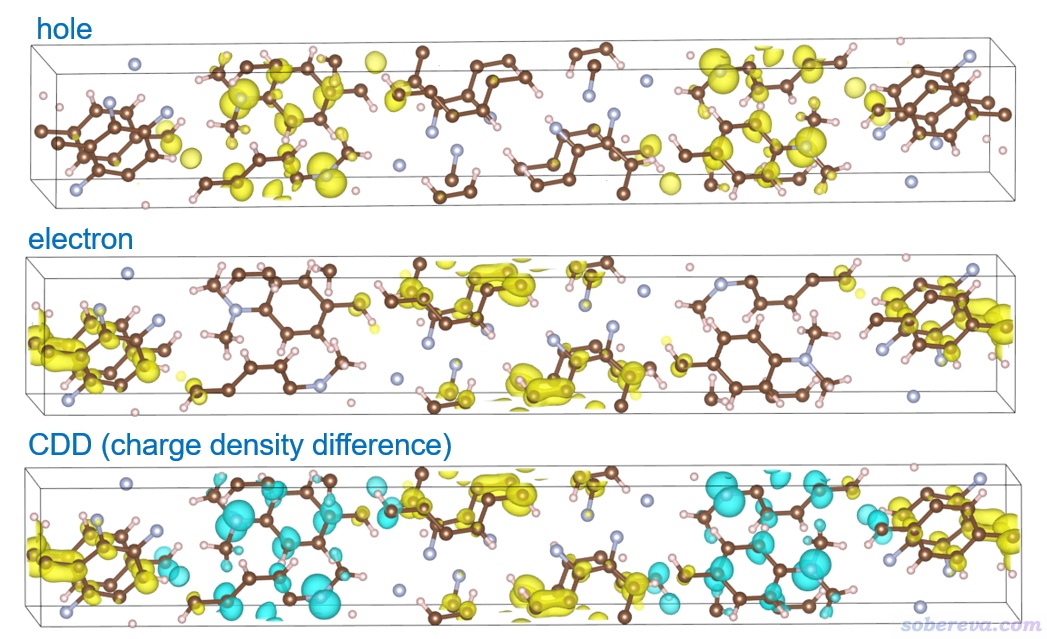
由圖可見,S0-S13激發也是個典型的電荷轉移激發,因為空穴和電子分布在明顯不同的分子上。從CDD圖上考察甚至更方便,只需一張圖就展現出了電子是怎么轉移的,青色和黃色分別是電子激發時電子減少和增加的地方。
如果用VMD來顯示等值面,還可以直接在顯示的時候把周期鏡像顯示出來,由于此時能看到完整分子,明顯更加容易觀察,如下所示
這里具體說一下用VMD怎么作上面的圖。在《在VMD里將cube文件瞬間繪制成效果極佳的等值面圖的方法》(http://www.shanxitv.org/483)中專門給了一個腳本用來十分快捷地顯示效果理想的等值面圖,先使用這個腳本把CDD.cub的0.001等值面顯示出來。然后在Graphics - Representation里把顯示分子結構的那個Representation(以下簡寫為Rep)刪掉,對于分別顯示CDD的正值和負值等值面的那兩個Rep,在Periodic標簽頁里都把-X和+Y都選上,這樣等值面就在X負方向和Y正方向各復制了一次,看上去像超胞的情況。之所以這里沒有把當前分子結構也這么操作來顯示相鄰的鏡像,是因為這么做之后跨盒子的鍵是顯示不了的,看起來略別扭。為了得到與當前等值面相對應的超胞結構文件,下面使用《Multiwfn中非常實用的幾何操作和坐標變換功能介紹》(http://www.shanxitv.org/610)文中介紹的做法。在Multiwfn載入bulk_TDDFT-MOS-1_0.molden后輸入
300 //主功能300
7 //對結構進行操作
19 //對體系進行平移復制
-1 //在第一個方向負方向復制一次
2 //在第二個方向正方向復制為原先兩倍
1 //在第三個方向保持不變
-2 //把當前結構導出為pdb文件
supercell.pdb
現在當前目錄下就有了supercell.pdb。將之拖到已顯示出CDD等值面的VMD圖形窗口里,把其顯示方式設為CPK,即得到上圖。
6 躍遷密度分析
前述的ACS Omega文章里通過所謂的induced charge density分布考察了分子晶體中不同分子在激發時的激子耦合對激發能的影響。實際上induced charge density就是《使用Multiwfn繪制躍遷密度矩陣和電荷轉移矩陣考察電子激發特征》(http://www.shanxitv.org/436)中筆者介紹的躍遷密度(transition density),文中的這類圖都可以用Multiwfn精確重復出來。這里我們也畫一下。
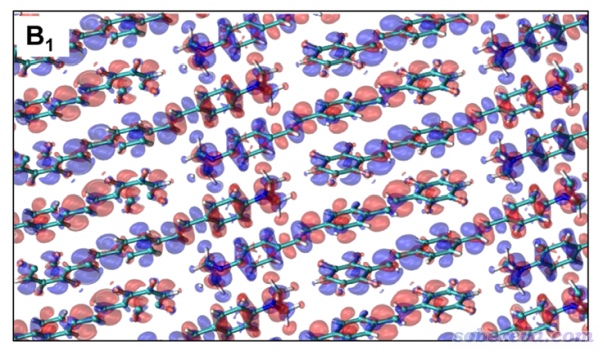
之前Multiwfn做完空穴-電子分析進入后處理菜單后,可以看到選項13是用來導出躍遷密度的cub文件的,選擇它,在當前目錄下就出現了transdens.cub,在本文的文件包里也提供了。按照上一節的方式對它繪制等值面圖,等值面數值為±0.0005的情況如下所示
讀者若仔細將上圖和ACS Omega文中圖2里的體相的induced charge density(下圖)相對比,會發現是完全一樣的,說明了計算的合理性。
至于怎么討論激子耦合本文就不詳述了,讀者可自行參看ACS Omega文中的討論,以及Multiwfn手冊4.A.9節中的計算激子耦合的介紹。
7 NTO分析
NTO(自然躍遷軌道)分析是很流行的激發態分析方法,在《使用Multiwfn做自然躍遷軌道(NTO)分析》(http://www.shanxitv.org/377)里有專門介紹和例子,本文就不再累述了。這個分析在Multiwfn中現在也可以用于周期性體系,這里我們就做一下。值得一提的是,CP2K自身也有NTO分析功能,但筆者發現很難用,一次只能對一個激發態輸出NTO軌道波函數,而且產生的非占據NTO軌道還明顯異常,因此很不推薦用CP2K自己的NTO功能,至少對目前的版本來說。
啟動Multiwfn,載入bulk_TDDFT-MOS-1_0.molden,然后輸入
18 //電子激發分析
6 //NTO分析
bulk_TDDFT.out //CP2K輸出文件
13 //考察S0-S13激發
立刻就在屏幕上給出了本征值最大的10個NTO對的本征值:
The highest 10 eigenvalues of NTO pairs:
0.311175 0.270759 0.215496 0.195281 0.000291
0.000248 0.000180 0.000177 0.000000 0.000000
Sum of all eigenvalues: 0.993608
由數據可見,NTO方法對當前這個電子激發表現極差!在本文第4節我們看到,對這個激發貢獻最大的四對MO躍遷貢獻分別是30.3%、25.6%、20.2%、17.6%,而在變換成NTO躍遷描述之后,依然還是有四對貢獻很大的躍遷,分別是31.1%、27.1%、21.5%、19.5%,比起原先的描述根本就沒什么改進。所以NTO分析在很多情況下是失敗、無用的,而空穴-電子分析則比NTO普適得多,對于任何情況都能很理想地使用。所以若無特殊情況(比如非要得到軌道相位信息等),不建議用NTO而應當用空穴-電子分析。雖然NTO對于分子體系多半情況表現得還行,但我發現對于周期性體系大多時候NTO派不上什么用處,即沒法將電子激發充分簡化描述成一對NTO躍遷所主導的情況。
雖然此例NTO分析沒用,但作為演示,還是看看NTO軌道。在Multiwfn里選擇3,再輸入S13_NTO.mwfn,在當前目錄下就出現了此文件。然后重新啟動Multiwfn,載入S13_NTO.mwfn,進入主功能0,然后在圖形窗口頂端的菜單中選擇Orbital info. - Show up to LUMO+10,此時在文本窗口可看到
...略
Orb: 292 Ene(au/eV): 0.000291 0.0079 Occ: 2.000000 Type:A+B (? )
Orb: 293 Ene(au/eV): 0.195281 5.3139 Occ: 2.000000 Type:A+B (? )
Orb: 294 Ene(au/eV): 0.215496 5.8639 Occ: 2.000000 Type:A+B (? )
Orb: 295 Ene(au/eV): 0.270759 7.3677 Occ: 2.000000 Type:A+B (? )
Orb: 296 Ene(au/eV): 0.311175 8.4675 Occ: 2.000000 Type:A+B (? )
Orb: 297 Ene(au/eV): 0.311175 8.4675 Occ: 0.000000 Type:A+B (? )
Orb: 298 Ene(au/eV): 0.270759 7.3677 Occ: 0.000000 Type:A+B (? )
Orb: 299 Ene(au/eV): 0.215496 5.8639 Occ: 0.000000 Type:A+B (? )
Orb: 300 Ene(au/eV): 0.195281 5.3139 Occ: 0.000000 Type:A+B (? )
Orb: 301 Ene(au/eV): 0.000291 0.0079 Occ: 0.000000 Type:A+B (? )
...略
這里以a.u.為單位的能量對應的是NTO本征值。可見占據和非占據NTO是配對的。比如296和297號軌道構成一個對電子激發貢獻31.1%的NTO對,其中296和297分別是占據和非占據的NTO。軌道圖形請讀者效仿前面第四節自行觀看,筆者就不多說了。
8 總結
本文介紹了如何將Multiwfn與CP2K相結合,繪制電子光譜,并對電子激發特征進行分析。可見二者結合使用,使得研究周期性體系的電子激發相關問題相當容易和便利,比起常見的Gaussian+Multiwfn的組合研究分子體系的電子激發并沒有額外的復雜性。希望讀者在本文的基礎上舉一反三,使用CP2K+Multiwfn研究更多類型的周期性體系的電子激發。
Multiwfn的電子激發分析分析功能非常多,遠遠不限于以上所述的幾種。但這些方法中目前只有部分明確支持周期性體系,有些分析在原理或程序上不支持周期性,而有些可能能支持但尚未做測試。明確支持周期性的分析在Multiwfn手冊2.9.2.2節說了。以后Multiwfn在周期性分析方面還會不斷做擴展和完善,使得更多方法完美兼容周期性體系。如有疑問歡迎在程序啟動時提示的Multiwfn論壇上發帖咨詢。
]]>文/Sobereva@北京科音 2021-Jun-2
0 前言
2021年4月29號,在人氣最高的計算化學專業論壇“計算化學公社”(http://bbs.keinsci.com)開展了為期一個月的四個投票:
你最常用的DFT泛函投票(http://bbs.keinsci.com/thread-22853-1-1.html)
你最常用的量子化學程序投票(http://bbs.keinsci.com/thread-22854-1-1.html)
你最常用的分子動力學程序投票(http://bbs.keinsci.com/thread-22855-1-1.html)
你最常用的第一性原理程序投票(http://bbs.keinsci.com/thread-22856-1-1.html)
現對投票結果進行總結和評論。未來預計每三年重新開展一次投票。要強調的是,這個投票只是體現流行程度,和方法/程序的好壞并沒必然關系。
1 你最常用的DFT泛函投票
本次可投的泛函有31種,雙雜化泛函不包括在內,明顯幾乎不會有人用的泛函也沒納入可投范圍。投票范疇僅限量子化學計算,不包含第一性原理計算。關系特別近的,比如M05-2X和M06-2X、wB97X和wB97XD當做同一個泛函來計。此次投票者共712人,比2018年投票人數299增加了很多,體現出計算化學公社論壇的影響力日益增強。本投票每個人最多選6項,且所投的泛函必須占平時全部研究工作的10%以上,帶不帶DFT-D校正算同一個泛函。按照得票率(票數除以總投票人數)繪制的圖如下,為便于對比2018年的結果也一起繪制了。此圖中諸如某泛函對應60%就代表有60%的人平時較多使用此泛函,后文的統計圖同理。
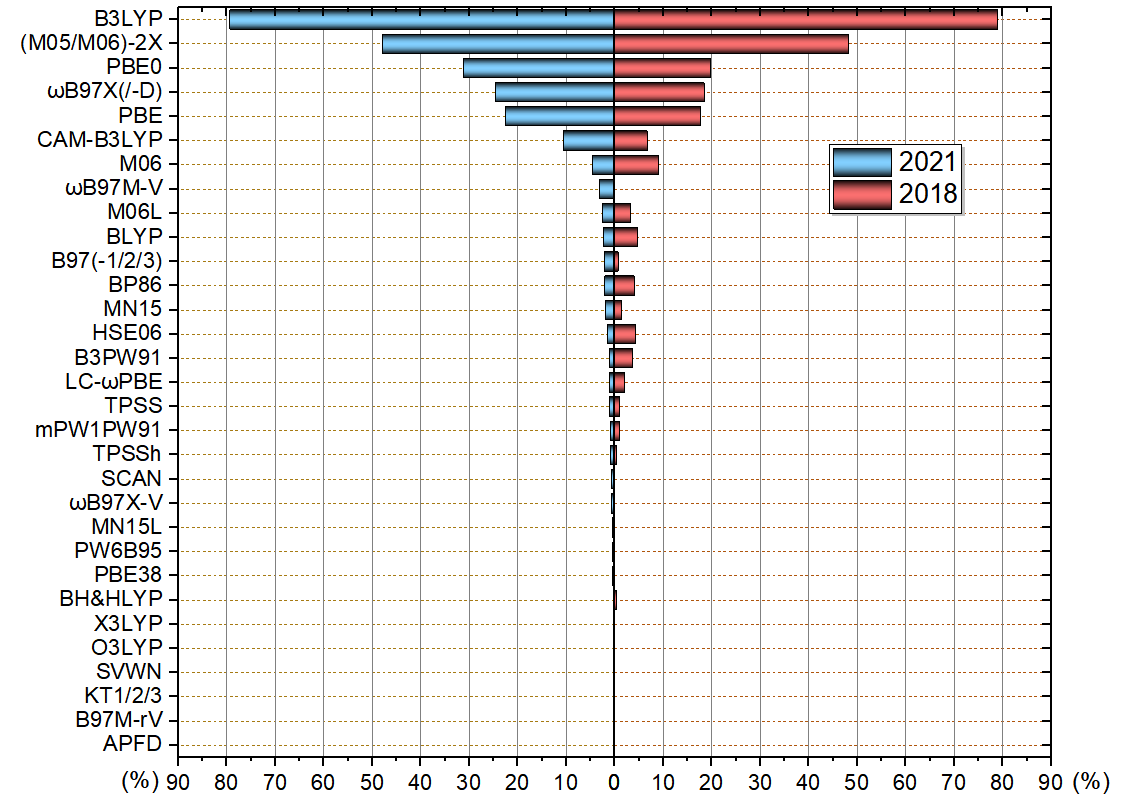
從結果來看,經歷了27年,B3LYP還是目前流行程度最高的泛函,而且加上DFT-D校正之后又給其續命了。估計在接下來的10年內,雖然其流行度No.1的地位也許不保,但至少還會是用得最多的泛函之一。雖然B3LYP算能量問題已經落伍了,如今用這泛函算能量很容易挨審稿人批,見比如《堅持使用B3LYP算有機體系的人的下場》(http://bbs.keinsci.com/thread-12773-1-1.html),但優化、振動分析方面B3LYP的地位相當穩固,多數情況都能用,而且結果不錯速度又快,這點在《談談量子化學研究中什么時候用B3LYP泛函優化幾何結構是適當的》(http://www.shanxitv.org/557)里專門說了。
M06-2X與B3LYP的得票率比例和2018年一樣依然是6:10,真是挺巧的。M06-2X的流行程度頂多也就穩定在這個程度了,不會再上漲了。雖然M06-2X算有機問題、弱相互作用能量問題方面比B3LYP都強得多得多,并且明白這些問題的人越來越多,理應相對于B3LYP的得票率會進一步上升,但M06-2X有耗時高、容易出小虛頻、容易收斂困難這些小問題,而且DFT-D3又令B3LYP在優化弱相互作用體系方面不落下風,而且比M06-2X算各種體系能量問題精度都更好的wB97M-V使用人數又逐漸增多,因此M06-2X的流行程度到現在這個程度基本算是達到瓶頸了。
PBE0、wB97X/X-D的得票率比2018年有所上漲,這有點令我意外,雖然有一些巧合因素,但也可能有一些必然因素。比如在我不少博文,以及計算化學公社論壇上不少帖子里,都提到PBE0-D3(BJ)算過渡金屬配合物、算局域激發態是很好的選擇,這有可能對PBE0的得票率上升有不可忽視的貢獻。
2016年提出的wB97M-V在2018年的時候還不怎么為人所知,當時沒有留給它選項。這次投票可見wB97M-V已經明顯展露頭角了,這有很多原因:在《簡談量子化學計算中DFT泛函的選擇》(http://www.shanxitv.org/272)以及計算化學公社論壇上不少討論中都已經推薦了wB97M-V,在免費而且流行度日益上升的ORCA中已支持了wB97M-V,而且近年來不斷有各種benchmark文章表明wB97M-V算能量的精度在普通泛函里是幾乎最頂尖的。估計再過3年,wB97M-V的得票率比現在能上升一倍左右。
M06、M06L的得票率如今比2018低了不少。這也是因為這倆泛函在如今看來沒什么用,它們在后來的許多benchmark文章里(哪怕是專門測過渡金屬配合物體系的)表現得都很平庸,M06能用的情況用MN15幾乎都是更好的選擇,而必須用M06L這樣純泛函的時候(如靜態相關很強的情況),也不如用改用更新的MN15L。
BLYP得票率降了很多,確實這如今也用處不大。在ORCA里為了計算快而用純泛函的話,基本上我都推薦用B97-3c。
MN15得票率和2018年相比幾乎沒變,主要也是這泛函顯得有點中庸,用戶對它的剛性需求不多。
B3PW91、LC-wPBE、mPW1PW91、HSE06得票率比2018年顯著降低,完全失勢了,它們在量化計算里如今基本也都沒多大用處,只要用戶清醒地知道什么時候用什么泛函最恰當,這些泛函幾乎在任何研究中都不會用到(也有一些例外情況,比如《使用Multiwfn預測晶體密度、蒸發焓、沸點、溶解自由能等性質》http://www.shanxitv.org/337里提到的Politzer等人擬合的預測公式里的靜電勢描述符是基于B3PW91算的)。BP86下降不少其實有點可惜,這老東西其實在計算過渡金屬體系方面還是挺有用的,比較魯棒。B97(-1/2/3)得票率有所上漲,一定程度上可能是因為每次北京科音量子化學培訓班的時候、網上答疑的時候我都建議用B97-2算NMR所致。
其它泛函就沒什么好評價的了,基本上2018年沒人用的泛函到現在還是沒人用。有些很少有人用的泛函,比如SCAN、TPSSh其實都是有用武之地的,但用戶基本都被其它更流行、表現也不錯的泛函吸走了。
有一個泛函極度悲催,2018年就是唯一0票的,到2021年還是唯一0票的,它 就 是:APFD!3年前我就說,不好的泛函怎么打廣告都沒用,縱使某本書第三版通篇都用APFD(并由此給初學者一種錯覺APFD是應當默認使用的),而且這書中文版也早已上市,但終究APFD還是完全不被接受。其實這么冷門的泛函在本次投票中理應不給其留出選項的,但我就想看看到底有沒有哪怕一個初學者用了APFD,結果令我欣慰。
2 你最常用的量子化學程序投票
可投程序有28種,投票者共642人,顯著超過2018年的233人。本投票每個人最多選三項,且所投的程序必須占平時全部研究工作的10%以上。按照得票率繪制的圖如下
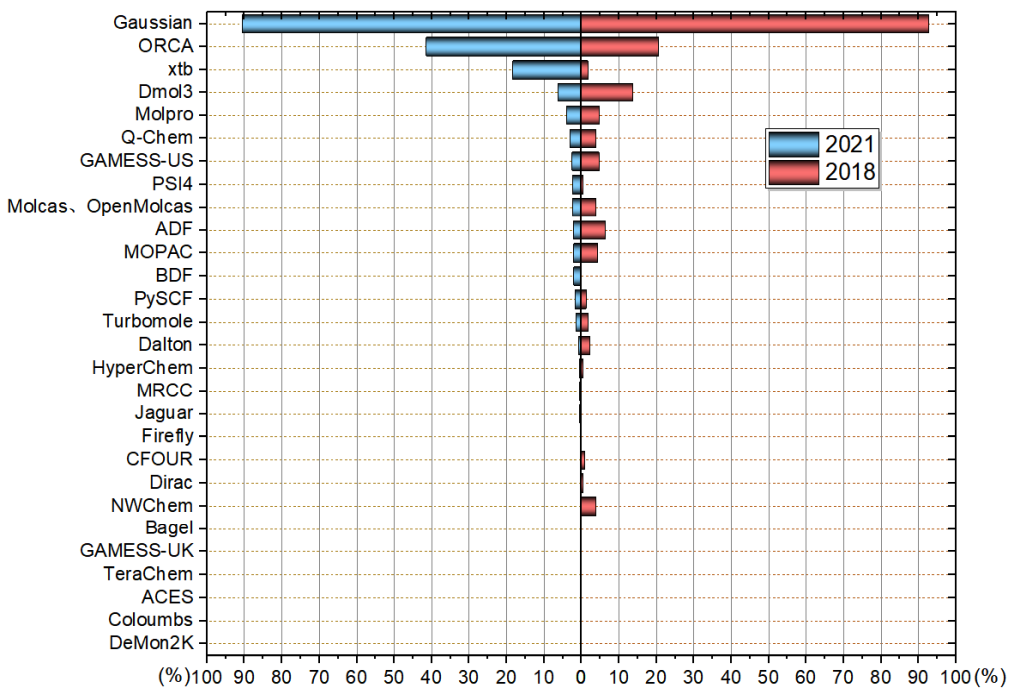
可見Gaussian依然是老大的位置,得票率穩居>90%。ORCA的用戶數提升幅度很大,這來自于近些年ORCA的迅速的發展、在計算化學公社論壇和思想家公社QQ群里口耳相傳。筆者近年也寫了不少ORCA相關的博文(見http://www.shanxitv.org/category/ORCA/),對于在國內普及ORCA起到了一定推動作用。雖然Gaussian的流行程度的地位在10年內難以動搖,ORCA若想充分代替Gaussian路還長得很,但明顯ORCA對Gaussian的威脅與日俱增。
xtb的用戶數是3年前的10倍有余!這是用戶數增速最快的程序。xtb支持的GFN-xTB對于粗略優化、跑要求不高的動力學、結合Molclus做構象搜索(見比如《使用Molclus結合xtb做的動力學模擬對瑞德西韋(Remdesivir)做構象搜索》http://bbs.keinsci.com/thread-16255-1-1.html)頗為有用,速度又快結果通常也定性合理,實用性頗高,在論壇和群里也經常有人討論xtb,因此其用戶數猛增是理所應當的,但增速實在是比我想象得更快。老師傅MOPAC有種要被xtb亂拳打死的感覺。
Dmol3和ADF用戶數猛跌!大勢已去!我作出上面的統計圖的時候,看到其流行度和3年前相差如此懸殊,我甚至噗~地噴出來。看來量子化學領域的用戶明顯逐漸趨于理性;如今信息越來越透明,計算化學公社論壇高質量的文章和討論也在選擇量子化學程序方面上起到了正確的導向,再加上免費的ORCA被越來越多的人認識,這都使得研究者們不再那么容易被又貴又*的程序所忽悠。Dmol3賊貴不說,程序還特弱,而且越來越顯得過時,比如到現在還不支持10年前就有的DFT-D3(這年頭基本沒有主流程序不支持這個),Dmol3用戶若發弱相互作用問題的文章只得尷尬地用DFT-D2,不被內行審稿人歧視才怪。如今在量子化學較高水平的交流群里,提問者一說要用Dmol3算xxx,大概率會被用異樣眼光看待,通常要么沒人理,或者被勸改用ORCA。對Dmol3還抱有幻想的初學者建議看看http://www.shanxitv.org/508。至于ADF,之前我在http://www.shanxitv.org/489里已經評述過,它相對于ORCA而言對于一般的研究真是沒什么使用價值,還按年收費賣得老貴,2018年投票的時候其得票率明顯高于其實用價值,今年的投票算是徹底打回原形了。
GAMESS-US越來越讓人覺得有點老古董了,發展慢,使用又麻煩。大多數國內的人用GAMESS-US基本都是沖著LMOEDA去的(從計算化學公社論壇的量子化學版的GAMESS-US分類的帖子里可見一斑)。由于賣點太少,使用門檻高,相關學習資源少,再加上ORCA來勢洶洶,GAMESS-US流行度不斷下降不難理解。
Molpro和阿Q流行度小降,也是對于大多數人缺乏剛性使用必要,又有其它程序的威脅所致。阿Q購買必要真是不大,以前泛函支持得多,特別是支持wB97M-V之類Gaussian沒有的泛函還算是對一般應用性研究用戶有價值的賣點,而如今ORCA不僅也支持了,而且掛著libxc還可以用各種泛函,折去了Q-Chem一個賣點(之前Q-Chem的TDDFT二階解析導數的賣點就被G16折去了)。雖然阿Q亂七八糟功能支持豐富,但那些功能大多過于偏學術,而且又缺乏教程來推廣,在大宗性研究中很少有機會用到。Q-Chem還有些相對有用的功能如今在ORCA、PSI4等其它免費程序里也都有替代品。Molpro的日子也不好過,MCSCF、多參考方法方面的老對手Molcas如今有了免費版OpenMolcas,這是挺致命的,而且ORCA這方面功能做得也越來越完善,以后Molpro的流行度可能還要降,更不好賣了。
PSI4用戶上漲不少,一方面是PSI4近年來發展很快,另一方面和這個博文可能也有不小關系:《使用PSI4做對稱匹配微擾理論(SAPT)能量分解計算》(http://www.shanxitv.org/526)。估計目前國內用PSI4大多數都是沖著SAPT去的。
其它程序用戶都很少。其中NWChem很慘,3年前流行度還能排個中游,如今流行度暴跌,沒什么人用了。也的確對于普通應用性研究者來說,這個程序如今使用必要性很低,發展慢,也受到ORCA的嚴重擠壓。
TeraChem是今年新加入投票的,只有慘淡的1票。這程序賣點是GPU加速,但這程序功能很少,賣得也不便宜,而且如今挖礦鬧得GPU價格暴漲,TeraChem相對于用其它程序靠CPU計算(尤其是ORCA里開RI)來說真是沒什么明顯好處了。
3 你最常用的分子動力學程序投票
共有397人參與了“你最常用的分子動力學程序投票”,共有19個程序可投,都是基于經典力場的分子動力學程序。每個用戶可以投3個,并且要求平時使用幾率超過10%。結果如下
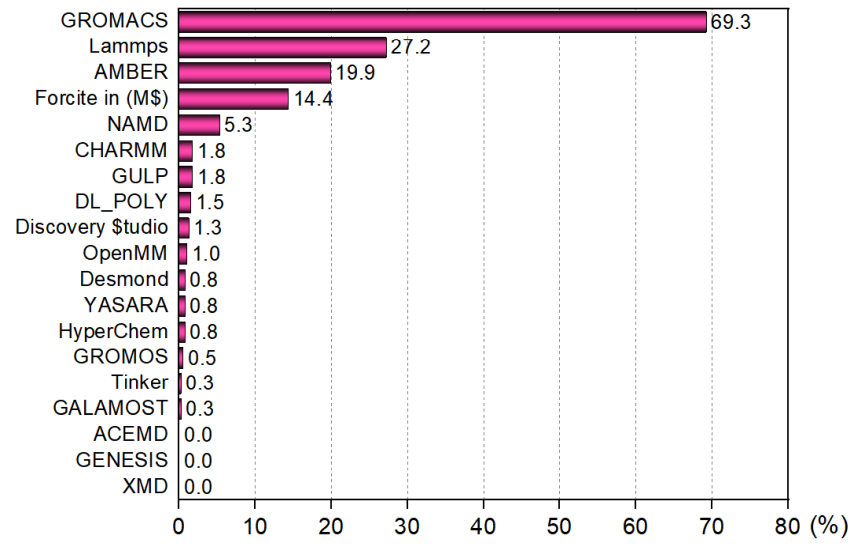
GROMACS用戶數遠超任何其它程序,其它動力學程序用戶數目的總和才差不多等于GROMACS,這和我預期的完全一致。Tinker的用戶數遠低于我的預計,這也是個挺知名的程序,但在國內實在不人氣。我不知道為啥有人會用HyperChem做動力學,這速度也太慢了,功能也太弱了,還收費,而且這個程序都已經停止開發很多年了。
4 你最常用的第一性原理程序投票
共有442人參與了“你最常用的第一性原理程序投票”的投票,共有26個程序可投,每個用戶可以投3個,并且要求平時使用幾率超過10%。結果如下
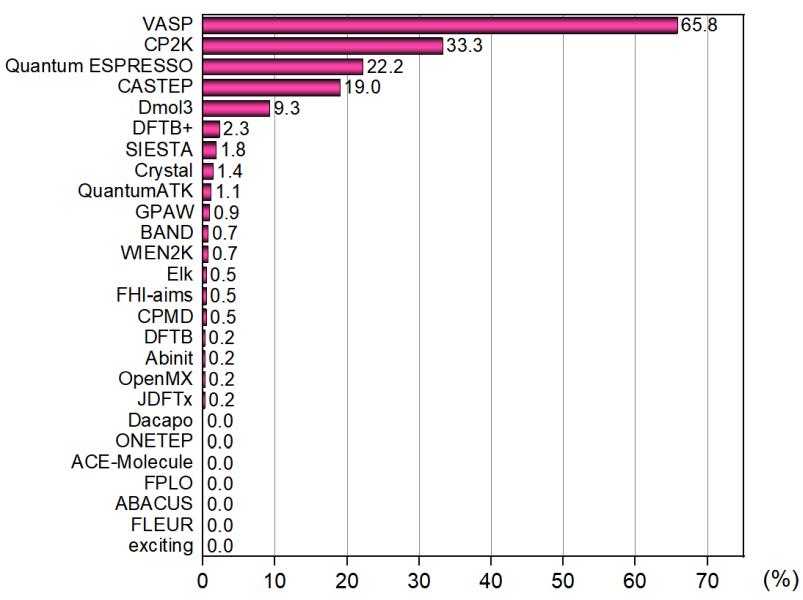
VASP流行度排第一,這個沒有什么懸念,但遠達不到Gaussian那樣在量化領域里有無法撼動的地位。稍微出乎我意料的是這次摸底調查中體系出CP2K和Quantum ESPRESSO的用戶數其實很多,比我想象中的多多了,這是令我比較欣慰的。我很希望看到有越來越多的人使用這樣又強大又免費的程序代替賣得越來越貴的VASP。
CP2K能排到第二真是令我很意外,因為從這個程序原文的引用次數來看,比Quantum ESPRESSO要少得多得多,占VASP用戶數可能也就是個零頭。投CP2K的人這次這么多,一方面是CP2K用戶群體確實增長較快,也略微有一定可能是因為今年Multiwfn支持了創建CP2K輸入文件的功能(見《使用Multiwfn非常便利地創建CP2K程序的輸入文件》http://www.shanxitv.org/587),一下子把CP2K輸入文件編寫門檻極大地降低,并導致一些之前做量化而不做第一性原理計算的人開始嘗試著使用CP2K算點東西了。CP2K這個大殺器以后必會被越來越多的人青睞,越來越流行,前途光明。也同時祝愿Quantum ESPRESSO的用戶數與VASP的差距能越來越小。相比紅紅火火的CP2K,從投票結果看歷史更久的CPMD幾乎已經算是涼了,近年來也都不怎么發展了,快要退出歷史舞臺。
Abinit的用戶數真是少得可憐,遠低于我的預期,以這個免費又功能豐富的程序的體量來看用戶數理應不該這么少。CASTEP和Dmol3目前雖然還有一些市場,但我估計過三年再次投票的話,得票率肯定會降低。Dmol3很多人用是為了圖快,但把CP2K用好的話,多數情況下真沒必要花大把銀子買Dmol3。
]]>文/Sobereva@北京科音
First release: 2021-Feb-27 Last update: 2022-Mar-8
1 前言
2010年提出的Noncovalent interaction (NCI)分析,也即Reduced density gradient (RDG)分析,是如今使用極為普遍的圖形化考察化學體系中弱相互作用的方法,筆者寫過諸多相關文章介紹原理和實現,還錄了操作演示視頻,如下所示,故本文對基本原理不再累述,并假定讀者已經具備了背景知識
? 《使用Multiwfn圖形化研究弱相互作用》(http://www.shanxitv.org/68)
? 《用Multiwfn+VMD做RDG分析時的一些要點和常見問題》(http://www.shanxitv.org/291)
? 《通過鍵級曲線和ELF/LOL/RDG等值面動畫研究化學反應過程》(http://www.shanxitv.org/200)
? 《使用Multiwfn做NCI分析展現分子內和分子間弱相互作用》(演示視頻。https://www.bilibili.com/video/av71561024)
? Multiwfn手冊3.23.1節
? 《使用Multiwfn研究分子動力學中的弱相互作用》(http://www.shanxitv.org/186)
筆者開發的Multiwfn是做NCI分析最流行、最好用的程序。但以前的版本主要面向分子體系,基于Gaussian、ORCA等量子化學程序產生的波函數進行分析。對于周期性體系往往也可以通過團簇模型結合Multiwfn做NCI分析,比如《使用量子化學程序基于簇模型計算金屬表面吸附問題》(http://www.shanxitv.org/540)里對銀團簇表面吸附苯做了NCI分析、在Multiwfn原文J. Comput. Chem., 33, 580 (2012)里截取了尿素團簇做了NCI分析,但是還是有很多情況不便于或者幾乎不適合用簇模型描述。為了讓周期性體系的NCI分析可以非常容易地實現、使這個方法能考察更廣泛的體系,從2021年2月更新的Multiwfn開始,Multiwfn也支持了結合CP2K程序產生的波函數做NCI分析,這使得圖形化考察固體內部和表面的弱相互作用極其容易,將在本文進行詳細示例。看了第3節的例子后你會發現計算過程真是巨簡單。本文也會提及很多讀者應了解的注意事項,以便在實際研究中遇到問題時恰當變通。
另外,Independent gradient model (IGM)也是非常重要的圖形化研究弱相互作用的方法,相對于NCI的關鍵好處是可以很靈活地自定義片段,能只顯示片段間的弱相互作用而不會受到片段內的干擾。此外,Multiwfn做IGM分析時還可以給出諸多定量指標便于討論。Multiwfn做IGM分析在此文有詳述:《通過獨立梯度模型(IGM)考察分子間弱相互作用》(http://www.shanxitv.org/407)。如今Multiwfn也將IGM分析擴展到了周期性體系,將在下文示例。本文還會順帶提及筆者提出的IGM方法的改良,即IGM based on Hirshfeld partition (IGMH),比IGM圖像效果明顯好得多,詳見《使用Multiwfn做IGMH分析非常清晰直觀地展現化學體系中的相互作用》(http://www.shanxitv.org/621)。
值得一提的是,Multiwfn還支持筆者提出的IRI方法,可以同時把弱相互作用和化學鍵作用區域直觀地展現,很有實用價值,在《使用IRI方法圖形化考察化學體系中的化學鍵和弱相互作用》(http://www.shanxitv.org/598)中有專門的介紹,并且給了計算孤立體系和計算周期性體系的例子,強烈建議一看!
對一般情況,筆者強烈建議用后來提出的IRI代替NCI,能明顯展現更多有價值的信息;也強烈建議用后來提出的IGMH代替IGM,圖像效果好得多,且物理意義更嚴格。IRI和NCI的分析操作幾乎完全相同,而IGMH和IGM的分析操作幾乎完全相同。 在《一篇最全面介紹各種弱相互作用可視化分析方法的文章已發表!》(http://www.shanxitv.org/667)中介紹的筆者的綜述文章中對上述可視化分析方法有非常全面深入的介紹,里面也展示了用于周期性體系的例子,推薦閱讀和引用。
Multiwfn可以在官網http://www.shanxitv.org/multiwfn免費下載,不熟悉Multiwfn的話注意參看《Multiwfn FAQ》(http://www.shanxitv.org/452)和《Multiwfn入門tips》(http://www.shanxitv.org/167)。請讀者務必使用2021-Feb-25日及之后更新的Multiwfn以確保情況和下文內容相符。本文例子中周期性體系的DFT計算部分使用CP2K 8.1版實現,安裝方法見《CP2K第一性原理程序在CentOS中的簡易安裝方法》(http://www.shanxitv.org/586)。CP2K結合Multiwfn可以非常容易地使用,見《使用Multiwfn非常便利地創建CP2K程序的輸入文件》(http://www.shanxitv.org/587)。本文繪圖使用VMD 1.9.3版,可以在http://www.ks.uiuc.edu/Research/vmd/免費下載。
下文涉及的所有文件都可以在http://www.shanxitv.org/attach/588/file.rar下載。cub文件比較大所以就沒有提供了。
2 輸入文件
在給出具體例子之前先介紹一下Multiwfn做NCI、IGM分析都需要用什么輸入文件。
后記:關于這部分知識,更多、更詳細的介紹見《詳談使用CP2K產生給Multiwfn用的molden格式的波函數文件》(http://www.shanxitv.org/651)。
2.1 普通NCI分析用的輸入文件
普通形式的NCI分析需要基于電子波函數計算電子密度及其導數。CP2K產生的molden文件記錄了Gaussian型基函數描述的電子波函數,Multiwfn可以基于它做各種波函數分析,也包括NCI分析。CP2K產生molden文件的方法在《使用Multiwfn非常便利地創建CP2K程序的輸入文件》(http://www.shanxitv.org/587)的3.1節已經示例過。如果你習慣自己寫CP2K輸入文件的話,是在&DFT字段里加上以下內容
&PRINT
&MO_MOLDEN
NDIGITS 8
GTO_KIND SPHERICAL
&END MO_MOLDEN
&END PRINT
這樣CP2K計算完畢后就會在當前目錄下產生一個.molden文件。
CP2K產生的molden文件還不能直接用于做周期性體系的NCI分析,因為標準的molden格式里沒有晶胞信息。為了能讓molden文件給Multiwfn提供晶胞信息,我給molden格式做了擴展,定義了[Cell]字段。用文本編輯器打開molden文件后,需手動在第二行插入[Cell]并在下面三行定義晶胞的三個平移矢量(埃),下面是個例子
[Molden Format]
[Cell]
7.13358000 0.00000000 0.00000000
0.00000000 7.13358000 0.00000000
0.00000000 0.00000000 7.13358000
[Atoms] AU
C 1 4 0.000000 0.000000 0.000000
C 2 4 1.685064 1.685064 1.685064
C 3 4 0.000000 3.370128 3.370128
...略
讓Multiwfn載入這樣修改過的molden文件后,晶胞信息就會被讀入,文件載入完畢后屏幕上就可以看到晶胞信息的提示了:
Cell information:
Translation vector 1 X= 13.480512 Y= 0.000000 Z= 0.000000 Bohr
Translation vector 2 X= 0.000000 Y= 13.480512 Z= 0.000000 Bohr
Translation vector 3 X= 0.000000 Y= 0.000000 Z= 13.480512 Bohr
Cell volume: 2449.7350 Bohr^3 ( 363.0134 Angstrom^3 )
有一點需要注意的是,如今CP2K計算普遍使用MOLOPT基組,這是個完全廣義收縮的基組,而Multiwfn的計算代碼并未對廣義收縮的基組進行專門的優化,因此使用MOLOPT基組得到的波函數在Multiwfn里計算耗時會比較高。如果你有個像樣的服務器那無所謂,但如果只有個個人PC,算NCI的格點數據的時候對稍大的體系會比較吃力。我一般建議用比如6-31G*、6-311G**之類基本像樣的而且是片段收縮的全電子基組在CP2K里做個GAPW單點計算來得到NCI分析用的molden文件,比起用DZVP-MOLOPT-SR-GTH基組做GPW計算得到的molden文件在NCI分析耗時上能降低好幾倍。另外,用全電子基組比起用MOLOPT這樣的贗勢基組還有個好處是內核電子被明確表達了。NCI分析對基組不敏感,一般用6-31G*就足夠了,用更大基組也不會發現結果有什么可查覺的改變。
如果由于特殊情況,你就是想用諸如MOLOPT贗勢基組在Multiwfn里做NCI等波函數分析,那么應當再手動改一下molden文件,使得Multiwfn明確知道各個原子的價電子數是多少(指的是使用此贗勢時原子在孤立存在時的價電子數。和原子在當前體系里的實際價電子數沒任何關系),這有兩個必要性:
? molden文件里并沒有明確記錄原子的價電子數,這導致Multiwfn算原子電荷的時候不知道實際上當前原子的核電荷數是多少,因此給出的原子電荷是不合理的。
? 讓Multiwfn知道被贗勢贗化的電子數的話,Multiwfn會通過自帶的EDF內核電子密度數據庫提供內核電子密度,此時做純粹基于電子密度的各種分析時結果會較接近于全電子基組的情況,即更為真實。關于這點詳見《在贗勢下做波函數分析的一些說明》(http://www.shanxitv.org/156)。
讓Multiwfn知道各個原子價電子數是多少,可以通過以下兩種修改molden文件的做法實現
(1)把元素名后頭的元素序號改成價電子數。比如molden文件的[Atoms]字段某一行原本是
C 3 6 0.000000 0.000000 0.000000
如果贗勢贗化了碳的1s的兩個電子,即價電子數為4,則改為
C 3 4 0.000000 0.000000 0.000000
(2)更方便的做法是直接在[Atoms]字段之前加入[Nval]字段,這是我對molden格式的擴展。例如插入以下內容
[Nval]
C 4
O 6
就告訴Multiwfn所有的碳都是4個價電子,氧是6個價電子。加入了[Nval]和[Cell]字段的一個例子見Multiwfn自帶的examples\PBC\CP2K_diamond_2x2_DZVP-MOLOPT.inp。
注意CP2K在考慮k點的時候不能產生molden文件,Multiwfn本身也不支持考慮k點,所以對于小晶胞體系必須用足夠大的超胞來計算。
實際上Multiwfn還可以基于Gaussian程序的PBC計算產生的fch文件做周期性體系的波函數分析。但對于二維、三維周期性體系,Gaussian的這個功能幾乎什么都算不動,毫無實用性,這里就不提了。Multiwfn也可以基于含有晶胞平移矢量信息的mwfn格式(https://doi.org/10.26434/chemrxiv.11872524)做周期性體系的波函數分析,但CP2K目前版本產生不了。
2.2 準分子近似的NCI分析以及IGM分析用的輸入文件
還有一種NCI分析是基于準分子近似(promolecular approximation)實現的,即使用原子自由狀態電子密度簡單疊加產生的近似的分子電子密度(準分子密度,promolecular density)進行計算。對于周期性體系的準分子近似的NCI分析,只需要給Multiwfn提供含有原子坐標和晶胞信息的文件作為輸入文件即可,比如cif文件、Gaussian的PBC任務的gjf文件、含有CRYST1字段的pdb文件等,詳見《使用Multiwfn非常便利地創建CP2K程序的輸入文件》(http://www.shanxitv.org/587)的第2節。
準分子近似的NCI計算耗時只是普通NCI分析的一個零頭。而且由于只需要結構信息,用任何第一性原理程序優化坐標/晶胞都可以,也可以直接用實驗的cif文件。但其結果合理性比普通的NCI略差,盡管通常來說定性一致。一般來說能算得動普通的NCI分析的時候就不建議用準分子近似。
IGM分析需要的輸入文件和準分子近似的NCI分析完全一樣,因為IGM分析也是完全依賴于幾何結構和Multiwfn內置的自由狀態原子密度實現的。
3 周期性體系的NCI分析示例
3.1 金剛石中的位阻作用
3.1.1 普通NCI分析
此例用NCI分析考察金剛石中的位阻作用。首先基于晶體結構,用Multiwfn產生一個做PBE/6-31G*單點計算并輸出molden文件的CP2K輸入文件,使用2*2*2的超胞。為此,用Multiwfn載入本文文件包里的金剛石的conventional cell的文件diamond.cif,然后輸入
cp2k //創建CP2K輸入文件
diamond_222.inp //要產生的CP2K輸入文件名
2 //選擇基組
10 //6-31G*
-2 //要求產生molden文件
-11 //幾何操作
19 //構建超胞
2 //第1個方向重復的次數
2 //第2個方向重復的次數
2 //第3個方向重復的次數
-10 //返回
0 //產生輸入文件
現在diamond_222.inp文件被產生在了當前目錄下,CP2K算完后就有了diamond_222-MOS-1_0.molden。用文本編輯器打開它,然后把diamond_222.inp里的晶胞信息復制進去,此時此文件開頭部分內容為
[Molden Format]
[Cell]
7.13358000 0.00000000 0.00000000
0.00000000 7.13358000 0.00000000
0.00000000 0.00000000 7.13358000
[Atoms] AU
C 1 6 0.000000 0.000000 0.000000
C 2 6 0.000000 3.370128 3.370128
...略
啟動Multiwfn,載入diamond_222-MOS-1_0.molden,然后輸入
20 //圖形化分析弱相互作用
1 //NCI分析
9 //借助晶胞信息定義格點
[按回車] //使用0,0,0作為盒子原點。這里說的盒子是指被計算的格點均勻分布的區域,后同
[按回車] //使用晶胞的三個平移矢量的長度作為盒子三個邊的長度
0.2 //格點間距
現在Multiwfn就開始計算NCI分析用的RDG和sign(λ2)rho這兩個函數的格點數據了。在筆者的2*E5-2696v3 36核服務器上僅僅11秒就算完了。之后選3把這兩個函數的格點數據分別導出為當前目錄下的func2.cub和func1.cub。將這兩個cub文件以及Multiwfn自帶的examples目錄下的RDGfill.vmd都拷到VMD目錄下,然后啟動VMD,在VMD的文本窗口輸入source RDGfill2.vmd執行此繪圖腳本,再輸入pbc box來繪制出盒子。之后在Graphics - Representation里選擇正處于CPK風格的那個representation,通過界面上的Sphere Scale把原子球尺寸調成0.7。此時的sign(λ2)rho著色的RDG等值面圖,也即一般說的NCI圖,如下所示
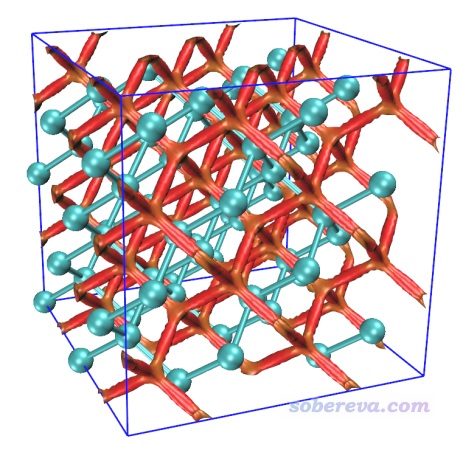
可見圖像效果非常理想。紅色的等值面在金剛石中呈網狀分布,體現出金剛石里面有顯著的位阻作用。讀者有興趣的話可以通過相同的方法去對硅也繪制這種圖,你會發現硅當中也有網狀的等值面,但顏色是綠色的,體現出由于硅的孔洞相對較大,孔洞中的位阻互斥就沒那么強了。
通常做周期性體系NCI分析時格點都按照此例方式來設即可,這樣格點會均勻分布在整個盒子里,從而把體系所有地方的弱相互作用都圖形化展現出來。格點間距應根據需要恰當設置,想要更光滑的等值面就把格點間距設得更小。注意在盒子范圍固定的情況下,計算耗時、cub文件的尺寸都和格點間距的三次方呈反比。此例如果把格點間距設成更小的0.13,你會發現得到的等值面上完全看不出任何鋸齒,但耗時會增加為原先的(0.2/0.13)^3=3.6倍。
NCI分析中經常涉及到繪制RDG與sign(λ2)rho之間的散點圖,做法在第1節里提到的筆者的RDG博文中,以及《繪制有填色效果的用于弱相互作用分析的RDG散點圖的方法》(http://www.shanxitv.org/399)中都做了介紹。周期性體系的NCI分析也可以這么繪制散點圖,這里就不再演示了,這和分析孤立體系的操作沒有任何區別。
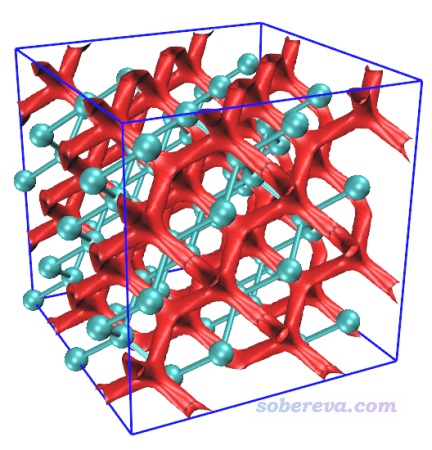
3.1.2 準分子近似的NCI分析
現在再演示一下如何對金剛石超胞做準分子近似的NCI分析。用上一節的diamond_222.inp或者diamond_222-MOS-1_0.molden當做Multiwfn的輸入文件都可以,因為它們都能給Multiwfn提供這種分析所需要的原子坐標和晶胞信息。這里用原胞diamond.cif作為輸入文件進行演示,請大家舉一反三。
啟動Multiwfn,載入diamond.cif,然后輸入
300 //其它功能(Part 3)
7 //對體系進行幾何操作
19 //構建超胞
2
2
2
-10 //返回
0 //返回主菜單。現在內存里記錄的金剛石的原胞已經成了2*2*2的超胞了
20 //圖形化分析弱相互作用
2 //準分子近似的NCI分析
9 //設置格點。同前,不再解釋
[按回車]
[按回車]
0.2 //格點間距(Bohr)
你會充分體會到準分子近似的NCI分析極快,一眨眼的功夫格點數據就算完了。之后選3導出cub文件,將導出的func1.cub、func2.cub以及Multiwfn自帶的examples目錄下的RDGfill_pro.vmd都復制到VMD目錄下。啟動VMD,在文本窗口里輸入source RDGfill_pro.vmd,然后輸入pbc box顯示盒子。之后在Graphics - Representation界面里改一下CPK原子球尺寸,然后選Isosurface那個representation,把Isovalue改成0.2并按回車。此時圖像如下,可見和普通NCI分析得到的圖像特征定性一致。
3.2 共價有機框架化合物(COF)中的弱相互作用
此例將對下面這個COF晶胞中的弱相互作用通過NCI方法進行圖形化展現。
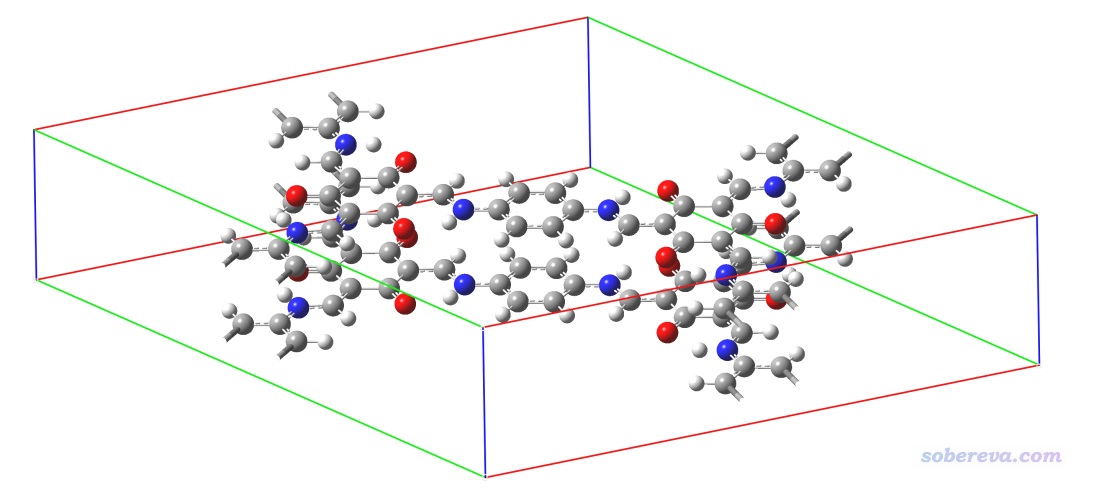
此體系的cif文件是本文文件包里的COF_12000N2.cif。這個晶胞已經很大了,顯然沒必要再弄成超胞。如《實驗測定分子結構的方法以及將實驗結構用于量子化學計算需要注意的問題》(http://www.shanxitv.org/569)一文所述,X光衍射得到的晶體結構中氫的位置通常是不可靠的,所以本例我們在做NCI分析前用CP2K先優化一下氫原子的位置。
啟動Multiwfn并載入COF_12000N2.cif,然后輸入
COF_12000N2.cif
cp2k
COF_opt.inp
-2 //要求產生molden文件
-1 //設置任務類型
3 //優化結構但不優化晶胞
9 //設置原子凍結
optH //只優化氫原子
2 //選擇基組
10 //6-31G*
0 //產生輸入文件
這樣就得到了PBE/6-31G*只優化COF中氫原子位置的CP2K輸入文件COF_opt.inp。運行之,在36核服務器上十來分鐘就算完了。之后可以看到一批以COF_opt-MOS-1_為開頭的molden文件,它們記錄了優化過程中各幀的波函數。其中序號最大的是COF_opt-MOS-1_4.molden,這是最后優化完的結構下的波函數文件。將COF_opt.inp里的晶胞信息手動加入到COF_opt-MOS-1_4.molden文件里作為[Cell]字段。
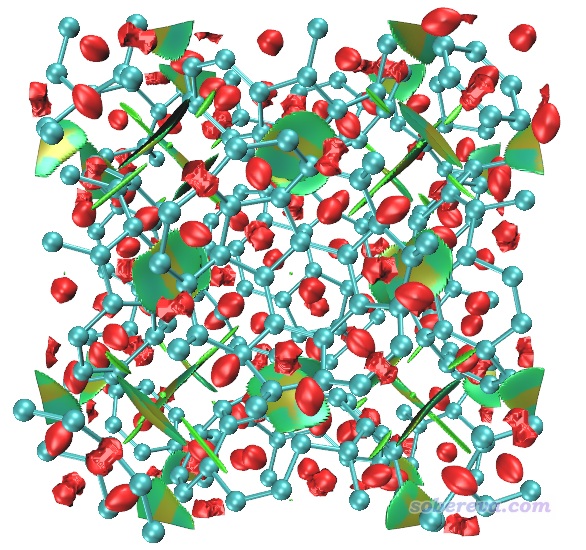
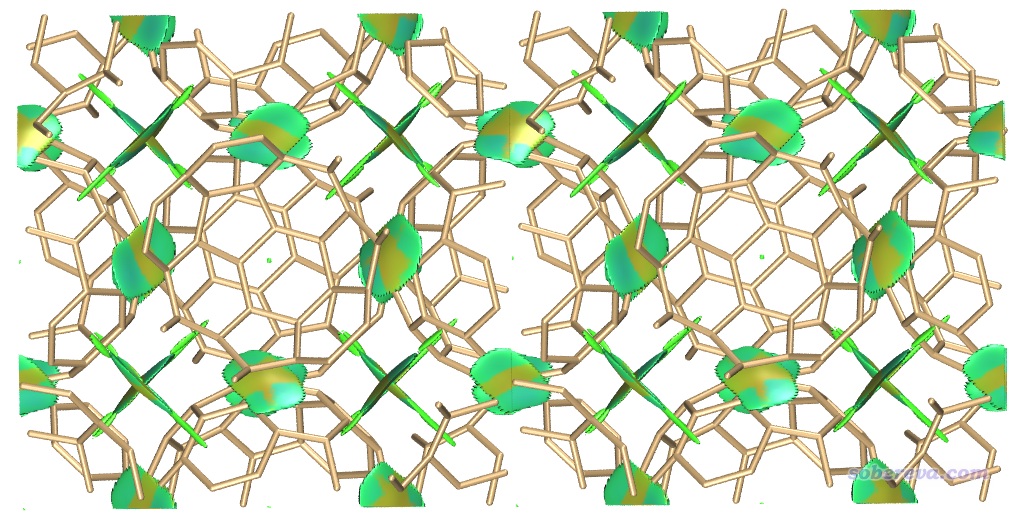
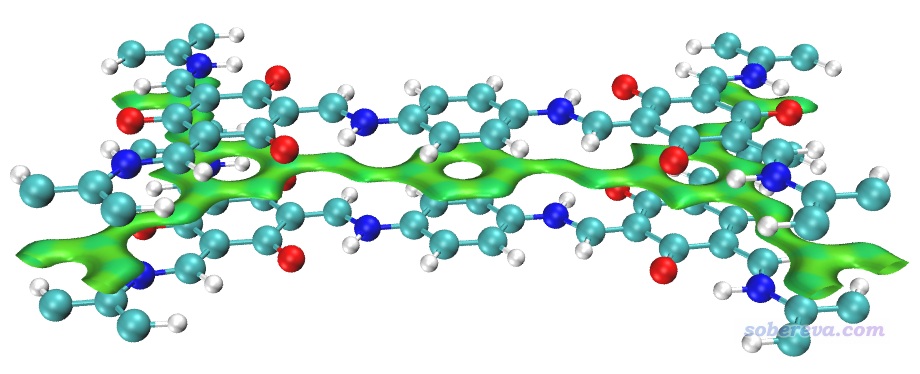
用Multiwfn載入COF_opt-MOS-1_4.molden,然后按照與3.1節完全相同的操作步驟讓Multiwfn做普通的NCI分析。使用0.2 Bohr格點間距時,在筆者的36核服務器上花了不到一分鐘算完。然后用前例說的RDGfill2.vmd腳本在VMD里對Multiwfn導出的func1.cub和func2.cub作圖,結果如下
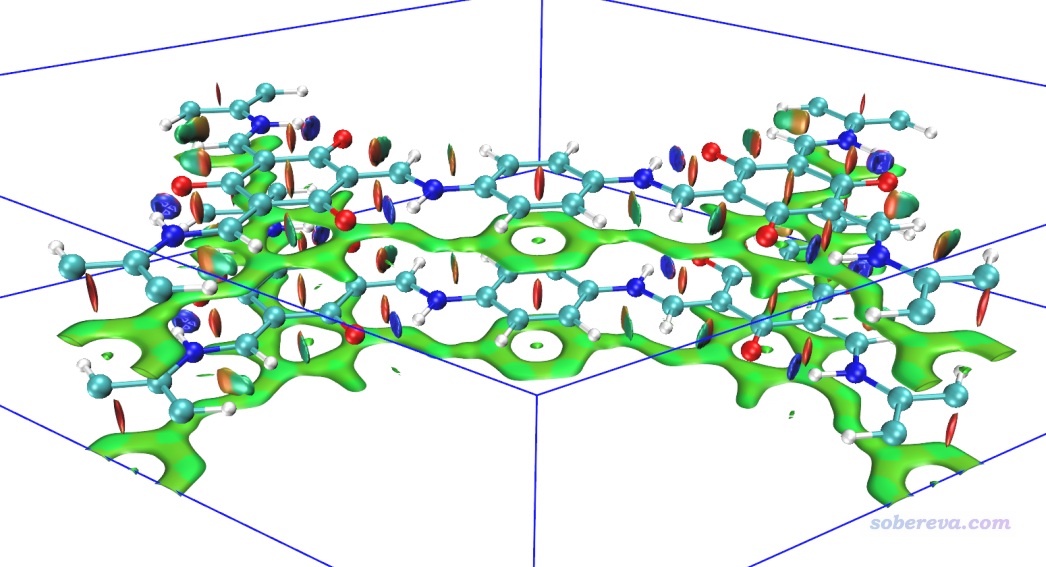
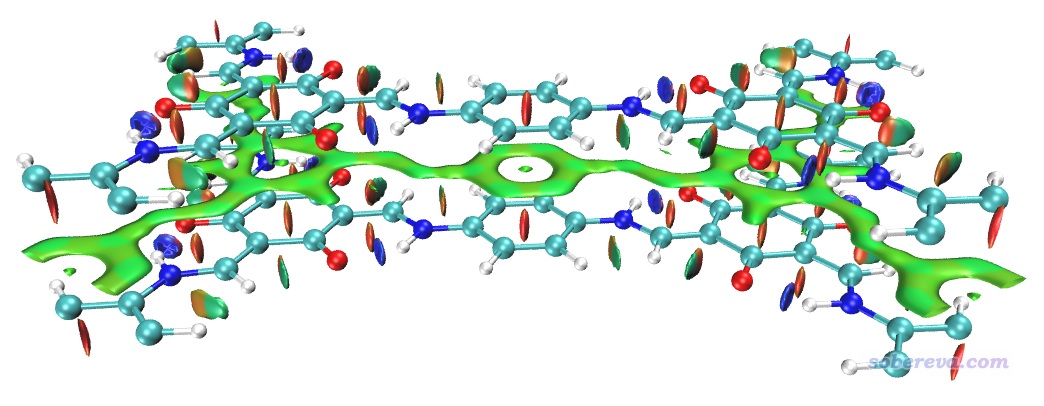
上圖非常清晰直觀地將COF晶胞中的兩層結構間的大面積pi-pi堆積展現了出來。苯環中央的紅色錐形等值面展現了環中的位阻作用。當前體系中強烈的N-H...O內氫鍵被非常藍的圓片型等值面表現了出來。
上圖一些等值面還不算特平滑,有的等值面邊緣鋸齒有點明顯,將格點間距降到0.15會明顯更好看,但耗時會多一倍多,每個cub文件增大到90兆。另外,上圖左側和右側的N-H...O內氫鍵的圓片型等值面中間凹陷了一些,像缺了一塊,這是因為這個內氫鍵很強,在作用區域有的地方電子密度超過了0.05 a.u.,而Multiwfn默認會把電子密度超過0.05 a.u.區域的RDG設為100來避免顯示出強作用區域(如化學鍵)的等值面。為解決這個問題,可以把Multiwfn目錄下的settings.ini中的RDG_maxrho從默認0.05設為改大到0.06 a.u.,這樣重新計算和繪制后就不會看到這個強內氫鍵的RDG等值面中間凹陷(被屏蔽掉)一塊了。
如果你想把上圖中下方的相鄰晶胞(-Z晶胞)的結構和RDG等值面也顯示出來,就進入Graphics - Representation界面,對每個Representation都進入Periodic標簽頁并勾選上-Z,即可看到下圖
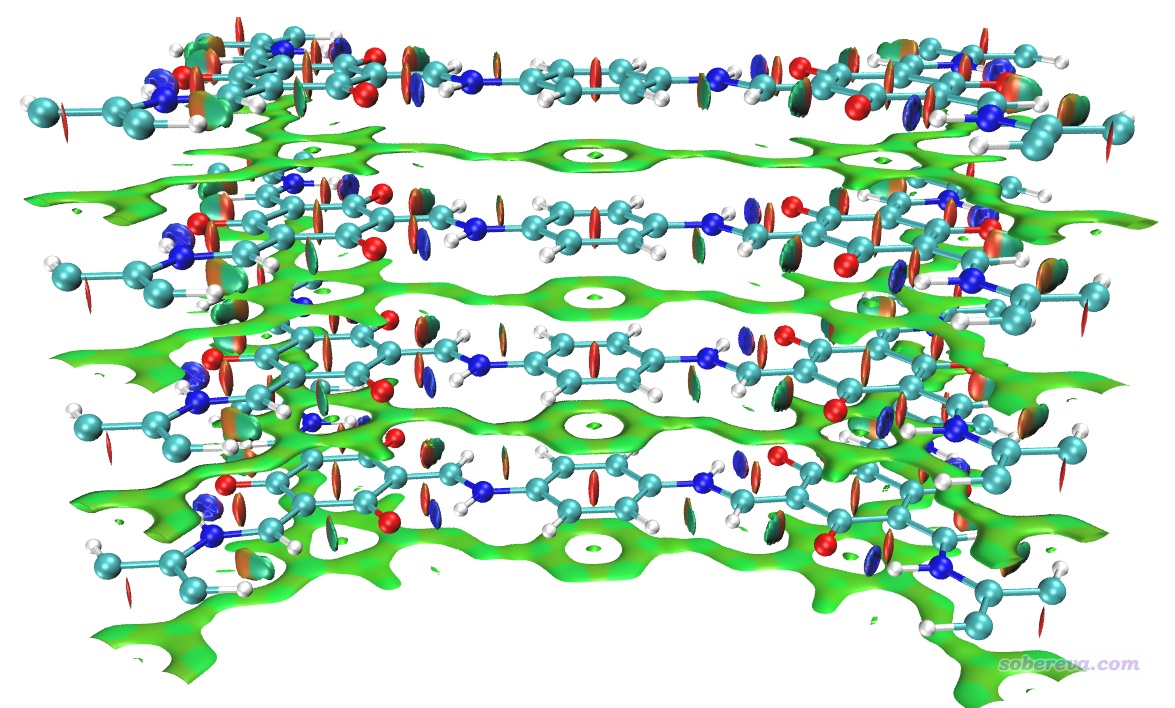
在之前不顯示相鄰鏡像的NCI圖中,我們看到在當前晶胞的底端也出現了大面積綠色等值面,這是這部分結構與-Z鏡像晶胞的上層部分的相互作用所導致的,看起來很礙眼,怎么避免顯示出來呢?在Multiwfn中有兩種做法可以實現:
(1)讓盒子起點位置的Z坐標稍微大于0,并讓盒子Z方向的尺寸適度小于晶胞的Z尺寸。具體來說,選擇做NCI分析,進入設置格點界面后,還是選擇模式9,然后輸入以下內容
0,0,1 //盒子原點的X、Y坐標都為0,Z坐標從1 Bohr開始,使得格點不會在晶胞最底層出現
0,0,10.85 //讓盒子的前兩個邊長等同于晶胞的前兩個平移矢量長度,而令盒子的Z尺寸比晶胞Z尺寸小2 Bohr,使得格點不會在晶胞頂端分布(注:晶胞尺寸在Multiwfn載入文件后屏幕上直接顯示了,Z尺寸是12.85 Bohr,減去2就是這里寫的10.85 Bohr)
0.2 //格點間距,同前
用這樣得到的cub文件在VMD里作圖如下所示,可見很理想,只保留了晶胞中間兩層結構間的層間和層內相互作用。
(2)另一種避免顯示與鏡像晶胞間的RDG等值面的方法是在Multiwfn計算時要求不考慮相應方向的周期性,我最推薦這種做法。比如對此例不想考慮Z方向的周期性,就把Multiwfn的settings.ini里的ifdoPBCxyz從原先的1,1,1改成1,1,0,這里0就代表第三個晶胞方向不考慮周期性(由于當前的晶胞第三個方向就是Z方向,所以等于不在Z方向考慮周期性)。這么改過之后照常計算和繪圖即可,格點還是按原先方式設置,結果和上圖完全相同。這種做法不僅方便,省得特意考慮格點分布范圍,而且由于忽略了一個方向的周期性,計算耗時還下降了百分之幾十,一舉兩得。
下面是對當前體系按照3.1.2節的過程繪制的準分子近似的NCI圖的結果,忽略了Z方向周期性。計算耗時極低,只花了1秒就算完了,圖像效果和普通NCI定性一致。
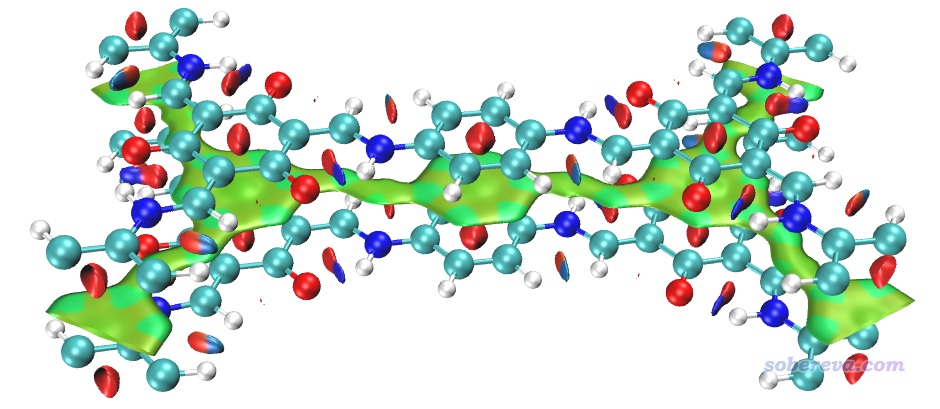
雖然準分子近似版NCI多數情況下和普通NCI定性一致,但是如果你對準確度要求比較高,比如考察氫鍵時想根據等值面藍色的深淺程度判斷強度,那么不要用準分子近似的版本,否則可能得到一些誤導性結論,畢竟用準分子近似相當于沒有考慮原子間相互作用引起的電子轉移、極化效應對NCI分析結果的影響。
3.3 氧化石墨烯與水的相互作用
在《使用Multiwfn非常便利地創建CP2K程序的輸入文件》(http://www.shanxitv.org/587)文中曾使用PBE-D3(BJ)/TZVP-MOLOPT-GTH對氧化石墨烯+一個水分子的表面吸附體系做了幾何優化,計算后得到了opt-1.restart文件,這其實是個CP2K的輸入文件,里面記錄了優化的最后一幀的信息。現在我們對這個結構做個NCI分析。首先基于這個文件創建一個PBE/6-311G**級別下的單點任務文件,用于得到molden文件。
啟動Multiwfn后輸入
opt-1.restart
cp2k
SP.inp
-2 //要求產生molden文件
2 //修改基組
11 //6-311G**
0 //產生輸入文件
用CP2K計算產生的SP.inp,算完后就有了SP-MOS-1_0.molden,然后把SP.inp里的晶胞信息手動寫入到這個molden文件作為[Cell]字段。
啟動Multiwfn,載入SP-MOS-1_0.molden。在計算前我們先進入主功能0看一下這個體系的結構特征,并且選圖形窗口菜單欄的Other settings里的Toggle showing cell frame把晶胞邊框顯示出來,
此時看到的圖像如下所示
做這個體系的NCI分析時要注意,由于石墨烯板緊貼著盒子最下方,因此如果直接將0,0,0作為盒子原點位置,屆時你會看到石墨烯里六元環中的位阻區域的RDG等值面被截斷,不好看。為避免這個問題,一個做法是用Multiwfn的主功能300的主功能7里的選項1先把整個體系往Z的正方向挪比如2 Bohr(約1埃),另一個辦法是定義格點的時候,讓盒子原點Z位置適當小于0,比如-2.0 Bohr。另外,當前體系水分子上方有大量的真空區,為了節約時間,可以讓Z方向盒子尺寸明顯小于晶胞的Z尺寸(設成其一半就足夠包圍感興趣的弱相互作用區域,可以節約一倍的計算時間)。下面計算時會考慮這兩點。
在當前圖形窗口的右上角點擊RETURN關閉窗口,然后輸入
20 //弱相互作用圖形化分析
1 //NCI分析
9 //借助晶胞信息定義格點
0,0,-2 //盒子原點位置(Bohr)
0,0,9 //當前晶胞Z方向尺寸是18.897 Bohr,令盒子Z尺寸取其一半,其它方向和晶胞尺寸相同
0.15 //格點間距
在筆者36核機子上13秒算完。算完后用選項3導出cub文件,和3.1.1節一樣在VMD里結合RDGfill2.vmd腳本繪制NCI圖,如下所示。為了讓大家了解當前盒子范圍,即格點數據計算的區域,在Graphics - Representation界面里我把對應Isosurface的那個representation里的Show下拉框從默認的Isosurface改為了Box+Isosurface。
從上圖可見,石墨烯每個環中央的位阻區域被清楚地通過紅色等值面展現了出來。水與氧橋之間有一塊淡藍色圓片型等值面,體現出氫鍵的存在,但顏色不算很深,說明不算很強的氫鍵。在水分子與碳的接觸區域還有一塊綠色的等值面,體現出水和氧化石墨烯之間還有明顯的范德華作用區域。上圖白色方框展現的是盒子范圍,可見盒子取得比較恰當,既沒有截斷任何等值面,也沒有明顯的浪費。由于當前體系與Z方向的鏡像距離比較遠,不會與其相互作用導致出現額外的RDG等值面,此例就沒有刻意把settings.ini里的ifdoPBCxyz像前例一樣改成1,1,0,但如果這么改一下的話倒是可以通過忽略Z方向周期性節約一些耗時。
3.4 C60晶體
此例考察一下C60富勒烯分子晶體中的弱相互作用,本文文件包里C60.cif是此體系的實驗結構。這個體系的晶胞里原子數很多,有240個,若用普通NCI分析耗時會較高,而且此體系主要是pi-pi堆積作用,這種作用靠準分子近似的NCI就可以合理地展現出來,所以此例就只用準分子近似的NCI了。
啟動Multiwfn,然后輸入
C60.cif
20
2 //準分子近似的NCI
9
[回車]
[回車]
0.15 //讓圖像較平滑,用較小的格點間距
計算完后導出cub文件并用RDGfill_pro.vmd在VMD里作圖,如下所示
這個圖像還不是很令人滿意,主要是紅色的表現位阻區域的等值面太多了,弄得圖像很凌亂,不便于觀察富勒烯之間的相互作用。為解決此問題,一方面可以用后文說的IGM,另一方面可以把sign(λ2)rho數值明顯大于0的區域手動屏蔽掉。具體做法是在Multiwfn做NCI分析的后處理菜單里選-1先把RDG vs sign(λ2)rho的散點圖繪制出來,如下所示
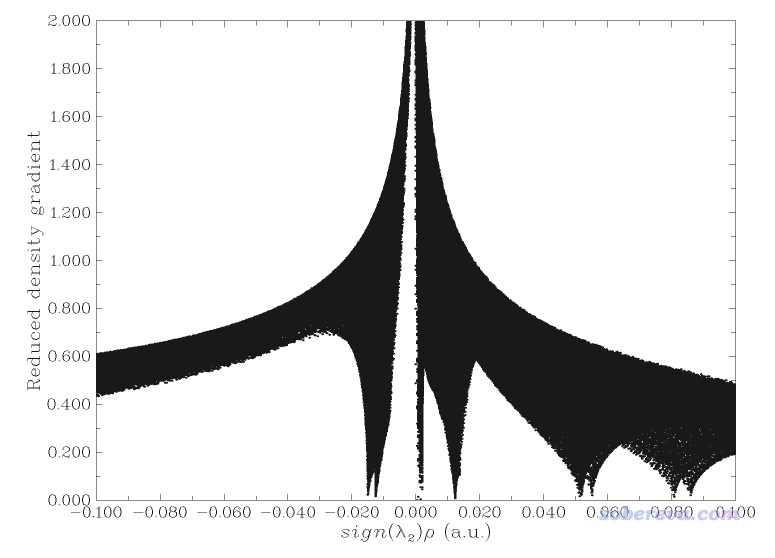
由圖可見在sign(λ2)rho>=0.05的區域有很多戳到底的spike,它們是導致分子內位阻作用的等值面出現的原因。為了屏蔽掉它們,在Multiwfn后處理菜單中選-2 Set RDG value where value of sign(λ2)rho is within a certain range,輸入0.02,999,然后輸入100。此時sign(λ2)rho數值大于0.02的區域的格點的RDG值就都被設為一個很大的數值100了,如果再重新繪制散點圖會看到下圖,可見sign(λ2)rho>0.02區域的spike都已經沒了。
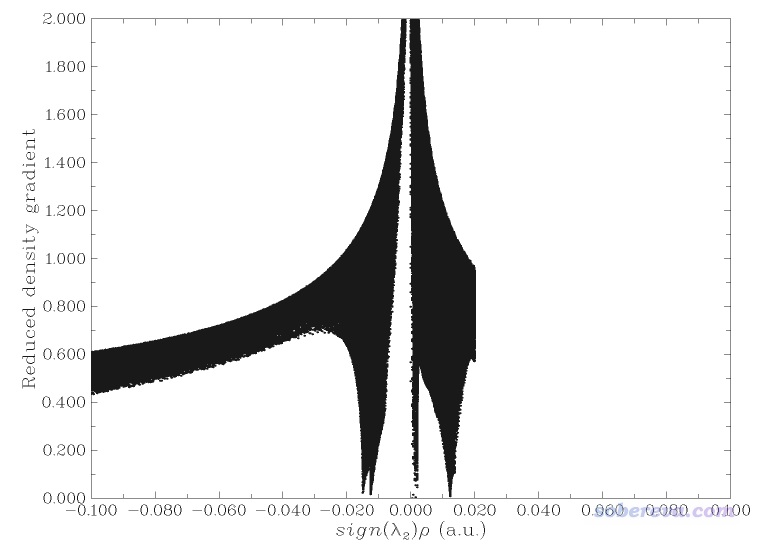
現在重新選擇導出cub文件,然后再次用RDGfill_pro.vmd在VMD里作圖。并且在VMD里把+X的鏡像也顯示出來便于考察。然后在VMD文本窗口里輸入
color Name C tan
color change rgb tan 0.700000 0.560000 0.360000
這會把碳的顏色改成黃褐色,因為原本的青色與等值面顏色的對比度不夠強烈。再在Graphics - Representation里把分子結構顯示方式改為Licorice,并把Bond Radius改為0.1,之后在VMD Main窗口里選Display - Orthographic改為正交視角。此時的圖像如下
可見當前的等值面清楚地把每個富勒烯與相鄰富勒烯之間的pi-pi堆積展現了出來。肯定有人注意到,上圖中央的富勒烯中間有些C-C鍵是斷開的,而且圖像中央的RDG等值面中間有個黑色的細縫。這是因為VMD沒法識別和顯示當前晶胞里的原子與相鄰鏡像晶胞里的原子間的鍵,而且等值面在跨盒子的地方不可避免地會顯示出個縫隙。如果想避免這倆問題讓圖像更完美,就只能在Multiwfn做這個準分子近似的NCI分析前,先用主功能300的子功能7的選項19把體系構造成2*1*1的超胞,然后再照常做NCI分析。此時計算耗時會明顯增高,cub文件大小也會增加一倍。
3.5 Ih晶型的冰
此例用普通NCI方法考察Ih晶型的冰中的弱相互作用。Ih是常壓下不是特別低溫的情況時最穩定的冰的晶型,也是日常生活中我們接觸到的冰的晶型。大家有興趣也可以類似地考察其它晶型的冰中的弱相互作用。
先產生優化冰晶胞中氫位置的CP2K輸入文件。啟動Multiwfn,載入本文文件包里的H2O-Ice-Ih.cif,然后輸入
cp2k
opt.inp
-2 //產生molden文件
2 //選擇基組
11 //6-311G**
3 //開啟色散校正
2 //DFT-D3(BJ)
-1 //設置任務類型
3 //優化結構但不優化晶胞
9 //設置凍結
optH //只優化氫
0 產生輸入文件
現在用CP2K進行計算,很快就算完了。把晶胞信息手動寫入對應優化好的結構的opt-MOS-1_5.molden文件里作為[Cell]字段。由于冰當中的氫鍵作用相對較強,為了避免氫鍵對應的RDG等值面被截斷,把RDG_maxrho設為0.07。然后照常做NCI分析。由于當前晶胞不大,用很小格點間距要算的格點數也不很多,因此為了圖像效果盡量精甚細膩,此例用了0.13 Bohr這樣很小的格點間距。
照常使用RDGfill2.vmd腳本在VMD里對Multiwfn做NCI分析后導出的cub文件作圖。為了讓冰當中的相互作用顯得比較完整,在Graphics - Representation界面里把顯示分子結構和顯示等值面的representation都設成+X、+Y、-Y、+Z、-Z鏡像都顯示,并且設置霧化效果使得遠處的物體被一定程度遮掩而避免擾亂視線。具體來說就是確保VMD Main的Display里的Depth cueing已經打開了,然后在Display - Display settings里把Cue Mode設成Linear,并把Cue Start和Cue End分別設為1.5和3.0。此時看到的圖像如下。
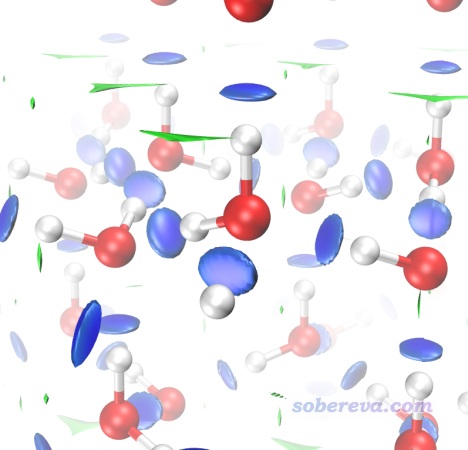
上圖非常清楚地展現出在冰當中,每個水與周圍四個水形成了氫鍵,而且RDG等值面非常藍,體現出氫鍵強度較大。零星的綠色等值面體現出冰當中還有一些主要算是范德華作用的區域,但可以忽略不計。
3.6 NaCl表面吸附CO
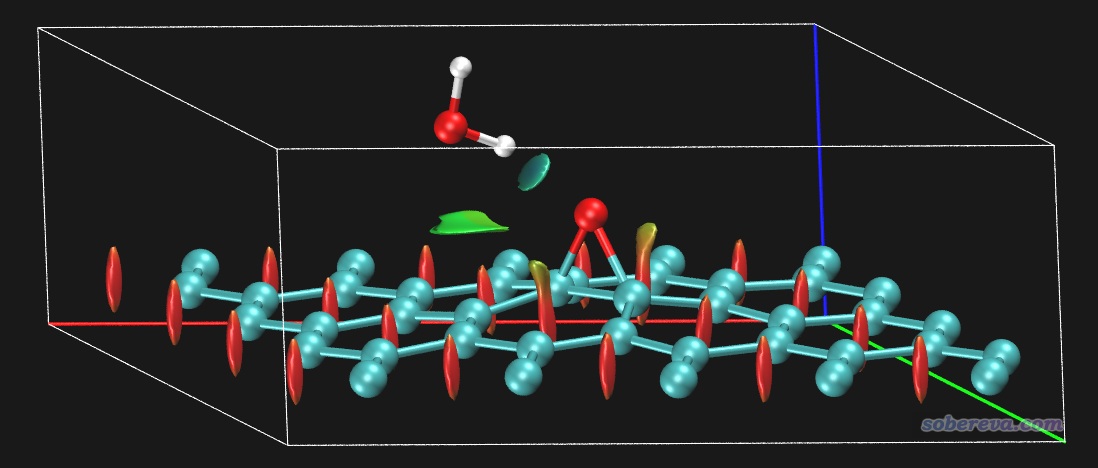
本例繪制NaCl表面吸附一氧化碳的NCI圖。根據諸多文章的研究,最穩定的吸附結構應當是一氧化碳的碳沖著Na+,此例也優化的是這種結構。用的是兩層厚度3*3的氯化鈉板,底層原子凍結,上層和CO都在優化過程中自由弛豫。先通過PBE-D3(BJ)/DZVP-MOLOPT-SR-GTH優化結構,然后PBE/6-31G*算單點得到波函數文件。此模型可以用GaussView很容易地搭建,保存成gjf并載入后通過Multiwfn很容易地就可以創建相應任務的CP2K輸入文件,相關過程這里就不累述了。單點計算后得到的SP-MOS-1_0.molden在本文文件包里提供了。之后對此體系做普通NCI分析,用0.15 Bohr格點間距,圖像如下所示。此體系計算耗時很高,為了節約時間,應恰當設置盒子Z方向尺寸,避免讓格點分布在根本沒原子出現的真空區。
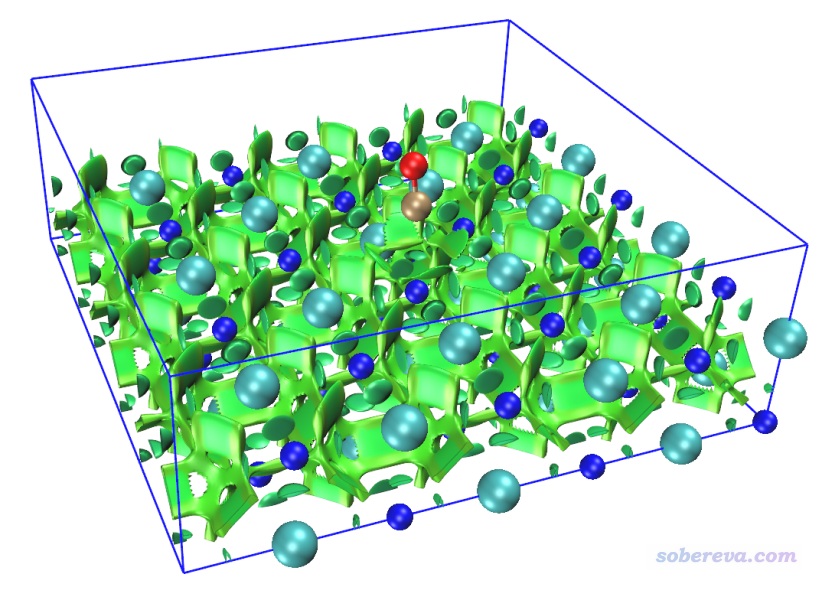
為了看得清楚,俯視圖和側視圖也在下面給出
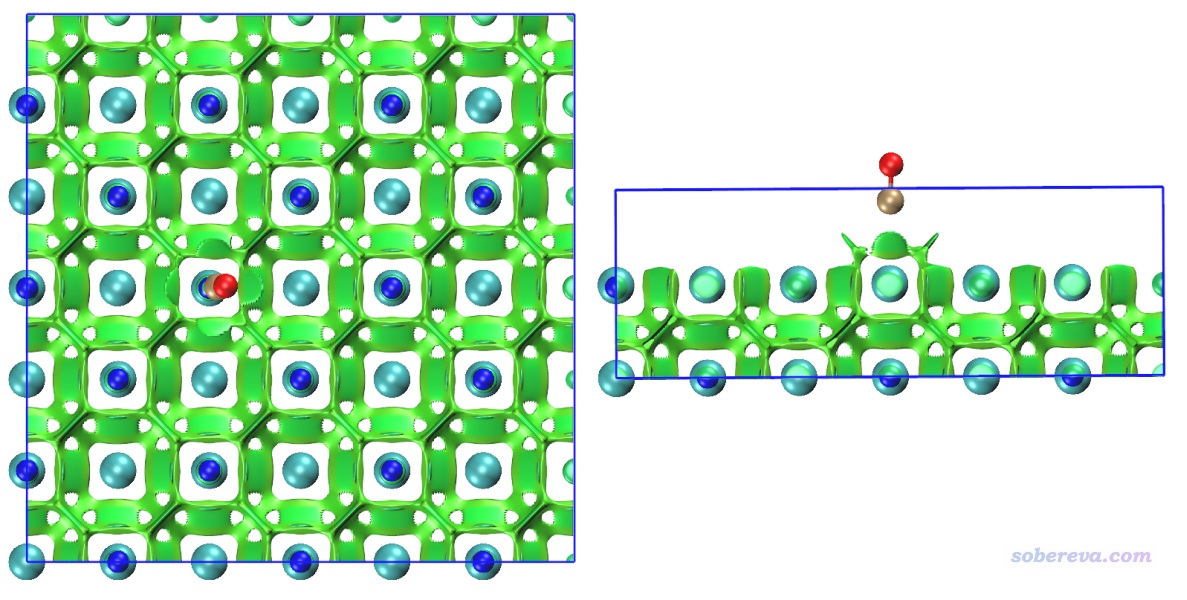
圖中青色圓球是Cl-,藍球是Na+,褐球是碳,紅球是氧,藍色方框展現的是計算的格點數據的分布范圍。從此圖可以清楚地看到一氧化碳的碳與Na+有明顯的相互作用,與旁邊的四個Cl-也有明顯作用,并且Na+和Cl-之間也有顯著相互作用。Na+帶顯著正電,它與碳的鍵軸末端的孤對電子明顯會形成顯著的靜電主導的吸引作用(文獻中的吸附能約為20 kJ/mol,不算小了),且Na+和Cl-之間毫無疑問是挺強的離子鍵,為何RDG等值面全都是綠色而不是藍色?這是因為鈉的半徑較大,而且價層孤立狀態下就只有一個電子,在當前體系中價電子就更少了,故在它與其它原子形成相互作用的區域必定電子密度(rho)比較低,所以sign(λ2)rho數量級很小,故著色是綠色。記住切勿盲目用RDG等值面顏色衡量作用強度,要正確理解NCI方法的本質和局限性。
上面的圖有個明顯的不足之處就是Na+和Cl-之間作用的等值面嚴重妨礙了觀看氯化鈉和一氧化碳之間的相互作用。既可以使用后文所述的IGM方法避免此問題,也可以使用Multiwfn的格點數據處理功能(主功能13)中的子功能14解決,它可以用來將NaCl板與一氧化碳交疊區域以外的區域的格點的RDG設成一個特定值,比如100,從而達到屏蔽NaCl內部相互作用的目的。下面就來這么屏蔽一下。NCI分析后導出的func2.cub是RDG的格點數據文件,將之載入Multiwfn,然后輸入
13 //主功能13
14 //設置兩個片段交疊區域以外區域格點的數值
1.3 //原子球半徑設為范德華半徑乘以1.3(片段占據的區域是片段內所有原子球的疊加)
100 //交疊區域以外的格點數值設為100
2 //手動輸入兩個片段里的原子序號
1-72 //片段1的原子序號,即NaCl板中的原子
73,74 //片段2的原子序號,即CO的原子
0 //將當前的格點數據導出為cub文件
[按回車] //格點數據導出為當前目錄下的func2.cub
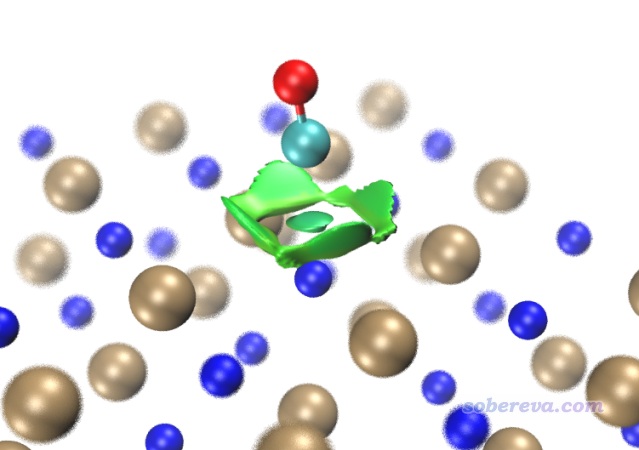
現在,用新得到的func2.cub替換原先的func2.cub,再用RDGfill2.vmd腳本作圖,得到如下圖像,可見非常理想,確實NaCl內部的等值面都被屏蔽掉了。為了效果更好,作圖時筆者還設了景深霧化,并且把RDG等值面數值從默認的0.5稍微設大到了0.6,這樣等值面更膨脹一些,邊緣鋸齒看起來也更小一點。
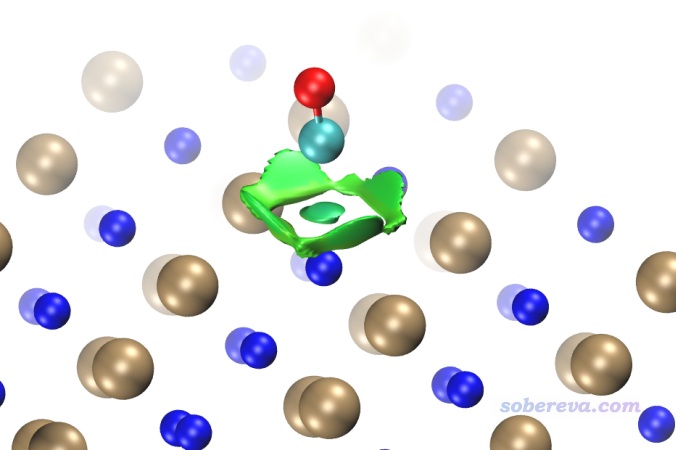
順帶一提,還可以在VMD中使用焦距虛化效果,如下所示,這樣層次更清楚。也可以通過把原子球調大來提升層次感。
像上面這樣只需要對一小部分區域作NCI圖的情況,在Multiwfn里設置計算的格點范圍的時候可以只讓盒子框住感興趣的區域(即感興趣的等值面可能出現的區域),耗時能節約非常多。這通過恰當設置盒子原點和邊長即可實現,也可以在格點設置界面里通過選項10 Set box of grid data visually using a GUI window在圖形窗口里直觀設置盒子的位置和尺寸,正如此文2.6節所示的情況:《使用量子化學程序基于簇模型計算金屬表面吸附問題》(http://www.shanxitv.org/540)。
4 周期性體系的IGM分析
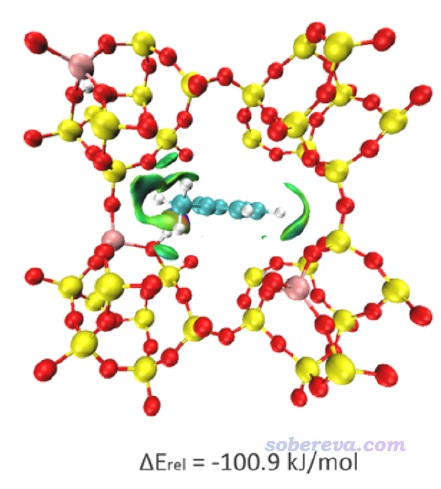
IGM分析只需要原子坐標。如果體系雖然是周期性的,但是被考察的區域與晶胞邊界有一定距離,那么完全可以按照《通過獨立梯度模型(IGM)考察分子間弱相互作用》(http://www.shanxitv.org/407)所述的當成孤立體系考察。比如Mater. Today Commun., 26, 102028 (2021)一文中,在筆者的建議下作者對他們研究的MOF吸附芳香化合物的問題做了IGM分析,如下所示,可見很好地展現出了MOF對分子的吸附時主要的作用區域。像這種情況就當成孤立體系計算做IGM計算就可以,考慮周期性的話耗時會明顯更高。
但也有一些情況IGM等值面要涉及到體系邊緣,這種情況就必須當成周期性做IGM分析了,否則盒子邊緣處的等值面就不正常了,比如3.2節的COF體系就是如此。這里我們就用IGM方法對COF這個體系做一下分析,如果沒讀過《通過獨立梯度模型(IGM)考察分子間弱相互作用》一文的話請仔細閱讀,基本知識本文不再累述。為了不顯示出中心晶胞和相鄰晶胞的相互作用對應的IGM等值面,還是把settings.ini里的ifdoPBCxyz設成1,1,0忽略掉Z方向的周期性。
啟動Multiwfn,然后輸入
COF_opt-1.restart //本文文件包里優化COF中氫原子位置任務產生的記錄最終結構的restart文件
20 //圖形化分析弱相互作用
10 //IGM
2 //定義兩個片段
1-72 //第一個片段的原子序號,即兩層COF其中一層中的原子
73-144 //第二個片段的原子序號,即另一層COF中的原子
9 //借助晶胞信息設置格點
[按回車] //用0,0,0作為盒子原點
[按回車] //用晶胞邊長作為盒子邊長
0.4 //格點間距。IGM等值面不容易像RDG的那么容易有鋸齒,所以可以用明顯大得多的格點間距節約時間
本身IGM分析就很快,而且格點間距又大,在普通4核機子上不到半分鐘就算完了。之后選3導出格點數據,在當前目錄下就有了dg_inter.cub、dg_intra.cub、dg.cub和sl2r.cub,將它們都挪到VMD目錄下。并且把Multiwfn目錄下的examples文件夾中的IGM繪圖腳本IGM_inter.vmd和IGM_intra.vmd都復制到VMD目錄下。
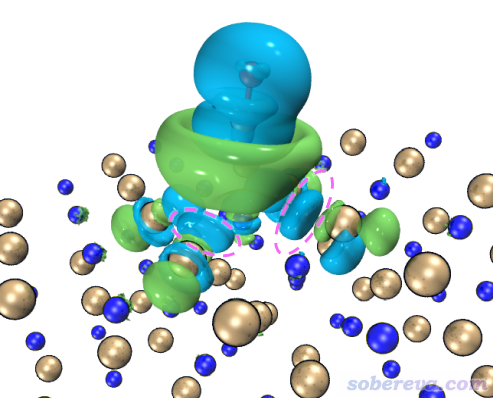
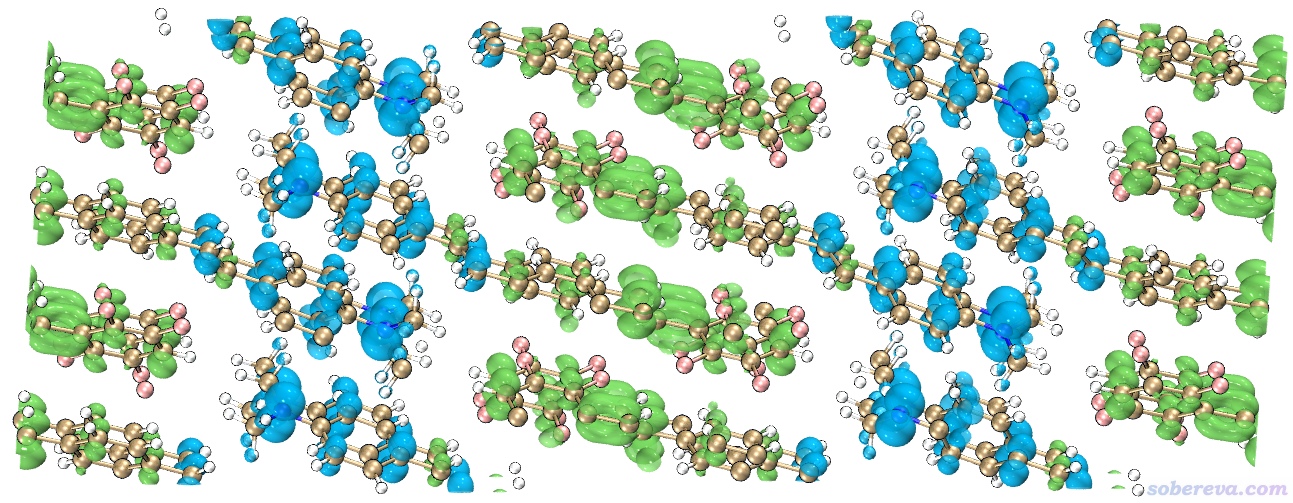
啟動VMD,然后在命令行窗口輸入source IGM_inter.vmd,這會繪制出IGM方法中定義的sign(λ2)rho著色的δg_inter函數的等值面。但是直接顯示出來的等值面非常窄,不能充分展現出兩層MOF間的pi-pi堆積,這是因為默認的δg_inter等值面數值偏大。因此進入Graphics - Representation界面,把Isovalue改成更小的0.005,此時圖像如下所示。可以看到此圖非常清楚地展現出了層間pi-pi堆積作用,而且片段內的相互作用不像NCI那樣被顯示出來,同時等值面很平滑,可見Multiwfn做IGM分析又簡單又快效果又非常好。IGM分析還有一個好處是可以把各個原子的貢獻給出來,并可以由此給原子球著色凸顯較重要的原子,詳見前述的《通過獨立梯度模型(IGM)考察分子間弱相互作用》。
如果在VMD里輸入source IGM_intra.vmd,可以把COF層內的相互作用,包括化學鍵作用和內氫鍵都展現出來(展現后者需要調小isovalue)。由于這不是此例我們感興趣的,就不說了。
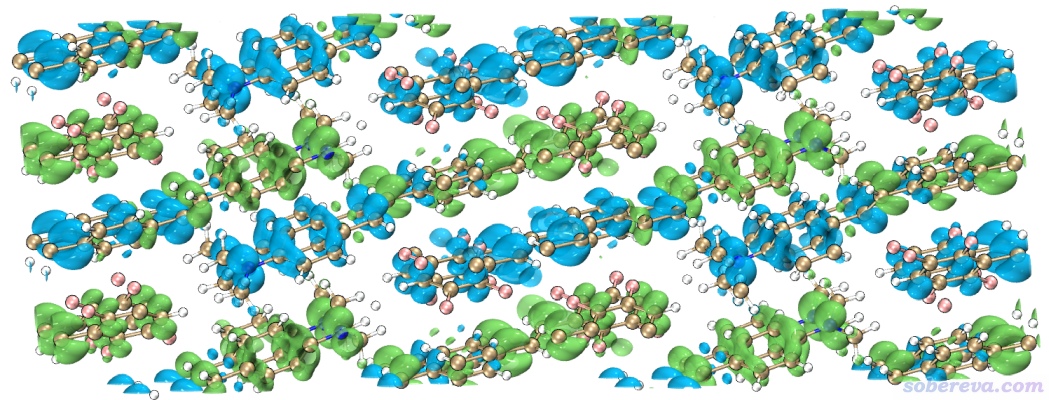
下面對本文3.6節考察過的NaCl吸附CO的體系做IGM分析,將NaCl和CO分別作為兩個片段,操作方式同上。下圖左側和右側對應0.005和0.003的δg_inter等值面。
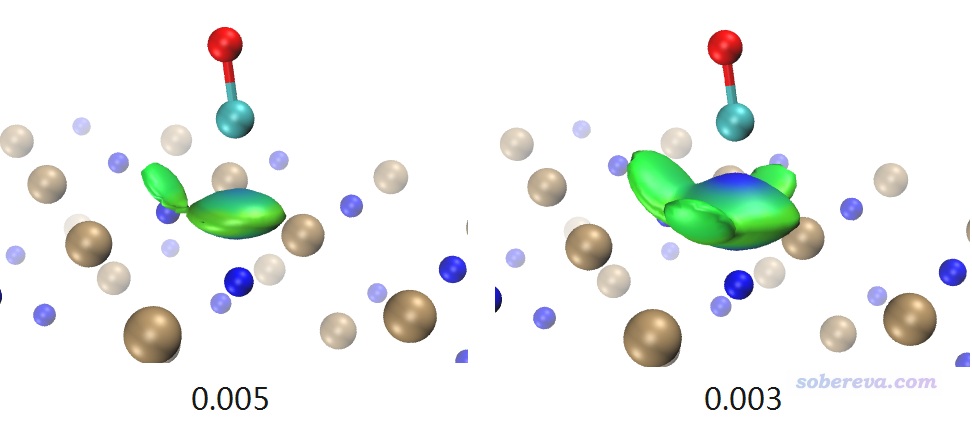
δg_inter數值取得越小,其等值面就可以把越弱的相互作用展現出來。如上所示,在0.005等值面的時候,C和Na+的相互作用區域很明顯,但C和周圍Cl-的范德華作用對應的等值面只顯示出了CO稍微歪過去方向的那一小塊。而將等值面數值降到0.003后,更多相互作用區域被等值面展現了出來,并且此時C和Na+接觸的那部分等值面中央顏色已經很藍了。為什么此時顏色這么藍而不像RDG等值面那樣是綠色的?這是因為此時的δg_inter等值面特別肥厚,已經延展到了比較靠近C的部分,此處電子密度已經不是很小了,sign(λ2)rho可以達到負零點零幾了,因此對應的著色明顯發藍。
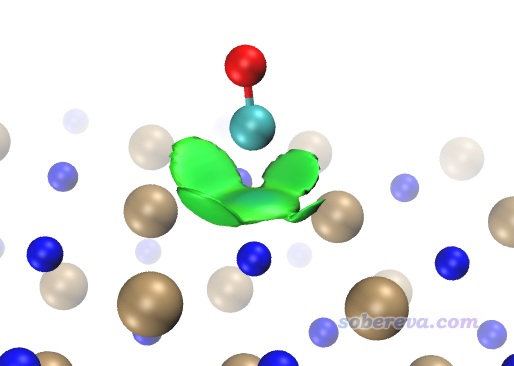
IGM等值面比較肥厚是IGM方法的十分明顯的弊端,而且上面的顏色往往還會誤導研究者,在筆者2022年提出的IGMH方法中被完美解決,而且IGMH比IGM原理更嚴格。讀者務必看《使用Multiwfn做IGMH分析非常清晰直觀地展現化學體系中的相互作用》(http://www.shanxitv.org/621),里面有IGMH非常全面的介紹和與IGM的對比。在Multiwfn中的使用上,IGMH和IGM的區別僅僅在于主功能20里應選擇11而非10,并且需要載入和NCI分析一樣的含有波函數信息的輸入文件。IGMH由于是基于波函數的分析,故耗時顯著高于IGM,而且和NCI分析一樣格點間距不宜太大以避免鋸齒。對當前體系,為節約耗時,建議設置格點時讓盒子僅框住感興趣的作用區域,并且把ifdoPBCxyz設為0,0,0當孤立體系處理(因為CO與NaCl作用的位置離盒子邊緣很遠,這么做不會有不良影響)。基于IGMH方法繪制的δg_inter為0.002的等值面圖如下所示,可見等值面和RDG的等值面很相似,都比較薄,而且和IGM一樣都完全避免了NaCl板內的等值面妨礙考察CO的吸附,圖像效果極度理想。我建議凡是能得到波函數的情況,都應當用IGMH代替IGM。
5 總結
本文詳細介紹了如何使用Multiwfn基于CP2K第一性原理程序產生的波函數做NCI分析,還介紹了怎么對周期性體系做IGM分析,還對筆者提出的比IGM強得多的IGMH分析也做了簡要示例。Multiwfn靈活方便的創建CP2K輸入文件的功能,結合Multiwfn非常簡單易用的NCI/IRI/IGM/IGMH分析功能,真是使得固體和表面的弱相互作用圖形化分析異乎尋常地簡單。研究周期性體系中的弱相互作用不要再只是算算結構、算算能量、只靠數據討論了,結合本文介紹的圖形化分析方法可以令文章大為增光添彩、明顯更吸引眼球,令枯燥抽象的分析討論變得直觀生動傳神。
Multiwfn對周期性體系的分析絕不限于本文所介紹的。目前的版本已可以對周期性體系計算原子電荷、計算鍵級、做AIM拓撲分析、計算和繪制上百種實空間函數(如ELF、LOL、IRI、電子密度拉普拉斯、能量密度、自旋密度等等)、做范德華勢分析(《談談范德華勢以及在Multiwfn中的計算、分析和繪制》http://www.shanxitv.org/551),等等。Multiwfn對周期性體系的支持詳情看Multiwfn手冊2.9節的說明。筆者在未來會陸續寫更多的Multiwfn對周期性體系波函數分析的相關教程。讀者也別忘了看《使用IRI方法圖形化考察化學體系中的化學鍵和弱相互作用》(http://www.shanxitv.org/598),其中有IRI研究周期性體系的介紹,比NCI分析能展現更豐富的信息,建議代替NCI。
順帶一提,如果你之前不會用CP2K的話,也完全不用擔心什么。如本文所示,靠Multiwfn可以非常簡單地創建本來特別繁瑣復雜的CP2K輸入文件,而且如前文提到的我的CP2K安裝教程所寫的,用CP2K預編譯版都完全不需要自己編譯,下載就能用。再加上CP2K又快又免費,結合Multiwfn做周期性體系的波函數分析簡直不能更好用。所以即便你以前用的是Quantum ESPRESSO、CASTEP等其它第一性原理程序,也可以毫無壓力地過度到Multiwfn+CP2K的分析組合上。
]]>文/Sobereva@北京科音
First release: 2021-Feb-22 Last update: 2023-Nov-7
1 前言
CP2K(https://www.cp2k.org)是速度超級快,功能又非常全面的第一性原理程序,受到越來越多的重視,有很多無可替代的重要價值。CP2K的一個問題是輸入文件寫起來極其繁瑣,一個簡單的任務的輸入文件就要動輒上百行。而且有時用戶還需要了解很多算法和數值實現細節、查閱繁復的手冊,甚至還要google半天、參考不少自帶測試文件才能最終寫出能用的輸入文件。這些問題使得CP2K這個優秀的程序不容易普及開來,初學者往往看到其極度復雜的輸入文件就打退堂鼓了。
筆者的Multiwfn程序(主頁&下載:http://www.shanxitv.org/multiwfn)的主功能100的子功能2能產生大量量子化學程序的輸入文件,諸如其中產生ORCA輸入文件的功能在此文專門介紹過《詳談Multiwfn產生ORCA量子化學程序的輸入文件的功能》(http://www.shanxitv.org/490),給很多人帶來了極大便利。為了讓CP2K這個利器對于常見計算問題用起來盡可能方便、盡可能把手動擋變得像自動擋、令使用門檻盡可能低,使之能更充分地發揮其巨大價值,筆者在Multiwfn里加入了產生CP2K輸入文件的功能,在此文將進行簡要介紹。這個功能雖然注定不可能讓完全零基礎的CP2K用戶毫無壓力地使用CP2K,但至少已經把使用難度和復雜度降到極限了,用戶只需要選擇干什么、怎么干,即可產生一個完成度>95%的輸入文件,之后一些必須根據實際情況調整的地方,比如平面波截斷能大小等,再自行改一下即可,這使得CP2K用戶得到了極大的解放!
如果Multiwfn的這個功能對你的研究產生了實際幫助,請在寫文章時順帶提及輸入文件是借助Multiwfn產生的(比如寫the CP2K input files were generated with help of Multiwfn program),并按程序啟動時顯示的要求恰當引用Multiwfn。
Multiwfn產生CP2K輸入文件的流程是:
1 啟動Multiwfn,載入一個Multiwfn可以識別的至少含有結構信息的文件
2 進入主功能100的子功能2,選擇產生CP2K輸入文件(即選項25)。也可以直接在Multiwfn主菜單里輸入cp2k進入此功能
3 輸入要產生的CP2K輸入文件的路徑
4 通過各種選項設置如何進行計算
5 選擇0,得到CP2K輸入文件
下面第2節先說載入到Multiwfn的輸入文件的要求。第3節給出一些例子演示產生CP2K輸入文件的過程。Multiwfn的這個功能始終不斷打磨并且迎合CP2K最新版本的改變,本文內容對應Multiwfn官網上最新版本的Multiwfn(不是今天剛下載的就不要覺得是最新的),一定不要用老版本!Multiwfn產生CP2K輸入文件的功能在未來還會不斷擴展,盡量做到更強大更完美,并且盡量迎合未來版本CP2K發生的各種變化,屆時本文也會相應地修改。
一定要注意,盡管靠Multiwfn創建CP2K輸入文件非常方便,也是絕對絕對不能把CP2K當黑箱用的!!!因為真正正確、準確、高效率地使用CP2K做計算需要掌握的理論背景知識真是超級多。“北京科音CP2K第一性原理計算培訓班”是從頭一次性系統、完整學習第一性原理計算和CP2K程序的最好途徑,是CP2K用戶絕對不應錯過的培訓,介紹見http://www.keinsci.com/workshop/KFP_content.html。此培訓深入淺出,能非常全面學習到第一性原理計算各種問題的背景知識、深入透徹掌握CP2K,比起自己在網上找零七八碎的往往還是有錯誤或過時的資料學習能少走無數彎路。培訓內容涵蓋面極廣,ppt多達兩千多頁,對不同主題都給了精心準備的大量例子使得學員能舉一反三。其中講解利用CP2K做各類問題的研究時都會使用Multiwfn創建輸入文件,學員從中會深切體會到Multiwfn對于CP2K的使用起到的重要價值。
2 給Multiwfn用的輸入文件
Multiwfn支持什么輸入文件這里都詳細說了:《詳談Multiwfn支持的輸入文件類型、產生方法以及相互轉換》(http://www.shanxitv.org/379)。對于產生CP2K的輸入文件,顯然載入到Multiwfn的文件中必須含有體系的結構才行。對于此類目的,Multiwfn支持一大波格式,如pdb/gro/xyz/mol/mol2/mwfn/wfn/wfx/molden/fch/cub/gjf/gro等等等等。
如果載入到Multiwfn的文件不包含晶胞信息,那么Multiwfn會默認將此體系視為孤立體系來設置CP2K里的計算參數,CP2K輸入文件里的晶胞信息是在分子周圍做一定距離的延展來得到的。
多數情況CP2K都是拿來算周期性體系的。若想讓Multiwfn產生的CP2K輸入文件里有實際的晶胞信息,載入到Multiwfn的文件里就必須有晶胞信息。用以下格式作為輸入文件時,Multiwfn會既讀取原子坐標也讀取晶胞信息。
(1)cif文件
(2)含有CRYST1字段的pdb或pqr文件
(3)CP2K的輸入文件(必須以.inp或.restart為后綴)
(4)GROMACS的gro文件
(5)含有@<TRIPOS>CRYSIN字段的mol2文件
(6)含有Ndim且其數值>0的mwfn文件(mwfn是Multiwfn私有的波函數文件格式,專門留出了字段記錄晶胞信息,見mwfn介紹文章https://doi.org/10.26434/chemrxiv.11872524)
(7)帶有Tv(平移矢量)信息的Gaussian輸入文件,即Gaussian做PBC任務的輸入文件,可以通過GaussView直接創建
(8)Gaussian做PBC任務產生的fch/fchk文件
(9)含有晶胞信息的xyz文件。這是我自己定義的,如果xyz文件的注釋行(第二行)包含諸如以下形式的內容(以埃為單位的平移矢量),就會被Multiwfn讀取
Tv_1: 7.426 0.0 0.0 Tv_2: 3.6 6 6.40 0.0 Tv_3: 0.0 0.0 10.0
也可以按照extended xyz格式的方式在xyz文件的第二行記錄晶胞信息,例如:
Lattice="7.426 0.0 0.0 -3.66 6.40 0.0 0.0 0.0 10.0"
(10)含有[Cell]字段的molden文件。此字段是我對molden格式做的擴展,這使得記錄波函數的molden文件也能記錄晶胞信息。大家可以直接手動往已有的molden文件里插入[Cell]信息,詳見《詳談使用CP2K產生給Multiwfn用的molden格式的波函數文件》(http://www.shanxitv.org/651)
(11)VASP的POSCAR、CHGCAR、CHG、ELFCAR、LOCPOT文件。文件名里必須包含相應字樣才能識別成相應格式,例如要作為POSCAR載入,文件名可以是POSCAR_miku或miku.POSCAR
(12)Quantum ESPRESSO的輸入文件
3 產生CP2K輸入文件的一些例子
下面就通過不同體系、不同類型計算演示一下Multiwfn產生CP2K輸入文件的功能,這些例子也可以作為CP2K入門例子。這些例子只用到了一部分Multiwfn提供的選項,其它的請自行嘗試,選項的含義都非常易于理解所以就不一一說了。下文用到的文件都在http://www.shanxitv.org/attach/587/file.zip中提供了。筆者假定讀者已經以正當方式安裝了CP2K,安裝方法見《CP2K第一性原理程序在CentOS中的簡易安裝方法》(http://www.shanxitv.org/586)。由于本文只是舉例,為了省計算時間,有些任務的k點取得比實際研究時應取的明顯要少。
3.1 做COF(共價有機框架)化合物的單點任務
本文文件包里COF_12000N2.cif是個COF的晶體結構文件,本例用Multiwfn對它產生一個最簡單的任務的輸入文件,即PBE泛函結合DZVP-MOLOPT-SR-GTH基組做單點計算。
啟動Multiwfn,然后依次輸入
COF_12000N2.cif //寫實際的路徑。也可以把此文件拖到Multiwfn文本窗口里自動產生路徑,省得手動輸入了
cp2k //進入產生CP2K輸入文件的功能
[按回車] //新產生的CP2K輸入文件將為當前目錄下的COF_12000N2.inp。也可以自己輸入路徑
0 //直接生成輸入文件
現在COF_12000N2.inp就在當前目錄下出現了,可以直接用CP2K跑了。這樣在默認設置下產生的輸入文件對應PBE/DZVP-MOLOPT-SR-GTH算單點,只考慮gamma點。可以打開.inp看一下有哪些自己需要改的,一般來說自己根據實際需要改的也就是影響精度的設置,比如影響平面波部分計算精度的&MGRID里的CUTOFF和REL_CUTOFF、控制密度矩陣收斂限的&SCF里的EPS_SCF,這些問題本文就不多說了。
Multiwfn產生的CP2K輸入文件里面有很多注釋,即#后面的內容,這有助于對CP2K還不是特別熟的人了解選項的含義、明白怎么設置。輸入文件里有些選項的值是根據我的使用經驗和習慣設的,也有一些設置雖然是默認的,卻也出現在了輸入文件里,這個考慮是便于大家之后自己手動修改(要不然想改一個默認設置的話還得照著手冊寫一堆字段實在太費事了)。
下面再用COF舉個例子,使用B97M-rV泛函結合6-31G*基組做GAPW形式的單點計算,并且要求算完后產生記錄此體系波函數的.molden文件,可用于之后在Multiwfn中做波函數分析,見比如《使用IRI方法圖形化考察化學體系中的化學鍵和弱相互作用》(http://www.shanxitv.org/598)和《使用Multiwfn結合CP2K通過NCI和IGM方法圖形化考察固體和表面的弱相互作用》(http://www.shanxitv.org/588)。關于molden文件更多信息見《詳談使用CP2K產生給Multiwfn用的molden格式的波函數文件》(http://www.shanxitv.org/651)。本例用較小的基組僅作為演示目的,對精度有要求的計算至少用6-311G**。值得一提的是,在純泛函中,B97M-rV是對有機體系能量相關問題計算得最好之一,并且這個泛函自帶rVV10色散校正。
啟動Multiwfn后依次輸入
COF_12000N2.cif
cp2k
COF_GAPW.inp
1 //設置理論方法
11 //B97M-rV(通過libxc庫實現)
2 //設置基組
10 //6-31G*
-2 //要求產生.molden文件
0 //產生輸入文件
現在當前目錄下就有了COF_GAPW.inp,直接就可以跑了。跑完之后當前目錄下會出現COF_GAPW-MOS-1_0.molden。
順帶一提,在Multiwfn的主功能0里可以直接看體系的結構和晶胞,由此可以確認一下結構和晶胞信息有沒有被Multiwfn從輸入的文件里正常載入。啟動Multiwfn并載入COF_12000N2.cif后,選0,就會進入圖形界面。關閉標簽和坐標軸,并且點擊菜單欄的Other settings里的Toggle showing cell frame,當前窗口將為下圖所示的狀態。可見原子位置和晶胞都正確。點擊Save picture按鈕可以在當前目錄下得到線條明顯更平滑的大尺寸圖像文件。
也可以用Multiwfn載入CP2K的.inp輸入文件或者CP2K計算過程中產生的.restart文件,在主功能0里可以看其中的結構是什么樣。還可以載入它們后,進入主功能100的子功能2,選擇導出gjf文件,這樣得到的gjf文件里會有Tv描述的晶胞信息,可以放到GaussView里進行觀看和編輯修改。還可以用主功能100的子功能2里的一堆選項轉換成其它一大堆常見格式、一大堆計算程序的輸入文件(包括Quantum ESPRESSO、VASP的POSCAR),賊靈活。
3.2 對SiC超胞做結構優化
這一節演示對SiC的3*3*3超胞用PBE/DZVP-MOLOPT-SR-GTH做晶胞參數不變的結構優化,并且開OT。
啟動Multiwfn后輸入
SiC.cif //在本文文件包里
cp2k
[按回車] //產生的輸入文件名為默認的SiC.inp
-1 //選擇任務
3 //結構優化(不變胞)
-11 //進入幾何操作界面。這個界面有豐富的功能,見《Multiwfn中非常實用的幾何操作和坐標變換功能介紹》(http://www.shanxitv.org/610)
19 //構建超胞
3 //方向1為原先的3倍
3 //方向2為原先的3倍
3 //方向3為原先的3倍
如果你想看一下當前結構的話,可以選0進入圖形界面,會看到下圖,可見結構沒問題
接著輸入
-10 //返回到之前創建CP2K輸入文件的界面
4 //開啟OT
0 //產生輸入文件
現在當前目錄下的SiC.inp就可以直接跑了。
實際上,還有一種等價的創建SiC 2*2*2超胞輸入文件的做法。進入Multiwfn的CP2K輸入文件創建界面后,輸入
4 //開啟OT
-1 //選擇任務
3 //結構優化(不變胞)
-9 //其它設置
3 //設置三個方向晶胞重復次數
3,3,3
0 //返回
0 //產生輸入文件
在得到的輸入文件中可以看到坐標部分只記錄了原胞,但有兩處MULTIPLE_UNIT_CELL 3 3 3,體現出是對3*3*3超胞進行計算。
3.3 優化Cu的晶胞
這一節創建的輸入文件對應于在PBEsol/DZVP-MOLOPT-SR-GTH級別下對Cu晶體做原子坐標和晶胞的優化。由于是導體,所以用了smearing。由于用的是原胞算的,所以此例設了k點。
啟動Multiwfn后依次輸入
Cu.cif //在本文文件包里,是銅晶體的cif文件
cp2k
Cu_cellopt.inp //在當前目錄下產生Cu_cellopt.inp
1 //設置理論方法
-3 //PBEsol,很適合算固體晶格常數的泛函
8 //設置k點
3,3,3
-1 //設置任務
4 //優化原子坐標和晶胞
6 //使用占據數smearing
0 //產生輸入文件
現在得到的Cu_cellopt.inp就可以直接算了。
3.4 氧化石墨烯+水分子的結構優化
這一節我們做一個氧化石墨烯+水分子的結構優化。具體來說,是在單層石墨烯的一個C-C鍵上加一個氧橋,然后放一個水分子到合適位置,與氧橋形成氫鍵。構建這個初始結構可以利用GaussView之類的程序。由于文字+圖片方式表達起來太費事,我把GaussView里的操作錄了個短小的視頻:http://www.shanxitv.org/attach/587/gview_build.mp4,其中旋轉水分子的時候按住alt鍵,平移水分子的時候按住alt+shift鍵。按照視頻建模完畢后,保存成gjf文件,就是此文文件包里的graphite_oxide_H2O.gjf,可見里面有Tv(平移矢量)信息。
此例的計算將使用PBE-D3(BJ)/TZVP-MOLOPT-GTH完成。由于當前體系有弱相互作用,所以加了DFT-D3(BJ)校正,相關知識見《談談“計算時是否需要加DFT-D3色散校正?”》(http://www.shanxitv.org/413)。
啟動Multiwfn,然后依次輸入
graphite_oxide_H2O.gjf
cp2k
opt.inp //新產生的文件名
-1 //設置任務
3 //優化結構(晶胞尺寸不變)
3 //設置色散校正
2 //DFT-D3(BJ)
2 //設置基組
3 //TZVP-MOLOPT-GTH
-7 //設置周期性
XY //當前體系是XY周期性,Z方向不考慮周期性
0 //產生輸入文件
之后就可以直接跑了。
順帶一提,在CP2K輸入文件生成界面里選擇優化任務后,可以通過選項9設置對哪些原子坐標進行凍結,可以通過諸如1,5,9-12,14-18形式方便地輸入。這種選擇語句還可以在GaussView里方便地生成,即把某些原子選中成黃色后,在Tools - Atom selection界面里就可以直接拷出來粘貼到Multiwfn窗口里。另外,Multiwfn讓你輸入被凍結的原子序號時若輸入optH還可以直接實現凍結重原子而只優化氫原子的目的。
3.5 用GFN1-xTB方法跑含有216個水的盒子的動力學
GROMACS程序自帶了一個做NPT動力學預平衡好的含有216個水的結構文件spc216.gro,在本文文件包里也給了。此例基于這個文件,創建一個用GFN1-xTB級別做NVT系綜的從頭算動力學的輸入文件。GFN1-xTB是半經驗層面的DFT,簡要介紹見《盤點Grimme迄今對理論化學的貢獻》(http://www.shanxitv.org/388)和《將Gaussian與Grimme的xtb程序聯用搜索過渡態、產生IRC、做振動分析》(http://www.shanxitv.org/421)。
啟動Multiwfn然后輸入
spc216.gro
cp2k
spc216_AIMD.inp
-1 //設置任務
6 //分子動力學
10 //設置熱浴
2 //CSVR熱浴,亦即GROMACS里的V-rescale
1 //設置理論方法
30 //GFN1-xTB
0 //創建輸入文件
在得到的spc216_AIMD.inp中,應結合注釋根據實際需要做恰當修改,比如控制模擬步數的&MD里的STEPS、控制初始溫度和控溫溫度的TEMPERATURE。諸如此類直接就可以手動很容易地改的參數,在Multiwfn的CP2K輸入文件創建界面里一般就沒有留出相應的選項了,因為完全沒必要。
注:實際上 GFN1-xTB模擬純水效果很差,密度明顯偏高。此例只不過是個例子而已。
值得一提的是,在CP2K輸入文件創建界面里選擇動力學任務后,會看到還有幾個相關選項:
選項12:設置壓浴
選項-5:設置動力學軌跡格式。不用控壓時默認是xyz,用控壓時默認是尺寸明顯更小而且記錄盒子信息的二進制格式dcd。也可以用pdb格式,記錄盒子信息,文件尺寸很大
選項-6:設置每多少步把坐標寫入一次軌跡文件
3.6 對HCN氫轉移找過渡態、做振動分析
此例對HCN->NCH氫轉移用dimer方法優化過渡態,之后再做個振動分析考察虛頻情況。此例只是做個演示,對這種分子體系找過渡態的問題CP2K遠遠不如Gaussian,又不好用效率又極低。
首先用GaussView創建個看上去像這個異構化反應的過渡態的結構,保存為H2CO_TS.gjf。然后啟動Multiwfn,輸入
H2CO_TS.gjf
cp2k
dimer.inp
-1 //選擇任務
7 //搜索過渡態
0 //產生輸入文件
然后用CP2K運行產生的dimer.inp。算完后會看到有一個dimer-1.restart文件,這是CP2K輸入文件,包含的是dimer任務最后一步的結構。基于這個結構,我們創建一個振動分析的輸入文件。
啟動Multiwfn,然后輸入
dimer-1.restart
cp2k
freq.inp
-1 //選擇任務
5 //振動分析
0 //產生輸入文件
然后用CP2K運行產生的freq.inp。跑完之后可以看到有且只有一個虛頻,目錄下還出現了freq-VIBRATIONS-1.mol,里面用molden格式記錄了振動矢量,可以用Molden程序觀看振動模式。
3.7 計算氮化硼二維材料的REPEAT原子電荷
做涉及固體表面的經典力場的分子動力學模擬,需要有固體部分的原子電荷,REPEAT電荷對于這個目的是不錯的選擇,對于擬合固體表面的原子電荷比RESP更理想。此例創建一個CP2K算氮化硼板的REPEAT電荷計算的輸入文件。
本文文件包里有個氮化硼晶胞文件BN.cif,用GaussView打開,刪掉兩層BN中的任意一層,并把盒子Z方向尺寸設為10埃(真空區不夠大的話會令REPEAT電荷不合理),之后保存為BN.gjf。
啟動Multiwfn,然后輸入
BN.gjf
cp2k
BN_REPEAT.inp
-4 //輸出原子電荷
7 //REPEAT
8 //設置k點
6,6,6
0 //產生輸入文件
為了讓結果更好,手動打開.inp文件,把ELEMENT N那行下面的基組改為TZV2P-MOLOPT-GTH。這個基組對B沒有定義,所以對B還是用原先默認的DZVP-MOLOPT-SR-GTH。
然后用CP2K運行得到的BN_REPEAT.inp即可,算完后會得到一個.resp后綴的文件,里面就是各個原子的電荷值。B為0.478,很合理。當前目錄下還有個.xyz文件,拖入VMD里可以看擬合點的分布。Multiwfn產生的CP2K算REPEAT電荷的擬合點分布設置是根據我的經驗已調整到了比較合理的情況,一般不需要再做修改。
3.8 用TDDFT算激發態
請看專門的文章:《使用CP2K結合Multiwfn對周期性體系模擬UV-Vis光譜和考察電子激發態》(http://www.shanxitv.org/634)。
3.9 做雜化泛函計算
請看專門的文章:《CP2K做雜化泛函計算的關鍵要點和簡單例子》(http://www.shanxitv.org/690)。
4 總結
Multiwfn創建CP2K輸入文件的功能使得的CP2K程序使用變得相當簡單,大大降低了使用門檻,不用再照著手冊和例子一個輸入文件寫半天(而且還很容易寫錯、寫得不合理),而只需要在Multiwfn里敲幾下鍵盤就可以產生出一個基本能用的輸入文件,而且借助此功能還可以通過GaussView方便地為CP2K計算來建模。這個功能也很有助于初學者學習CP2K的輸入文件的編寫。
Multiwfn創建CP2K輸入文件的界面在未來還會加入更豐富的選項、讓CP2K的更多的功能用起來變得更簡單。在發表文章時提及輸入文件是借助Multiwfn創建的并恰當引用Multiwfn程序原文,是對Multiwfn這個功能繼續開發最好的鼓勵與支持。
Multiwfn創建CP2K輸入文件能做的任務遠遠不止上面演示的那些,還有NEB、NMR、路徑積分分子動力學、X吸收光譜,支持的方法還有MP2、雙雜化、GW、分子力場等等,在北京科音CP2K第一性原理計算培訓班(http://www.keinsci.com/workshop/KFP_content.html)里都有非常全面深入講解。
]]>文/Sobereva@北京科音
First release: 2021-Feb-16 Last update: 2024-Jan-30
1 前言
CP2K(https://www.cp2k.org)是非常好的第一性原理程序,開源免費,跑中、大周期性體系比起主流的基于平面波的程序如Quantum ESPRESSO、VASP等速度快得多。筆者開發的Multiwfn(http://www.shanxitv.org/multiwfn)的創建CP2K輸入文件的功能使得CP2K做常見任務用起來非常簡單(見《使用Multiwfn非常便利地創建CP2K程序的輸入文件》http://www.shanxitv.org/587),而且筆者開設了十分系統深入講解CP2K使用的北京科音CP2K第一性原理計算培訓班(http://www.keinsci.com/workshop/KFP_content.html),以后用CP2K的人肯定會越來越多。本文介紹一下CP2K怎么在非常適合做科學計算的Linux發行版CentOS(包括與之沒實質區別的Rocky Linux和Red Hat)上進行安裝,對于其它Linux系統在一定程度上也適用,至少可以作為參考。
最初寫本文的時候CP2K最新的是8.1版。后來以同樣的方法,經測試也可以順利編譯8.2、9.1、2022.1、2023.1、2023.2、2024.1。最初寫本文的時候我用的是CentOS,后來我改用Rocky Linux后實測也可以用本文完全相同的方式安裝。
CP2K有sopt、ssmp、popt、psmp四種版本,如果你不了解的話,先看一下本文文末附錄中的介紹。
CP2K有三種安裝方式:
(1)先依次手動編譯CP2K所需要的各個庫,然后再編譯CP2K,具體過程見官方說明https://github.com/cp2k/cp2k/blob/master/INSTALL.md。我不推薦這種做法,因為CP2K涉及的庫特別多,一個一個手動編譯頗為麻煩。如果你有經驗和耐心可以這么鼓搗。
(2)使用CP2K自帶的toolchain腳本。toolchain可以自動把CP2K依賴的各種庫都一一下載并且自動編譯,最后輸入幾行命令再把CP2K編譯出來就OK了。整個過程非常簡單省事,下文第2節就介紹這種做法。
(3)直接用官方預編譯的ssmp版,下載后直接就能用,極為方便,下文第3節會說具體做法。
2 基于toolchain安裝CP2K
2.1 相關知識
CP2K是基于Fortran的程序,但它依賴的一堆庫很多都是C/C++寫的,所以Fortran和C/C++編譯器都得有。CP2K的編譯對于編譯器有明確的要求,兼容情況見https://www.cp2k.org/dev:compiler_support。可見如果用gcc+gfortran來編譯的話,gcc必須>=5.5版。CentOS 7.x自帶的gcc是4.8.5版,因此沒法直接編譯,要么升級gcc編譯器(具體做法自行google,有把系統弄出毛病的風險),要么用CentOS >=8.0版。CentOS 8.0自帶的gcc是8.3.1,可以非常順利地結合toolchain編譯CP2K。下面筆者所述的情況和編譯方式對于CentOS 8.0和CentOS 8 Stream是完全適用的,不適用于更老的CentOS。(PS:常有人問我裝CentOS 8的時候應該選什么,建議Base Environment選Workstation,組件選GNOME Applications、Legacy UNIX Compatibility、Development Tools、Scientific Support)
用合適版本的Intel的icc和ifort編譯器來編譯CP2K及相關的庫也可以,這樣CentOS 7.x的用戶也省得升級gcc或者換系統了。但19.0.1版結合toolchain用的時候筆者發現有的庫編譯不過去,筆者不打算深究,本文就不說這種做法了。
toolchain運行過程中會自動下載很多庫的壓縮包,所以必須聯網,而且網速還不能太慢,否則有的庫半天也下不下來,甚至最終失敗。如果你在大陸,強烈建議通過科學的方式加速對外部網絡的訪問。
關于MKL數學庫
CP2K會利用到BLAS和LAPACK標準庫中的子程序。默認情況下會用OpenBLAS庫提供的這部分子程序,但據說OpenBLAS的LAPACK子程序的效率不如Intel的MKL數學庫好,因此改用MKL可能計算速度更快,不過筆者的一些簡單的對比測試并未發現用MKL時速度有顯著優勢。如果你想用MKL的話,那么運行下一節的toolchain前先把MKL裝到系統里,即運行下列命令:
dnf config-manager --add-repo https://yum.repos.intel.com/mkl/setup/intel-mkl.repo
rpm --import https://yum.repos.intel.com/intel-gpg-keys/GPG-PUB-KEY-INTEL-SW-PRODUCTS-2019.PUB
dnf install intel-mkl
此時MKL會被裝到/opt/intel/目錄下,被裝到筆者機子里的是MKL 2018版,占2.2GB硬盤。把下面的語句加入到~/.bashrc文件中,然后重新進入終端使之生效
source /opt/intel/parallel_studio_xe_2018.0.033/bin/psxevars.sh
上面這行語句會使得MKLROOT環境變量指向MKL庫的目錄。當運行toolchain的時候發現MKLROOT環境變量被指定了,就會自動利用MKL數學庫(無需額外寫--with-mkl選項)。
如果你想離線安裝MKL,可以去Intel官網免費注冊并下載Intel OneAPI base toolkit離線安裝包。安裝的時候只選擇安裝其中的MKL就夠了。裝好后把下面這句加入~/.bashrc文件里,然后重新進入終端使之生效
source /opt/intel/oneapi/mkl/[實際版本號]/env/vars.sh
但這樣裝MKL的情況下,筆者遇到過CP2K 8.1的toolchain在裝COSMA庫的時候編譯不過去的問題。如果你也遇到了此問題,把toolchain的命令后頭加上--with-cosma=no避免裝COSMA庫即可,這對性能影響甚微。
2.2 通過toolchain安裝CP2K依賴的庫
去https://github.com/cp2k/cp2k/releases/下載CP2K壓縮包cp2k-8.1.tar.bz2,運行tar -xf cp2k-8.1.tar.bz2命令解壓之。下文假設解壓后的目錄是/sob/cp2k-8.1/。
運行以下命令
cd /sob/cp2k-8.1/tools/toolchain/
./install_cp2k_toolchain.sh --with-sirius=no --with-openmpi=install --with-plumed=install
之后這個toolchain腳本就會依次下載各個庫的壓縮包到toolchain/build目錄下并解壓和自動編譯,編譯產生的可執行文件、庫文件、頭文件等都自動裝到了toolchain/install目錄下。在筆者的普通四核機子結合非常暢通的網絡下,整個過程耗時約兩個小時。其中最耗時的是編譯libint那一步,花了一個小時有余,一定要耐心。
以下信息應了解一下
? 如果運行上述腳本后提示ERROR: (./scripts/install_gcc.sh) cannot find gfortran,應當運行dnf install gcc-gfortran先把gfortran編譯器裝上,然后重新運行腳本。
? 運行./install_cp2k_toolchain.sh --help可以查看toolchain的幫助。可見有的庫默認是安裝的,有的默認不裝,通過選項來決定,可以按需調整。toolchain腳本的使用細節見https://github.com/cp2k/cp2k/blob/master/tools/toolchain/README.md
? --with-sirius=no選項代表不裝本來自動會裝的SIRIUS庫。這個庫使得CP2K可以像Quantum ESPRESSO那樣完全基于平面波+贗勢做計算,但一般這用不上,想做這種計算的人一般直接就用Quantum ESPRESSO了,其安裝見《Quantum ESPRESSO在Linux中的安裝方法》(http://www.shanxitv.org/562)。
? --with-openmpi=install代表安裝OpenMPI庫,這使得編譯出來的CP2K可以通過MPI方式并行計算。CP2K也支持其它MPI庫如Intel MPI和MPICH。我個人比較習慣用OpenMPI,這也是目前最主流的。重要提示:如果你的機子里已經有了OpenMPI,應當用--with-openmpi=system,這使得CP2K直接用機子里現成的OpenMPI,否則額外再自動裝一個OpenMPI可能造成一些沖突。
? --with-plumed=install代表安裝默認不自動裝的PLUMED庫,這使得CP2K可以結合PLUMED做增強采樣的從頭算動力學。如果你不需要此功能的話可以不加這個選項,可以節約少量編譯時間。
? toolchain默認用所有CPU核心并行編譯,可以自行加上-j [并行核數]來明確指定用多少核。編譯的耗時和CPU核數關系很大。筆者在36核機子上也就花了不到20分鐘就把toolchain過程全都完成了。
? toolchain默認自動下載和編譯cmake。如果你通過yum或dnf已經裝過cmake而且其版本較新,可以再加上--with-cmake=system用當前系統里的cmake,能節約編譯時間。
? 注意硬盤的空余空間應當足夠。對于CP2K 8.1在上述命令執行完畢后,toolchain/build目錄約占9GB,toolchain/install目錄占約2GB。如果硬盤吃緊,建議toolchain運行成功后把這個build目錄刪掉,里面的文件之后用不著。
? 如果toolchain運行過程中某個庫編譯失敗,可以去toolchain/build目錄下的那個庫的目錄中去找編譯過程輸出的log文件,在里面搜error根據報錯試圖分析原因。toolchain運行失敗后可以重新運行,它會根據根據toolchain/build目錄的內容做判斷,之前已經下載和編譯成功的庫會自動跳過,而從失敗的庫繼續編譯。如果把build和install目錄都刪了,則toolchain會從頭執行。
? 如果在安裝某個庫的過程中一直卡著,通過top命令發現CPU也沒在編譯庫,那么幾乎一定是因為網速太慢導致那些庫的壓縮包老也下載不完(在大陸區域不可描述的訪問國際互聯網的條件下尤為常見)。可以去tools/toochain/build目錄下看看正在裝的這個庫的壓縮包,如果尺寸特別小,而且尺寸增加得特別緩慢,說明就是這個問題所致。解決方法是開微屁恩加速訪問國際互聯網。還一個辦法是找個訪問國際互聯網通暢的機子或者拜托朋友,在那里安裝一次當前版本的CP2K,然后把build目錄下的.tgz、.tar.gz、.tar.bz2那些庫的壓縮包拷到之前那個機子的build目錄下,這樣那個機子在安裝CP2K過程中就會直接用這些包而不會試圖下載了,對于沒法訪問Internet的機子也可以這樣離線安裝CP2K。
2.3 編譯CP2K
接著上一節,現在把/sob/cp2k-8.1/tools/toolchain/install/arch/下所有文件拷到/sob/cp2k-8.1/arch目錄下。這些文件定義了編譯不同版本的CP2K所需的參數,其內容是toolchain腳本根據裝的庫和當前環境自動產生的。
然后運行以下命令
source /sob/cp2k-8.1/tools/toolchain/install/setup
cd /sob/cp2k-8.1
make -j 4 ARCH=local VERSION="ssmp psmp"
-j后面的數字是并行編譯用的核數,機子有多少物理核心建議就設多少。在筆者的普通4核機子上花了40分鐘編譯完。
注:如果編譯中途報錯,并且從后往前找error的時候看到cannot find -lz的報錯提示,則運行dnf install zlib-devel命令裝上zlib庫,再重新運行上面的make那行命令即可。
編譯出的可執行程序現在都產生在了/sob/cp2k-8.1/exe/local目錄下,共1.1GB。這里面cp2k.popt、cp2k.psmp、cp2k.sopt、cp2k.ssmp就是我們所需要的CP2K的可執行文件了(popt和sopt其實分別是psmp和ssmp的符號鏈接)。
把以下內容加入到~/.bashrc文件里:
source /sob/cp2k-8.1/tools/toolchain/install/setup
export PATH=$PATH:/sob/cp2k-8.1/exe/local
重新進入終端后,就可以通過cp2k.ssmp等命令運行cp2k了。運行諸如cp2k.ssmp -v可以查看CP2K的版本、編譯時用的庫和參數信息。
注1:上面source這行必須有,因為有的庫提供的.so文件是CP2K啟動時所需的,source這個腳本使得相應的庫的目錄被加入到動態庫的搜索路徑中。而且用了這個之后toolchain過程中裝的OpenMPI的可執行文件mpirun等才能直接用。
注2:cp2k-8.1目錄下的lib和obj目錄分別存的是CP2K編譯過程產生的靜態庫文件和.o文件,總體積不小。由于之后用不著,因此如果想省硬盤可以把這倆目錄刪掉。
3 直接用官方的預編譯版CP2K
對于CP2K 8.1,官方預編譯版只提供了ssmp的,并且為了兼容性考慮,編譯選項比較保守,沒有根據CPU內核進行優化、沒有利用SIMD指令集、用的是-O2而非更激進優化的-O3選項,也沒用MKL。不過這并不代表官方預編譯版的就很慢,筆者對簡單任務測試過發現在速度上和自己編譯的ssmp版差異不太大。不過,如果對某些類型任務發現ssmp版的CPU占用率普遍較低,吐血建議自己編譯popt版,此時有可能二者速度差異超大、用ssmp完全發揮不出CP2K本來的代碼效率,甚至可能ssmp版幾乎算不動。
使用官方的預編譯的ssmp版CP2K的做法如下:
和2.2節所述相同,先下載cp2k-8.1.tar.bz2壓縮包并解壓到比如/sob/cp2k-8.1目錄。
去https://github.com/cp2k/cp2k/releases/下載CP2K的預編譯版可執行文件cp2k-8.1-Linux-x86_64.ssmp,改名為cp2k.ssmp并隨便放到一個位置,假設放到了/sob/cp2k-8.1目錄下。
將下面兩行加入到~/.bashrc文件中:
export PATH=$PATH:/sob/cp2k-8.1
export CP2K_DATA_DIR=/sob/cp2k-8.1/data
保存后重新進入終端,CP2K就可以通過cp2k.ssmp命令使用了(ssmp版CP2K不是必須叫cp2k.ssmp,也可以改名為cp2k,這樣運行更方便)。
為什么設CP2K_DATA_DIR環境變量這里說一下。在CP2K輸入文件中,如果諸如BASIS_SET_FILE_NAME、POTENTIAL_FILE_NAME等關鍵詞只定義了文件名而沒有給路徑,程序默認先在當前目錄下搜索相應文件,找不到的話去CP2K_DATA_DIR搜索。CP2K_DATA_DIR對應的是編譯的時候CP2K目錄下的data目錄的路徑,但開發者在編譯的時候其對應的路徑顯然跟我們當前情況不符,因此這里通過export來將CP2K_DATA_DIR環境變量改成自己機子里實際的data目錄的路徑。
4 運行和測試CP2K
這里提供一個簡單的輸入文件用于測試:http://www.shanxitv.org/attach/586/test.inp。這是Multiwfn生成的2*2*2金剛石超胞做PBE/DZVP-MOLOPT-SR-GTH單點計算的輸入文件。
先測試ssmp版。將test.inp放到當前目錄下,運行:cp2k.ssmp test.inp |tee test.out。輸出信息會在屏幕上顯示,也同時寫入到了test.out里。默認情況下所有CPU核心都會被用于OpenMP并行計算,如果比如想只用4核,就先運行export OMP_NUM_THREADS=4命令然后再運行CP2K,此時運行過程中CP2K進程的CPU占用率應當在300~400%。
再測試popt版。假設用4核通過MPI方式并行,就執行:mpirun -np 4 cp2k.popt test.inp |tee test.out。在top中看到會有4個cp2k.popt在運行,占用率皆接近100%。
如果你是自己編譯的CP2K,建議默認用popt版而不要用ssmp版,因為在某些情況下后者運行效率遠不及popt版(但也有些任務二者速度差異不大,看具體情況)。為了運行popt版省事,建議在~/.bashrc里面加入一行alias cp2k='mpirun -np 4 cp2k.popt'。重新進入終端后,只要輸入cp2k test.inp |tee test.out就等價于輸入mpirun -np 4 cp2k.popt test.inp |tee test.out了,用起來省事多了。
注:跑sopt、popt版時,不管設不設OMP_NUM_THREADS、設多少,OMP_NUM_THREADS都會被強行視為1。
附:CP2K的并行以及四種版本
CP2K支持MPI方式并行也支持OpenMP方式并行。最初CP2K是完全基于MPI并行的,但每個核心對應一個MPI進程來并行跑CP2K的話,對某些任務、較大體系消耗內存較高。CP2K如今很多代碼也利用OpenMP方式實現了并行化,OpenMP并行的好處是很多數據可以在不同線程之間共享而不用保存副本,從而比MPI并行明顯更節約內存。但由于有些CP2K代碼仍只能通過MPI方式并行,因此單純靠OpenMP并行的話某些任務的速度可能明顯不及MPI并行,而且并行核數很多時純OpenMP的并行效率比純MPI并行的略低是很多科學計算程序中常見的現象。CP2K也支持MPI和OpenMP混合并行,比如CPU有36核,那么可以比如用9個MPI進程,每個MPI進程下屬4個OpenMP線程,這樣9*4把36個核都利用上,比直接用36個OpenMP線程并行效率可能明顯更高,而比用36個MPI進程則明顯更省內存(這對于雜化泛函計算比較重要。雜化泛函耗內存遠高于純泛函,如果借助OpenMP節約內存,使得有足夠內存儲存所有雙電子積分,即in-core方式做SCF,就可以避免每次迭代過程中重算這些積分,令SCF迭代過程耗時低得多)。
MPI或MPI+OpenMP可以跨節點并行(OpenMP限于節點內),而純OpenMP只能單機并行,因為OpenMP是基于共享內存的并行技術。
根據支持的并行方式的不同,CP2K分為四個版本:
sopt:只能單機單核計算,無法并行。s意為single
ssmp:OpenMP并行,可以單機多核運行。smp意為Symmetric multiprocessing
popt:MPI并行,可以單機并行也可以跨節點并行。p意為parallel
psmp:MPI+OpenMP混合并行,可以單機并行也可以跨節點并行
sopt版嚴格等價于ssmp版結合OMP_NUM_THREADS=1,popt版嚴格等價于psmp版結合OMP_NUM_THREADS=1。
實際上還有sdbg和pdbg版,前者相當于ssmp結合debug設置,后者相當于psmp結合debug設置,但這對于開發者調試程序才有意義,所以本文2.3節我們沒有編譯這倆版本。
]]>文/Sobereva@北京科音 2020-Jul-22
1 前言
XCrySDen(http://www.xcrysden.org)是非常流行的第一性原理程序Quantum ESPRESSO用戶經常用的重要工具,可以觀看輸入輸出文件、觀看軌跡、設置k點等,而且作圖效果挺不錯。XCrySDen在Ubuntu上比較容易運行,但對于做計算化學的人用得非常多的CentOS就不那么容易了。XCrySDen很老的版本提供了semishared版,在CentOS里解壓后就能運行,但較新的XCrySDen官方只提供了shared版(至少是對于撰文時最新的1.6.2版而言),里面涉及的一些動態庫在CentOS里沒有相應的源。為了讓CentOS用戶用XCrySDen毫無障礙,筆者在CentOS下編譯了XCrySDen,并且同時提供了傻瓜式編譯的源代碼包。
2 預編譯版XCrySDen的安裝
下載http://www.shanxitv.org/attach/564/xcrysden-1.6.2_sobereva.tar.gz。解壓后,進入此目錄,運行./xcrysden就可以啟動了。如果在~/.bashrc目錄下加上export PATH=$PATH:[XCrySDen的目錄名],則重新進入終端后就可以在任意目錄下直接啟動XCrySDen了。
這個筆者編譯的XCrySDen 1.6.2在CentOS 7系列各個版本上都可以運行。如果讀者裝系統的時候裝的方式和《在VMware 15中安裝CentOS 7.6的完整過程視頻演示》(http://www.shanxitv.org/454)里演示的相同,不需要裝額外的庫就可以直接運行。如果運行時提示缺庫,Google一下報錯提示,用yum安裝相應的包即可。
對于CentOS 8.0,筆者發現沒法直接運行,但只要把解壓后目錄下的tcl目錄下的xcInit.tcl里的兩處0m都改為0就可以運行,并且關閉程序的時候必須點擊右上角強行關閉。
3 XCrySDen的編譯
下面是基于筆者修改的XCrySDen 1.6.2的源代碼包的編譯過程。前面說的筆者的預編譯版如果能正常用就沒必要自己編譯。在CentOS 7.x和8.0下按以下方法都能編譯通過。
運行以下命令安裝編譯過程要用的庫
yum install libGL-devel libGLU-devel libXmu-devel
機子里應當已經裝了gcc和gfortran,如果沒裝的話運行yum install gcc-gfortran來安裝。
下載筆者修改的源代碼包:http://www.shanxitv.org/attach/564/xcrysden-1.6.2_src_sobereva.tar.gz。解壓后進入其中,運行make all即可,大約5分鐘就能編譯完畢。之后直接運行./xcrysden即可啟動。
對于某些CentOS版本,比如CentOS 7.4,編譯中途可能失敗,需要在解壓目錄下的Makefile中的X_LIB=后面加入-lXss選項,然后重新make all。經測試至少對于CentOS 7.7不用加這個。
關于筆者修改的XCrySDen源代碼包的一些細節:Make.sys文件是在system/Make.sys-shared基礎上修改的,原先的這個文件完全沒法用,筆者改了許多地方才終于令編譯能成功。具體改了哪些,自行對照Make.sys-shared就知道了。Makefile文件也做了修改,把all:后面的mesa去掉了,因為CentOS的源直接就有這個,通過前述的yum步驟已經安裝了,因此就沒必要再在make all的時候編譯了。其實對于CentOS 8,由于源里的tcl/tk已經升為了XCrySDen 1.6.2要求的8.6版,因此不編譯tcl/tk而直接通過源來裝也不是不可以。另外,原本make all的時候會自動下載tcl、tk、Togl、fftw、bwidget包,但在大陸地區由于網速問題,很容易中途下載失敗。因此筆者直接將這些壓縮包放到了external/src目錄下,這樣編譯過程中就會自動利用,而不自動下載這些包了。
]]>文/Sobereva@北京科音
First release: 2020-Jul-10 Last update: 2020-Jul-23
1 前言
Quantum ESPRESSO (QE)是用戶非常多、極為流行的第一性原理程序,而且完全開源免費。本文介紹一下QE在Linux下的安裝過程。本文對于CentOS 7.x系列系統下安裝QE 6.5是完全適合的,對于其它QE版本或其它Linux系統請自行嘗試、隨機應變。本文使用root賬戶,對于普通用戶請安裝到自己有讀寫權限的目錄。本文基于OpenMPI庫+MKL庫+gfortran/gcc編譯器進行編譯。編譯的是純CPU版本,不支持GPU加速(GPU加速還需要有PGI Fortran編譯器)。如果機子里還沒裝gcc和gfortran,應先用yum install gcc命令進行安裝。
關于編譯的更多細節可以看QE的手冊https://www.quantum-espresso.org/Doc/user_guide/node10.html。
在CentOS下使用yum也可以不通過編譯來安裝,但有一些弊端,見此文第6節。
2 安裝OpenMPI
為了讓QE能基于MPI并行計算,需要先裝MPI庫,一般就用OpenMPI。筆者用的是OpenMPI 4.0.3,經測試與QE 6.5完全兼容。
去https://www.open-mpi.org下載最新的OpenMPI包,解壓后進入此目錄,運行以下命令將之編譯并安裝到指定目錄下。這里筆者安裝到了/sob/openmpi目錄下。
./configure --prefix=/sob/openmpi
make all install
在~/.bashrc文件中加入以下內容
export PATH=$PATH:/sob/openmpi/bin
export LD_LIBRARY_PATH=$LD_LIBRARY_PATH:/sob/openmpi/lib
然后重新打開終端,以上設置就生效了。可以運行mpiexec -V,如果正常顯示出了OpenMPI的版本,說明已經裝好了。之后可以刪掉OpenMPI壓縮包和解壓出的目錄。
注:如果機子里之前還有其它MPI庫,應當運行which mpiexec來看看是否確實指向的是新裝的OpenMPI,如果指向的是其它的,則并行運行可能失敗。比如如果你之前在機子里裝過Intel編譯器的時候順帶裝了Intel MPI,則應當在編譯QE以及運行QE前將~/.bashrc里的相應配置語句注釋掉后重新進入終端,免得被利用的是Intel MPI而非新裝的OpenMPI。
3 安裝MKL庫
為了讓QE能利用效率很高的MKL數學庫來提升計算速度,應當在編譯QE前先把MKL裝上。MKL庫目前是免費的,CentOS下可以運行以下兩行命令安裝。期間會下載幾百兆的文件,文件會被安裝到/opt/intel目錄下,占3GB多(對于2020-Jul-10時下載的版本而言)。如果你之前機子里裝過Intel編譯器,且在裝的時候已經選擇裝了MKL,就不需要再這么裝一遍了。
添加intel的源:
yum-config-manager --add-repo https://yum.repos.intel.com/mkl/setup/intel-mkl.repo
下載并安裝MKL:
yum install -y intel-mkl
4 編譯QE
在https://github.com/QEF/q-e/releases下載QE最新版源代碼包,比如qe-6.5-ReleasePack.tgz。
解壓并進入其中,運行以下命令。這里-enable-openmp使得QE也可以利用OpenMP來并行,如果不打算以OpenMP并行的話就不寫這個。
./configure --prefix=/sob/qe65 -enable-openmp
make all install -j
四核機子上經過幾分鐘編譯完畢,可執行文件都被裝到了/sob/qe65/bin目錄下。解壓出的目錄和壓縮包此時雖然可以刪掉,但我建議還是留著解壓出的目錄,里面有些文件以后還用得著。
注:QE在編譯過程默認調用gfortran、gcc和mpif90。如果想改默認的編譯器,應對QE目錄下的make.inc文件里的編譯器設置進行修改,而且在之前編譯OpenMPI的時候也用相應的編譯器。
在~/.bashrc文件中加入以下內容(如果你用的不是root的話,前兩行不用加)
export OMPI_ALLOW_RUN_AS_ROOT=1
export OMPI_ALLOW_RUN_AS_ROOT_CONFIRM=1
export PATH=$PATH:/sob/qe65/bin
export LD_LIBRARY_PATH=$LD_LIBRARY_PATH:/opt/intel/compilers_and_libraries_2020.2.254/linux/mkl/lib/intel64_lin
這里往LD_LIBRARY_PATH環境變量添加的Intel MKL庫的目錄名應當與你當前機子里的實際路徑一致。
之后重新進入終端,QE的可執行文件就可以在任意目錄下直接運行了。
以上述方式編譯出來的QE沒有包含EPW、PLUMED、Wannier90、WanT、YAMBO、GIPAW程序,如果需要編譯的話,看官網上User's Guide for Quantum ESPRESSO文檔的2.5節。
5 測試QE
下面對QE最關鍵的PWscf模塊做簡單測試。下載http://www.shanxitv.org/attach/562/diamond.zip并解壓,此任務是對金剛石做SCF計算。
QE是MPI和OpenMP混合方式并行的程序,實際并行核數是MPI進程數與每個下屬的OpenMP線程數的乘積。
先測試純MPI并行方式運行。進入diamond目錄后,運行以下命令,使用4個MPI進程計算,每個MPI進程下屬只有一個線程。
export OMP_NUM_THREADS=1
mpirun -n 4 pw.x < pwscf.in |tee pwscf.out
如果任務能正常完成,末尾顯示JOB DONE,就說明已經裝好了。注:如果不設置OMP_NUM_THREADS環境變量的話,機子有多少核,OpenMP就會用多少個線程。
然后再測試純OpenMP并行方式運行。運行以下命令,將使用一個MPI進程下屬4個OpenMP線程進行計算
export OMP_NUM_THREADS=4
pw.x < pwscf.in |tee pwscf.out
也可以使用QE自帶的測試集進行測試。做法是進入QE解壓后的目錄的test-suite子目錄,在里面運行make run-tests-parallel命令,就會在并行運算下對所有任務進行測試,每一項對應的含義見此目錄下的README。如果只想測試比如PWscf模塊,則運行make run-tests-pw-parallel。
6 在CentOS下使用yum安裝QE
下面文字適用于CentOS,使用root的情況。會安裝基于OpenMPI并行但不支持OpenMP并行的QE 6.5版。至少對于對于CentOS 7.7和8.0而言,被安裝的是QE 6.5版。Ubuntu下也可以用apt-get裝,本文就不提了。
運行以下命令:
yum install epel-release
yum install quantum-espresso-openmpi
期間會自動安裝OpenMPI、OpenBLAS、ScaLapack等包。如果你是用yum install quantum-espresso,則安裝的是只能串行計算的版本,沒實際價值。
之后在~/.bashrc文件里加入以下內容:
export PATH=$PATH:/usr/lib64/openmpi/bin/
如果你用的是root的話同時加入
export OMPI_ALLOW_RUN_AS_ROOT=1
export OMPI_ALLOW_RUN_AS_ROOT_CONFIRM=1
之后重新進入終端即可使用。
QE的文件被安裝到了/usr/lib64/openmpi/bin/目錄下,可執行文件都帶著_openmpi后綴。比如可以運行mpirun -n 4 pw.x_openmpi < pwscf.in。
對于計算密集型程序,像QE這種編譯不麻煩的話,我鼓勵自行編譯,因為yum裝的CentOS軟件源里的預編譯版為了兼容性、減少庫的依賴,在一些地方可能會打一些折扣,比如沒用MKL(對QE提供高質量的BLAS、ScaLapack和FFT)、用的編譯選項比較保守,故性能可能遜于自己編譯的。而且自己編譯的話可以自定義文件產生的位置,雖然yum也可以用--installroot=...選項指定安裝在哪,但是有些程序可能不能運行。
另外,QE的文檔信息很零散,比如支持的泛函完整列表甚至還得去看源代碼包里的func.f90里的注釋等等,所以最好有源代碼包。之前若自己用源代碼包編譯過之后還可以直接用make epw、make w90、make gipaw等命令編譯安裝與QE有關的程序。
]]>