使用Multiwfn非常便利地創建CP2K程序的輸入文件
使用Multiwfn非常便利地創建CP2K程序的輸入文件
文/Sobereva@北京科音
First release: 2021-Feb-22 Last update: 2023-May-11
1 前言
CP2K(https://www.cp2k.org)是速度非常快,功能又比較全面的第一性原理程序,受到越來越多的重視,有很多無可替代的重要價值。CP2K的一個問題是輸入文件寫起來極其繁瑣,一個簡單的任務的輸入文件就要動輒上百行。而且有時用戶還需要了解很多算法和數值實現細節、查閱繁復的手冊,甚至還要google半天、參考不少自帶測試文件才能最終寫出能用的輸入文件。這些問題使得CP2K這個優秀的程序不容易普及開來,初學者往往看到其極度復雜的輸入文件就打退堂鼓了。
筆者的Multiwfn程序(主頁&下載:http://www.shanxitv.org/multiwfn)的主功能100的子功能2能產生大量量子化學程序的輸入文件,諸如其中產生ORCA輸入文件的功能在此文專門介紹過《詳談Multiwfn產生ORCA量子化學程序的輸入文件的功能》(http://www.shanxitv.org/490),給很多人帶來了極大便利。為了讓CP2K這個利器對于常見計算問題用起來盡可能方便、盡可能像自動擋、令使用門檻盡可能低,使之能更充分地發揮價值,筆者在Multiwfn里加入了產生CP2K輸入文件的功能,在此文將進行介紹。這個功能雖然注定不可能讓完全零基礎的CP2K用戶毫無壓力地使用CP2K,但至少已經把使用難度降到極限了,用戶只需要選擇干什么、怎么干,即可產生一個完成度>95%的輸入文件,之后一些必須根據實際情況調整的地方,比如平面波截斷能大小等,再自行改一下即可,這使得CP2K用戶得到了極大的解放。
如果Multiwfn的這個功能對你的研究產生了實際幫助,請在寫文章時順帶提及輸入文件是借助Multiwfn產生的(比如寫the CP2K input files were generated with help of Multiwfn program),并按程序啟動時顯示的要求恰當引用Multiwfn。
Multiwfn產生CP2K輸入文件的流程是:
1 啟動Multiwfn,載入一個Multiwfn可以識別的至少含有結構信息的文件
2 進入主功能100的子功能2,選擇產生CP2K輸入文件(即選項25)。也可以直接在Multiwfn主菜單里輸入cp2k進入此功能
3 輸入要產生的CP2K輸入文件的路徑
4 通過各種選項設置如何進行計算
5 選擇0,得到CP2K輸入文件
下面第2節先說載入到Multiwfn的輸入文件的要求。第3節給出一些例子演示產生CP2K輸入文件的過程。本文內容對應Multiwfn官網上最新版本的Multiwfn(不是今天剛下載的就不要覺得是最新的),一定不要用老版本。Multiwfn產生CP2K輸入文件的功能在未來還會不斷擴展,盡量做到更強大更完美,并且盡量迎合未來版本CP2K發生的各種變化,屆時本文也會相應地修改。
順帶一提,如果你希望從頭系統性學習CP2K的使用、掌握第一性原理計算,非常強烈建議參加北京科音CP2K第一性原理計算培訓班,介紹見http://www.keinsci.com/workshop/KFP_content.html。通過這個培訓能全面學習到第一性原理計算各種問題的知識、完整透徹掌握CP2K,其中講解利用CP2K做各類問題的研究時都會使用Multiwfn創建輸入文件,學員會深切體會到Multiwfn對于CP2K的使用起到的重要價值。
2 給Multiwfn用的輸入文件
Multiwfn支持什么輸入文件這里都詳細說了:《詳談Multiwfn支持的輸入文件類型、產生方法以及相互轉換》(http://www.shanxitv.org/379)。對于產生CP2K的輸入文件,顯然載入到Multiwfn的文件中必須含有體系的結構才行。對于此類目的,Multiwfn支持一大波格式,如pdb/gro/xyz/mol/mol2/mwfn/wfn/wfx/molden/fch/cub/gjf/gro等等等等。
如果載入到Multiwfn的文件不包含晶胞信息,那么Multiwfn會默認將此體系視為孤立體系來設置CP2K里的計算參數,CP2K輸入文件里的晶胞信息是在分子周圍做一定距離的延展來得到的。
多數情況CP2K都是拿來算周期性體系的。若想讓Multiwfn產生的CP2K輸入文件里有實際的晶胞信息,載入到Multiwfn的文件里就必須有晶胞信息。用以下格式作為輸入文件時,Multiwfn會既讀取原子坐標也讀取晶胞信息。
(1)cif文件
(2)含有CRYST1字段的pdb或pqr文件
(3)CP2K的輸入文件(必須以.inp或.restart為后綴)
(4)GROMACS的gro文件
(5)含有@<TRIPOS>CRYSIN字段的mol2文件
(6)含有Ndim且其數值>0的mwfn文件(mwfn是Multiwfn私有的波函數文件格式,專門留出了字段記錄晶胞信息,見mwfn介紹文章https://doi.org/10.26434/chemrxiv.11872524)
(7)帶有Tv(平移矢量)信息的Gaussian輸入文件,即Gaussian做PBC任務的輸入文件,可以通過GaussView直接創建
(8)Gaussian做PBC任務產生的fch/fchk文件
(9)含有晶胞信息的xyz文件。這是我自己定義的,如果xyz文件的注釋行(第二行)包含諸如以下形式的內容(以埃為單位的平移矢量),就會被Multiwfn讀取
Tv_1: 7.426 0.0 0.0 Tv_2: 3.6 6 6.40 0.0 Tv_3: 0.0 0.0 10.0
也可以按照extended xyz格式的方式在xyz文件的第二行記錄晶胞信息,例如:
Lattice="7.426 0.0 0.0 -3.66 6.40 0.0 0.0 0.0 10.0"
(10)含有[Cell]字段的molden文件。此字段是我對molden格式做的擴展,這使得記錄波函數的molden文件也能記錄晶胞信息。大家可以直接手動往已有的molden文件里插入[Cell]信息,見Multiwfn手冊2.9.2節的格式示例。
(11)VASP的POSCAR、CHGCAR、CHG、ELFCAR、LOCPOT文件。文件名里必須包含相應字樣才能識別成相應格式,例如要作為POSCAR載入,文件名可以是POSCAR_miku或miku.POSCAR
(12)Quantum ESPRESSO的輸入文件
3 產生CP2K輸入文件的一些例子
下面就通過不同體系、不同類型計算演示一下Multiwfn產生CP2K輸入文件的功能,這些例子也可以作為CP2K入門例子。這些例子只用到了一部分Multiwfn提供的選項,其它的請自行嘗試,選項的含義都非常易于理解所以就不一一說了。下文用到的文件都在http://www.shanxitv.org/attach/587/file.zip中提供了。筆者假定讀者已經以正當方式安裝了CP2K,安裝方法見《CP2K第一性原理程序在CentOS中的簡易安裝方法》(http://www.shanxitv.org/586)。由于本文只是舉例,為了省計算時間,有些任務的k點取得比實際研究時應取的明顯要少。
3.1 做COF(共價有機框架)化合物的單點任務
本文文件包里COF_12000N2.cif是個COF的晶體結構文件,本例用Multiwfn對它產生一個最簡單的任務的輸入文件,即PBE泛函結合DZVP-MOLOPT-SR-GTH基組做單點計算。
啟動Multiwfn,然后依次輸入
COF_12000N2.cif //寫實際的路徑。也可以把此文件拖到Multiwfn文本窗口里自動產生路徑,省得手動輸入了
cp2k //進入產生CP2K輸入文件的功能
[按回車] //新產生的CP2K輸入文件將為當前目錄下的COF_12000N2.inp。也可以自己輸入路徑
0 //直接生成輸入文件
現在COF_12000N2.inp就在當前目錄下出現了,可以直接用CP2K跑了。這樣在默認設置下產生的輸入文件對應PBE/DZVP-MOLOPT-SR-GTH算單點,只考慮gamma點。可以打開.inp看一下有哪些自己需要改的,一般來說自己根據實際需要改的也就是影響精度的設置,比如影響平面波部分計算精度的&MGRID里的CUTOFF和REL_CUTOFF、控制密度矩陣收斂限的&SCF里的EPS_SCF,這些問題本文就不多說了。
Multiwfn產生的CP2K輸入文件里面有很多注釋,即#后面的內容,這有助于對CP2K還不是特別熟的人了解選項的含義、明白怎么設置。輸入文件里有些選項的值是根據我的使用經驗和習慣設的,也有一些設置雖然是默認的,卻也出現在了輸入文件里,這個考慮是便于大家之后自己手動修改(要不然想改一個默認設置的話還得照著手冊寫一堆字段實在太費事了)。
下面再用COF舉個例子,使用B97M-rV泛函結合6-31G*基組做GAPW形式的單點計算,并且要求算完后產生記錄此體系波函數的.molden文件(可用于之后在Multiwfn中做波函數分析,見比如《使用IRI方法圖形化考察化學體系中的化學鍵和弱相互作用》http://www.shanxitv.org/598和《使用Multiwfn結合CP2K通過NCI和IGM方法圖形化考察固體和表面的弱相互作用》http://www.shanxitv.org/588)。本例用較小的基組僅作為演示目的,對精度有要求的計算至少用6-311G**。值得一提的是,在純泛函中,B97M-rV是對有機體系能量相關問題計算得最好的,并且這個泛函自帶rVV10色散校正。
啟動Multiwfn后依次輸入
COF_12000N2.cif
cp2k
COF_GAPW.inp
1 //設置理論方法
11 //B97M-rV(通過libxc庫實現)
2 //設置基組
10 //6-31G*
-2 //要求產生.molden文件
0 //產生輸入文件
現在當前目錄下就有了COF_GAPW.inp,直接就可以跑了。跑完之后當前目錄下會出現COF_GAPW-MOS-1_0.molden。
順帶一提,在Multiwfn的主功能0里可以直接看體系的結構和晶胞,由此可以確認一下結構和晶胞信息有沒有被Multiwfn從輸入的文件里正常載入。啟動Multiwfn并載入COF_12000N2.cif后,選0,就會進入圖形界面。關閉標簽和坐標軸,并且點擊菜單欄的Other settings里的Toggle showing cell frame,當前窗口將為下圖所示的狀態。可見原子位置和晶胞都正確。點擊Save picture按鈕可以在當前目錄下得到線條明顯更平滑的大尺寸圖像文件。
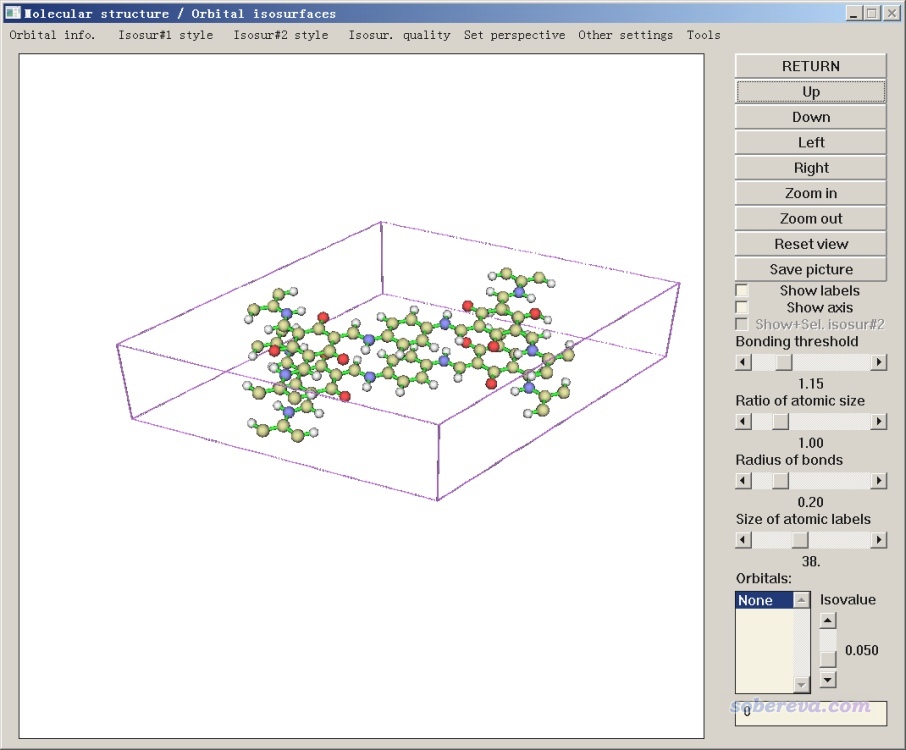
也可以用Multiwfn載入CP2K的.inp輸入文件或者CP2K計算過程中產生的.restart文件,在主功能0里可以看其中的結構是什么樣。還可以載入它們后,進入主功能100的子功能2,選擇導出gjf文件,這樣得到的gjf文件里會有Tv描述的晶胞信息,可以放到GaussView里進行觀看和編輯修改。還可以用主功能100的子功能2里的一堆選項轉換成其它一大堆常見格式、一大堆計算程序的輸入文件(包括Quantum ESPRESSO、VASP的POSCAR),賊靈活。
3.2 用雜化泛函計算SiC超胞
這一節創建的輸入文件對應于用雜化泛函PBE0結合DZVP-MOLOPT-SR-GTH基組對SiC的2*2*2超胞算一個單點。
啟動Multiwfn后輸入
SiC.cif //在本文文件包里
cp2k
[按回車] //產生的輸入文件名為默認的SiC.inp
1 //設置理論方法
-6 //PBE0,并且利用ADMM方法巨幅降低HF交換部分的耗時
-11 //進入幾何操作界面。這個界面有豐富的功能,見《Multiwfn中非常實用的幾何操作和坐標變換功能介紹》(http://www.shanxitv.org/610)
19 //構建超胞
2 //方向1為原先的兩倍
2 //方向2為原先的兩倍
2 //方向3為原先的兩倍
如果你想看一下當前結構的話,可以選0進入圖形界面,會看到下圖,可見結構沒問題

接著輸入
-10 //返回到之前創建CP2K輸入文件的界面
0 //產生輸入文件
現在當前目錄下的SiC.inp就可以直接跑了。跑之前建議用文本編輯器打開SiC.inp,根據機子實際情況,將其中的&HF/&MEMORY下面的MAX_MEMORY恰當設大,比如50000(單位是MB)。因為做雜化泛函計算時,內存給得夠大的話,就可以令四中心雙電子積分只在第一輪迭代的時候算一次,之后就存到內存里,用不著每輪SCF迭代都計算,之后每輪SCF耗時就非常低了。
實際上,還有一種等價的創建SiC 2*2*2超胞輸入文件的做法。進入Multiwfn的CP2K輸入文件創建界面后,輸入
1 //設置理論方法
-6 //PBE0-ADMM
-9 //其它設置
3 //設置三個方向晶胞重復次數
2,2,2
0 //返回
0 //產生輸入文件
在得到的輸入文件中可以看到坐標部分只記錄了原胞,但有兩處MULTIPLE_UNIT_CELL 2 2 2,體現出是對2*2*2超胞進行計算。
有CP2K使用常識的人都知道,在做雜化泛函計算之前應當先做純泛函計算,以其收斂的波函數作為雜化泛函計算的初猜波函數。額外做的純泛函計算耗時只占雜化泛函計算的一個零頭,卻能令雜化泛函容易收斂得多得多得多,并因此巨幅節約耗時。為實現這個目的,先用Multiwfn產生一個PBE泛函的輸入文件PBE.inp并進行計算(基組和PBE0計算時相同),算完后會得到PBE-RESTART.wfn文件,然后在PBE0的輸入文件里把# WFN_RESTART_FILE_NAME to_be_specified這一行改為WFN_RESTART_FILE_NAME PBE-RESTART.wfn,然后把# SCF_GUESS RESTART這一行的#去掉。這樣PBE0計算開始時就會把PBE-RESTART.wfn里的PBE波函數當初猜了。
3.3 優化Cu的晶胞
這一節創建的輸入文件對應于在PBEsol/DZVP-MOLOPT-SR-GTH級別下對Cu晶體做原子坐標和晶胞的優化。由于是導體,所以用了smearing。由于用的是原胞算的,所以此例設了k點。
啟動Multiwfn后依次輸入
Cu.cif //在本文文件包里,是銅晶體的cif文件
cp2k
Cu_cellopt.inp //在當前目錄下產生Cu_cellopt.inp
1 //設置理論方法
-3 //PBEsol,很適合算固體晶格常數的泛函
8 //設置k點
3,3,3
-1 //設置任務
4 //優化原子坐標和晶胞
6 //使用占據數smearing
0 //產生輸入文件
現在得到的Cu_cellopt.inp就可以直接算了。
3.4 氧化石墨烯+水分子的結構優化
這一節我們做一個氧化石墨烯+水分子的結構優化。具體來說,是在單層石墨烯的一個C-C鍵上加一個氧橋,然后放一個水分子到合適位置,與氧橋形成氫鍵。構建這個初始結構可以利用GaussView之類的程序。由于文字+圖片方式表達起來太費事,我把GaussView里的操作錄了個短小的視頻:http://www.shanxitv.org/attach/587/gview_build.mp4,其中旋轉水分子的時候按住alt鍵,平移水分子的時候按住alt+shift鍵。按照視頻建模完畢后,保存成gjf文件,就是此文文件包里的graphite_oxide_H2O.gjf,可見里面有Tv(平移矢量)信息。
此例的計算將使用PBE-D3(BJ)/TZVP-MOLOPT-SR-GTH完成。由于當前體系有弱相互作用,所以加了DFT-D3(BJ)校正,相關知識見《談談“計算時是否需要加DFT-D3色散校正?”》(http://www.shanxitv.org/413)。
啟動Multiwfn,然后依次輸入
graphite_oxide_H2O.gjf
cp2k
opt.inp //新產生的文件名
-1 //設置任務
3 //優化結構(晶胞尺寸不變)
3 //設置色散校正
2 //DFT-D3(BJ)
2 //設置基組
3 //TZVP-MOLOPT-GTH
-7 //設置周期性
XY //當前體系是XY周期性,Z方向不考慮周期性
0 //產生輸入文件
之后就可以直接跑了。
順帶一提,在CP2K輸入文件生成界面里選擇優化任務后,可以通過選項9設置對哪些原子坐標進行凍結,可以通過諸如1,5,9-12,14-18形式方便地輸入。這種選擇語句還可以在GaussView里方便地生成,即把某些原子選中成黃色后,在Tools - Atom selection界面里就可以直接拷出來粘貼到Multiwfn窗口里。另外,Multiwfn讓你輸入被凍結的原子序號時若輸入optH還可以直接實現凍結重原子而只優化氫原子的目的。
3.5 用GFN1-xTB方法跑含有216個水的盒子的動力學
GROMACS程序自帶了一個做NPT動力學預平衡好的含有216個水的結構文件spc216.gro,在本文文件包里也給了。此例基于這個文件,創建一個用GFN1-xTB級別做NVT系綜的從頭算動力學的輸入文件。GFN1-xTB是半經驗層面的DFT,簡要介紹見《盤點Grimme迄今對理論化學的貢獻》(http://www.shanxitv.org/388)和《將Gaussian與Grimme的xtb程序聯用搜索過渡態、產生IRC、做振動分析》(http://www.shanxitv.org/421)。
啟動Multiwfn然后輸入
spc216.gro
cp2k
spc216_AIMD.inp
-1 //設置任務
6 //分子動力學
10 //設置熱浴
2 //CSVR熱浴,亦即GROMACS里的V-rescale
1 //設置理論方法
30 //GFN1-xTB
0 //創建輸入文件
在得到的spc216_AIMD.inp中,應結合注釋根據實際需要做恰當修改,比如控制模擬步數的&MD里的STEPS、控制初始溫度和控溫溫度的TEMPERATURE。諸如此類直接就可以手動很容易地改的參數,在Multiwfn的CP2K輸入文件創建界面里一般就沒有留出相應的選項了,因為完全沒必要。
注:實際上 GFN1-xTB模擬純水效果很差,密度明顯偏高。此例只不過是個例子而已。
值得一提的是,在CP2K輸入文件創建界面里選擇動力學任務后,會看到還有幾個相關選項:
選項12:設置壓浴
選項-5:設置動力學軌跡格式。不用控壓時默認是xyz,用控壓時默認是尺寸明顯更小而且記錄盒子信息的二進制格式dcd。也可以用pdb格式,記錄盒子信息,文件尺寸很大
選項-6:設置每多少步把坐標寫入一次軌跡文件
3.6 對HCN氫轉移找過渡態、做振動分析
此例對HCN->NCH氫轉移用dimer方法優化過渡態,之后再做個振動分析考察虛頻情況。此例只是做個演示,對這種分子體系找過渡態的問題CP2K遠遠不如Gaussian,又不好用效率又極低。
首先用GaussView創建個看上去像這個異構化反應的過渡態的結構,保存為H2CO_TS.gjf。然后啟動Multiwfn,輸入
H2CO_TS.gjf
cp2k
dimer.inp
-1 //選擇任務
7 //搜索過渡態
0 //產生輸入文件
然后用CP2K運行產生的dimer.inp。算完后會看到有一個dimer-1.restart文件,這是CP2K輸入文件,包含的是dimer任務最后一步的結構。基于這個結構,我們創建一個振動分析的輸入文件。
啟動Multiwfn,然后輸入
dimer-1.restart
cp2k
freq.inp
-1 //選擇任務
5 //振動分析
0 //產生輸入文件
然后用CP2K運行產生的freq.inp。跑完之后可以看到有且只有一個虛頻,目錄下還出現了freq-VIBRATIONS-1.mol,里面用molden格式記錄了振動矢量,可以用Molden程序觀看振動模式。
3.7 計算氮化硼二維材料的REPEAT原子電荷
做涉及固體表面的經典力場的分子動力學模擬,需要有固體部分的原子電荷,REPEAT電荷對于這個目的是不錯的選擇,對于擬合固體表面的原子電荷比RESP更理想。此例創建一個CP2K算氮化硼板的REPEAT電荷計算的輸入文件。
本文文件包里有個氮化硼晶胞文件BN.cif,用GaussView打開,刪掉兩層BN中的任意一層,并把盒子Z方向尺寸設為10埃(真空區不夠大的話會令REPEAT電荷不合理),之后保存為BN.gjf。
啟動Multiwfn,然后輸入
BN.gjf
cp2k
BN_REPEAT.inp
-4 //輸出原子電荷
7 //REPEAT
8 //設置k點
6,6,6
0 //產生輸入文件
為了讓結果更好,手動打開.inp文件,把ELEMENT N那行下面的基組改為TZV2P-MOLOPT-GTH。這個基組對B沒有定義,所以對B還是用原先默認的DZVP-MOLOPT-SR-GTH。
然后用CP2K運行得到的BN_REPEAT.inp即可,算完后會得到一個.resp后綴的文件,里面就是各個原子的電荷值。B為0.478,很合理。當前目錄下還有個.xyz文件,拖入VMD里可以看擬合點的分布。Multiwfn產生的CP2K算REPEAT電荷的擬合點分布設置是根據我的經驗已調整到了比較合理的情況,一般不需要再做修改。
3.8 用TDDFT算激發態
請看專門的文章:《使用CP2K結合Multiwfn對周期性體系模擬UV-Vis光譜和考察電子激發態》(http://www.shanxitv.org/634)。
4 總結
Multiwfn創建CP2K輸入文件的功能使得的CP2K程序使用變得相當簡單,大大降低了使用門檻,不用再照著手冊和例子一個輸入文件寫半天(而且還很容易寫錯、寫得不合理),而只需要在Multiwfn里敲幾下鍵盤就可以產生出一個基本能用的輸入文件,而且借助此功能還可以通過GaussView方便地為CP2K計算來建模。這個功能也很有助于初學者學習CP2K的輸入文件的編寫。
Multiwfn創建CP2K輸入文件的界面在未來還會加入更豐富的選項、讓CP2K的更多的功能用起來變得更簡單。在發表文章時提及輸入文件是借助Multiwfn創建的并恰當引用Multiwfn程序原文,是對Multiwfn這個功能繼續開發最好的鼓勵與支持。