談談“計算時是否需要加DFT-D3色散校正?”
談談“計算時是否需要加DFT-D3色散校正?”
文/Sobereva @北京科音
First release: 2018-Apr-21 Last update: 2022-May-29
0 前言
筆者經常被問及的一個問題就是“我要計算xxx,是否需要加DFT-D色散校正?”。關于DFT-D,在以下博文里已經有充分的介紹:
亂談DFT-D
http://www.shanxitv.org/83
DFT-D色散校正的使用
http://www.shanxitv.org/210
大體系弱相互作用計算的解決之道
http://www.shanxitv.org/214
其實把以上博文所述內容都弄明白,什么時候該加DFT-D色散校正就已經應該心里很有數了。但是鑒于還是有大量初學者犯糊涂,我覺得有必要在此文里再強調一下,也算是對上面文章的補充。下文第一節是介紹一些常識,如果想對什么時候該加DFT-D這個問題了解更深刻可以看看,如果只是急于獲得答案,可以略過。
1 化學體系中的相互作用常識
化學體系中的相互作用主要分為化學鍵作用和弱相互作用兩大類。從相互作用能上看,前者一般強度比較強,共價鍵和離子鍵都屬于此類,也可以叫強相互作用;而后者一般強度比較弱,比前者通常弱一個數量級。弱相互作用既可以是分子間的,也可以是分子內的。弱相互包括范德華作用、pi-pi堆積作用、氫鍵、二氫鍵、鹵鍵,以及后來很多人炒作概念而提出來的碳/硫/磷/金/銀/銅...鍵等等。還有個詞叫做非共價相互作用(noncovalent interaction),這個詞的范疇相當于弱相互作用和離子鍵的并集。
弱相互作用如上所列,形式多樣,但主要本質只有兩個:
(1)靜電相互作用。可以起到互斥作用也可以起到吸引作用,看具體情況(體系,以及相對位置)。這里說的靜電相互作用也把極化作用包含進去了。
(2)色散作用。它起到吸引作用。必須從量子力學角度才能予以解釋。而從量化理論角度來講,它對應于電子的長程的庫侖相關作用。
一般強度的氫/二氫/鹵/硫/磷鍵等等是以靜電吸引主導,色散作用為輔構成的,這點通過能量分解也可以體現出來。而一般說的范德華相互作用,以及pi-pi堆積作用的本質都是色散作用。
注1:還有個作用叫交換互斥,它起到互斥作用,不管算什么類型相互作用都要考慮這個,只有在較近距離(比如小于兩個原子的范德華半徑和)的時候才開始凸顯出來,且原子間距離越近此作用越大。正是這個作用使得弱相互作用的勢能曲線總是有極小點,而不會令原子間距離因為靜電吸引和色散作用而無限減小。
注2:“范德華作用”這個詞描述的是電中性的兩個原子之間的非共價相互作用,故本質包含了交換互斥和色散吸引作用兩個部分。這個詞可以囊括比如兩個氬原子間的相互作用的全部內涵。這個詞也可以用來描述無極性分子間的全部相互作用(但實際上,沒有絕對嚴格意義的無極性分子,比如就連一般說的無極性分子氮氣也有四極矩,可以認為是局部極性,因此氮氣之間也有靜電相互作用,討論見http://www.shanxitv.org/209。但本文就不咬文嚼字了)。至于一些初級的教材里說的范德華作用包括“誘導力、色散力、取向力”,這種歪曲的說法應該徹底從量化研究者的腦海中刪除。
注3:弱相互作用實際上也伴隨著一些共價作用,但共價作用此時僅僅是個“陪襯”而已,對弱相互作用能貢獻程度極小,故在弱相互作用范疇里不需要專門特意考慮(但是弱相互作用范疇里面也有強度很大的情況,比如能到100kJ/mol數量級的氫鍵,此時共價作用就不能忽視了,此文說的弱相互作用不考慮此情況)。
注4:筆者強烈建議仔細看一下此文和其中提到的原文,對于正確認識氫鍵的作用本質極有益處:《透徹認識氫鍵本質、簡單可靠地估計氫鍵強度:一篇2019年JCC上的重要研究文章介紹》(http://www.shanxitv.org/513)。
不管是什么DFT泛函,什么理論方法,對上面提到的靜電相互作用和交換互斥作用總是可以至少定性正確地表現。然而老一代泛函對于色散作用描述極糟(如SVWN、PBE、PW91),或者根本不能描述(如BLYP、B3LYP),因此這些泛函完全沒法描述范德華結合、pi-pi堆積這樣本質來自于色散吸引的弱相互作用,結果會定性錯誤;而考察氫鍵等靜電主導的弱相互作用時,雖然多數情況定性正確,但肯定定量明顯不準。這些泛函有這些問題一方面是擬合參數的時候沒有考慮弱相互作用體系,一方面是泛函的相關勢的長程漸進行為明顯不對。
為什么Grimme要提出DFT-D?出發點就是解決老一代泛函計算弱相互作用糟糕的情況。更確切來說,解決的是對色散作用描述糟糕的問題。因此,DFT-D叫做色散校正。但要注意,色散校正,或者解決傳統泛函描述色散作用爛的方法絕對不止DFT-D這一種,還有很多其它做法,比如vdW-DF、XDM、TS、LRD、DFT-ulg、dDSC、OBS等等,由于這些方法要么實現復雜、要么會導致計算耗時增加許多、要么效果不好、要么還沒有被主流程序普遍支持等原因,時下流行程度和DFT-D相比微不足道,以至于DFT-D簡直成了色散校正的代名詞。
DFT泛函不加色散校正就不能描述好弱相互作用了?非也。由于弱相互作用越來越受重視,從21世紀00年代中期開始提出的泛函越來越多地注重對弱相互作用的描述。比如2006年的M05-2X描述各種弱相互作用就已經挺不錯了,因為在擬合參數的訓練集中已經納入了弱相互作用體系,之后08年的M06-2X在這方面又得到了一定進步。而比如08年提出的ωB97XD,在提出當初就已經帶了DFT-D2校正以使得計算各類弱相互作用能夠有比較好的能力。至于從2006年開始提出的雙雜化泛函,由于里面已經包含了MP2成份,而MP2本身就已經能夠定性正確描述色散作用,因此雙雜化泛函對各類弱相互作用都有比較好的描述能力,哪怕其中有的不及M06-2X,但也總比較老一代的泛函要強得多得多。
DFT-D第一代是2004年提出的,2017年提出了第四代(DFT-D4)。截止到2019年初,最流行的還是2010年提出的DFT-D3,它純粹基于幾何結構來計算色散校正能,加到原有泛函的能量上就是色散校正后的能量,比如加到B3LYP能量上就成了B3LYP-D3能量。更多信息見前面提到的《DFT-D色散校正的使用》和《亂談DFT-D》,這里不再累述。DFT-D1/2/3都只利用幾何信息而不考慮體系電子結構,計算都很迅速,幾乎不增加任何計算量,故是“免費”的。而到了DFT-D4,校正能才考慮電子結構(依賴于原子電荷),帶來的好處就是可以合理體現電子結構對色散校正的影響(比如同一個結構下,激發態和基態是不同的,單重態和三重態是不同的,中性和離子狀態是不同的...特別是對于計算諸如金屬氧化物等原子電荷數值較大的情況,由于考慮了實際電子結構,結果會比用DFT-D3更好)。關于DFT-D4,筆者寫過專門的文章介紹:《DFT-D4色散校正的簡介與使用》(http://www.shanxitv.org/464)。
對于該不該、有無必要加DFT-D3這個問題,下面對計算弱相互作用的情況和計算其它任務的情況分別說。
2 計算弱相互作用的情況加不加DFT-D3?
這里說的計算弱相互作用,顯然包括計算分子間相互作用能、優化分子二聚體/多聚體幾何結構(以及隨后的振動分析)、研究物理吸附等場合。對于很多柔性大分子,存在顯著的分子內相互作用,因此弱相互作用對其不同構象間的相對能量、構象結構影響極大,故研究它們的構象問題也屬于下述的“計算弱相互作用”的范疇里。是否存在明顯分子內/分子間相互作用其實一看結構心里就有數,只要非鍵連的原子之間距離挨得較近,就應當考慮弱相互作用。如果化學直覺不夠而不好判斷,可以用Multiwfn做IGM分析,見《通過獨立梯度模型(IGM)考察分子間弱相互作用》(http://www.shanxitv.org/407);或者做RDG/NCI分析,見《使用Multiwfn圖形化研究弱相互作用》(http://www.shanxitv.org/68),操作都十分簡單,敲幾下鍵盤的事。
當你研究的問題涉及到弱相互作用,而且當前用的理論方法不足以合理描述色散作用,色散校正是一定要加的!鑒于DFT-D3是免費的而且是最流行的色散校正方法,我們一般首選加DFT-D3。比如說B3LYP描述色散作用完全失敗,當拿它計算pi-pi堆積這種完全由色散作用所控制的弱相互作用時,不加色散校正結果是荒唐的。而用B3LYP計算氫鍵這種靜電主導而色散作用為輔的弱相互作用時,加色散校正后的結果也才可能用來發文章(可能有人看到這句話會突然跳出來說:我看那誰誰誰,還挺有名的,以前用B3LYP算氫鍵不是也發文章了?要知道,弱相互作用的計算在最近十幾年發展極其迅速,05年之前DFT算弱相互作用的文章完全沒法看,05~10年的文章也只能有選擇性地參考,10年之后的DFT算弱相互作用的文章才算是進入成熟期。研究者們對DFT做弱相互作用計算的認識在這期間也發生了翻天覆地的變化。除非碰上外行審稿人或者投極爛的期刊,這年頭用B3LYP算弱相互作用能發表才怪。)
當研究弱相互作用相關的問題時,如果目前用的DFT泛函已經可以不錯地描述色散作用了,比如用的是M06-2X,那顯然DFT-D3不是必須加的,加了不會有什么壞處,但帶來的改進甚微。如果用的是雙雜化泛函,由于對色散作用也已經描述不錯了,因此不加D3算各種弱相互作用的精度也不賴,至少不至于因不加D3而被審稿人拒,但考慮到從統計結果來看,加了D3后精度改進還是有不少油水的,所以建議總是加上。很多后來提出的雙雜化泛函,比如PWPB95、DSD-PBEP86,由于在提出時就已經帶著D3了,所以用的時候更是強烈建議帶著D3(值得一提的是,Gaussian16里寫DSD-PBEP86關鍵詞的時候自動就是帶DFT-D3的)。
以下給出Grimme在Phys. Chem. Chem. Phys., 19, 32184 (2017)中列出的幾個泛函的弱相互作用平均計算誤差,D3用的是BJ阻尼:
BLYP:18.56 kcal/mol
BLYP-D3:3.70 kcal/mol
B3LYP:15.43 kcal/mol
B3LYP-D3:2.94 kcal/mol
PWPB95:5.74 kcal/mol
PWPB95-D3:2.27 kcal/mol
可見,BLYP、B3LYP加D3后改進甚大,不加D3誤差大到根本沒法用。PWPB95雙雜化泛函加了D3之后也有很大改進。
下面是不同明尼蘇達系列泛函加D3前后的平均誤差情況。明尼蘇達系列泛函只能用零阻尼形式的D3校正。
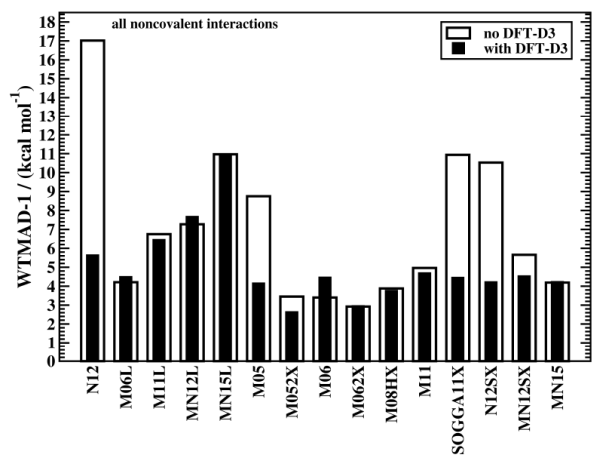
可見,對于計算弱相互作用常用的M06-2X,加D3的意義不大,改進甚微。僅對于M06-2X計算pi-pi堆積,我建議還是盡量加上D3,因為對這種情況M06-2X有時誤差可能略大,加D3后有不可忽略的改進。并非所有明尼蘇達系列泛函算弱相互作用都不錯,有的實在是非常糟糕,而且加D3都救不活,比如MN15L這種。關于某些明尼蘇達系列泛函加D3校正后結果仍較爛原因的一些細致的探討,可以看J. Phys. Chem. Lett., 6, 3891 (2015)。
對于后HF、多參考方法等等,是沒有色散校正這一說的。DFT-D僅能用于DFT、DFTB、HF、半經驗。高檔次后HF方法,比如MP4、CCSD(T),已經可以很準確計算各類弱相互作用體系了,也沒什么系統性誤差,且誤差已明顯小于DFT泛函結合DFT-D校正后的誤差了,顯然對這些精細的方法再試圖通過相對來說形式較為粗俗的DFT-D來校正是沒有任何意義的,因此也沒人對這些方法去擬合DFT-D參數。而至于MP2,由于耗時和雙雜化差不多,而算弱相互作用精度又不及雙雜化(氫鍵除外),因此也沒人專給它搞DFT-D參數。
(后記:2018年有人提出了給MP2加DFT-D3的方法,叫MP2D。雖然比MP2在計算色散作用上有明顯改進,但相比于帶色散校正的雙雜化泛函在精度上完全沒有優勢,所以完全沒存在意義。2022年有有人給SCS-MP2加上DFT-D3,叫SCS-MP2D,也同樣沒有什么實際價值)
有人問:“我用xxx泛函,我知道計算弱相互作用能的時候有必要加D3,那優化和振動分析有沒有必要?”顯然有必要!D3校正能是依賴于幾何結構的,因此,D3校正本質上校正的是體系的勢能面,會令所有依賴于勢能面的問題都受到影響,自然也包括優化出的結構(勢能面極小點的位置)、振動頻率(取決于勢能面極小點處Hessian矩陣)、IRC(質權坐標下的勢能面上的能量極小路徑)等。你算能量時候用D3,就代表你已經知道考慮D3校正對于研究當前體系是有意義的,如果幾何優化等任務不加D3,怎么說得通?必然會糟至審稿人吐槽,何況D3還是免費的。所以,優化、振動分析、單點、IRC等等,要加D3都加,如果確信沒必要加,那就都不加。
另外,如果你研究的是一系列體系,用的是同一個泛函,只要其中一個加了D3,其它所有的計算全都得加D3,否則沒法橫向比較。如果涉及到能量直接求差的情況,比如復合物能量減去各個片段能量、一個構象減去另一個構象的能量,那所有涉及的體系加不加D3就更得統一,否則能量求差毫無意義。
可能有時候研究的問題并不直接牽扯弱相互作用,但是體系內又存在弱相互作用,這時候如果你用的泛函描述色散能力不足,那照樣也得加D3。比如說研究某個大分子的局部異構化過程,這個過程本身不怎么涉及到弱相互作用特征的變化,但大分子其余部分存在分子內弱相互作用(比如有一些內氫鍵),對這種情況,你若是用M06-2X算的則D3可加可不加,但用B3LYP算就應當加D3。或許你不覺得對此問題加D3有什么必要,但畢竟加D3又不花錢又基本沒壞處,還省得之后審稿人找碴又讓你加D3重算,何故不加?而且還完全排除了不考慮色散作用會對結果產生不可忽略的誤差的風險。
3 與弱相互作用無關的情況加不加DFT-D3?
因為D3校正僅僅是基于幾何結構計算校正能,與電子態無關,而且只改變能量而不影響體系的波函數,因此對這些問題,加不加D3結果都一樣:計算某個結構下的gap、軌道、偶極矩、極化率、NMR、原子電荷、鍵級、靜電勢分布等等。因為這些問題不涉及體系的能量或者能量對核坐標的導數。D3也不會影響這些問題的計算結果:垂直激發能、理論電子光譜(不考慮振動耦合時)、垂直電離能、垂直電子親合能、垂直單-三重態能量差等等,因為計算不同電子態時用的幾何結構相同,把D3校正能精確抵消了。但是,D3可以間接地對上述問題產生影響。比如一個二聚體,用B3LYP和B3LYP-D3優化出來的結構可能相差很大,那顯然最終計算的偶極矩、吸收光譜等性質也會有明顯不同。
諸如有人問過我,計算激發態用不用加D3,那顯然得看你算激發態的什么。算垂直吸收/發射能和振子強度、計算激發態與基態的密度差、計算NTO等等,前面說了,D3絲毫不可能影響結果,因為結構沒變,而且D3不影響波函數。而對于激發態幾何優化,D3顯然會影響結果,而且如果優化基態時加了D3則優化激發態也得加,得保持統一。
對于主要只體現化學鍵作用的體系,加D3是沒必要的,雖然加了也沒壞處。諸如優化有機小分子、簡單配合物(如二茂鐵)、簡單陰陽離子體系(比如乙酸鈉、硫酸鈣)等等,加D3基本不會對結果產生任何影響。一方面是D3里面有阻尼函數,阻尼函數會使得校正函數在近程區域衰減為0,從而避免影響原先DFT泛函就已經能描述良好的范疇,另一方面是對于這些較強的相互作用,微小的色散作用的影響本身就是可以忽略的。但如果是有機大分子、配體比較大的配合物(此時配體間容易有弱相互作用)、比較大的陰陽離子對等等情況,弱相互作用的存在不可忽略,當使用對色散作用描述爛的泛函的時候建議加上D3。
對于計算小體系化學反應能(也包括原子化能、生成焓等),沒有太大必要加D3,但是如果體系比較大,對于描述色散作用能力不足的泛函還是建議加上D3。畢竟體系原子數一多,體系內弱相互作用總量就會較大,更嚴謹地來說,體系中的中、長程電子相關總效應會較大。D3雖然原本目的是校正色散作用的描述能力,但實際上也等效地一定程度改進了很多老泛函在中、長程區域對電子相關描述能力的不足。
下面的圖來自Phys. Chem. Chem. Phys., 19, 32184 (2017),可見D3對于反應能的計算精度是有一定改進的,而且對大體系改進得比小體系更多。
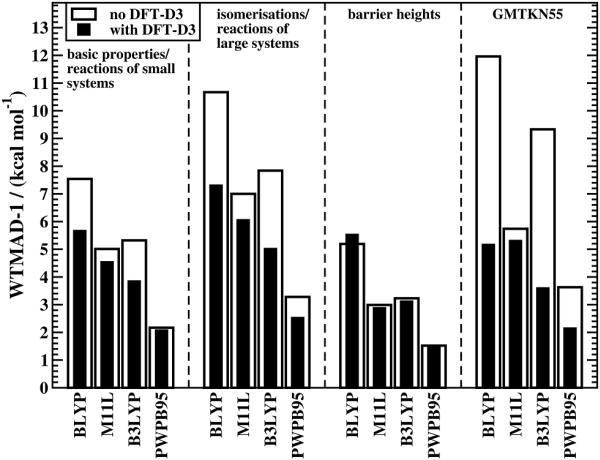
但加D3后結果變差也不是不可能,要想找反例總能找得到。比如JCTC, 13, 3537 (2017)報道PBE0-D3算生成焓比PBE0精度有所下降。不過,相對于一些負面消息,更多的文獻測試還是體現出D3會帶來積極的效果。
從上圖看D3對能壘計算結果并無改進,但實際研究工作往往是研究一整套化學反應過程,若優化極小點、算極小點能量用D3的話,對過渡態也一定得用D3來保持統一。
雖然上面已經講了很多,但肯定有些初學者由于基礎知識不足,面對一些情況對于加不加D3還是會迷糊。簡單一句話:不知道該不該加就加。反正又免費用著又方便,且有益無害。