談談原子間是否成鍵的判斷問題
談談原子間是否成鍵的判斷問題
文/Sobereva @北京科音
First release: 2018-Apr-27 Last update: 2021-Sep-25
0 前言
經常有人在網上問類似這種問題:“我優化出的結構,在gview里怎么顯示A原子和B原子沒有成鍵啊,應該成鍵才對啊,我是不是算錯啦?”這種問題每次都解釋一遍比較麻煩,遂寫此文專門談一談。
如果對于化學體系中的相互作用類型缺乏基本的了解,建議看一下《談談“計算時是否需要加DFT-D3色散校正?》(http://www.shanxitv.org/413)第1節的簡要介紹。對于“鍵”這個主題感興趣的讀者,有興趣可以看看Chemistry is about energy and its changes: A critique of bond-length/bond-strength correlations(Coord. Chem. Rev., 344, 355 (2017)),其中有不少有益的討論,有助于加深對“鍵”這個概念的認識。如果大家想全面了解關于化學鍵的分析手段,務必要看《Multiwfn支持的分析化學鍵的方法一覽》(http://www.shanxitv.org/471)。本文涉及到一些Multiwfn相關內容,若對此程序不了解,看《Multiwfn入門tips》(http://www.shanxitv.org/167)和《Multiwfn FAQ》(http://www.shanxitv.org/452)。此程序可以在官方主頁http://www.shanxitv.org/multiwfn免費下載。
討論原子間是否成鍵這個問題前,首先要明確,原子間是否成鍵,絕對沒有一刀切的標準。鍵本來就是個人為定義的抽象的概念,從實驗上也沒法直接觀測到“鍵”這個東西,只能通過實驗或理論計算得到的一些可觀測性質去試圖反映,或者通過一些純理論的分析方法去試圖具象化“鍵”這個概念。因此原子間是否成鍵,絕對不是一個非黑即白的問題。對明顯成鍵的情況,無論用什么方法去分析討論,結論都會是“成鍵了”;對明顯沒成鍵的情況,無論用什么方法去分析討論,結論都會是“沒成鍵”;而對于非典型、模棱兩可的“灰色地帶”,非要討論是否成鍵,是沒有太大意義的,而且從不同角度去討論,結論可能大相徑庭。雖然對“灰色地帶”非要判斷倆原子間是否成鍵沒什么意思,但從實用角度來說,有些人還是希望做個一刀切式的判斷。下面就依次說一些常見的用來判斷是否成鍵的做法。
1 根據幾何關系判斷
用可視化程序打開包含結構信息的文件,程序會自動顯示哪些原子是鍵連的,哪些沒有鍵連。如果輸入文件里有鍵連關系信息(比如pdb文件的CONECT字段、mol或mol2文件的記錄拓撲關系的字段),那么有的可視化程序可能會直接根據這些信息來設定成鍵方式。可視化程序也往往會自動根據原子間的距離結合原子半徑等信息判斷是否成鍵。例如在默認情況下,Multiwfn在顯示體系結構時,判斷原子間是否顯示化學鍵是看原子間距離是否小于兩個原子的CSD共價半徑和的1.15倍(關于原子半徑,詳見《簡談原子半徑》http://www.shanxitv.org/255)。Multiwfn以這種方式判斷是否成鍵雖然并不嚴格(本來也不可能存在嚴格的辦法),但從直覺上就知道是基本靠譜的,因為如果兩個原子間形成了非常典型的化學鍵,那么鍵長和兩個原子的共價半徑之和應當相差不多;而如果實際鍵長超過共價半徑和都高達15%了,那肯定有什么因素導致他們的成鍵被極大地削弱,說它們沒成鍵也不至于很冤枉。
對于常用的可視化程序gview,有這么幾種情況:
(1)載入gjf:如果gjf里面用geom=connectivity定義了原子間連接關系,則會根據連接關系顯示成鍵。否則自動判斷成鍵
(2)載入chk/fch:會直接按照其中的連接關系顯示成鍵(chk/fch文件中有專門的字段記錄原子連接關系)
(3)載入out/log:自動判斷連接關系(Gaussian輸出文件里沒有原子連接關系信息)
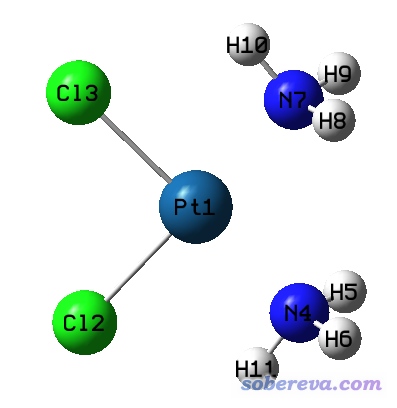
gview判斷成鍵的規則并沒有公開,但應當和Multiwfn用的思想比較類似。gview判斷成鍵的標準往往過于苛刻,導致本來按理說100%可以算是成鍵的情況卻往往判斷成不成鍵。比如下面的二氯二胺合鉑是在B3LYP/SDD/6-31G*下優化的,這是非常常用且合理的計算配合物的級別,結果gview愣是認為Pt和NH3沒有成鍵。然而稍有化學常識的人都知道,它倆肯定應當算是形成了配位鍵。
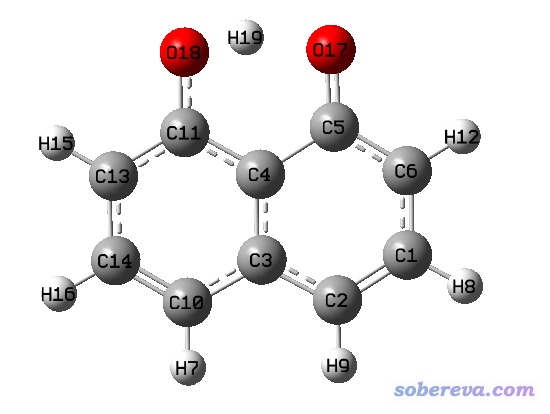
下面的體系是C10O2H7-陰離子,用M06-2X/6-311G**優化的。從結構上就能知道,O18-H19距離并不遠,應該當成成鍵才是,而且有不少方法可以論證這一點,而gview居然顯示它們沒有成鍵。
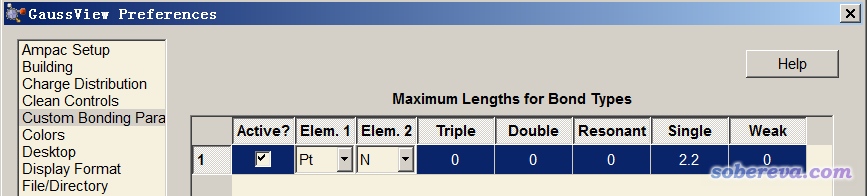
上述兩個例子暴露出gview根據幾何距離判斷成鍵的明顯不足,所以千萬別把gview是否判斷為成鍵太當回事。而上面提到的兩類鍵,用Multiwfn顯示的時候都會被判斷成成鍵,說明Multiwfn用的判斷標準更好一些。盡管如此,利用原子間距離一刀切來判定成鍵終究是很粗鄙的做法。值得一提的是,gview從6.0開始,已經支持了自定義成鍵規則。進入File - Preferences,例如按照下圖設置,就可以讓Pt-N之間距離小于2.2就被判斷成單重鍵。還可以增加判斷Resonant鍵(介于單重鍵和二重鍵之間)、二重鍵、三重鍵的距離閾值上限。
如上修改后,重新載入二氯二胺合鉑的文件,或者選Edit-Rebond重新判斷成鍵,就會發現Pt已經和N連上了。如果你是用的gview 5.0.x及以前的版本,則成鍵判斷標準是沒法改的,只能自己用Modify bond工具手動連上你覺得本應該被視為成鍵的原子了。
在Multiwfn里有個計算原子連接性指數的功能,用主功能100的子功能9直接計算,見手冊3.100.9節的介紹,可以用.fch、.wfn、.xyz、.mol等各種支持的含有原子坐標信息的文件作為輸入文件。當原子間距離大致等于或小于兩個原子的共價半徑和的時候,這個函數接近1.0。當鍵長進一步增加,此函數會逐漸平滑降低,到距離達到共價半徑和兩倍時,此函數幾乎為0。這個原子連接性指數可以視為當前結構下某個鍵存在的百分比,這比起用一刀切方式判斷“存在”還是“不存在”明顯更有意義。
得一提的是,經常有初學者對量子化學最基本知識缺乏了解,以為在gview里是否連鍵會影響結果。對于量化計算來說,在gview里怎么連鍵的,換句話說,在gjf文件里geom=connectivity提供的原子間連接關系是什么樣的,對于量化計算完全不影響結果,因為沒有哪個量化理論方法在計算過程中是依賴于用戶設定的成鍵關系的。僅對于涉及到分子力學的計算,連接關系才必須指定,因為這直接影響到計算時勢函數的設定。
也經常有人問我另一個著名、流行的可視化程序VMD中的鍵的判斷問題,我專門有一篇文章說明:《談談VMD可視化程序的連接關系的判斷和設置問題》(http://www.shanxitv.org/534)。
2 根據鍵級大小判斷
鍵級是非常有用的討論化學鍵的手段,鍵級的定義有非常多,有經驗性地通過可觀測量(諸如鍵長、電子密度等)定義的,也有利用波函數來定義的,在《Multiwfn支持的分析化學鍵的方法一覽》(http://www.shanxitv.org/471)一文中對鍵級有全面的介紹和討論。筆者之前的文章J. Phys. Chem. A, 117, 3100 (2013)里對常見的鍵級也有充分的分析對比。目前最常用的,也是比較適合判斷成鍵問題的,是Mayer鍵級,從形式上看它也算是60年代末提出的Wiberg鍵級用于現代量子化學計算的廣義化形式。Multiwfn程序可以十分方便地基于.fch、.molden等文件計算Mayer鍵級。Multiwfn也可以算模糊鍵級,物理意義上相當基于模糊原子空間定義的Mayer鍵級。
Mayer鍵級是基于量子化學計算產生的波函數計算的。對于同類鍵(比如C-O鍵和C-O鍵之間比較),其數值通常和鍵的強度有正相關性,也因此,隨著鍵的強度變化其數值會平滑變化。比如通過《通過鍵級曲線和ELF/LOL/RDG等值面動畫研究化學反應過程》(http://www.shanxitv.org/200)一文中的例子可以看到,隨著鍵的長度增加/降低,相應的Mayer鍵級會逐漸減小/增加。這也體現出化學鍵內在的特征,即鍵是一個“強度”問題,并沒有一個一刀切式的判斷“是否”存在的標準。Mayer鍵級從物理意義上可以理解為原子間共享的電子對數,因此對于單/雙/三重鍵,由于基本上是共享一/兩/三對電子,Mayer鍵級應比較接近1.0/2.0/3.0,而沒有或幾乎沒有成鍵的原子間Mayer鍵級應當很接近0。顯然,Mayer鍵級不直接告訴你化學鍵是否存在,只不過給你一個反映鍵的存在性的定量數值,一刀切的標準怎么設,只能由研究者在理解Mayer鍵級物理意義和基本特征的基礎上自行定奪。
用Multiwfn計算Mayer鍵級超級容易。啟動Multiwfn,把.fch等文件拖入到Multiwfn窗口,輸入9,回車,1,回車,程序即會把數值大于0.05的鍵級都輸出出來。比如對于前面C10O2H7-陰離子的例子,在M06-2X/6-311G**波函數下,兩個O-H鍵的結果為
17(O ) 19(H ) 0.32208317
18(O ) 19(H ) 0.71849440
可見O18-H19鍵級比較大,雖然離1.0有一定距離,但算是成鍵毫不為過。而O17-H19的鍵級較小,遠小于0.5,因此還不足以算是成鍵,但是其數值又明顯大于0,因此可以認為在當前體系中,O17-H19存在一定的化學鍵作用特征,但若說是“成鍵了”則太過了。
順帶提醒一下,算Mayer鍵級的時候用的基組千萬不能有彌散函數,否則結果毫無意義(雖然大家知道對于計算陰離子體系的能量等問題加彌散函數是很有必要的,但在計算Mayer鍵級的時候可以把彌散函數砍掉再算個單點來產生波函數),而前面提到的模糊鍵級則不怕彌散函數。計算Mayer鍵級也不要用過大的基組,在諸如6-31G*、def2-SVP、6-311G**這種檔次計算的結果已經合理了,如果非要用比如cc-pVQZ等很大的基組,結果可能反倒更差。
像NaCl這種明顯是離子鍵的體系,按照一般化學觀念來說,應該沒有共享電子,Mayer鍵級似乎應該接近于0,但實際中Mayer鍵級從數值上看更類似于“總鍵級”。對NaCl在B3LYP/6-31G*下算的Mayer鍵級數值約0.8,因此無疑算是成鍵的。這某種程度上相當于把Na-Cl鍵當成了極性極高的共價鍵來看待,雖然極性極高,但還是共享電子了。再比如乙酸鈉,在B3LYP/def2-SVP下算出來Na與每個氧之間的Mayer鍵級為0.31,看似小到已不算是成鍵,但由于有兩個氧,因此相當于Na+與乙酸根總鍵級已經超過了0.6,故也算是成鍵的。因此借助Mayer鍵級判斷鍵的存在性比較普適,不局限于特定類型。
3 根據是否存在BD型NBO判斷
有些人使用NBO程序做NBO分析,認為如果NBO程序給出了對應A-B鍵的BD型NBO軌道,就可以算作A-B之間成鍵了。這種做法非常不科學,我強烈不建議使用這種做法!用NBO者,必懂NBO的基本原理、基本思想,否則必被坑。NBO的相關資料在這里有匯總:http://bbs.keinsci.com/thread-102-1-1.html。NBO程序在產生NBO軌道的時候,是通過特定的算法自動進行搜索產生的,而搜索過程中引入了大量人為的、存在任意性的、缺乏物理意義的設定,其中關鍵的是NBO軌道的占據數。在兩個原子間搜索BD型NBO軌道時,若相應的NBO的占據數稍微低一些,則這個NBO就會被pass掉,不呈現在最終NBO列表中。用這種算法產生的NBO軌道來判斷成鍵是非常不合適的。比如前面看到的C10O2H7-陰離子的例子,用Gaussian自帶的NBO 3.1計算,本該算作成鍵的O18-H19卻沒有出現對應的BD型NBO軌道,可見十分誤導。
4 根據是否存在對應的定域化分子軌道判斷
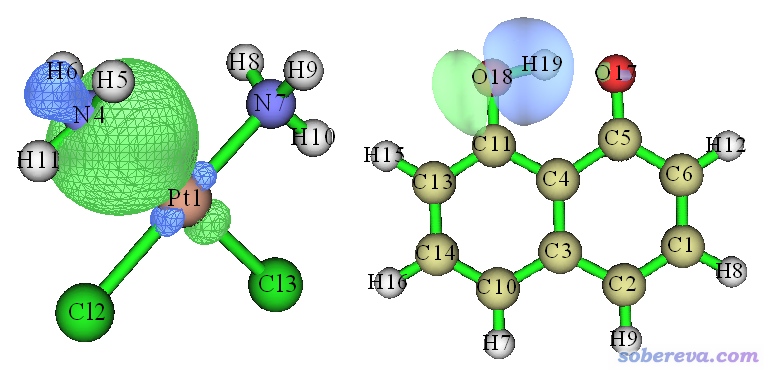
使用軌道定域化方法判斷是否成鍵是很好的做法。軌道定域化方法在《Multiwfn的軌道定域化功能的使用以及與NBO、AdNDP分析的對比》(http://www.shanxitv.org/380)文中有十分詳細的論述和示例,這里不再多說和演示。軌道定域化方法的目的是產生定域化軌道,有很多不同算法,這些算法中不管是哪個,都比通過搜索來產生NBO軌道的處理優雅得多得多,也明顯更有物理意義。只要產生出的占據的定域化軌道中有主要對應于某兩個原子的軌道,就可以說這兩個原子是成鍵的。而且由于與此同時還能給出成鍵軌道的圖形,放在文章當中會成為證明原子間成鍵很好的論據。比如前面提到的二氯二胺合鉑里的Pt-N鍵以及C10O2H7-陰離子中的O18-H19鍵,它們所對應的通過Pipek-Mezey方法產生的定域化分子軌道如下所示:
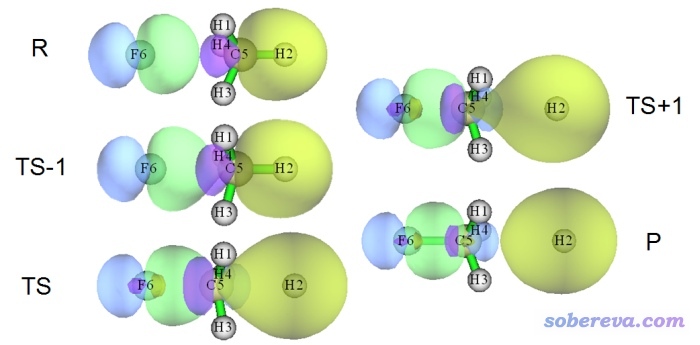
是否存在BD型NBO軌道對于判斷處于平衡結構的體系的成鍵問題都經常不合理,更別提用于成鍵特征模棱兩可的過渡態了。而通過定域化分子軌道討論則完全沒這個問題,因為在整個化學過程中定域化軌道的變化都是平滑、連續的。比如下圖是一個SN2反應過程中幾個關鍵的點的定域化分子軌道的圖形,順序是R→TS-1→TS→TS+1→P,要形成的新鍵對應的軌道用綠/藍色表示,要斷裂的鍵對應的軌道以黃/紫色顯示。由圖可見定域化分子軌道很好地描述了這個SN2反應過程中成鍵特征的變化。
順帶一提,對定域化分子軌道做軌道成分分析,還可以了解鍵的極性。比如B3LYP/6-31G**下對乙醇的O-H鍵對應的定域化分子軌道做SCPA軌道成分分析,O貢獻68.5%,H貢獻27.5%。而在同樣級別下,對NaCl的Na-Cl鍵對應的定域化軌道做軌道成分分析,結果是Na貢獻89.4%,Cl貢獻10.6%,由于其極性極高,因此被視為離子鍵。
5 根據勢能曲線拐點判斷
有一種在數學上比較嚴格,但化學意義并不明確的一刀切判斷成鍵與否的做法是尋找鍵解離勢能曲線的拐點。所謂拐點就是曲線的曲率(即曲線的二階導數)變號的位置。如果鍵長小于拐點,可認為成鍵;如果大于拐點,則認為不成鍵。下圖的藍色曲線是通過UB3LYP/6-311G*以對稱破缺方式對乙烷碳碳鍵進行柔性掃描產生的勢能曲線,紅色曲線是它的二階導數曲線。在2.28埃的位置紅色曲線與Y=0的橫線正好相交,因此2.28埃就是乙烷C-C鍵勢能曲線的拐點。
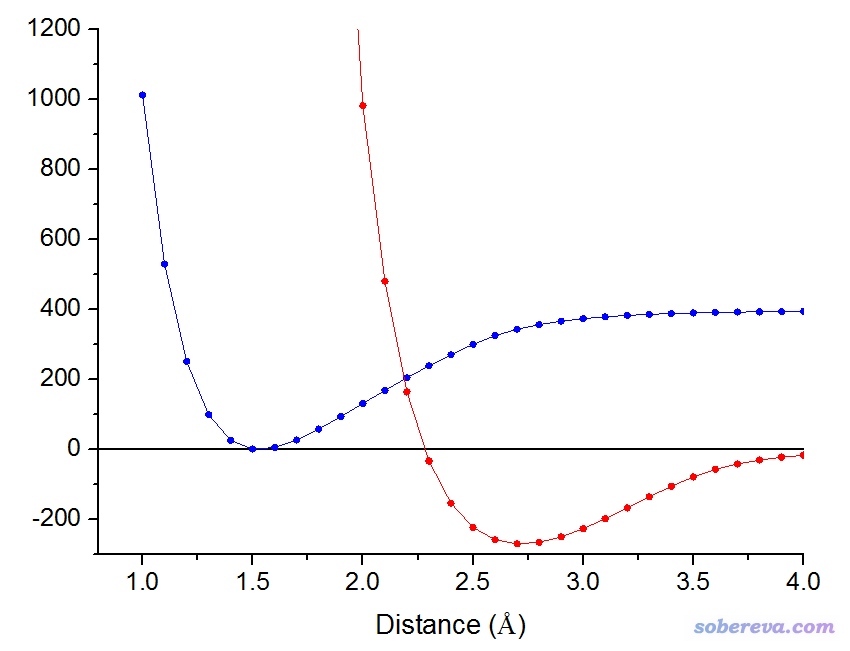
此體系平衡結構下C-C鍵鍵長大約是1.5,如果以拐點作為判斷成鍵與否的標準,相當于超過平衡鍵長52%以內都算作是成鍵的。相比于本文第1節談到的幾何結構判斷標準,拐點這個標準應該說是極其寬松的。這個判斷方式是否有意義,應自行定奪。基于勢能曲線,我們也完全可以定義其它的判斷成鍵的標準,比如說,可以將勢阱深度一半的位置作為一刀切的標準,對于當前體系差不多是r(C-C)=2.17埃的位置。
基于勢能曲線來判斷成鍵的做法普適性差,只對于簡單體系,尤其是雙原子分子比較適用。而對于復雜體系里的某些鍵,往往根本沒法定義恰當的掃描方式(比如環中的),或者即便掃出來了,可能結果也與周圍其它原子存在明顯耦合而沒法被用來判斷成鍵。
PS:想獲得本節的圖其實很簡單。用量化程序做掃描,將結果導入到Origin里,A列是鍵長,B列是電子能量。然后新增一列C,設為每個點相對于勢能曲線最低點的能量差。然后選Analysis-Mathematics-Differentiate,X和Y列分別選A和C,Derivative Order選2,選中Savitzky-Golay Smooth,然后點OK。此時就會出現D列,就是二階導數曲線。把A列數據作為X軸,C、D列數據作為Y軸,繪制折線圖,即可獲得上圖。本文使用的是Origin 9。
6 利用ELF、價層電子密度、變形密度判斷共價鍵的存在
這一節提及的幾個方法主要是用來判斷共價鍵的存在性的,對于討論離子鍵沒直接用處。
電子定域化函數(ELF)是一種十分重要、筆者十分推薦的、普適性非常強的考察什么地方存在共價鍵的方法,在“ELF綜述和重要文獻小合集”(http://bbs.keinsci.com/thread-2100-1-1.html)中有豐富的學習資源,故這里不再詳談。在ELF分析方面Multiwfn是所有程序中最強大、最易用的。十分常用的對ELF進行分析的做法是繪制填色平面圖,繪制方法在上述合集中提到的博文中以及Multiwfn手冊4.4節都介紹了。對于C10O2H7-陰離子的例子,其分子平面上的ELF圖如下
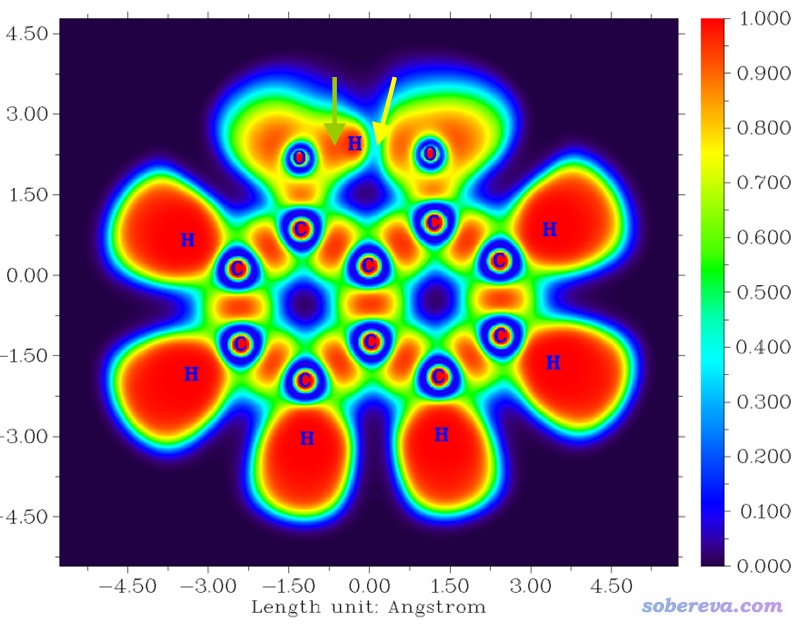
在綠色箭頭所指位置,ELF數值很大,近乎達到了ELF值域上限1.0,因此認為形成了共價鍵是一點問題也沒有的。而黃色箭頭所指位置,ELF數值較小,甚至連0.5都沒到,因此不適合判斷為形成了共價鍵。
對體系的總電子密度繪圖對于討論成鍵問題沒什么用處,而筆者在物理化學學報, 34, 503(http://www.whxb.pku.edu.cn/EN/10.3866/PKU.WHXB201709252)中提出可以利用價層電子密度判斷是否形成共價鍵。價層電子密度在Multiwfn中也可以很便利地繪制,即載入含有波函數信息的文件后,先進入主功能6,選擇選項34把內核軌道占據數清零,然后再照常繪制電子密度圖即可。對于C10O2H7-陰離子,價層電子密度填色圖如下
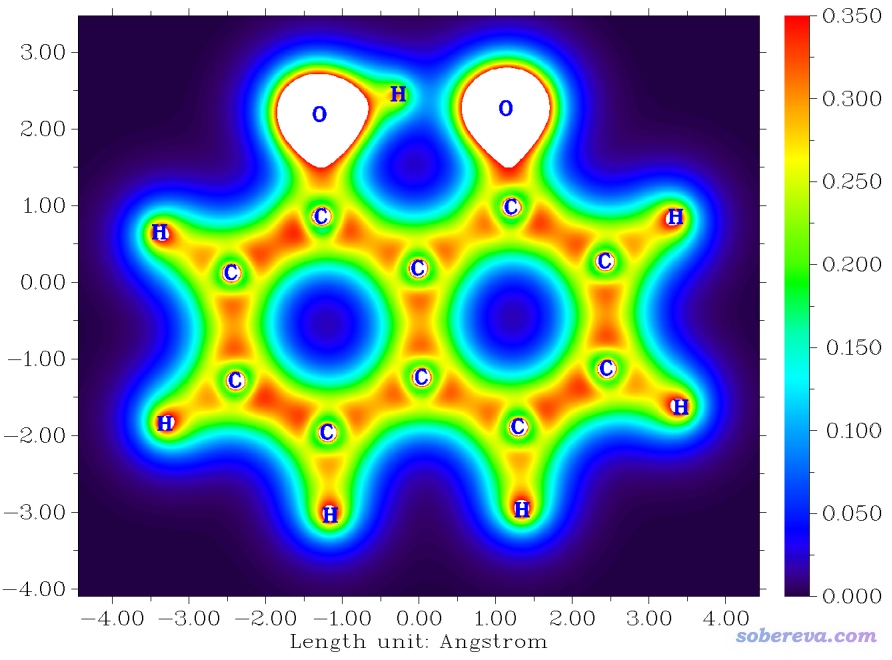
可見,形成共價鍵的原子間價層電子密度都相對來說比較大,在當前色彩刻度設定下呈現橙色或紅色,距離較小的那個O-H鍵也符合這個特點。而距離比較遠的O...H之間價層電子密度明顯非常低,顏色呈青藍色,因此相較之下不適合判斷為形成了共價鍵。
變形密度體現的是構成體系的原子在產生相互作用前后體系電子密度的變化,在《使用Multiwfn作電子密度差圖》(http://www.shanxitv.org/113)中對其有詳細的介紹并介紹了繪制方法。C10O2H7-陰離子的變形密度的等值線圖如下
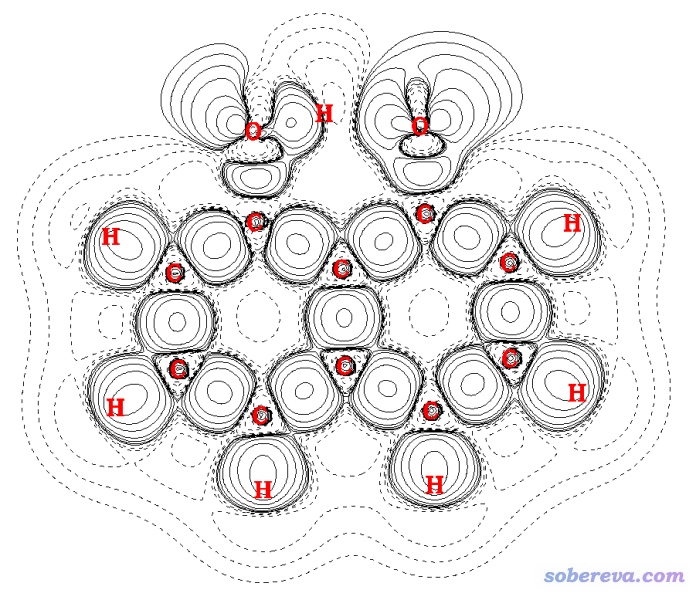
圖中實線是數值為正(電子密度增加)的區域,虛線是數值為負(電子密度減小)的區域。距離近的O-H之間是實線區域,體現出這個區域形成了共價鍵。因為眾所周知,共價鍵的形成總伴隨著兩個原子間電子密度的顯著增加。而距離遠的O...H之間則并非都是實線,因此沒法說形成了典型的共價鍵。
還有很多其它的實空間函數也可以用來判斷是否存在共價鍵問題,比如SCI(J. Phys. Chem. A, 122, 3087)、電子能量密度(Angew. Chem. Int. Ed. Engl., 23, 627)、LOL(J. Mol. Struct. (THEOCHEM), 527, 51)、eta指數(J. Phys. Chem. A, 114, 552)、|勢能密度|/拉格朗日動能密度(J. Chem. Phys., 117, 5529)等等,這里不再多說,它們都可以通過Multiwfn非常方便地分析,在《Multiwfn支持的分析化學鍵的方法一覽》(http://www.shanxitv.org/471)里都有介紹。
7 根據IRI方法圖形化判斷
IRI(interaction region indicator)是筆者在Chemistry—Methods, 1, 231 (2021) DOI:10.1002/cmtd.202100007中提出的方法,在《使用IRI方法圖形化考察化學體系中的化學鍵和弱相互作用》(http://www.shanxitv.org/598)中專門做了介紹,圖文并茂,例子豐富,簡單易懂,讀者務必一看。IRI分析方法可以將化學體系里所有的相互作用,包括弱相互作用和化學鍵作用,同時通過圖像清晰地展現出來,因此通過IRI圖可以一目了然判斷原子間有沒有成鍵,筆者強烈推薦!由于博文寫得特別詳細,本文就不累述了。
8 根據AIM理論的鍵臨界點(BCP)判斷
AIM理論在《AIM學習資料和重要文獻合集》(http://bbs.keinsci.com/thread-362-1-1.html)中有豐富的學習資源。AIM中有個概念叫鍵臨界點(BCP),有的人用兩個原子間是否有BCP作為它們是否成鍵的依據。BCP是基于電子密度從數學角度嚴格定義的概念,其實化學意義說不上多明確。從實際角度來看,BCP算是存在化學鍵的必要非充分條件。化學鍵屬于強相互作用,這一般總會導致BCP的出現,而BCP的出現未必代表兩個原子形成了化學鍵,比如就連兩個Ne原子形成的復合物中也會發現Ne之間存在BCP。不存在BCP時通常可以說相應兩個原子間沒有化學鍵程度的作用,但是不能說兩個原子不存在明顯的弱相互作用。比如筆者在Chemistry—Methods, 1, 231 (2021) DOI:10.1002/cmtd.202100007中所指出的,有的分子內氫鍵(甚至不弱)明明存在,但卻沒有對應的BCP。
對于C10O2H7-陰離子,通過Multiwfn找出的BCP如下圖的桔色小球所示。雖然H19-O17之間算不上化學鍵,但由于已經產生了十分顯著的內氫鍵(甚至由于這個內氫鍵的存在,使得O18-H19的距離比一般羥基中O-H鍵長大不少),因此在H19-O17之間也發現了BCP。
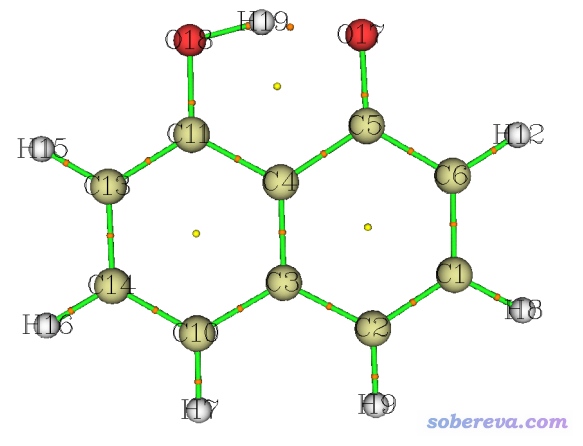
總之,BCP的出現條件比化學鍵的存在要寬松得多。因此BCP的存在不是化學鍵存在的證據,而不存在BCP則可作為否認兩個原子間存在化學鍵的強有力的依據。另外,通過BCP位置上各種實空間函數的數值,還可以判斷原子間相互作用的特征,在《Multiwfn支持的分析化學鍵的方法一覽》(http://www.shanxitv.org/471)和《AIM鍵臨界點處電子密度拉普拉斯值符號判斷相互作用類型失敗原因的圖形分析》(http://www.shanxitv.org/161)文中也有很多討論。