使用Multiwfn繪制紅外、拉曼、UV-Vis、ECD、VCD和ROA光譜圖
注1:筆者后來又撰寫了《使用Multiwfn繪制構象權重平均的光譜》(http://www.shanxitv.org/383),演示了如何使用Multiwfn超級便利地繪制構象權重平均的光譜,強烈建議在讀完本文后閱讀!
注2:筆者后來又撰寫了《使用Multiwfn一鍵批量產生各類光譜圖》(http://www.shanxitv.org/479),配有演示視頻,介紹了如何通過批處理腳本,實現一鍵或一條命令就把當前目錄下所有文件的光譜圖繪制出來,超級方便!
注3:Multiwfn支持基于理論模擬或者實驗測定的UV-Vis光譜預測化學物質的顏色的功能,極具實用性且十分方便!見此文的詳細介紹:《通過量子化學計算和Multiwfn程序預測化學物質的顏色》(http://www.shanxitv.org/662)
文/Sobereva @北京科音
First release: 2014-Mar-19 Last update: 2023-Jun-16
1 前言
Multiwfn是功能最為強大的波函數分析程序,雖然光譜圖的繪制并不是Multiwfn分內的事,但是由于光譜的繪制在計算化學中經常要涉及,為了方便量化研究者,筆者也給Multiwfn加入了繪制光譜的功能,目前此功能已經被很多研究文章所使用。Multiwfn的這個功能比起常用的GaussView繪制光譜的功能靈活、強大得多,可以滿足高端用戶的需求,而且支持的程序不限于Gaussian。目前也有其它一些繪制光譜的程序,和Multiwfn相比又弱又難用,毫無使用價值。此文將介紹在Multiwfn中繪制各類光譜的方法。由于很多人并不清楚理論計算產生光譜的基本原理,比如經常見到有人搞不懂振子強度和峰高、吸光度的關系,因此在第二節將首先介紹一些基本原理。如果急著馬上就作出圖來,可以直接看3.1節對輸入文件的說明和第5節的例子。
除了本文介紹的這些光譜外,Multiwfn還具備靈活強大的繪制NMR譜的功能,見《使用Multiwfn繪制NMR譜》(http://www.shanxitv.org/565)。Multiwfn還可以非常方便地繪制光電子譜,見《使用Multiwfn繪制光電子譜》(http://www.shanxitv.org/478)。
Multiwfn可以在主頁http://www.shanxitv.org/multiwfn上免費下載。如果是第一次接觸Multiwfn程序,建議先閱讀《Multiwfn入門tips》(http://www.shanxitv.org/167)、Multiwfn波函數分析程序的意義、功能與用途(http://www.shanxitv.org/184)了解一些基本信息。
使用Multiwfn繪制各種光譜用于發表文章時請務必記得按照要求恰當引用Multiwfn程序,在程序主頁、程序啟動時的提示以及《Multiwfn FAQ》(http://www.shanxitv.org/452)里都明確說了。
PS:由于Multiwfn在繪制光譜方面的強大、靈活和便利,目前已經有很多文章都利用了Multiwfn繪制了光譜,比如Chem. Asian J., 16, 56 (2021)、Org. Lett., 15, 3526 (2013)、Org. Chem. Front., 6, 1619 (2019)、Inorg. Chem., 58, 4253 (2019)、Tetrahedron, 75, 2797 (2019)、Mar. Drugs, 18, 58 (2020)、Anal. Chem., 91, 4780 (2019)、ChemPhysChem, 19, 2995 (2018)、J Raman Spectrosc., 50, 1405 (2019)、Anal. Chim. Acta, 1106, 88 (2020)。
2 理論光譜產生的原理
2.1 IR、VCD、ECD、UV/Vis光譜
物質的實驗光譜是一條連續的吸收曲線,包含一大堆吸收峰,表現了對不同頻率的光的吸收度的不同,一般用摩爾吸收系數(epsilon,ε)來衡量,含義是溶液濃度為1mol/L、液層厚度為1cm時的吸光度,單位是L/mol/cm。
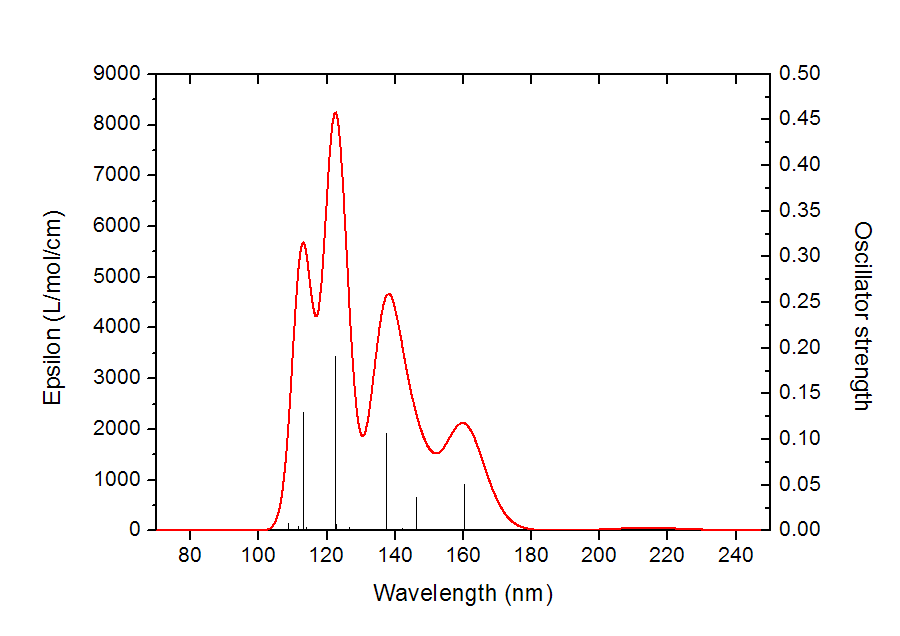
理論光譜計算給出的是離散的躍遷數據。比如做電子激發計算,程序會給出基態到各個電子激發態的躍遷能以及振子強度。比如下圖,黑色的一條條豎線的橫坐標位置就是電子激發能,豎線高度就是振子強度(對應右側坐標軸)。
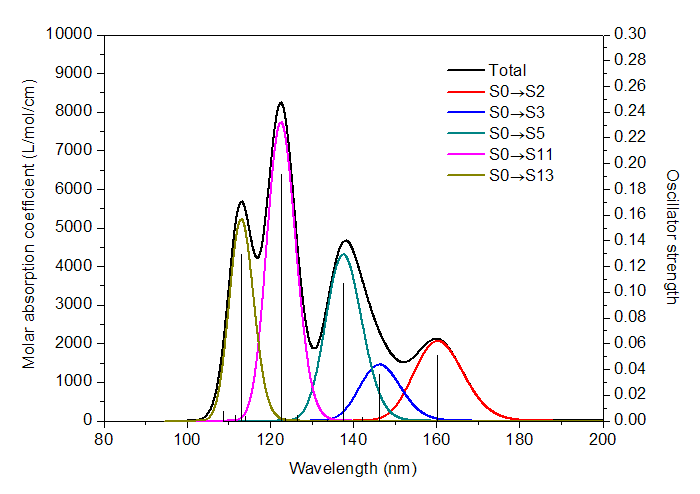
顯然理論計算給出的離散的躍遷數據和實驗給出的連續的吸收曲線完全不同,若想和實驗光譜對應上,以預測實際光譜或者對實驗光譜進行解釋,就需要對理論得到的躍遷數據進行“展寬”成為峰形。先對每個躍遷進行展寬,比如圖中160nm處的豎線對應S0->S2的躍遷,展寬后就是紅色曲線,S0->S11展寬后就粉色曲線(由于躍遷方式很多,為了避免太亂,圖中只示意了把振子強度大于0.01的躍遷展寬后的曲線)。把所有躍遷都這么進行展寬成為X-Y曲線后,再把所有曲線的Y值相加,所得到的總的曲線,即圖中的黑色曲線,這就是理論預測或者說理論模擬出的光譜圖。
吸收曲線的每個峰往往對應于一個強度較大的躍遷。比如圖中113nm左右有個峰,很顯然就是對應于S0->S13的躍遷(振子強度為0.129)。但是峰的位置和具有較大強度的躍遷的能量卻并不總是對應的,無論是實際光譜還是上述方式理論模擬出的光譜都是如此,因為所有躍遷都對附近范圍的吸收曲線有貢獻。例如上圖中S0->S3的躍遷振子強度不算非常小,為0.036,但是黑色曲線在相應位置處(146.3nm)卻沒有峰。其原因從上圖中容易理解,這是因為在S0->S3附近有個振子強度更大的躍遷S0->S5(振子強度為0.107),它對光譜的貢獻(青色曲線所示)比起S0->S3的貢獻(藍色曲線所示)大得多,這導致S0->S3沒有對應的峰,而被淹沒進了最大值在138.2nm處的吸收峰了。這個例子也說明了考察每個躍遷對光譜的各自的貢獻的用處,假設我們不把這些振子強度大的躍遷的貢獻分別畫出來,光譜圖的內在結構是不容易搞清楚的,甚至導致對峰的本質的錯誤指認。這種繪制各個躍遷的獨立的貢獻的功能是GaussView這樣的非專業程序所不具備的,在Multiwfn里則能實現。
上面這個簡單的例子討論的是紫外光譜,這個產生光譜的過程對于紅外、ECD(電子圓二色譜)、VCD(振動圓二色譜)也是一致的。這些光譜的理論計算都會給出一個個躍遷模式的能量和強度值,都得把它們各自展寬成曲線并相加才能得到能夠和實驗結果相對比的光譜。不同類型光譜計算給出的強度有不同的叫法,紅外叫紅外強度,UV-Vis叫振子強度,VCD/ECD計算叫轉子強度(有正有負,對應于左、右兩種圓偏振光)。在下文中我們統稱為躍遷的“強度”。
PS:這里順帶說一下單位。對于紅外、拉曼和VCD譜,常用的單位是cm^-1;對于UV-Vis和ECD,常用的有1000cm^-1、eV和nm。1eV = 8.0655*1000cm^-1。eV為單位的能量的倒數乘以1240.7011就是nm為單位的能量,nm為單位的能量的倒數乘以1240.7011就是eV為單位的能量,因此nm和eV、cm^-1這樣的能量單位并不是線性的轉換關系。振子強度是沒有量綱的,紅外強度單位通常是km/mol(千米每摩爾),有時候也用1 esu^2*cm^2 = 2.5066 km/mol為單位。Gaussian輸出的ECD轉子強度單位是cgs (10^-40 erg-esu-cm/Gauss),VCD轉子強度單位是10^-44 esu^2 cm^2。
給定一個躍遷的能量和強度,怎么把它像前面的圖所示的那樣展寬成峰形的曲線?這需要兩個條件,一是展寬函數,它定義了曲線的函數形式;二是半高全寬FWHM(full width at half maximum),它決定了展寬出的峰在高度為一半的位置的峰的寬度。很多地方也用HWHM,是指峰高一半處寬度的一半,HWHM=FWHM/2。顯然FWHM越大,峰看起來也就越寬,FWHM越小,則峰看起來就越窄,如果FWHM是一個無限小的值,那么這就不是峰了,而是一個豎線了,相當于沒做展寬。下面的圖展示了FWHM為不同值的時候的吸收曲線
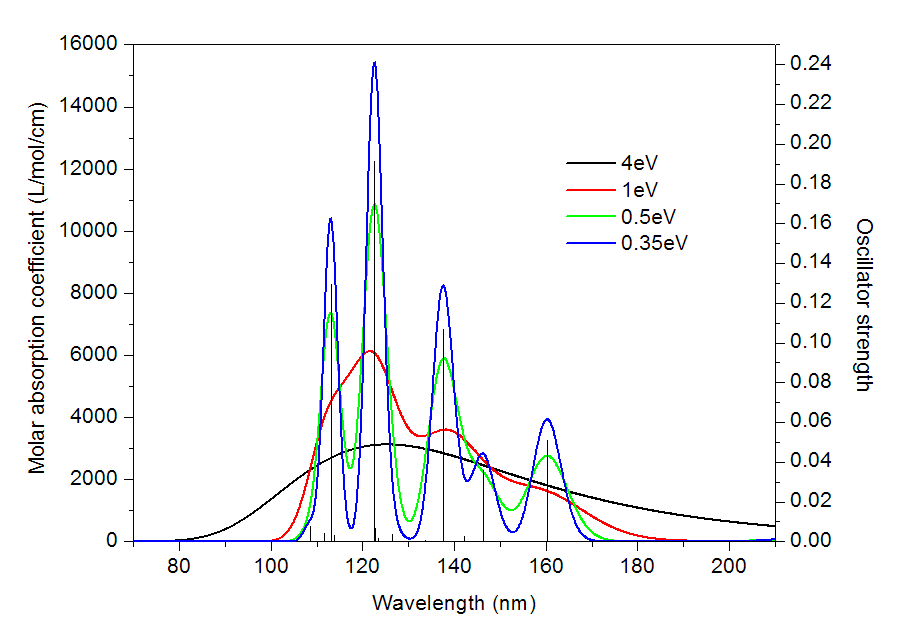
可見,FWHM=4eV的時候,峰的特征根本看不見,整體就是一個大弧線,顯然這樣的光譜是沒有意義的。FWHM減小為1eV的時候,峰的輪廓已經出來了,但還是比較含糊,或者說此時的光譜分辨率太低,連S0->S11和S0->S13躍遷對應的兩個吸收峰都分辨不開。FWHM=0.5eV時,每個強度較大的峰都能看出來了,但是情況和之前一樣,S0->S3和S0->S5躍遷區分不開,合并成了一個峰。FWHM進一步降低到0.35eV,這時候光譜分辨率足夠高了,S0->S3和S0->S5躍遷都能看到各自的吸收峰了。實驗光譜的分辨率是有限的,難以做到非常精細,所以理論模擬光譜的時候FWHM應當選擇合適,以便于能夠和實驗比對。FWHM的選擇帶有一定任意性,如果不知道怎么設就用程序默認的就好了。順帶一提,對于UV-Vis光譜,如果分辨率特別高,不僅能看出電子態的躍遷,還同時可以看到不同振動態的躍遷,這稱為振動分辨的電子光譜,有興趣者可參考《振動分辨的電子光譜的計算》(http://www.shanxitv.org/223)。
說完了FWHM,再來說展寬函數。常用的展寬函數有下面三個,高斯函數、洛倫茲函數和Pseudo-Voigt函數。
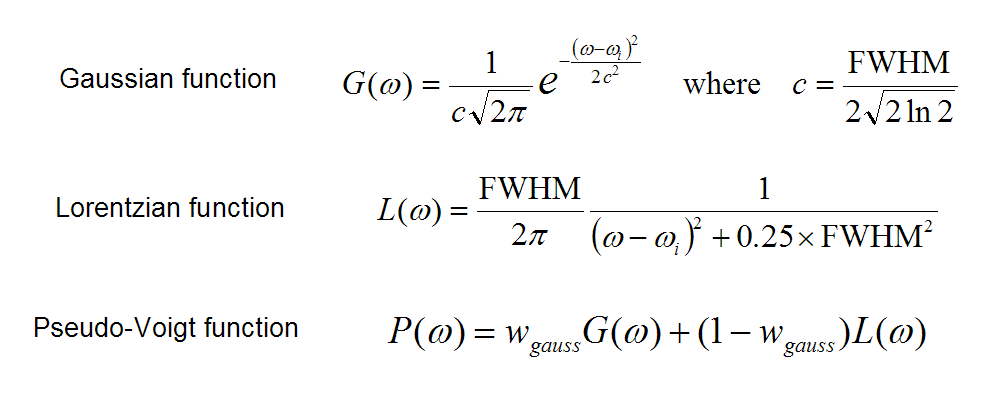
通常,電子光譜(UV-Vis、ECD)用高斯函數來展寬,振動光譜(紅外、拉曼、VCD)用洛倫茲函數來展寬,洛倫茲函數衰減得比高斯函數要緩慢。Pseudo-Voigt函數則是高斯函數和洛倫茲函數的線性組合,組合系數是可調的。上面的公式中,ω就是光譜的橫坐標,在給定了躍遷能量ω_i和FWHM之后,就立刻有了這個躍遷對應的吸收曲線的表達式。在ω=ω_i的位置處峰最高,而隨著ω偏離ω_i函數值逐漸衰減。
上面給出的函數都是歸一化的,也就是說函數的積分值為1。展寬時當然也要把躍遷的強度考慮進去。這里有個重點務必記住:躍遷的強度正比于它對應的峰的積分面積。因此,如果A躍遷的強度是0.5,B躍遷的強度是0.1,那么A展寬出的峰的面積也就應當為B的5倍,所以我們要把振子強度乘到展寬函數前面去。值得一提的是,當FWHM相同時峰高正比于峰面積,所以A的峰高也相應地為B的5倍。
我們有了展寬函數的數學形式,也知道了峰面積和躍遷強度的正比關系,展寬出來的吸收曲線怎么才能和實驗光譜在定量上對應上?對于紅外譜,通過比較紅外強度單位和摩爾吸收系數的單位(見Multiwfn手冊3.13.1節),可以推得如果以cm^-1為橫坐標單位,L/mol/cm為縱坐標單位,則如果一個躍遷的紅外強度為p,那么展寬出的曲線面積應當為100*p,也就是說把展寬函數再乘上100*p即可。對于其它類型的光譜則沒有這樣的形式上的關系,我們只能模擬出光譜的形狀,其數值和實際光譜相差一個系數因子,只有通過將大量實驗光譜和模擬光譜相對比才能得到這個因子。好在對于UV-Vis有人做過這件事,結論是:如果將1000cm^-1單位用于光譜的橫軸,將L/mol/cm單位用于光譜的縱軸,那么1個單位的振子強度展寬出的曲線的積分面積應當為1/4.32*10^6。如果把eV作為橫軸單位,則1單位振子強度應當展寬出面積為28700的曲線。有了這個關系,理論模擬的UV-Vis光譜就和實驗光譜在定量上就有一定可比性了。不過,對于拉曼、VCD、ECD都缺乏這樣的關系,所以只能簡單地讓p強度的躍遷展寬出積分面積為p的峰,得自己調刻度系數來使模擬的譜和實驗譜吻合。
量子化學理論計算出的躍遷都是一個個離散的,為什么實測的光譜是連續的,而不是僅在入射頻率恰當與激發能精確一致的位置才有才有吸收,從而觀測到離散的譜線?這個問題在一些分子光譜書里都有介紹,導致譜線具有有限寬度的原因有很多,比如(1)不確定原理導致的加寬,即激發態壽命是有限的,故而激發態能量有不確定性(2)由于分子的運動產生的多普勒效應導致的加寬(3)分子間碰撞產生能級位移導致的加寬(4)高輻射強度導致低能態的布居數耗盡導致的飽和加寬(5)柔性分子具有大量可及構象。
理論模擬的光譜和實驗光譜常有一定整體的偏差,為了能夠盡量相符,我們往往需要一些調節。一是對光譜的高度進行scale,即乘上刻度系數,使模擬光譜的峰高能和實驗光譜有較好的對應。通常想算準光譜的強度比起算準峰的位置更為困難,能定性符合就不錯了,而且如上所述模擬光譜和實驗光譜的本來就缺乏理論上的對應關系,所以做這樣的高度的scale完全是合理且也是必要的。另外就是對模擬光譜的橫坐標也進行scale或整體加減一個數值,以消除躍遷能量計算的系統性的偏差。比如CIS算的激發能通常偏高,一些研究中會被乘上0.72來修正,而振動光譜眾所周知也需要乘上頻率校正因子以解決計算方法的系統誤差并同時等效地考慮非諧振效應,這點可參見《談談諧振頻率校正因子》(http://www.shanxitv.org/221)。另外,有時候還需要調節FWHM和展寬函數使結果更好地接近實驗譜。這類調節并不算是弄虛作假,因為涉及到的問題是難以克服或者根本不可能克服的,這只是采取一些技巧以便于更好地分析和解釋實驗光譜。
2.2 拉曼光譜
拉曼光譜是散射光譜,橫軸是散射光相對于入射光的頻率,縱軸是散射光的強度。量子化學程序直接算出來的是每個振動模式的拉曼活性(Raman activity),單位一般是?4/amu(amu是原子質量單位)。拉曼活性是每個振動模式自身的特征,和入射光頻率及溫度無關。原理上來說,獲得模擬的拉曼光譜應當先把每個振動模式的拉曼活性轉換為每個振動模式的拉曼強度(Raman intensity),拉曼強度是依賴于入射光頻率和溫度的。轉換關系如下:
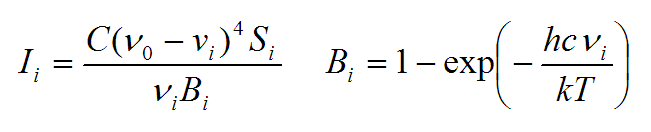
其中S_i、I_i、νi是第i個振動模式的拉曼活性、拉曼強度和振動頻率(波數)。ν0是入射光頻率(波數)。T是溫度。h是普朗克常數,c是光速,k是玻爾茲曼常數。C是個常數,這無關緊要,我們感興趣的并非吸收峰的絕對高度,故隨便選取個值來讓譜圖縱軸數量級合適即可。使用上面這個公式時可以引用筆者的這篇深入研究各種尺寸碳環的振動光譜的文章:Chem. Asian J., 16, 56 (2021) DOI: 10.1002/asia.202001228,文中末尾的Computational Section就給出了這個公式。這篇文章在《揭示各種新奇的碳環體系的振動特征》(http://www.shanxitv.org/578)里有深入淺出的介紹,看看里面的細節對于經常研究振動譜的人很有好處。
這樣轉換成拉曼強度后,再按照上一節所述方式通過洛倫茲函數進行展寬,就得到了可以和實驗對照的拉曼光譜。注意,GaussView等程序繪制拉曼光譜時并不先轉換出拉曼強度,是直接基于拉曼活性進行展寬,這么做顯然是不嚴格的。不過,基于拉曼強度和活性展寬出的圖的峰的位置都一樣,而且每個峰的拉曼強度正比于拉曼活性,因此直接基于拉曼活性展寬出的譜圖倒也并非沒意義,和實際拉曼光譜在基本特征上還是相似的。但顯然,基于拉曼活性和拉曼強度展寬出的譜圖的具體形狀還是有所不同,峰高不同。
還有一種拉曼光譜叫做預共振拉曼光譜,就是指入射光波長和體系的電子激發能非常接近時,拉曼信號會顯著增強,利用這種技術可以使檢出限明顯降低。從理論計算角度來說,計算拉曼活性需要計算極化率對正則坐標的導數。常規拉曼計算時極化率用的是靜態極化率,而計算預共振拉曼時極化率用的是含頻極化率。獲得嚴格的預共振拉曼光譜,不僅極化率要用含頻極化率,而且之后還是要按照上述方式把拉曼活性轉化成拉曼強度才行,即入射光頻率是從兩個方面影響最終的預共振拉曼光譜。
2.3 ROA光譜
ROA(拉曼光學活性)測的是散射的右旋圓偏振光與左旋圓偏振光強度的差值,即I_R - I_L。這是一種振動譜,因此橫坐標的位置和IR是相同的。有手性的物質才有ROA信號,對映異構體的ROA譜正好呈鏡像。ROA譜有不同形式,SCP (scattered circular polarization)是指入射光是線偏振光,散射光是圓偏振光;而DCP (dual circular polarization)是指入射光和散射光全都是圓偏振光。比較常研究的是SCP(180)形式的ROA譜,也叫做SCP backscattered ROA譜,即測量的散射的圓偏振光與入射的線偏振光的方向夾角是180度。Gaussian還能算SCP(90)和DCP(180)譜。ROA譜和Raman一樣也是依賴于入射光頻率的,繪制嚴格的ROA譜,也是要基于Gaussian給出的數據將ROA活性,使用與Raman活性->Raman強度相同的公式轉換為真正意義的ROA強度。Gaussian在計算ROA的時候還順帶會給出當前入射光波長下的Raman SCP(180)、Raman SCP(90)、Raman DCP(180)數據,由Raman活性轉換為Raman強度后對應散射的右旋圓偏振光與左旋圓偏振光強度的總和,即I_R + I_L。由于這時候的入射光波長肯定遠遠偏離電子激發能,無法引發共振,故這時得到的Raman譜可以叫做far from resonance拉曼譜。
3 Multiwfn繪制光譜的輸入文件
3.1 Gaussian的輸出文件
Multiwfn能夠直接從Gaussian的輸出文件中讀取躍遷能和強度數據用于作圖。繪制不同的光譜圖所要求的關鍵詞如下(1)對于紅外光譜,需要寫freq關鍵詞。從G09 D.01開始,freq=anharm非諧振計算不僅給出非諧振頻率還給出對應的強度,對于這樣的Gaussian輸出文件,Multiwfn在讀入時會提示你要載入非諧振的數據還是諧振的數據。
(2)對于拉曼光譜,寫freq=raman關鍵詞就可以了(由于長期以來以訛傳訛,無數人居然誤以為Gaussian的freq任務默認就計算raman,還每次都特意寫上noraman以為這樣會節省時間,這是彌天大誤,Gaussian只對HF的freq任務才默認計算raman!!!)。若要繪制非諧振的Raman光譜,用freq(raman,anharm)的輸出文件。
如果要繪制預共振拉曼,應當還同時寫上CPHF=rdfreq關鍵詞,并在輸入文件末尾空一行寫上所有要算的入射光頻率,比如300nm 400nm 500nm。如果不寫單位,則默認是原子單位。
(3)對于VCD光譜,寫freq=VCD關鍵詞就可以了。Gaussian從G16開始支持非諧振的VCD計算,用freq(VCD,anharm)的輸出文件可以在Multiwfn里繪制非諧振的VCD譜。
(4)對于UV-Vis和ECD光譜,直接用普通的TDDFT、TDHF、CIS、ZINDO、EOM-CCSD的輸出文件即可,不需要再加其它關鍵詞。不會做TDDFT者參看《Gaussian中用TDDFT計算激發態和吸收、熒光、磷光光譜的方法》(http://www.shanxitv.org/314)。ECD的轉子強度有兩種表象,一種是長度表象,另一種是速度表象,前者依賴于原點而后者則不依賴,在完備基組下它們是一致的,但是在有限基組下則有些偏差,盡管定性一致,Gaussian會同時給出二者,在Multiwfn中可以選擇讀取哪種轉子強度(GaussView作ECD圖時用的是速度表象的轉子強度)。
(5)對于ROA光譜,寫freq=ROA,并在輸入文件末尾空一行寫上所有要算的入射光頻率,比如450nm 532nm 600nm。
對于上述非諧振的情況,Gaussian不僅會算出基頻的數據,還會算出泛音和合頻的數據,當你讓Multiwfn載入非諧振數據的時候,基頻總是會載入的,而泛音或合頻的數據是否載入可以自由選擇。
對于入射光頻率不止一個的情況,載入預共振拉曼或者ROA數據的時候,可以選擇載入哪個頻率的。
3.2 ORCA的輸出文件
Multiwfn能夠直接從ORCA的輸出文件中讀取數據繪制IR、Raman、UV-Vis和ECD光譜。需要用到關鍵詞參看Multiwfn手冊3.13.2節的詳細說明。對于UV-Vis和ECD,不僅支持ORCA的TDDFT、TDHF、CIS、ZINDO、EOM-CCSD、(DLPNO-)STEOM-CCSD的輸出,還支持內嵌在ORCA里的能夠快速計算很大體系的sTDA和sTD-DFT任務的輸出。從ORCA 4.1開始ORCA支持了在TDDFT計算過程中考慮旋軌耦合(SOC)效應,只要在%tddft里加入dosoc true即可,非常容易。基于這樣的輸出文件,Multiwfn可以繪制考慮SOC后的UV-Vis和ECD光譜,例子見《使用ORCA在TDDFT下計算旋軌耦合矩陣元》(http://www.shanxitv.org/462)。3.3 sTDA輸出的文件
sTDA是Grimme提出的一種超級快速地計算大體系激發態的方法,Grimme也給出了同名的sTDA程序(https://www.chemie.uni-bonn.de/pctc/mulliken-center/software/stda/stda),此程序輸出的stda.dat文件也可以作為輸入文件來繪制UV-Vis和ECD光譜。
3.4 xtb輸出的文件
GFN-xTB是半經驗層面的DFT方法,可以快速計算幾百甚至上千原子的大體系的能量,以及做優化和振動分析。GFN-xTB方法可以通過xtb程序實現(https://github.com/grimme-lab/xtb/),使用簡介見《將Gaussian與Grimme的xtb程序聯用搜索過渡態、產生IRC、做振動分析》(http://www.shanxitv.org/421)。xtb 6.5及以后版本做振動分析任務(例如運行xtb test.xyz --ohess)后在當前目錄下產生的vibspectrum文件可以作為Multiwfn的輸入文件用來繪制紅外光譜圖。
3.5 CP2K輸出的文件
可以使用第一性原理程序CP2K的振動分析的輸出文件作為Multiwfn的輸入文件用于繪制孤立或周期性體系的紅外光譜和拉曼光譜。按照《使用Multiwfn非常便利地創建CP2K程序的輸入文件》(http://www.shanxitv.org/587)里所述,這樣的CP2K輸入文件敲幾下鍵盤就能產生。也可以使用CP2K做TDDFT的輸出文件當Multiwfn的輸入文件來繪制UV-Vis譜,過程極為簡單,此文有完整的例子:《使用CP2K結合Multiwfn對周期性體系模擬UV-Vis光譜和考察電子激發態》(http://www.shanxitv.org/634)。
3.6 文本文件
考慮到很多人不是以上程序的用戶,也為了靈活起見,以便于自定義,Multiwfn也支持從文本文件里直接讀取躍遷能和強度,并且以這種方式輸入數據還有個額外的好處,就是可以定義每個躍遷各自的FWHM,而不強制要求對所有躍遷在展寬時都用相同的FWHM。這樣的文本文件的格式如下:numdata inptype
energy strength [FWHM] //1號躍遷的能量、強度和FWHM。FWHM不是必需要寫的
energy strength [FWHM] //2號躍遷
energy strength [FWHM] //3號躍遷
...
energy strength [FWHM] //numdata號躍遷
其中numdata是躍遷的數目,也就是條目數。intype為1時只讀取躍遷的能量和強度,FWHM由程序自動設定;intype為2時還同時讀取每個躍遷的FWHM。躍遷信息應當按照能量從小到大排列。
對于繪制紅外、拉曼和VCD光譜,此文件里的能量和FWHM的單位必須都是cm^-1,而對于UV-Vis和ECD則必須以eV為單位。此文件里的強度單位對于紅外、拉曼、ECD和VCD必須分別是km/mol, ?^4/amu, cgs (10^-40 erg-esu-cm/Gauss), 10^-44 esu^2 cm^2,這也正是Gaussian程序輸出的默認的單位。而UV-Vis振子強度本身就是無量綱的。
這種文本文件的一個例子如下。將它作為Multiwfn啟動時載入的文件即可。
6 2
81.32920 0.72170 8.0
417.97970 3.58980 8.0
544.67320 21.06430 8.0
583.12940 41.33960 8.0
678.66900 91.47940 8.0
867.37410 2.94480 8.0
4 使用方法和選項
啟動Multiwfn并載入輸入文件后,進入主功能11,然后選擇要繪制的光譜類型,然后就能看見一個菜單,選0就可以立刻繪制出光譜。圖中既有模擬出的吸收曲線,對應左側坐標軸;也顯示出了各個躍遷,以一條條離散的豎線表示,數值對應于右側坐標軸。另外還包含很多其它選項,下面依次介紹,對于不同類型的光譜選項的具體文字往往會不同。
-2 Export transition data to plain text file:把躍遷數據導出到當前目錄下的transinfo.txt,格式和3.3節介紹的格式一樣,因此用戶可以自行修改里面的數值然后直接作為Multiwfn的輸入文件。
-1 Show transition data:這個功能把躍遷數據直接顯示到屏幕上,例如
Index Freq.(cm^-1) Intens.( km/mol esu^2*cm^2)
1 81.32870 0.72170 0.28792
2 417.97970 3.58980 1.43214
3 544.67310 21.06430 8.40353
4 583.12940 41.33960 16.49230
5 678.66890 91.47940 36.49541
很多人想把Gaussian輸出文件里的躍遷數據給批量提取出來,但自己又不會寫腳本去實現,那么就可以用Multiwfn的這個功能了。躍遷能量、強度整整齊齊地列出來,將它們從屏幕上直接拷貝下來,就可以很方便地進一步處理了。如果不知道怎么從屏幕上把數據拷貝下來,參見手冊5.4節。
1 Save picture:把光譜圖輸出到當前目錄下,和選項0看到的圖一樣。圖像格式可以通過-4 Set format of saved graphical file選項來修改,默認格式也可以通過settings.ini里的graphformat來設。圖像的寬和高由settings.ini里的graph1Dsize參數決定。吐血建議使用pdf格式保存圖像,這樣線條和文字都比用png等位圖格式清晰平滑得多!雖然pdf格式一般沒法嵌入文章里,但你把pdf用觀看程序打開然后截圖轉成png格式,效果也比Multiwfn直接導出的png格式要好很多。
2 Export X-Y data set of lines and curves to plain text file:把模擬出的光譜曲線以X-Y數據點形式導出到當前目錄下的spectrum_curve.txt中,而離散的豎線則導出到spectrum_line.txt里。把這兩個文件導入到Origin之類的程序里作圖,就可以得到和Multiwfn里產生的圖同樣的效果。由于Origin這樣的程序可調選項更為豐富,因此如果你覺得Multiwfn直接給出的圖的效果不滿意,可以基于這兩個文件在自己擅長的繪圖軟件中作出完全符合自己要求的圖。
3 Set lower and upper limit of X-axis:設定光譜圖的X軸上、下限和刻度間隔,默認是自動確定。自行設定的上下限可以反過來,比如既可以輸入0,1000,100也可以輸入1000,0,100,只不過圖像左右反轉了。
4 Set left Y-axis、5 Set right Y-axis:設定左、右側Y坐標軸。需要輸入起始值、終止值和刻度間隔。
6 Select broaden function:選擇展寬函數,包括Gaussian、Lorentzian和Pseudo-Voigt。
7 Set scale ratio for curve:光譜圖的刻度因子,設為k的話,那么最后光譜圖的數值就會被乘以k。
8 Input full width at half maximum (FWHM):設定FWHM。
9 Toggle showing discrete lines:在繪制的光譜圖中是否把表示躍遷的離散的線顯示出來。
10 Switch the unit of infrared intensity / Set the unit of excitation energy:對于紅外,在km/mol和esu^2*cm^2兩個紅外強度單位之間切換。對于UV-Vis和ECD,設定光譜圖中的能量單位,可以為eV、nm或1000cm^-1。
11 Set Gaussian-weighting coefficient:當展寬函數被選為Pseudo-Voigt時,就可以用這個選項來設定高斯函數的權重。
12 Set shift value in X:設定模擬的光譜圖的X坐標的位移值。
13 Set colors of curve and discrete lines:設定圖中的曲線以及離散的豎線的顏色。
14 Set scale factor for transition energies (or vibrational frequenices):對所有或特定的躍遷能量乘以一個因子,主要用于實現施加頻率校正因子的目的。
15 Output contributions of individual transitions to the spectrum:需要輸入一個閾值,比如k。然后就像選項2一樣會輸出spectrum_curve.txt和spectrum_line.txt,只不過這次輸出的spectrum_curve.txt里面還包含了所有強度絕對值大于k的躍遷產生的貢獻。如果你輸入0,然后輸入一個光譜橫坐標位置,則對此處貢獻最大的10個躍遷的貢獻值和貢獻百分比會被輸出。
16 Find the positions of local minima and maxima:列出光譜圖中的極大值和極小值的位置和數值,這對于確定峰的準確位置很有用。
17 Other plotting settings:其它一些零碎的作圖設置,例如切換是否顯示譜圖上的虛線網格、是否顯示坐標軸上的標簽、修改坐標軸標簽和文字的尺寸、設置圖例的位置、設置坐標軸的數值類型(浮點數、指數、科學計數法)、設置坐標軸保留的小數位等。
19 Convert Raman (or ROA) activities to intensities:對于繪制拉曼光譜,默認情況下繪制的是基于拉曼/ROA活性展寬的光譜。如果選了這個選項,并且輸入溫度和入射光波數,就會按前述公式把拉曼活性轉換為拉曼強度。之后再作圖看到的就是基于拉曼強度展寬的結果了。若再次選這個選項,并且輸入的頻率和溫度和之前一樣,則會轉換成原先的拉曼活性,相當于逆操作。如果是ROA,用這個選項可以把Gaussian直接輸出的ROA強度轉換為真正意義的ROA強度。
20 Modify strengths:手動設定指定躍遷的強度數據。
22 Set thickness of curves/lines/texts/axes/grid:用于設置曲線、離散豎線、文字、坐標軸、虛線網格的粗細。
23 Set status of showing spikes to indicate transition levels:繪制光譜時可以在作圖區域底部顯示豎線來指示各個躍遷能級的位置(這和上述的離散豎線不一樣,因為高度都相同),還可以通過設置顏色區分不同的躍遷類型。這個選項就是用來做具體設定的,見本文第8節的例子。
在光譜繪制界面里經常要進行諸多設置才能達到最滿意的效果。為了避免日后重新繪制完全相同的圖(或在此基礎上進一步修改)時還得再重新輸入一遍設置,在光譜繪制界面里允許用戶輸入s(意為save)來將作圖設定保存到一個用戶指定的文本文件里。日后再次啟動Multiwfn,載入相同體系并進入光譜繪制界面后,只需輸入l(意為load),然后選擇之前保存的繪圖設置文件,就能立刻恢復之前的作圖設定。
Multiwfn繪制光譜的時候實際上是計算一系列點上的值,在當前的X軸范圍內總共計算num1Dpoints個點。num1Dpoints是settings.ini文件里的參數,默認是3000。這個參數設定得越大,光譜越平滑,導出的X-Y數據點也就越多。通常默認數值足夠了,再增加也看不出什么變化。
5 實例
下面給出Multiwfn繪制各種類型光譜的實例。請閱讀完整,因為有不少用法和技巧是對各種類型光譜繪制都共通的,它們穿插在這些例子中介紹。
5.1 紅外光譜
這個實例繪制NH3BF3的紅外光譜。啟動Multiwfn,然后依次輸入
examples\spectra\NH3BF3_freq.out //程序自帶的例子文件,是B3LYP/6-31G*下的振動分析
11
1 //紅外光譜
0 //在屏幕上顯示光譜
立刻可以看到下圖
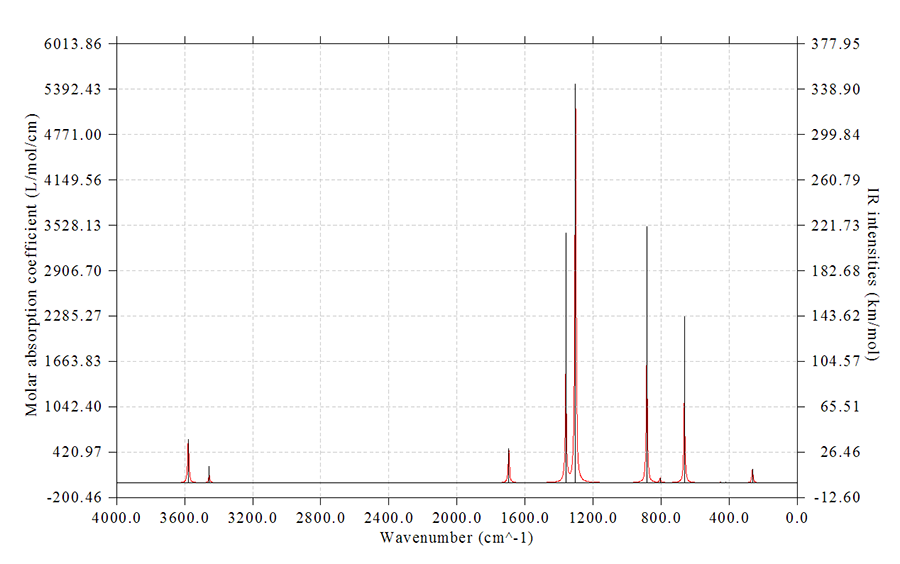
在圖上點右鍵就可以關閉圖像。
在光譜顯示出來的同時我們在文本窗口里可以看到光譜曲線的極值點位置和數值也被顯示了出來:
Extrema on the spectrum curve:
Maximum 1 X: 3578.5262 Value: 585.2897
Maximum 2 X: 3454.4848 Value: 110.4777
Maximum 3 X: 1695.2317 Value: 460.3222
Maximum 4 X: 1359.1197 Value: 1717.7809
Maximum 5 X: 1303.1010 Value: 5467.1462
Maximum 6 X: 884.2948 Value: 1738.5775
...略
注意極值點的位置的定位精度和前面提到的num1Dpoints參數有關,此參數值越大,數據點的間距越小,對于特別是尖銳的峰的描述越精確,峰的最大值的位置的定位精度也就越高。
接下來我們隨意地對圖像進行一些調整,都不是必要的,純粹只是示例一下而已
14 //對頻率乘上校正因子
按回車選擇所有模式
0.9614 //對這些模式的躍遷能乘上B3LYP/6-31G*下的頻率校正因子0.9614(低頻區域的校正因子和這個值是顯著不同的,這里為了省事就沒有單獨對高頻和低頻部分單獨乘上各自的校正因子)。實際上這一步直接按回車也行,會自動用0.9614
9 //不顯示離散的豎線
8 //修改FWHM
20 //改為20cm^-1
4 //修改左側坐標軸
2400,-200,200 //Y軸下端為2400,上端為-200,步長為200。此時得到的圖是上下反轉的,可以與縱軸為透過率的實驗IR譜圖對應
y //對應地修改右側坐標軸的設定使得左右坐標軸的零點位置一致(選y或n無所謂,反正這里我們不繪制出離散的豎線)
0 //重新作圖
結果如下所示
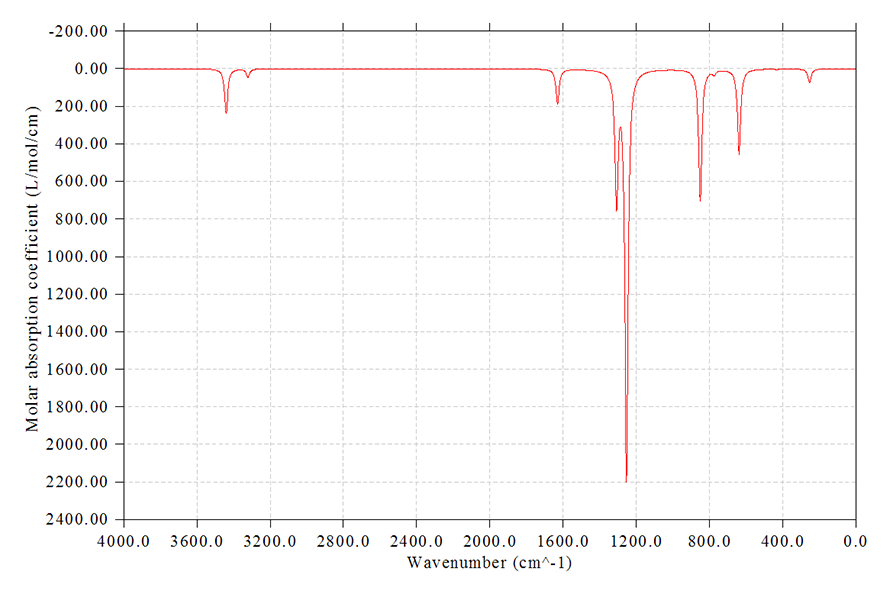
不僅當前的光譜圖已經是考慮頻率校正因子后的了,如果你現在用選項-1看躍遷數據,給出的值也已經是考慮校正因子后的情況了。如果你不再施加校正因子了,那么再次進入選項14,輸入y(代表把頻率恢復成原先的),然后校正因子輸入為1即可。
你也可以對不同的振動模式設置不同的頻率校正因子。進入選項14后對特定的一批模式設一個校正因子后,可以再次進入選項14,然后輸入n(不把已校正過的頻率恢復成原先的頻率),然后再選另一批模式并設置校正因子。可以反復多次這樣操作,效果會疊加。
5.2 拉曼光譜
和繪制紅外光譜過程沒什么區別。用第3節方式得到的Gaussian/ORCA的輸出文件或文本文件作為Multiwfn的輸入文件,然后進主功能11,然后選Raman,再選0繪圖即可。比如苯胺的圖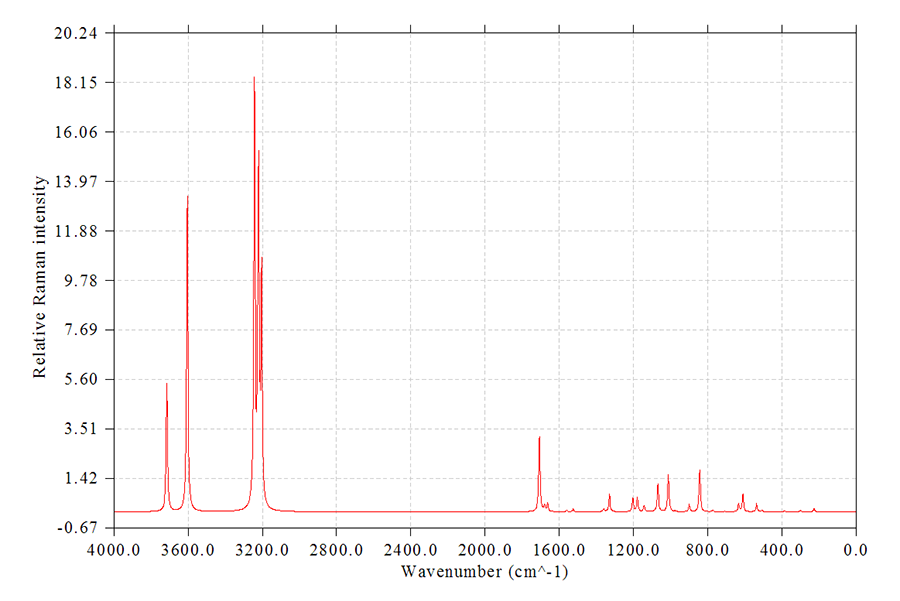
上面的圖是直接基于拉曼活性展寬產生的,若要更嚴格地繪制拉曼光譜,則應當先選擇19,輸入入射光波數和溫度來轉換成拉曼強度,然后再選0看到的就是基于拉曼強度展寬出的拉曼光譜了,此時峰的高度明顯更為合理,更接近于實驗。
如果要繪制預共振拉曼光譜,就把預共振拉曼計算的輸出文件載入Multiwfn即可,進入繪制光譜界面前程序會讓你選擇載入哪個激發波長時的拉曼活性信息。然后,最好再按照如上方式把拉曼活性轉化成拉曼強度,之后選0繪圖。
對于諧振近似下計算的拉曼,強烈建議在繪圖前乘上當前級別的頻率校正因子以修正諧振近似對頻率帶來的系統性誤差。
5.3 UV-Vis光譜
這一節我們我們用乙酸的TDDFT的輸出文件為例子繪制UV-Vis譜,這也正是本文第二節討論的那個光譜,用的是程序自帶的例子文件,是B3LYP/cc-pVDZ TD(nstates=15)的輸出。如果需要得到更完整的譜,nstates應該更大,但這里不追求得到準確的譜,只是示例罷了。啟動Multiwfn后輸入examples\spectra\acetic_acid_TDDFT.out
11 //繪制光譜
3 //UV-Vis
0 //繪圖
然后就看到了圖。
這里介紹下怎么把數據導出來用Origin程序來重新繪制,這樣專業的繪圖程序在繪圖時擁有更多更靈活的可調選項。選擇2,曲線數據就導出到了當前目錄下spectrum_curve.txt下面,屏幕上也明確提示了其中每一列數據是什么含義。這里第一列是波長(nm),第二列是摩爾吸收系數。將spectrum_curve.txt直接拖到Origin窗口里(本文用的Origin 8),選擇繪制曲線圖,把A、B兩列分別當成X和Y軸數據即可。Multiwfn還同時輸出了spectrum_line.txt,把這里面的兩列數據分別當成X和Y軸在Origin里繪制成曲線圖,就得到了一條條離散的豎線表示出躍遷能和強度(實際上,每個豎線對應于一個往返的折線,比如對(5,0),(5,3.2),(5,0)這三個點繪制成曲線圖,看起來就是在X=5的位置上出現了高度為3.2的豎線)。若在曲線圖的那張圖上點右鍵選New layer-(Linked) Right Y,在新增的這個Layer上繪制spectrum_line.txt的數據,經過一些細微的調整,就能得到下面的圖
如果想繪制出本文第二節給出的那種顯示每個躍遷各自貢獻的圖,需要選15,然后輸入閾值,強度絕對值大于這個閾值的躍遷對每個點的貢獻會被輸出到spectrum_curve.txt的第三列及之后的列當中,從屏幕上可以看到列的編號和躍遷模式的編號的對應關系:
Column# Transition#
3 2
4 3
5 5
6 11
7 13
例如第6列的數據對應于基態到第11激發態的躍遷展寬出的曲線。而spectrum_curve.txt的前兩列數據,以及同時輸出的spectrum_line.txt,和選項2輸出的完全一樣。把這兩個文件里的數據都放到Origin里作圖,就能得到第二節的那種圖了。examples\spectra目錄下的acetic_acid_TDDFT.opj是那幅圖的Origin 8的.opj文件,如果不知道怎么繪制可以直接參考這個文件。
在Multiwfn中還可以特別方便地計算出特定位置由各個躍遷的貢獻量。使用Multiwfn顯示UV-Vis光譜時,在文本窗口看到了如下光譜極大點信息:
Maximum 1 X: 113.0588 Value: 5680.9864
Maximum 2 X: 122.5085 Value: 8239.4308
Maximum 3 X: 138.0728 Value: 4667.7944
Maximum 4 X: 159.7516 Value: 2123.3506
Maximum 5 X: 213.7324 Value: 48.5312
假設這里我們要考察138.0728 nm的那個峰的主要貢獻來源,就選擇選項15,輸入0,然后輸入138.0728,之后對此處貢獻最大的10個躍遷的貢獻量和貢獻百分比就被輸出了,如下所示
Sum of absolute values of all transitions: 4667.79444
The individual terms are ranked by magnitude of contribution:
#Transition Contribution %
5 4273.59504 91.555
3 309.47511 6.630
4 68.05085 1.458
6 11.67756 0.250
11 2.33209 0.050
8 2.13478 0.046
7 0.22284 0.005
2 0.14507 0.003
10 0.10019 0.002
9 0.06091 0.001
可見貢獻最大的是S0->S5躍遷,S0->S3也有一定貢獻但相對次要。
對于其它類型的光譜,如下述的ECD等,同樣可以以上述方式獲得各個躍遷產生的獨立貢獻曲線和對某個波長處的貢獻值。
用Multiwfn也能容易地繪制熒光光譜。根據Kasha規則熒光是從S1發射的(但也有違背Kasha規則的情況),因此和繪制吸收光譜的關鍵不同點是要把S1以上激發態的振子強度都設為0。具體來說是先做S1激發態優化任務,然后將輸出文件載入Multiwfn,依次選11、3。然后進選項20,選擇除了第一激發態以外的所有的態(比如算了5個態,就輸入2-5),然后輸入0使得它們的振子強度為0。然后再選0繪圖,看到的就是熒光光譜了。
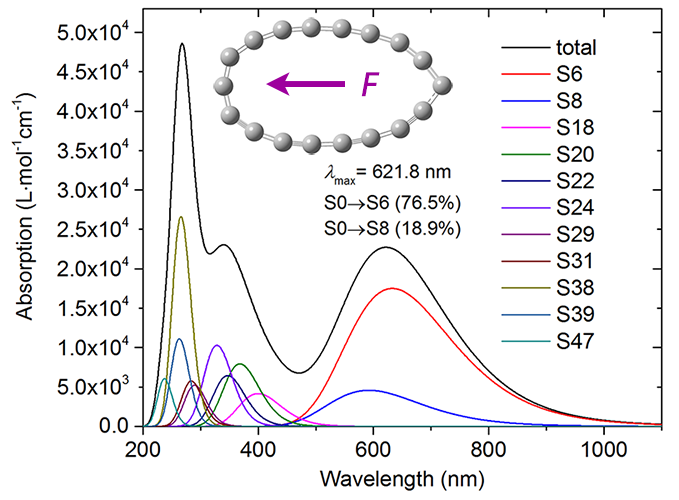
值得一提的是,在《一篇文章深入揭示外電場對18碳環的超強調控作用》(http://www.shanxitv.org/570)一文中,筆者研究了電場對18碳環電子吸收光譜的影響,發現在0.0275 a.u.這較強電場下體系在600多nm處出現了一個新的峰。為了研究其本質,筆者對光譜曲線以上述方式用Multiwfn做了分解,得到如下圖像。此圖清晰地展現出S0->S6激發是這個吸收峰的主要貢獻者,因此之后用Multiwfn分析這個激發的特征就能基本闡明這個新的峰是怎么來的了。具體分析見http://www.shanxitv.org/570這篇文章。
Multiwfn還可以對具有各向異性特征的體系繪制體系對不同方向打來的光的吸收光譜,對于深入認識這類體系的光譜本質特征非常有用,強烈建議看看此文:《使用Multiwfn計算特定方向的UV-Vis吸收光譜》(http://www.shanxitv.org/648)。
5.4 振動圓二色譜(VCD)
這里來繪制S-methyloxirane的VCD譜,用的是程序自帶的例子文件,是B3LYP/6-31G*下由freq=VCD關鍵詞計算得到的。啟動Multiwfn后輸入examples\spectra\S-methyloxirane_VCD.out
11
5 //繪制VCD
0 //顯示光譜
假設我們感興趣的是700~1700cm^-1部分,想只看這部分,于是選3修改橫坐標范圍,輸入1700,700,100,再選0重新作圖,得到下面的圖
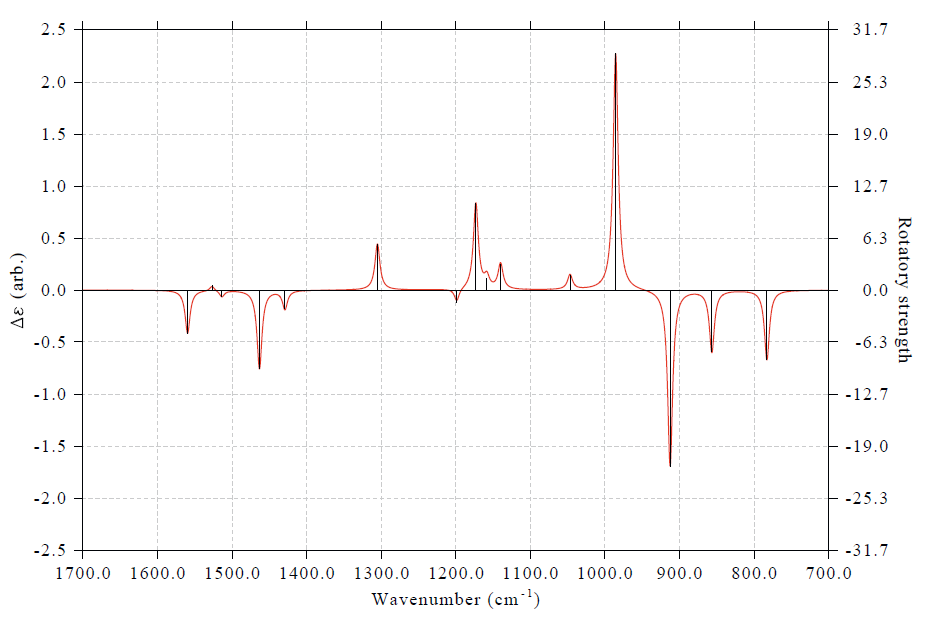
對于雜化泛函結合諧振近似計算的VCD,建議在繪圖前乘上當前級別的基頻頻率校正因子以修正系統性誤差。
5.5 電子圓二色譜(ECD)
這里用天冬酰胺為例子繪制ECD譜。啟動Multiwfn后輸入
examples\spectra\Asn_TDDFT.out //PBE1PBE/6-311G* TD(nstates=30)計算的輸出
11
4 //繪制ECD
2 //讀取速度表象的轉子強度
0 //繪制圖像
結果如下
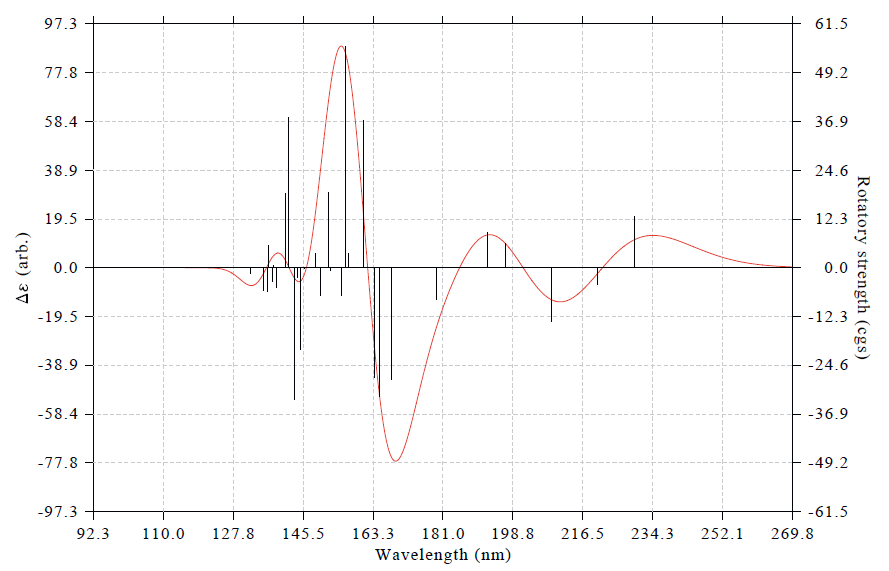
兩篇典型的使用Multiwfn繪制天然產物ECD光譜的文章見Org. Lett.,15,3526(2013)和J. Nat. Prod.,77,346(2014)。左側坐標軸里的arb.是arbitrary unit(任意單位)的縮寫。要知道VCD和ECD譜感興趣的只是曲線的形狀,而不是絕對數值,所以理論模擬的ECD譜的單位就應該標注為arb.(PS:以前有人問我怎么把坐標軸弄成和UV-Vis一樣的L/mol/cm,這是毫無意義的問題)。
借這個例子的機會,筆者展示一下如何將光譜極值點非常方便地標注在圖上。在繪圖界面里接著輸入
16 //修改極值點標簽顯示設置
1 //修改標簽顯示狀態
3 //顯示極大點和極小點標簽
0 //返回
4 //修改Y軸(因為顯示了標簽,為了避免標簽露在外頭,因此加大Y軸范圍)
-100,110,20 //Y軸下限、上限、刻度間隔
y //相應地縮放右側Y軸
0 //繪制光譜
此時看到下圖,可見極大和極小點的標簽都已經清楚出現在了圖上
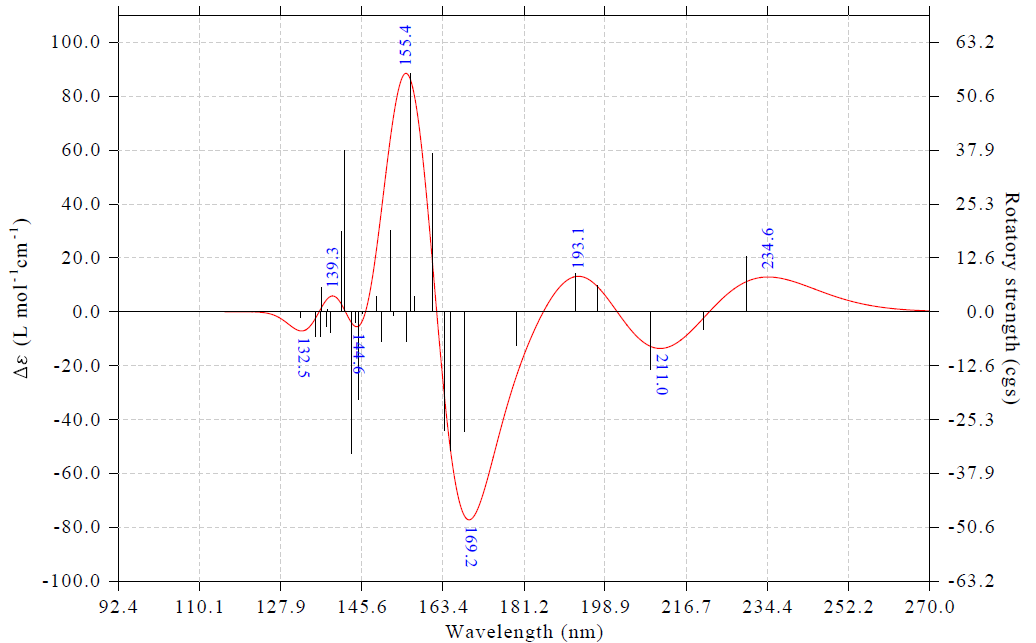
通過選項16里的子選項,可以對標簽的標注情況做很多自定義和調整,這里隨便做一些操作來進行演示。接著輸入
16 //修改極值點標簽顯示設置
6 //切換標簽內容為Y軸數值
4 //切換為不旋轉標簽
3 //設置標簽的小數位數
0 //0個小數位,即顯示為整數
2 //設置標簽尺寸
50 //令標簽比默認的更大(默認是30)
0 //返回
3 //設置橫坐標
120,280,20 //調整下限、上限和標簽間隔,使光譜主體特征充滿畫面
0 //作圖
此時看到下圖
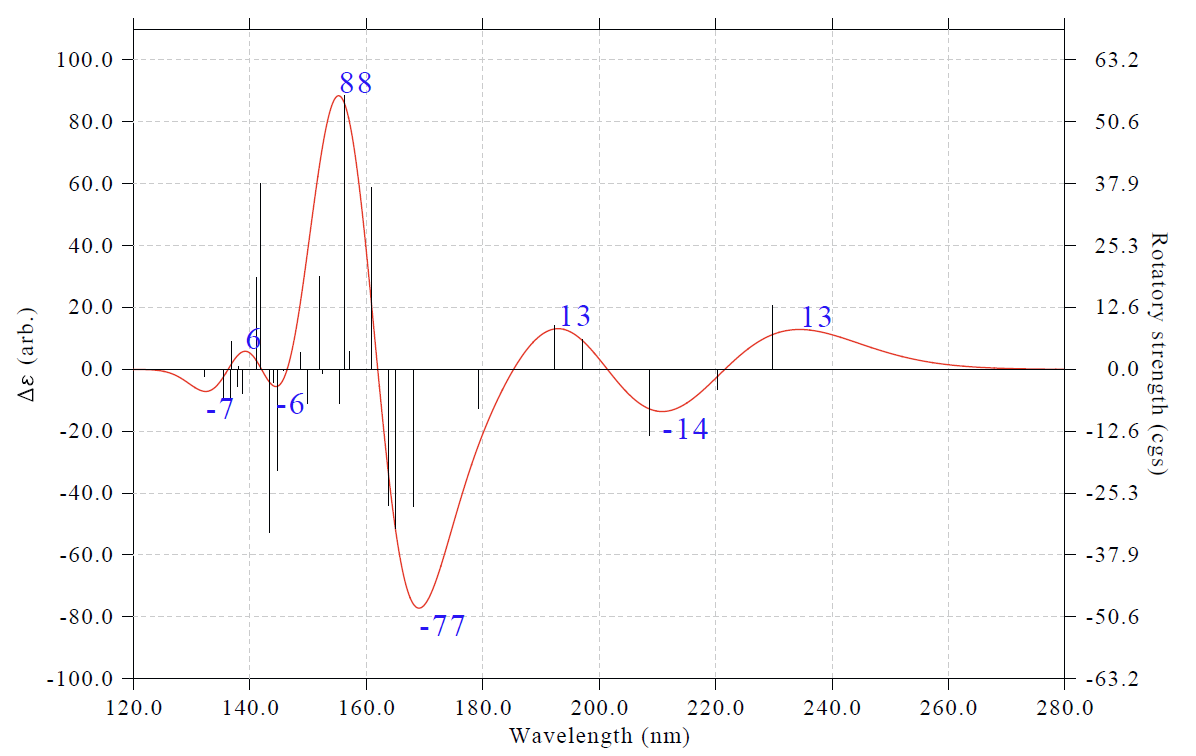
類似地,對其它類型光譜也都可以這樣把極值點標簽標注出來。我比較建議大家平時作圖時都把標簽標注出來,因為一般文獻里都沒有做這樣的標注,若在你的光譜圖里標注出來,相當于增添了一些亮點。
5.6 拉曼光學活性光譜(ROA)
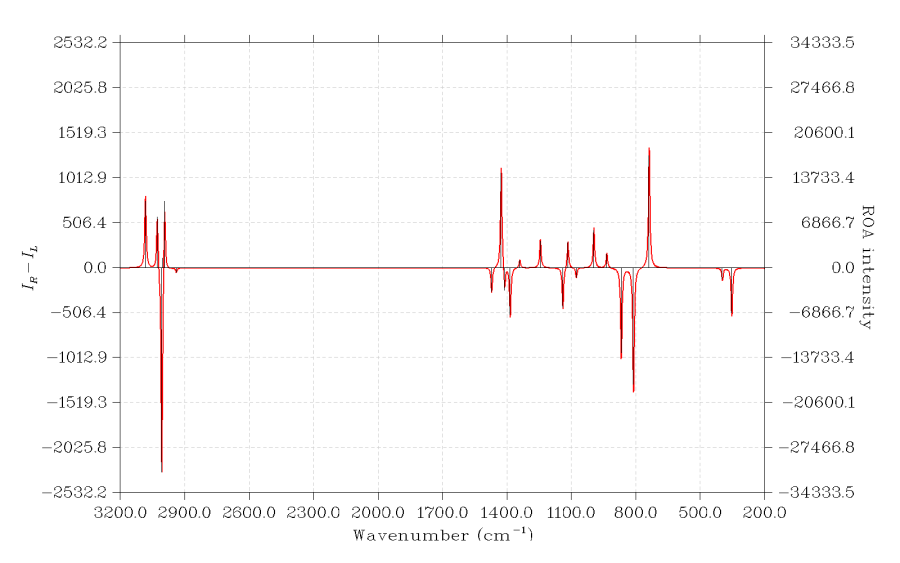
這里用S-環氧丙烷為例繪制它在532nm入射光波長下的ROA SCP(180)譜。用的是B3LYP/aug-cc-pVDZ做freq=ROA計算的輸出,考慮了500nm 532nm 600nm三種入射光波長。算ROA譜最好帶彌散函數,這樣才可能得到比較準確的ROA強度。
啟動Multiwfn后輸入
examples\spectra\S-methyloxirane_ROA.out
11
6 //繪制ROA
2 //讀取532nm的數據
2 //讀取ROA SCP(180)的數據
14 //對頻率乘上校正因子
直接按回車選擇所有頻率
0.97 //適合B3LYP/aug-cc-pVDZ的基頻校正因子
19 //把從輸出文件直接讀取的ROA活性轉化為ROA強度
532nm //入射光波長
按回車使用298.15K作為當前溫度
3 //修改橫坐標
3200,200,300 //橫坐標上限3200,下限200,步長300cm-1
0 //繪制圖像
此時會看到下圖。實際用在文章當中,建議把縱坐標的數值標簽給自行ps掉,因為對于ROA譜來說,數值的絕對大小并無意義,只有曲線形狀、峰的相對高低是有意義的。
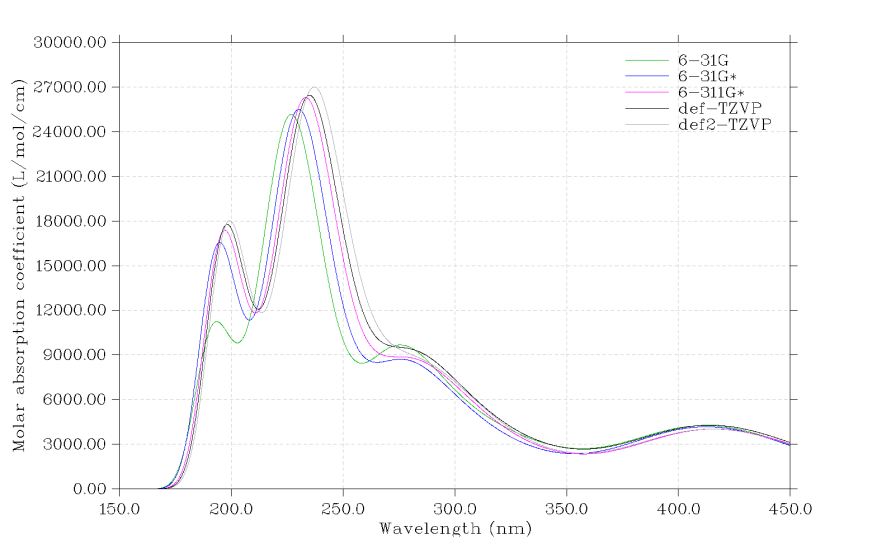
6 同時繪制多個體系的光譜
在Multiwfn中可以極為方便地同時繪制多個體系便于比較,只需要寫一個multiple.txt文件,每一行是一個輸入文件路徑,然后后面空一格寫對應的圖例文字,之后以multiple.txt作為輸入文件照常執行繪圖步驟即可。例如同時繪制不同基組下計算的茜素染料的UV-Vis光譜以檢驗基組對結果的影響,我們用不同基組分別計算完之后,寫一個multiple.txt,內容如下d:\basis_set\PBE0_6-31G.out 6-31G
d:\basis_set\PBE0_6-31Gx.out 6-31G*
d:\basis_set\PBE0_6-311Gx.out 6-311G*
d:\basis_set\PBE0_TZVP.out def-TZVP
d:\basis_set\PBE0_def2TZVP.out def2-TZVP
啟動Multiwfn,載入multiple.txt,然后按照常規步驟繪制UV-Vis圖,就看到以下結果,不同基組下的結果差異一目了然
7 在繪圖時考慮玻爾茲曼分布對光譜的影響
對于柔性分子,勢能面有很多極小點。在有限溫度下,分子并非只處于特定構象,而是在許多構象上都有一定的分布比例,當然能量越高的構象分布比例越低。分布比例可以通過玻爾茲曼分布公式來計算,具體方法見《根據Boltzmann分布計算分子不同構象所占比例》(http://www.shanxitv.org/165)。不同構象下分子的光譜往往是存在差異的,因此對于柔性分子若想較為準確地模擬出它的實際光譜,就必須把構象的權重考慮進去。考慮構象權重的光譜可以在Multiwfn中極為容易地繪制,方法在《使用Multiwfn繪制構象權重平均的光譜》(http://www.shanxitv.org/383)當中做了示例。
8 通過豎線指示躍遷能級和類型
Multiwfn允許在繪圖區域底部增加豎線來指示躍遷能級位置和類型。實際體系中,往往有很多躍遷的強度值非常低,在理論模擬出的光譜上難以看到它們的位置,在實驗上也難以觀測到,這叫做“暗態”,但是它們可以被理論計算出來。如果想在圖上也能體現出它們的存在,就可以在繪圖區域底部在每個躍遷能級位置繪制一條豎線(豎線高度都是統一的)。另外,我們還可以對不同的躍遷能級使用不同的顏色,這樣可以從圖上一目了然地區分不同類型躍遷的能級分布情況。注:Multiwfn的這個功能僅對于繪制單個體系有效。
下面看一個具體例子,18碳環。對這個十分特殊的體系筆者做了大量研究工作,匯總見http://www.shanxitv.org/carbon_ring.html,筆者對于它及其它尺寸的碳環做了深入的振動譜方面的研究,參看《揭示各種新奇的碳環體系的振動特征》(http://www.shanxitv.org/578)。18碳環的振動模式有平面內的振動,也有偏離平面的振動。我們希望繪制它的紅外光譜,并且在圖上體現出所有振動模式的位置,同時通過顏色區分開平面內和偏離平面的振動。用到的18碳環的振動分析輸出文件可以在這里下載:http://www.shanxitv.org/attach/224/C18_optfreq.out。
啟動Multiwfn,然后輸入
C18_optfreq.out
11 //繪制光譜
1 //紅外光譜
23 //用豎線指示躍遷能級的位置。之后用選項1~10可以分別設置10套豎線
1 //定義第一套豎線
1,2,5,6,9,10,13,14,15,18,20,21,24,25,26,27,32-48 //之前筆者根據GaussView顯示的振動模式判斷出的平面內振動模式的序號
14 //用棕色顯示
2 //定義第二套豎線
3,4,7,8,11,12,16,17,19,22,23,28,29,30,31 //偏離平面的振動模式的序號
3 //用藍色顯示
0 //返回
0 //繪圖
此時繪制出的圖和我們預期的一樣,但是由于坐標軸范圍還不是很理想,因此我們關閉圖像后輸入以下命令再做調整
3 //調整橫坐標
2500,0,-300 //從2800到0 cm^-1,每300 cm^-1顯示一次標簽
4 //設置左邊縱軸范圍
0,3600,500
y //自動相應地調整右邊縱坐標范圍
0 //重新繪圖
此時看到下圖
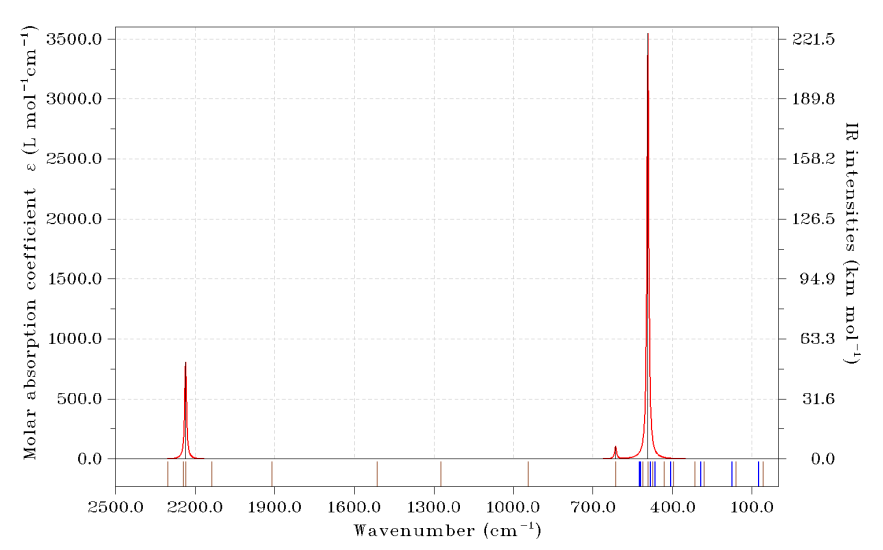
可見此體系有大量振動模式都是躍遷禁阻的,這是因為體系的高對稱性所致。通過圖下方的豎線,我們可以一目了然地看到實際的各個振動模式都在什么位置。棕色豎線是平面內振動模式,藍色豎線是偏離平面的振動模式,由圖可以清楚看出偏離平面的振動基本都在低波數范圍,而平面內的振動則有不少是高頻的。如果大家繪制的是電子光譜,還可以把比如n->pi*、pi->pi*躍遷,或者局域激發、電荷轉移激發的那些模式以不同顏色區分。
當前體系有D9h高對稱性,因此很多躍遷是簡并的。我們還可以讓豎線高度體現簡并度,這樣在寫文章的時候向讀者描述躍遷的簡并情況就很直觀方便了。接著上面的例子,輸入以下內容
23 //重新進入設置豎線的界面
-3 //用豎線高度體現簡并度
0.2 //判斷簡并度的閾值,對于振動譜單位是cm^-1。如果有一批軌道彼此間躍遷能量相距小于這個閾值,則它們被認為簡并,只有其中躍遷能最低的那個顯示豎線,高度對應簡并度
0 //返回
0 //重新繪圖
此時看到下圖(圖的上面部分同前)

圖的下方顯示了坐標刻度,高度達到第一個刻度說明這個躍遷是非簡并的(簡并度為1.0),達到第二個刻度說明是二重簡并的。可見當前大部分的豎線是全高的,只有少數是半高,因此當前體系絕大多數振動躍遷都是雙重簡并的。
上述這樣的圖用在文章里,對于展示躍遷特征、簡并情況非常方便直觀,使得比起普通的光譜圖信息明顯更豐富,非常鼓勵大家使用。上圖也正是筆者發表的論文Chem. Asian J., 16, 56 (2021) DOI: 10.1002/asia.202001228里的圖,非常建議讀者看看文章里的討論,此文可在http://www.shanxitv.org/carbon_ring.html里提及的網盤中下載。