使用Multiwfn繪制NMR譜
使用Multiwfn繪制NMR譜
文/Sobereva@北京科音
First release: 2020-Oct-8 Last update: 2023-Jan-16
摘要:流行的波函數分析程序Multiwfn(http://www.shanxitv.org/multiwfn)也有強大、靈活、易用的光譜繪制功能,支持繪制UV-Vis、ECD、IR、Raman、VCD、ROA,見《使用Multiwfn繪制紅外、拉曼、UV-Vis、ECD、VCD和ROA光譜圖》(http://www.shanxitv.org/224)。從Multiwfn 3.7版開始,還支持了NMR譜的繪制,在此文就結合具體例子進行介紹。至于NMR光譜計算方面的知識,在此文不做展開介紹,在北京科音的初級/基礎量子化學培訓班里都有非常詳細講解(見北京科音官網http://www.keinsci.com的“科研培訓”欄目)。
1 關于NMR譜和Multiwfn的繪制功能
NMR(核磁共振)譜是化學領域最重要的譜之一,能夠展現原子所處的化學環境。大多數主流量子化學程序都有計算各個原子核位置的磁屏蔽張量的功能。磁屏蔽張量的對角元的平均值對應于各向同性磁屏蔽值(σiso,以下簡稱為磁屏蔽值),令參考物質中相應元素的原子的σiso減去當前化學物質中的原子的σiso,就是一般所說的化學位移。將化學位移用洛倫茲函數進行展寬,就可以得到和實驗對照的NMR圖譜(本文不考慮核自旋-自旋耦合造成的峰的分裂問題)。如果體系有多個構象,而且構象間轉變速率較快,那么實際觀測到的NMR峰的位置將是各個構象的權重平均。另外,如果有n個同元素的原子的磁屏蔽值非常接近,應當將它們視為是簡并的,當做強度(簡并度)為n的一個信號來對待。
Gaussian是計算NMR最常用的量子化學程序,其御用的圖形界面GaussView可以載入Gaussian的NMR任務的輸出文件繪制NMR譜,但有明顯局限性:
(1)GaussView 6之前的版本無法得到NMR曲線圖,而從6開始,雖然能給出NMR曲線,但是卻沒法導出曲線數據(至少對6.0.16版而言),因此無法放到諸如Origin之類的程序里進一步調整作圖效果
(2)繪制NMR譜的時候不支持構象權重平均,也沒法對特定一批原子的磁屏蔽值取平均
(3)只能對Gaussian的輸出文件進行繪制
(4)收費,而且不便宜
而使用Multiwfn繪制NMR則有很多好處
(1)開源免費
(2)不僅支持Gaussian,還可以基于ORCA、CP2K、BDF輸出的文件繪制,輸入文件例子見Multiwfn手冊3.13.5節。而對于其它程序的用戶,還可以自行把計算結果整理成Multiwfn支持的通用的記錄磁屏蔽值的格式從而用Multiwfn繪圖(例子見自帶的examples\spectra\NMR\general.txt文件)
(3)支持繪制構象權重平均譜,也可以同時繪制多個體系
(4)可以對指定的一批原子的磁屏蔽值取平均(例如甲基的三個氫)
(5)各個峰對應的原子序號可以直接標在圖上,標注的風格可調
(6)可以基于標度法繪制NMR譜。標度法是極為重要的NMR的計算方法,詳見《談談如何又好又快地計算NMR化學位移》(http://www.shanxitv.org/354)
(7)曲線數據、離散豎線數據可以用選項2導出,便于在第三方程序如Origin里繪制以更靈活地調整作圖設置
(8)內置了很常用的NMR計算級別對應的TMS的C、H磁屏蔽參考值,以及標度法的參數
(9)作圖選項可以很靈活的控制,比如曲線的顏色和粗細、是否顯示格子/曲線/豎線、峰的半高全寬(FWHM)、判斷簡并的閾值、橫/縱坐標軸范圍等等
(10)作圖選項可以保存和導入,免得每次作圖都重新設置一遍。具體來說,在界面里輸入s代表保存設置到某文件,輸入l代表從某文件中載入設置
(11)可導出的圖片格式非常豐富,如tif、png、gif等位圖格式(尺寸通過settings.in文件里的graph2Dsize參數設置),以及pdf、svg、wmf、eps等矢量圖格式。筆者最建議用pdf,線條非常光滑、可無損縮放
一般來說,對單一結構,繪制NMR譜的流程是這樣:
(1)先用Gaussian等程序做NMR任務計算
(2)用Multiwfn載入輸出文件,進入主功能11,選擇NMR
(3)通過選項6選擇考慮的元素是什么
(4)通過選項7設置怎么得到化學位移,是基于參考物質的值求差計算,還是用標度法
(5)選擇0繪制光譜檢查效果。如果有不滿意的,再通過界面上的選項調整
(6)選擇1把光譜導出成圖像文件(導出的格式可通過界面里的選項-3選擇)
下面,筆者就通過一系列實例演示如何通過Multiwfn方便靈活地針對各種情況繪制NMR譜,請讀者務必舉一反三。本文只涉及最常見的1H和13C譜,對于其它元素的NMR譜也可以用和本文相同的做法繪制。下面例子中涉及的文件有的在Multiwfn自帶的examples\spectra\NMR目錄下直接提供了,其它的可以在這里下載:http://www.shanxitv.org/attach/565/file.zip。本文的計算都使用Gaussian 09 D.01版。Multiwfn對應于官網上最新版本的情況。
Multiwfn可以在官網http://www.shanxitv.org/multiwfn免費下載。如果不了解此程序,建議參看《Multiwfn入門tips》(http://www.shanxitv.org/167)和《Multiwfn FAQ》(http://www.shanxitv.org/452)。
2 例1:使用常規方法計算和繪制二乙烯酮的1H和13C NMR譜
二乙烯酮(diketene)的結構,以及它在氘代氯仿環境中的H和C的實驗化學位移如下所示
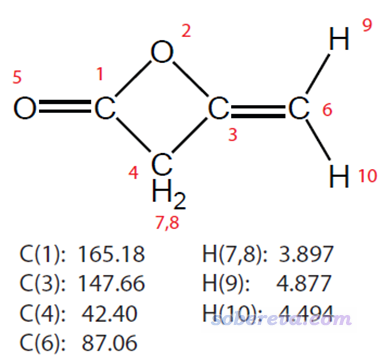
按照常規方法計算1H和13C NMR的話,需要先計算四甲基硅烷(TMS)的氫和碳的磁屏蔽值,并且其優化的級別和計算NMR的級別,包括方法、基組、溶劑模型,必須和計算自己的物質完全相同。J. Chem. Theory Comput., 10, 572 (2014)的有機體系的NMR測試體現出在Gaussian直接支持的普通泛函里B97-2表現較好,所以本文用它來算NMR,基組使用足夠好的def2-TZVP(實際上用pcSseg-1基組算NMR與之精度差不多但便宜得多,要用的話需要自行去BSE基組數據庫拷定義)。在NMR計算時都使用SMD表現氯仿環境,不熟悉溶劑模型的話看《談談隱式溶劑模型下溶解自由能和體系自由能的計算》(http://www.shanxitv.org/327)。優化不需要用大基組,見《淺談為什么優化和振動分析不需要用大基組》(http://www.shanxitv.org/387),此例就用def2-SVP。優化用的泛函就用常規的B3LYP,對于當前情況沒有任何問題,更多信息見《談談量子化學研究中什么時候用B3LYP泛函優化幾何結構是適當的》(http://www.shanxitv.org/557)。由于氯仿這樣極低極性溶劑對結構優化沒什么影響,因此為了省時在優化時就沒用。
TMS以及二乙烯酮的優化任務、NMR任務的輸出文件都在本文文件包里提供了。在TMS_NMR.out中可看到
2 C Isotropic = 186.8707 Anisotropy = 7.5367
和
3 H Isotropic = 31.5143 Anisotropy = 9.1687
即曰參考物質的13C和1H的磁屏蔽值分別為186.8707和31.5143 ppm,把這個值記住。由于TMS是Td點群的,計算前筆者也對稱化過結構,故所有氫都等價、所有碳都等價,所以不需要再看其它的氫和碳的值。
啟動Multiwfn,然后輸入
diketene_NMR.out //本文文件包里的二乙烯酮的NMR任務的輸出文件
11 //繪制光譜
7 //NMR
現在就進入了NMR繪制界面了。
我們先繪制碳譜。從屏幕上的選項6的提示可見,當前默認就是只考慮碳,所以不用改這項。如果現在直接選擇選項0繪圖,繪制的是各個碳的磁屏蔽值。但我們想獲得能與實驗相對比的化學位移譜圖,因此現在選擇選項7來設置將磁屏蔽值轉化為化學位移的方式。之后我們選1 Set reference shielding value to determine chemical shift,代表提供參考值來得到化學位移。之后程序提示你輸入參考物質的磁屏蔽值,我們輸入前面得到的TMS的C的磁屏蔽值186.8707即可(Multiwfn程序的設計超級貼心,已經把B97-2/def2-TZVP等常用的NMR計算級別在氯仿下的TMS參考值內置了,所以如屏幕所示,這一步如果我們直接輸入a來選擇與當前情況對應的B97-2/def2-TZVP G09的值也行,和手動輸入186.8707完全等價)。
現在磁屏蔽值已經轉化成了化學位移了,我們選擇選項0繪圖,得到下圖
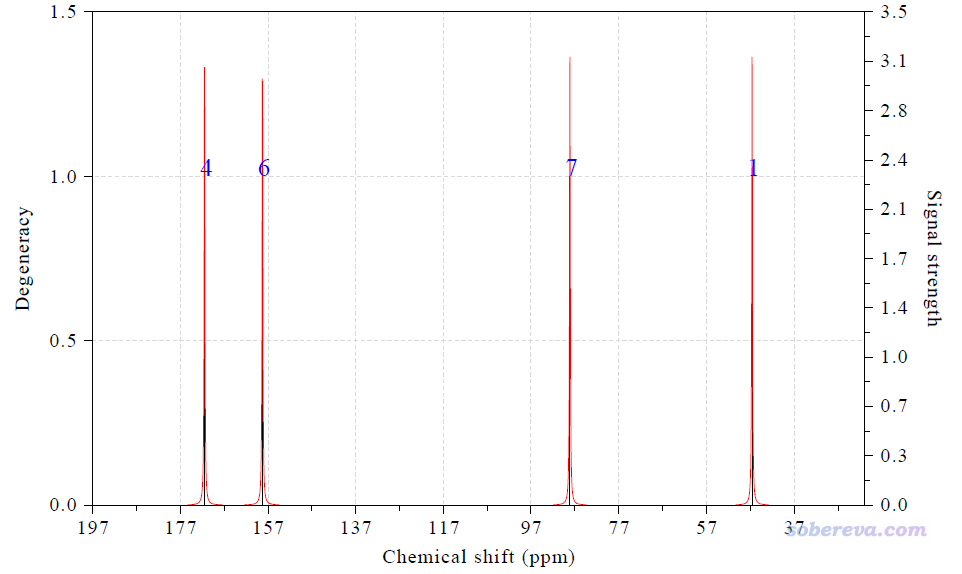
與此同時,在文本窗口還給出了具體數值:
Term: 1 Chemical shift: 46.583 ppm Atom: 1(C )
Term: 2 Chemical shift: 171.456 ppm Atom: 4(C )
Term: 3 Chemical shift: 158.244 ppm Atom: 6(C )
Term: 4 Chemical shift: 88.132 ppm Atom: 7(C )
上圖中,黑色豎線的橫坐標位置對應于化學位移值,豎線高度對應于左側的坐標軸,即簡并度。因為當前所有碳的化學位移相差都較明顯,所以簡并度都為1。圖中紅線是將黑色豎線用洛倫茲函數進行展寬得到的,數值對應于右邊的坐標軸,含義是NMR的信號強度。圖中藍色的數字是相應的峰對應的碳原子在當前體系中的序號(和GaussView里看到的序號一致)。
這四個碳的化學位移的實驗值從小到大是42.40、87.06、147.66、165.18 ppm,而上面給出的計算值是46.58、88.13、158.24、171.46 ppm,可見有的和實驗相符很好,有的差異大一些(有的原子就是難算準,想要更準的話,考慮用專為NMR優化的KT1泛函算,Dalton程序支持;也可以用耦合簇算NMR,Dalton和CFOUR程序支持)。
下面我們繪制1H NMR譜。輸入以下內容
6 //選擇被考慮的元素
H //氫
7 //選擇確定化學位移的方式
1 //輸入參考值
31.5143 //之前計算TMS得到的氫的磁屏蔽值。這里輸入a也是等效的,因為這本身就是內置的值
0 //繪制譜圖
現在看到下圖
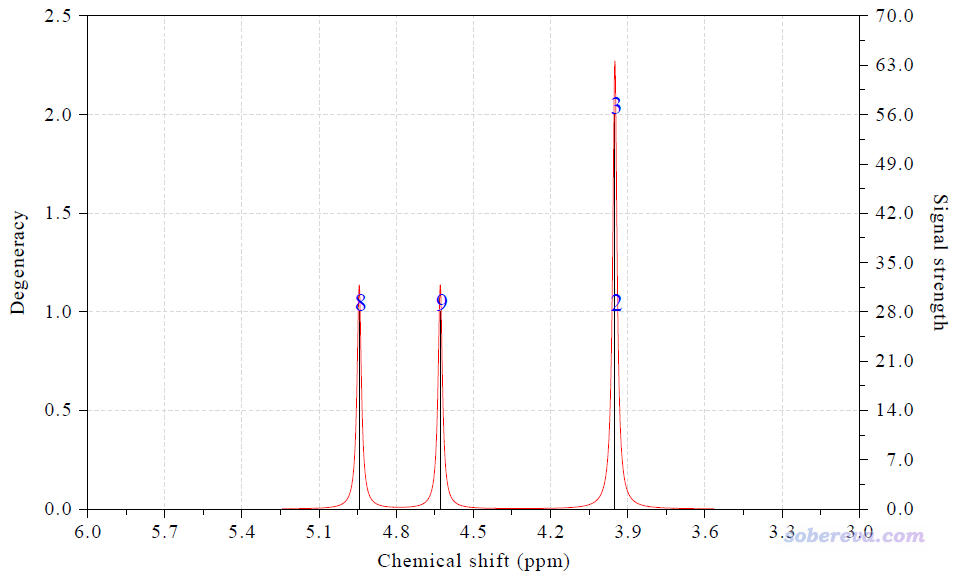
屏幕上顯示的具體化學位移值:
Term: 1 Chemical shift: 3.950 ppm Atom: 2(H ) 3(H )
Term: 2 Chemical shift: 4.944 ppm Atom: 8(H )
Term: 3 Chemical shift: 4.629 ppm Atom: 9(H )
由于體系是Cs對稱性的,在鏡面兩側的2H和3H化學環境完全相同,所以化學位移也精確相同,故被視為了簡并,因此上圖中黑色豎線高度是其它的兩倍。上面三類氫的實驗的1H的化學位移分別是3.897、4.877、4.494 ppm,計算值與之相比差異分別是+0.053、+0.067、+0.135 ppm,誤差不大,結果理想。
如果大家想修改作圖效果,用屏幕上的相應選項即可,文字提示得都很明白,若有看不明白的試試便知,有些在Multiwfn手冊的3.13.5節有詳細解釋。如果你想把各個原子的磁屏蔽值和以當前方式確定的化學位移都導出,可以選選項-2,會在當前目錄下輸出NMRdata.txt,內容如下,其中Shielding(iso)就是Gaussian輸出文件里的原子的磁屏蔽值。
Atom Shielding(iso) Chemical Shift
2(H ) 27.564 3.950
3(H ) 27.564 3.950
8(H ) 26.570 4.944
9(H ) 26.886 4.629
3 例2:乙醛的1H NMR譜
這一節我們繪制乙醛的1H NMR譜。這個例子主要是讓大家注意有些原子的磁屏蔽值要取平均的問題。使用上一節的計算方法產生的乙醛的NMR任務的輸出文件是examples\spectra\NMR\Acetaldehyde.out,體系結構如下
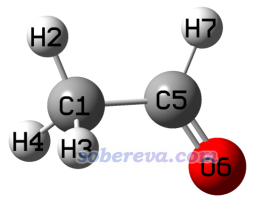
由于甲基的旋轉勢壘非常低,所以在實際環境中甲基的旋轉頻率非常高,1H NMR實驗分辨不了甲基上的三個氫的信號,它們在實驗譜上一起對應同一個峰。而由于我們計算乙醛用的結構是Cs對稱性的,所以H2與H3和H4的磁屏蔽值不同。因此我們在繪制NMR譜之前,必須將它們的磁屏蔽值取平均。
啟動Multiwfn,然后輸入
examples\spectra\NMR\Acetaldehyde.out
11 //繪制光譜
7 //NMR
6 //選擇被考慮的元素
H //氫
7 //選擇確定化學位移的方式
1 //輸入參考值
a //用內置的G09下用B97-2/def2-TZVP在SMD表現的氯仿環境下算的TMS的值,優化級別也和當前例子一樣
10 //對某些原子取平均
2-4 //甲基上的H2、H3、H4(序號也可以不連續)
0 //繪制譜圖
此時看到下圖
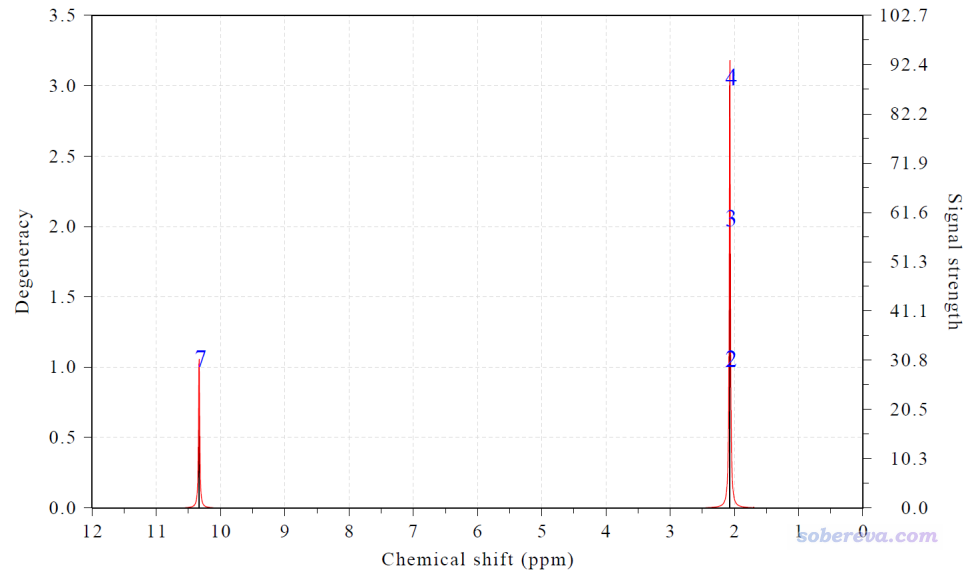
屏幕上輸出的信息為
Term: 1 Chemical shift: 2.070 ppm Atom: 2(H ) 3(H ) 4(H )
Term: 2 Chemical shift: 10.333 ppm Atom: 7(H )
氯仿下乙醛的甲基氫的化學位移實驗值為2.12 ppm,我們如上得到的是2.07 ppm,和實驗相符很好。
4 例3:基于標度法繪制2,5-降冰片二烯的NMR譜
這一節我們基于標度法(scaling method)得到的化學位移繪制2,5-降冰片二烯(2,5-norbornadiene)的1H和13C NMR譜。此體系的結構和氯仿下的實驗化學位移如下
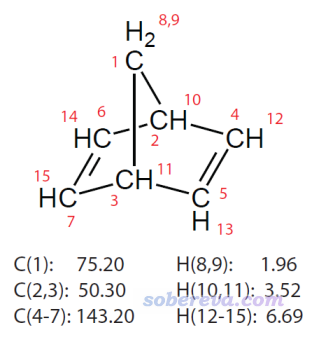
標度法引入了前人事先擬合的斜率和截距參數,在很便宜的級別下就能得到系統性誤差較小的化學位移值,多數情況下比用前面的標準方法得到的絕對誤差明顯更小,而且還不需要自行先計算參考物質的磁屏蔽值,因此還更為方便。如果沒看過此文的話一定要仔細看:《談談如何又好又快地計算NMR化學位移》(http://www.shanxitv.org/354)。此例用的計算級別是標度法中又好又便宜的一種,即用B3LYP/6-31G*在氣相下優化,然后用B3LYP/6-31G*結合SMD表現的氯仿環境算NMR。此情況對于1H,擬合的斜率為-1.0157、截距為32.2109,而對于13C,斜率為-0.9449、截距為188.4418。這個級別對應的2,5-降冰片二烯的NMR任務的輸出文件為本文文件包里的2,5-norbornadiene_NMR.out。
啟動Multiwfn,然后輸入
norbornadiene_NMR.out
11 //繪制光譜
7 //NMR
7 //選擇確定化學位移的方式
2 //設置標度法的斜率和截距
-0.9449,188.4418 //用于13C NMR的斜率和截距。這一步也可以直接輸入a來使用內置的當前級別的斜率和截距值(真貼心!)
0 //繪圖
得到的圖像如下
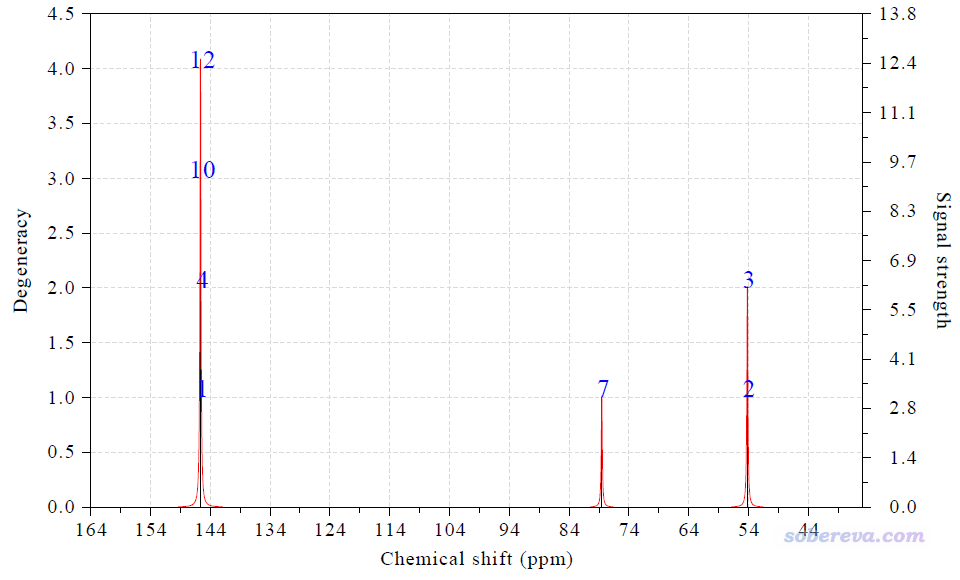
文本窗口顯示的化學位移值如下
Term: 1 Chemical shift: 145.622 ppm Atom: 1(C ) 4(C ) 10(C )
12(C )
Term: 2 Chemical shift: 54.211 ppm Atom: 2(C ) 3(C )
Term: 3 Chemical shift: 78.544 ppm Atom: 7(C )
和前面圖中的實驗值相比,三種碳的誤差分別為2.42、3.91、3.34 ppm,能有這樣的結果是比較理想的。
再來繪制1H的NMR譜,依次輸入
6 //選擇考慮的元素
H //氫
7 //選擇確定化學位移的方式
2 //設置標度法的斜率和截距
-1.0157,32.2109 //也可以直接輸入a
0 //繪圖
為避免啰嗦,結果就不在這里展示了。
5 例4:繪制纈氨酸的構象權重平均的1H NMR譜
本例演示如何在Multiwfn中繪制各個構象以及構象權重平均的NMR譜,體系的某些原子在不同構象下的化學位移往往明顯不同。構象的分布比例可以通過《根據Boltzmann分布計算分子不同構象所占比例》(http://www.shanxitv.org/165)介紹的做法計算,在需要獲得各個構象的自由能,自由能的計算看《談談隱式溶劑模型下溶解自由能和體系自由能的計算》(http://www.shanxitv.org/327)和《使用Shermo結合量子化學程序方便地計算分子的各種熱力學數據》(http://www.shanxitv.org/552)。如果你不知道體系都有什么可能的構象的話,可以用免費靈活的Molclus程序做構象搜索,見官網http://www.keinsci.com/research/molclus.html。
已知纈氨酸在水中有如下兩種構象,筆者之前計算出的構象分布比例也給出了。本例我們要繪制它的構象權重的1H NMR譜
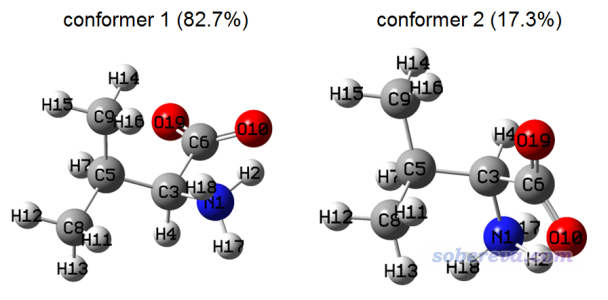
此體系是有現成的重水下的實驗NMR譜的,見https://hmdb.ca/spectra/nmr_one_d/1582,我們要與實驗譜進行對比。在重水中,質子化的氨基的氫會被重水中的重氫所取代,因此這些氫是沒有NMR信號的,在繪圖的時候需要消除掉,具體來說就是在Multiwfn中將這些氫的強度值設為0(默認是1)。另外,如前所述,還需要將每個甲基的氫的磁屏蔽值取平均。
Multiwfn的examples\spectra\NMR\valine目錄下的conf1.out和conf2.out是上面圖中的兩個構象在B97-2/def2-TZVP下做的NMR任務的輸出文件,幾何結構是在B3LYP-D3(BJ)/6-311G**下進行優化的,這兩個任務都用了IEFPCM模型表現了水環境。TMS也必須在這個級別下計算,相應的NMR任務輸出文件是此目錄下的TMS.out,可見其中氫的磁屏蔽值是31.8294 ppm。
為了繪制構象權重的NMR譜,需要創建一個含有每個構象NMR任務輸出文件的文本文件,里面也寫上構象的權重(0至1之間,總和必須為1),文件名要么是multiple.txt,要么末尾是_multiple.txt。比如我們創建valine_multiple.txt,內容如下
examples\spectra\NMR\valine\conf1.out 0.825
examples\spectra\NMR\valine\conf2.out 0.175
注意,如果你用的是Linux版Multiwfn,路徑應當用反斜杠,并且文件名必須用雙引號擴住,即寫為:
"examples/spectra/NMR/valine/conf1.out" 0.825
"examples/spectra/NMR/valine/conf2.out" 0.175
啟動Multiwfn后輸入
valine_multiple.txt
11 //繪制光譜
7 //NMR
6 //設置被考慮的元素
H //氫
7 //設置化學位移的計算方式
1 //設置參考值
31.8294 //來自TMS.out
10 //化學等價原子取平均
11-13 //甲基上的三個氫的序號
10 //化學等價原子取平均
14-16 //另一個甲基上的三個氫的序號
11 //設置某些原子的強度值
2,17,18 //質子化氨基上的三個氫的序號
0 //強度設為0,即它們在譜圖中將不可見
0 //繪制NMR
此時看到下圖
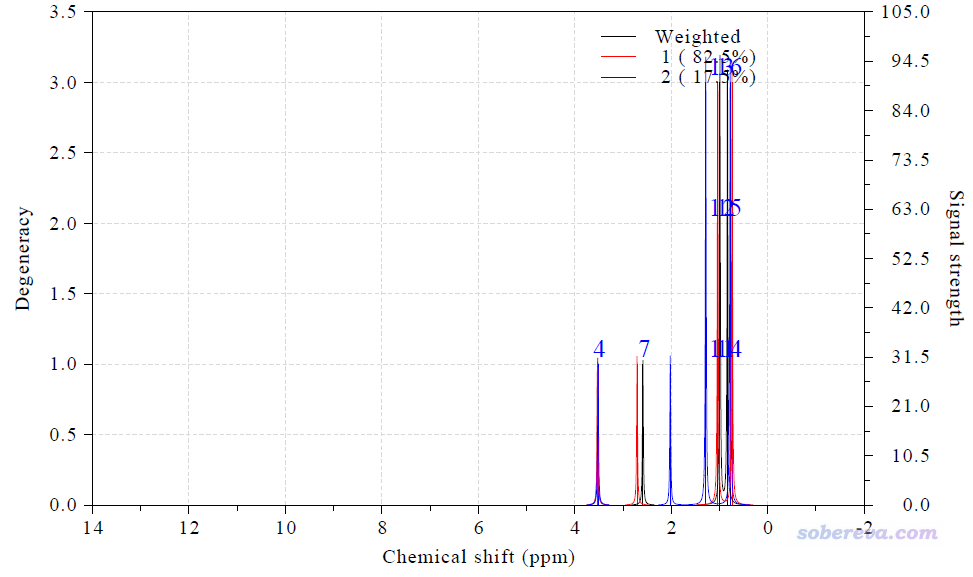
圖中黑線是權重的NMR譜,紅線和藍線分別是兩個構象的NMR譜。此時在文本窗口還輸出了兩個構象,以及構象權重平均的化學位移,如下所示。Strength是強度值,可見H2、H17、H18的強度已經被設為了0,而每個甲基的三個氫都被合并為了一項,強度為3,相當于簡并度為3。
System 1:
Term: 1 Chemical shift: 11.503 ppm Atom: 2(H ) Strength: 0.000
Term: 2 Chemical shift: 3.530 ppm Atom: 4(H ) Strength: 1.000
Term: 3 Chemical shift: 2.710 ppm Atom: 7(H ) Strength: 1.000
Term: 4 Chemical shift: 1.038 ppm Atom: 11(H ) 12(H ) 13(H ) S
trength: 3.000
Term: 5 Chemical shift: 0.741 ppm Atom: 14(H ) 15(H ) 16(H ) S
trength: 3.000
Term: 6 Chemical shift: 4.052 ppm Atom: 17(H ) Strength: 0.000
Term: 7 Chemical shift: 3.898 ppm Atom: 18(H ) Strength: 0.000
System 2:
Term: 1 Chemical shift: 11.223 ppm Atom: 2(H ) Strength: 0.000
Term: 2 Chemical shift: 3.519 ppm Atom: 4(H ) Strength: 1.000
Term: 3 Chemical shift: 2.023 ppm Atom: 7(H ) Strength: 1.000
Term: 4 Chemical shift: 0.780 ppm Atom: 11(H ) 12(H ) 13(H ) S
trength: 3.000
Term: 5 Chemical shift: 1.286 ppm Atom: 14(H ) 15(H ) 16(H ) S
trength: 3.000
Term: 6 Chemical shift: 4.400 ppm Atom: 17(H ) Strength: 0.000
Term: 7 Chemical shift: 3.789 ppm Atom: 18(H ) Strength: 0.000
Weighted data:
Term: 1 Chemical shift: 11.454 ppm Atom: 2(H ) Strength: 0.000
Term: 2 Chemical shift: 3.528 ppm Atom: 4(H ) Strength: 1.000
Term: 3 Chemical shift: 2.590 ppm Atom: 7(H ) Strength: 1.000
Term: 4 Chemical shift: 0.993 ppm Atom: 11(H ) 12(H ) 13(H ) S
trength: 3.000
Term: 5 Chemical shift: 0.836 ppm Atom: 14(H ) 15(H ) 16(H ) S
trength: 3.000
Term: 6 Chemical shift: 4.113 ppm Atom: 17(H ) Strength: 0.000
Term: 7 Chemical shift: 3.879 ppm Atom: 18(H ) Strength: 0.000
上面的NMR譜還不太理想,橫坐標范圍太寬,而且圖例擋住了譜圖。因此我們做一些調整。接著輸入
3 //修改橫坐標范圍
4,0,0.5 //范圍從4到0,每0.5 ppm繪制一個標簽
12 //不顯示豎線以使得圖像更簡潔
18 //其它繪圖設定
5 //設置圖例的橫坐標位置
1300 //讓圖例的橫坐標位置比默認的更靠左。默認值是2200,數值越小就越靠左
0 //返回
0 //重新作圖
此時圖像如下,非常干凈清楚
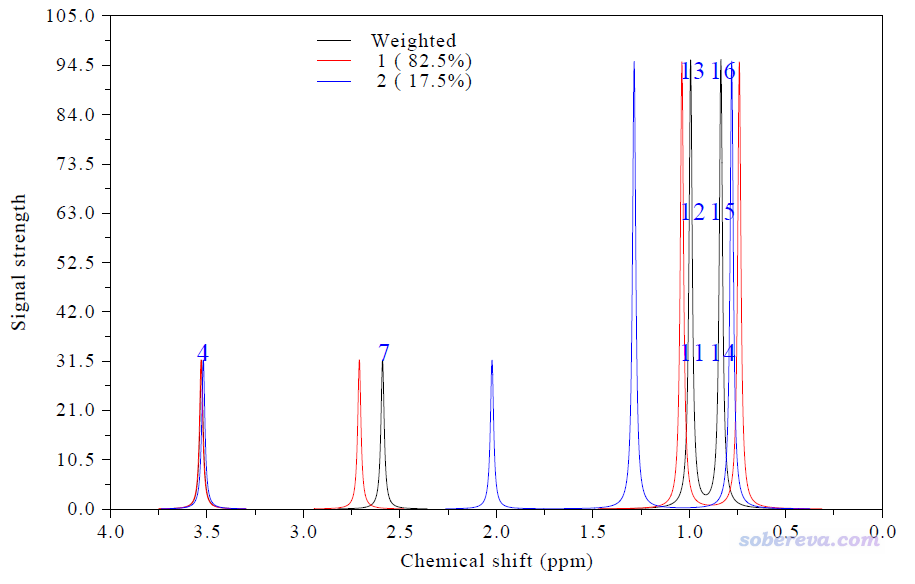
由圖可見,H4的化學位移幾乎完全不受構象影響,而H7,以及甲基氫,都受構象影響很大,故必須考慮構象的權重平均。
此體系的重水下的1H NMR實驗譜如下
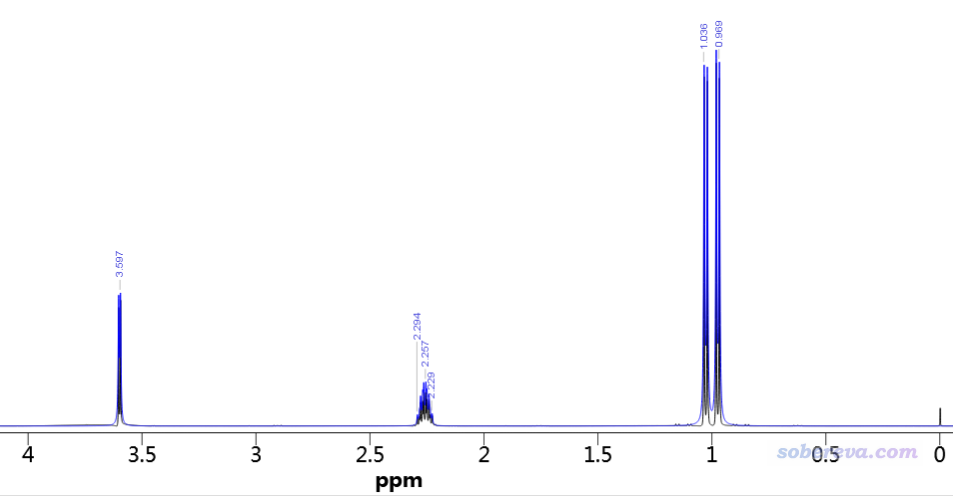
忽略實驗譜中核自旋-自旋耦合效應造成的峰的分裂現象,我們得到的構象權重譜和實驗譜相對照時基本特征都能吻合。特別是實驗譜在1 ppm的地方有兩個峰,我們的譜圖也有這個特征。不過,實驗譜在2.3 ppm有峰,而我們譜圖的相應的峰位置在2.59 ppm,相差稍微多一些。這較大可能是因為計算出的構象分布比例還不是特別準確,構象分布對這個峰的位置影響極大。構象1和構象2的相應的峰位置分別是2.71和2.02 ppm(從Multiwfn的文本窗口的信息可見),因此構象2占的比例越大,這個峰的化學位移就越低。
順帶一提,利用當前界面的選項17的子選項2,可以要求只顯示權重的譜,或者只顯示兩個構象各自的譜。
可以在當前界面里輸入s將作圖設置保存到一個文本文件中,以后重新作圖時進入主功能11后就可以輸入l從指定的文件中恢復作圖設定。但對原子屏蔽值求平均、設置原子的強度值的操作需要每次重新做。
6 例5:同時繪制多個體系的NMR譜
Multiwfn中還可以同時繪制多個體系的NMR譜,有個前提是所有體系的原子數必須相同。本例我們把上一節的兩個纈氨酸構象的NMR譜繪制到一起。
創建一個文本文件,文件名要么是multiple.txt,要么末尾是_multiple.txt。我們創建的multiple.txt的內容如下,每一行是一個NMR任務的輸出文件路徑,之后是圖例文字
examples\spectra\NMR\valine\conf1.out conformer 1
examples\spectra\NMR\valine\conf2.out conformer 2
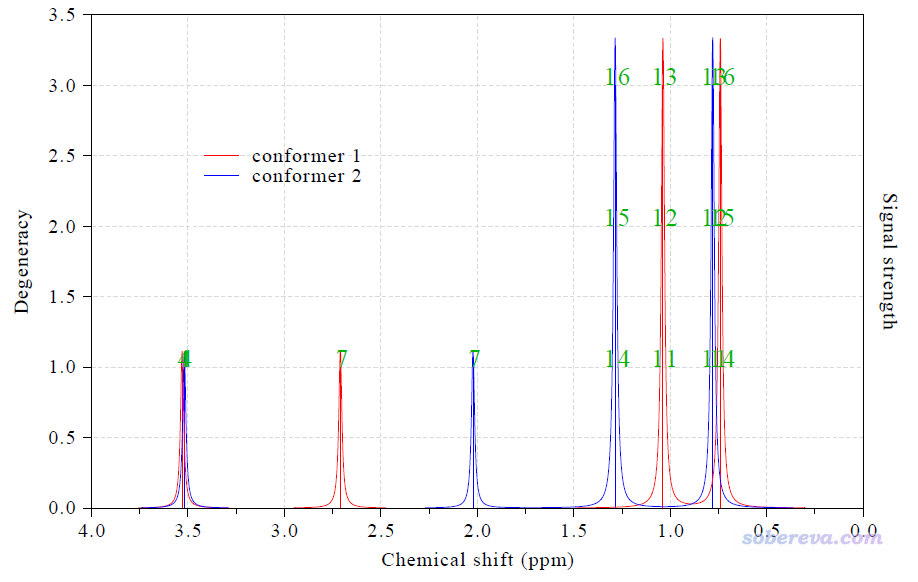
啟動Multiwfn,載入multiple.txt,然后按照前面例子的過程照常繪制,就可以得到下圖,可見每個構象的每個峰對應的原子序號都標注上了,具體的峰位置在文本窗口里也都給出了。繪制此圖所有要敲入的命令我在examples\spectra\NMR\valine\drawmulti.txt中也給出了,大家可以在載入輸入文件后把此文件里的命令直接復制到Multiwfn窗口里。
7 總結
本文介紹并演示了Multiwfn繪制NMR譜的功能,從本文的例子可見Multiwfn的這個功能非常方便靈活,一些地方設計得很貼心,考慮得很周到,鼓勵大家在以后量子化學研究NMR中使用。此功能還有很多其它選項在上面的例子里沒有提及,建議大家仔細把界面里的選項都仔細看一遍,以了解Multiwfn在繪制時都能做哪些設置。
本文只用Gaussian的輸出文件作為了示例,對于ORCA用戶,在Multiwfn中繪制NMR的操作是完全一樣的,用ORCA的NMR任務的輸出文件即可。ORCA做NMR任務的關鍵詞很簡單,比如! B3LYP/G 6-31G* NMR cpcm(chloroform)就代表用B3LYP/6-31G*在CPCM隱式溶劑模型表現的氯仿環境下做NMR計算。CP2K用戶通過Multiwfn繪制NMR譜要用NMR計算產生的.data文件作為輸入文件。也別忘了,如前所述,Multiwfn繪制NMR的功能支持通用的輸入格式,因此如果大家是Dalton、GAMESS-US、NWChem、Q-Chem、Molpro、Dirac等其它量子化學程序的用戶,乃至第一性原理程序的用戶,也是可以靠Multiwfn繪制NMR譜的。