談談如何又好又快地計算NMR化學位移
2021-Oct-24重要補充:本文中對泛函選擇的說法已經過時了。后來筆者發現用revTPSS泛函結合pcSseg-1計算NMR相當好而且很便宜,詳見《revTPSS泛函結合pcSseg-1基組是計算NMR很好的選擇》(http://www.shanxitv.org/623),性價比不輸于本文介紹的標度法,精度更明顯強于本文提到的其它方法,如B97-2、KT2、MP2。
談談如何又好又快地計算NMR化學位移
文/Sobereva @北京科音
First release: 2016-Nov-10 Last update: 2020-Oct-8
摘要:本文重點介紹一種又便宜又準確的計算有機分子13C和1H化學位移的方法,英文叫scaling,筆者把它翻譯成標度法。第一節首先會說一些和NMR計算有關的亂七八糟的知識,然后在第二節介紹標度法,最后在第三節給個實例對比標度法相對于一般計算方式的優勢。
如果想繪制NMR譜的話,最佳的做法是使用Multiwfn程序繪制,易用、靈活、強大而且免費,見《使用Multiwfn繪制NMR譜》(http://www.shanxitv.org/565)。而且在Multiwfn里還可以直接使用標度法得到化學位移,方便極了。
1 相關知識
量子化學程序計算NMR時會計算每個原子核位置的磁屏蔽張量,這是3*3矩陣,表現出原子核附近的電子對不同方向磁場的不同屏蔽,比如Gaussian輸出的(單位為ppm):11 H Isotropic = 31.8273 Anisotropy = 10.7602
XX= 30.2103 YX= 2.1740 ZX= -2.1740
XY= 2.2346 YY= 32.6358 ZY= -5.2595
XZ= -2.2346 YZ= -5.2595 ZZ= 32.6358
由于分子在溶液中會隨意旋轉,所以我們一般關注的是各項同性磁屏蔽值σiso,它對應于磁屏蔽張量的三個對角元的平均值,也即上面輸出的Isotropic = 31.8273。
量子化學計算NMR化學位移的最標準的做法是先計算參考物質四甲基硅烷(TMS)的碳、氫的σiso作為參考值,然后再在相同級別下計算自己要研究的物質的碳、氫的σiso,和參考值求差值即得到化學位移。優化顯然可以和計算NMR用不同級別。
對于這種標準計算方式,有大量文章研究了不同基組、理論方法下的計算精度,如JCTC,10,572(2014)、JCP,138,024111(2013)。精度和耗時都是:CCSD(T)>CCSD>MP2>DFT>=HF。DFT里面算NMR最好的泛函是KT2(Dalton支持),其次一般認為是B97-2(在Gaussian里寫為B972)。不過,這些測試都是用高精度理論計算的結果作為參考值,將結果拿去和實驗結果對比,往往又是另一番風景了。后面會看到MP2到了實際中就慫了。半經驗方法也可以算NMR。Walter Thiel等人搞的MB3和Hyperchem支持的TNDO是專門用來算NMR的半經驗方法,不過很少被使用。Gaussian09目前最高支持用MP2來算NMR,想用耦合簇級別算NMR得用Dalton、CFOUR等程序。
另外,JCTC,10,572中提出了GIAO-SCS-MP2方法,思路類似SCS-MP2,但專門用來算NMR,結果比MP2有顯著改進,甚至接近CCSD(T)。原理是把磁屏蔽張量分解成:HF貢獻+c1*自旋相同貢獻+c2*自旋相反貢獻,c1、c2向CCSD(T)精確計算值來擬合。可惜此方法目前沒有被公開的程序支持,可能以后的Q-Chem會支持吧。
與磁相關的屬性在量化程序中有GIAO、IGLO、CSGT、IGAIM這幾種計算形式,完備基組下結果相同,但有限基組下會有差異,隨基組增加收斂性快慢不同。GIAO是公認首選,也是Gaussian默認的。所以一些人做NMR計算時候在NMR后面寫個=GIAO純屬多余。
再說下計算NMR用的基組。6-31G**、def2-SVP這個檔很糙,6-311G(2d,p)還湊合,def2-TZVP比較理想,def2-QZVP或cc-pVQZ就完美了。使用彌散函數沒有任何意義,幾乎不會對結果有任何改進還暴增耗時。
計算NMR最最最最理想的基組是Jensen搞的pcS系列,原文見JCTC,4,719(2008)。后來為了節約pcS在主流量化程序里的耗時,Jensen在JCTC,11,132(2015)中把pcS從廣義收縮轉化成了片段收縮版本,稱pcSseg,它們在EMSL基組庫上都能下載到定義。pcSseg系列特別適合結合DFT計算磁屏蔽值。pcSseg-1大小和6-31G**相近,NMR計算精度卻和def2-TZVP、cc-pVTZ差不多。pcSseg-0大小和3-21G相仿佛,精度雖明顯不如pcS-1,但是卻比6-31G**整體還好一點。另外還有pcSseg-2,比pcSseg-1大不少,但對NMR改進有限,不算很劃算。
總的來說,在Gaussian下計算NMR,一般用B97-2即可,基組建議用pcSseg-1,如果懶得從EMSL上拷定義就用def2-TZVP。注意,算NMR的時候MP2比DFT的昂貴程度遠遠遠遠高于算單點的時候!切勿小看MP2算NMR的耗時,哪怕只是結合pcSseg-1這種不太大的基組用于中等體系!
溶劑環境是會對化學位移計算結果有一定影響的,因此NMR計算時應當通過PCM、SMD等隱式溶劑模型表現溶劑環境。不過,由于NMR大多是在(氘代)氯仿下測的,其極性比較小,因此是否用溶劑模型,對結果精度影響相對有限,而且由于一些誤差抵消方面的巧合因素,可能不帶溶劑模型反倒結果更好點。不過鑒于使用隱式溶劑模型并不會增加太多耗時,為了嚴謹起見,計算時還是用隱式溶劑模型表現實際溶劑環境為宜。幾何優化時用不用隱式溶劑模型無所謂,因為氯仿環境下和氣相下優化的結構一般不會有什么差異。
2 標度法與WC04、WP04泛函
按照以上方式計算,哪怕用了較好方法和較高質量基組,也考慮了溶劑效應,但算出來的化學位移往往還是和實驗值差異不小。一個原因是沒考慮構象分布的影響,這點本文不涉及,這對于柔性大分子一般才牽扯到,另一大原因就是理論計算的結果和實驗值有系統性誤差,應當通過某種形式的校正來消除。這其實和頻率校正因子(見《談談諧振頻率校正因子》http://www.shanxitv.org/221)的做法在某種意義上很相似。
標度法是用來計算化學位移的一種又準又便宜的方法,用這種方法計算化學位移(δ)的方式是
δ=(截距-σiso)/(-斜率)
其中截距和斜率都是事先擬合好的標度參數,擬合方式是對一批有機分子,讓特定級別下以這種方式算的化學位移和實驗化學位移盡可能相同。
不同文獻報道了不同級別、不同方式得到標度參數,在這個網站上有匯總:http://cheshirenmr.info。進入后點擊左側的Scaling Factors,可以看到一個索引,說了網頁中每個表格對應的是什么情況。表格#1、#2...對應不同數據來源,每個子表對應不同具體情況,如不同溶劑、不同程序。
表格里數據很多,這里作為例子,我們只關注G09下基于氣相優化的結構結合SMD溶劑模型表現氯仿環境時候算化學位移的情況,其對應的表是Table #1b(以后網站可能會更新,表格序號也許會變)。要找到最合用的級別,一方面是看RMSD值(相對于實驗值的均方根誤差),另一方面是看計算用的基組。RMSD又低,基組又小的級別是我們需要的。當前我們看到B3LYP/6-31G*在氣相優化結構,然后B3LYP/6-31G*結合SMD表現氯仿環境計算NMR的時候RMSD很低,甚至比一大批更大基組下的RMSD還低,所以這明顯是很理想的計算級別。此級別下算氫譜的截距是32.2109,斜率是-1.0157,因此我們直接把這個級別下算出來的氫的σiso代入這個公式就直接得到化學位移了:δ=(32.2109-σiso)/1.0157。對于算碳的化學位移也是用相應的標度參數即可。這個做法真是超簡單,不僅可以用小基組,連計算參考物質TMS的過程都省了。到底這個做法準不準?下一節我們通過實例測試一下便知。
可以注意到表中還有一個耗時低且RMSD也低的級別是M06-2X/6-31G*優化氣相結構,然后mPW1PW91/6-31G*結合SMD溶劑模型表現氯仿環境計算NMR。這個耗時和誤差與上面B3LYP/6-31G*那個半斤八兩,但考慮到M06-2X耗時還是高于B3LYP不少,而且這還得牽扯到用兩個泛函,所以我建議用上面B3LYP/6-31G*的級別來做標度法計算。不過,鑒于M06-2X描述弱相互作用比B3LYP好很多,所以如果弱相互作用可能明顯影響體系結構的話,我倒建議用mPW1PW91//M06-2X這個。
從表格中可以看到,用了大基組結果未必比小基組下的好,甚至結果精度可能不增反降。所以用標度法的時候切勿盲目用大基組,哪怕你的計算資源有地方沒處用。從一些RMSD上也看到,加了彌散函數后結果并沒改進,這也說明彌散函數對計算NMR從各種意義上都是毛用也沒有。所以切勿效仿一些人計算NMR時也用彌散的做法。
值得順帶提一下的是WC04和WP04泛函,這是在JCTC,2,1085(2006)中Cramer等人提出的專門算氯仿下化學位移的泛函。WC04專用于算碳,WP04專門算氫,P代表proton。它們在B3LYP基礎上對參數進行了優化,通過一批有機分子作為訓練集,使得在用6-311+G(2d,p)基組結合IEFPCM溶劑模型表現氯仿時的計算的化學位移盡可能接近氯仿下的實驗值。這兩個泛函在Gaussian中的用法是:
WC04:BLYP IOp(3/76=1000007400,3/77=0999900001,3/78=0000109999)
WP04:BLYP IOp(3/76=1000001189,3/77=0961409999,3/78=0000109999)
顯然,用這兩個泛函算化學位移時,TMS參考數據也得分別在這兩個泛函下算。
WC04和WP04泛函雖然在原文的測試中精度明顯勝于其它泛函,但是相對于其它一些泛函用標度法計算化學位移的時候在精度上就沒有什么優勢了。而且,這倆泛函參數化的時候還是結合的6-311+G(2d,p)基組,用它的代價比較高(而且其彌散函數純屬多余,但由于是在這個基組下擬合的參數,故用這倆泛函的時候不得不帶彌散函數)。所以,有了標度法,WC04和WP04就沒什么用武之地了。
3 實例
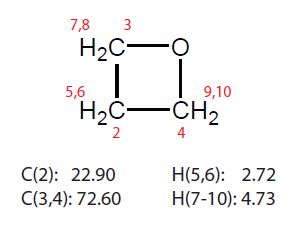
這里我們用Gaussian09程序,以兩個隨便找的有機小分子為例,對比一下MP2/pcSseg-1、MP2/def2-TZVP,以及B97-2/pcSseg-1用標準方法計算的氫和碳的化學位移,以及用標度法算的結果。標度法考慮前面提到的兩種:(1)B3LYP/6-31G*優化和計算NMR (2)M06-2X/6-31G*優化,mPW1PW91/6-31G*算NMR。另外,我們也用WC04和WP04結合標配的6-311+G(2d,p)基組計算一下結果。結構優化都在氣相下完成,算NMR的時候都用scrf(SMD,solvent=chloroform)關鍵詞表現氯仿環境。除了標度法(2)的情況外優化TMS和被研究的分子都在B3LYP/6-31G*下完成。測試分子1是惡丁環(oxetane),其氯仿下化學位移如下
測試分子2是異惡唑(isoxazole),其氯仿下化學位移如下
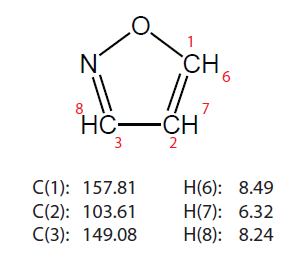
下面是化學位移計算結果。表中的MP2是指用的基組和B97-2一樣都是pcSseg-1的情況。
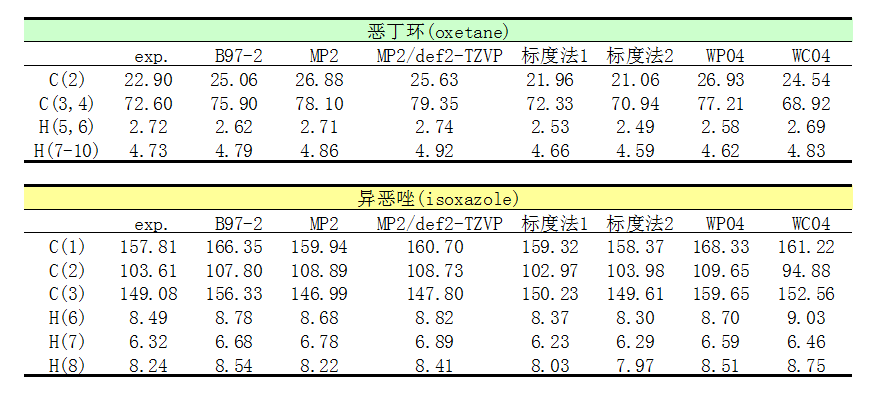
從表中可見,B97-2的結果差強人意。MP2雖然文獻里表明其比B97-2更準,但是實際上陣后發現,其精度,特別是碳的,并不比B97-2好,著實令人失望,多花那么多時間根本不值得。pcSseg-1和def2-TZVP結果差異不算太大,往往尺寸更小的pcSseg-1的結果比def2-TZVP還更準。標度法1計算代價很低,結果卻相當不錯,完虐B97-2和MP2,碳的化學位移誤差大多都在1ppm以內,最大相差1.5ppm,而氫的化學位移誤差都在0.21ppm以內。標度法2算異惡唑的碳的化學位移很準,但其它項目不如標度法1。專為算氫譜優化的WP04泛函算碳譜很差,算氫譜則和B97-2半斤八兩。專為算碳譜優化的WC04表現也不算多好,比標度法的精度還是差遠了。
通過以上數據看出,B3LYP/6-31G*氣相下優化,然后B3LYP/6-31G* scrf(SMD,solvent=chloroform)計算磁屏蔽值,再用標度法轉化成氫和碳的化學位移,不僅很便宜,而且結果和實驗相符相當理想,強烈推薦大家作為平時計算NMR的標準方法。