Gaussian中用TDDFT計算激發態和吸收、熒光、磷光光譜的方法
Gaussian中用TDDFT計算激發態和吸收、熒光、磷光光譜的方法
文/Sobereva @北京科音
First release: 2015-Dec-22 Last update: 2022-Mar-1
1 前言
總是有人問Gaussian中怎么算激發能之類的問題。Gaussian手冊的SCRF關鍵詞里有個所謂的“7步”過程,寫得本身沒問題,但是過程過于繁瑣、好幾個問題攪合在一起,對激發態計算、溶劑模型一竅不通的初學者看了之后99%都沒法正確理解其含義,會被嚴重誤導。還經常問有人問第幾步怎么操作怎么回事、讀什么數據,怎么手冊里用7步算的我才用了兩三步。感覺不得不寫個文章澄清一下以正視聽。由于筆者過于繁忙,所以沒時間在此文把原理將得很細,但只要理解本文介紹的原理,嚴格follow本文的例子,對自己的體系肯定能算對。關于激發態、電子光譜計算的非常全面、深入的內容和大量實例筆者在每年開辦的北京科音初級、基礎量子化學培訓班上會詳細講(關注北京科音主頁http://www.keinsci.com的“科研培訓”欄目)。
本文主要考慮用最常用的TDDFT方法算激發態的情況。下面這三篇沒看過的話一定要看看
使用Multiwfn繪制紅外、拉曼、UV-Vis、ECD、VCD和ROA光譜圖 http://www.shanxitv.org/224
亂談激發態的計算方法 http://www.shanxitv.org/265
Multiwfn支持的電子激發分析方法一覽 http://www.shanxitv.org/437
以下跟激發態研究有關的博文也強烈推薦看看
圖解電子激發的分類 http://www.shanxitv.org/284
使用Multiwfn便利地查看所有激發態中的主要軌道躍遷貢獻 http://www.shanxitv.org/529
電子激發任務中軌道躍遷貢獻的計算 http://www.shanxitv.org/230
使用Multiwfn做空穴-電子分析全面考察電子激發特征 http://www.shanxitv.org/434
使用Multiwfn繪制躍遷密度矩陣和電荷轉移矩陣考察電子激發特征 http://www.shanxitv.org/436
在Multiwfn中通過IFCT方法計算電子激發過程中任意片段間的電子轉移量 http://www.shanxitv.org/433
使用Multiwfn繪制電荷轉移光譜(CTS)直觀分析電子光譜內在特征 http://www.shanxitv.org/628
使用Multiwfn繪制構象權重平均的光譜 http://www.shanxitv.org/383
使用Multiwfn計算特定方向的UV-Vis吸收光譜 http://www.shanxitv.org/648
使用Multiwfn做自然躍遷軌道(NTO)分析 http://www.shanxitv.org/377
在Multiwfn中基于fch產生自然軌道的方法與激發態波函數、自旋自然軌道分析實例 http://www.shanxitv.org/403
使用Multiwfn考察軌道間重疊程度和質心距離 http://www.shanxitv.org/371
電子激發過程中片段間電荷轉移百分比的計算 http://www.shanxitv.org/398
通過量子化學計算和Multiwfn程序預測化學物質的顏色 http://www.shanxitv.org/662
使用Multiwfn計算odd electron density考察激發態單電子分布 http://www.shanxitv.org/583
振動分辨的電子光譜的計算 http://www.shanxitv.org/223
談談勢能面交叉對激發態優化的影響 http://www.shanxitv.org/468
基于背景電荷計算分子在晶體環境中的吸收光譜 http://www.shanxitv.org/579
使用ORCA在TDDFT下計算旋軌耦合矩陣元和繪制旋軌耦合校正的UV-Vis光譜 http://www.shanxitv.org/462
使用CP2K結合Multiwfn對周期性體系模擬UV-Vis光譜和考察電子激發態 http://www.shanxitv.org/634
CIS、TDDFT計算的CT激發能隨CT距離的漸進行為的總結 http://www.shanxitv.org/321
另外也建議讀一下這篇入門級綜述文章,會有幫助:Chem. Soc. Rev., 42, 845-856 (2013)。文中的說法凡是和本文有沖突的,一律以本文為準。
下文第2節先介紹一下基礎知識,第3節介紹TDDFT計算涉及到的關鍵詞,第4節給出一些例子。
2 原理
2.1 理論方法
各種計算激發態的方法,比如CIS、TDDFT、TDHF、ZINDO、EOM-CCSD、SAC-CI、LR-CC3、CASPT2等等,直接給出的都是在某個結構下,基態與各個激發態之間的電子能量差。這些方法也可以優化激發態和對激發態做振動分析,也能給出激發態波函數信息用于討論激發態原子電荷、偶極矩、電子定域性等問題。目前最流行的用得最多的是TDDFT,達到了精度與效率的較好平衡點。但TDDFT計算精度嚴重依賴于泛函,必須根據體系特征、激發類型選擇合適的泛函才能得到不錯結果。至于基組,若泛函選擇得當,對于大體系用6-31G*就能得到定性正確結果,如果計算量尚有余裕想改進定量精度,建議用def-TZVP(Gaussian里直接寫為TZVP)。若計算里德堡激發,則必須有彌散函數,至少aug-cc-pVDZ。泛函和基組的選擇的詳細討論見前述的《亂談激發態的計算方法》,判斷激發類型的方法見前述的《圖解電子激發的分類》。
2.2 電子躍遷過程
實際上,吸收、發射過程既涉及到電子態的改變也涉及到振動態的改變,完整考慮這樣的過程比較麻煩,需要優化基態結構、對基態做振動分析、優化激發態結構(挺耗時)、對激發態做振動分析(巨耗時)。
為了簡化計算和討論,人們一般忽略核振動的量子效應,只考慮電子勢能面。此時電子躍遷可用以下假想模型表示:
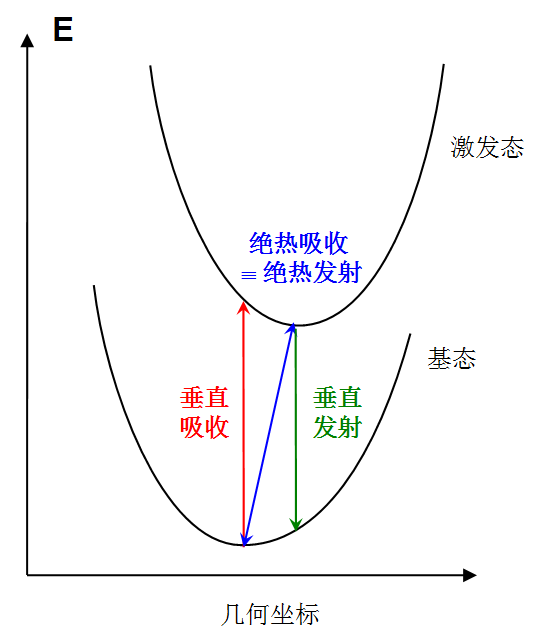
我們一般研究激發態計算的是以下三種理想化的躍遷方式:
(1)垂直吸收:電子從基態吸收光子而被激發到激發態,結構保持基態的極小點結構。計算垂直吸收能包含以下兩步
1.優化基態幾何結構
2.在1的結構下計算基態到感興趣的激發態的激發能
(2)垂直發射:電子從激發態發射光子而退激到基態,結構保持激發態的極小點結構。計算垂直發射能包含以下兩步
1.優化激發態幾何結構(初猜結構無所謂,如果不知道激發態結構什么樣,一般用基態極小點結構作為初猜結構)
2.在1的結構下計算基態到激發態的激發能(這便是這個激發態垂直發射到基態的能量)
(3)絕熱吸收:電子從基態被激發到激發態,結構也從基態極小點結構變化到激發態極小點結構。計算絕熱吸收能包含以下三步
1.優化基態幾何結構,在最后一步得到基態極小點能量
2.優化激發態幾何結構,在最后一步得到激發態極小點能量
3.將第二步得到的能量與第一步得到的能量求差值
絕熱發射是絕熱吸收的逆過程,躍遷能量相同。
以上躍遷方式是理想化的,對理論研究有用,對于研究實際問題也有用。實際吸收、發射光譜的最大峰位置一般分別比較接近于垂直吸收能和垂直發射能。而0-0躍遷,即兩個電子態的振動基態間的躍遷能,比較接近于絕熱激發能。
基態是單重態(S0)時,發射過程又分為兩類:
(1)熒光發射:是從單重態激發態發射光子退激到基態。根據kasha規則,熒光發射多數情況是從S1(能量最低的單重態激發態)退激到S0。因此,按照上述垂直發射方式計算熒光的時候,激發態一般選的是S1態。少數情況kasha規則不滿足,比如Azulene大多是從S2而非S1退激到S0的。
(2)磷光發射:是從三重態激發態發射光子退激到基態。磷光發射是從T1(能量最低的三重態激發態)退激到S0的。因此,按照上述垂直發射方式計算磷光的時候,激發態選的是T1態。
2.3 躍遷電偶極矩與振子強度
假設初始態電子波函數為|i>,末態為|j>,則兩個態之間的躍遷電偶極矩為<i|-r|j>。這里r是坐標矢量,負號是因為電子帶負電荷。躍遷偶極矩一般指的就是躍遷電偶極矩,但也有躍遷磁偶極矩、躍遷速度偶極矩等等。注意,躍遷電偶極矩和兩個態之間電偶極矩的差值,即<j|-r|j>-<i|-r|i>,根本沒直接關系,很多初學者都搞混了。
有了躍遷電偶極矩和兩個態能量差ΔE,就能得到兩個態之間躍遷對應的振子強度f:(2/3)ΔE*|<i|-r|j>|^2,是個無量綱的量。
基態體系與某個激發態之間的振子強度越大,就越容易吸收相應頻率的電磁波而躍遷到那個激發態上,因此吸收光譜中相應的吸收峰也越強。一般情況下振子強度小于1,但也完全可以大于1,極強吸收峰的振子強度甚至能達到接近7、8。振子強度小于0.01一般可以認為躍遷禁阻。
通過振子強度和兩個態之間的能量差就可以根據愛因斯坦公式計算激發態壽命,對于熒光和磷光發射都適用,往往和實驗對應得不錯。如果和實驗值差異很大,有可能發生了顯著的內轉換、外轉換等無輻過程。計算公式為:壽命τ=1.5/(f*ΔE^2)。這里壽命的單位為秒,ΔE以cm-1為單位。壽命的倒數就是自發輻射速率。
三重態和單重態之間是自旋禁阻的,僅當考慮旋軌耦合時振子強度才不為零,但數值依然很小,所以磷光壽命遠比熒光壽命長。
2.4 電子光譜的產生
激發能、振子強度都是純粹的理論數據,要把它轉化為能和實驗對比的UV-Vis(紫外-可見吸收光譜)、熒光、磷光光譜,需要把躍遷進行展寬。這里簡單說一下計算過程。
(1)UV-Vis光譜:在基態優化的結構下計算基態到一批激發態的激發能和振子強度,然后把每個躍遷根據這兩個量用高斯函數展寬,再把所有躍遷進行疊加,即得到UV-Vis光譜圖。Gaussian等程序還順便輸出基態到各激發態的轉子強度,用它代替振子強度進行展寬的話得到的是電子圓二色譜(ECD)。
(2)熒光光譜:假設kasha規則是滿足的,在優化的S1結構下計算S1->S0的垂直發射能以及對應的振子強度,然后用高斯函數展寬成峰形即是熒光光譜圖。
(3)磷光光譜:在優化的T1結構下計算T1->S0的垂直發射能以及對應的振子強度(需考慮旋軌耦合,否則必為0),然后用高斯函數展寬成峰形即是磷光光譜圖。
至于展寬的細節和實現,詳見前述的《使用Multiwfn繪制紅外、拉曼、UV-Vis、ECD和VCD光譜圖》。
由于電子激發實際上還涉及到不同振動態之間的躍遷,所以電子光譜是有精細結構的(但在極性溶劑下觀測不到,只能觀測到帶狀光譜)。如果不考慮核振動,即用前面介紹的計算方式,得到的理論光譜是沒有精細結構的,特別是熒光、磷光光譜圖上僅僅只有一個高斯峰形而已。如果要得到含有精細結構的電子光譜,即振動分辨(Vibrationally-resolved)的電子光譜,必須考慮核振動波函數。具體做法見前述的《振動分辨的電子光譜的計算》。算這個的成本比前述不考慮核振動效應時高得多。
2.5 溶劑效應
溶劑效應對激發態的影響有多方面,會影響激發能而導致譜峰位移,還會影響吸收/發射的強度、使譜峰加寬和變形、影響激發態結構、出現外轉換而退激等等。凡是實際是在溶液中發生的和電子激發有關的問題,計算時應當考慮溶劑,溶劑極性越強考慮的必要性越大。
量化計算激發能的時候一般都是用隱式溶劑模型,將溶劑環境當成具有一定介電常數的連續介質來考慮。這種方式考慮溶劑效應一般可以基本合理表現出它對激發能、振子強度的影響以及對激發態結構的影響。隱式溶劑模型種類很多,激發態計算的目的一般就用PCM,這也是Gaussian的SCRF關鍵詞默認的,其它程序有的只支持比PCM形式更簡單的COSMO,效果也差不多。
TDDFT級別下,隱式溶劑模型的溶劑場對激發態的響應有不同考慮方式。常用的有兩種:
(1)線性響應(Linear response, LR):溶劑對激發能的修正量是躍遷密度與躍遷密度導致的溶劑極化之間的相互作用。這種處理比較常用,相比氣相計算不會額外帶來太多耗時。
(2)態特定(State-specific, SS):令溶劑直接對指定的激發態的密度進行響應,通過反復迭代令溶劑場與激發態密度間完全自洽。每次迭代都要做電子激發計算求出激發態密度,因而也稱外迭代(External iteration)。這將使計算耗時增加一個數量級,還使得TDDFT失去解析梯度。因此僅當對結果要求精確的時候使用,且一般不用在優化激發態的過程中。而且一次只能考慮一個激發態,難以獲得同時涉及很多態的吸收光譜。一般來說,除非考察的是電荷轉移激發態,否則昂貴、麻煩的SS實際上并不比LR有多大優勢,甚至還可能結果更差。
溶質會使溶劑分子被極化,包括溶劑的電子結構被極化以及分子朝向被極化,分別對應于溶劑弛豫的快部分和慢部分。對于處于某個電子態的體系,若溶劑的兩部分都已經對其弛豫了,稱為平衡溶劑(eq);如果只是快部分弛豫了,稱為非平衡溶劑(neq)。垂直吸收、垂直發射的過程非常短暫,末態時溶劑分子只有其電子部分來得及被重新極化,而朝向來不及被重新極化還停留在初態的狀況,因此初態計算時應處于平衡溶劑下,而末態計算時應處于非平衡溶劑下。絕熱吸收/發射過程則可認為在初態和末態時溶劑都已完全弛豫,即都處在平衡溶劑下。在溶劑中計算吸收/發射能嚴格來說應當考慮非平衡溶劑效應(盡管是否考慮影響不很大),根據上面的討論,計算方法如下所示
垂直吸收能 = E_ES(R_GS, neq) - E_GS(R_GS, eq)
垂直發射能 = E_ES(R_ES, eq) - E_GS(R_ES, neq)
絕熱吸收能 = 絕熱發射能 = E_ES(R_ES, eq) - E_GS(R_GS, eq)
其中E_ES和E_GS分別代表激發態和基態能量。R_ES和R_GS分別代表激發態極小點結構和基態極小點結構。諸如E_ES(R_GS, neq)的含義就是在基態極小點結構下、非平衡溶劑下的激發態能量。
是否考慮非平衡溶劑效應,用線性響應還是態特定方法,是不同類別的問題,可以以不同方式組合。原理上說,用態特定方法,同時考慮非平衡溶劑效應,得到的激發能是最準確的,但也最耗時、繁瑣,Gaussian手冊上那個“7步”例子之所以搞得那么麻煩,就是因為用了這種方式考慮溶劑,而且從吸收到發射算了一個完整來回,于是把初學者們搞暈了。如果不要求那么高,用線性響應模型其實就夠了,由于誤差抵消之類因素,也不一定比態特定的結果差,還省了大量時間。
3. Gaussian中的TDDFT計算
3.1 關鍵詞
Gaussian中做TDDFT計算的關鍵詞基本格式是“# 泛函/基組 TD(選項)”。激發態優化、振動分析、找過渡態、產生IRC、勢能面掃描等依賴于勢能面的任務對于激發態也照樣可以做,關鍵詞和基態時完全一致,比如要優化激發態就加個opt,要算激發態頻率就加個freq。
TD里的常用選項有下
nstates=N:表明總共算能量最低的N個激發態的信息,如激發能、躍遷電/磁偶極矩、振子強度、組態系數等。默認為3。
root=i:選擇感興趣的態。如果當前任務是與某個激發態有關的,比如幾何優化、振動分析、獲得偶極矩等,則表明是對第i個激發態做的。只是計算激發態信息則不需要管此設定。默認為1。
singlet:要求計算的N個激發態都為單重態,此為默認。
triplet:要求計算的N個激發態都為三重態。
50-50:要求同時計算N個單重態和N個三重態。
注意singlet、triplet、50-50關鍵詞只對基態是閉殼層有效,基態是開殼層的話,沒法指定激發態的自旋多重度,而只能是算出什么態就是什么態,得根據激發態的<S**2>來判斷激發態的自旋多重度。<S**2>的理想值為S(S+1),S是體系的自旋量子數。常見的幾種情況如下:
單重態=0.0
二重態=0.75
三重態=2.0
四重態=3.75
五重態=6.0
...
因此,比如一個自由基,算出來某個激發態的<S**2>=0.85,相對來說比較接近于上表的0.75,因此可以認為是二重態激發態。
3.2 nstates的設置
原理上nstates越大結果越準確,但設得越大計算耗時越多,因此不能盲目設大。nstates應該設多大值得專門說一下,有兩種情況:
(1)研究特定激發態的情況
除非有對稱性,否則無論哪種激發態算法,都是按照激發能從低往高算的。即計算第i個激發態,就必須把能量小于i的激發態也算出來。所以,如果感興趣的是第i個激發態,則總共需要計算至少i個激發態。
對于TDDFT,研究第i個激發態時不要恰好只算i個態,建議算到i+3個態,算到i+5個態更穩妥,比起只算i個態時第i個態的能量會更準確。因為Gaussian是通過Davidson迭代方式近似求解TDDFT方程來得到最低的一批解,其中最高的幾個態的誤差會比更低的態更大。但也沒必要算的態數超過i太多,否則會浪費很多時間。
(2)計算電子光譜的情況,需要綜合考慮三點問題:
波長范圍(主要):對于同一個體系,算的態數越多,涉及的激發能范圍就越高,得到的理論光譜因此就能覆蓋到光譜圖越低的波長。
體系(主要):若要算到同樣的波長,體系共軛程度越大、雜原子越多,需要算越多的態。對于同類體系,原子數越多時需要算越多的態。
理論方法(次要):算到同樣的波長,HF成份越高的泛函做TDDFT由于激發能整體越高,因此需要算越少的態。
通常計算光譜需要覆蓋整個可見光區和部分UV區,一般感興趣的是2~7eV區間。算多少態需要經驗,往往還需要嘗試(可以先用低級別的方法如ZINDO或小基組測試一下)。比如有的小體系可能算10個足矣,但一些共軛大分子體系往往要算上百乃至幾百。態數算少了光譜范圍覆蓋不足需要重算,而算得太多則白算出來一堆能量過高的不感興趣的激發態,這都會浪費大量時間。
Gaussian的TDDFT支持一個叫DEmin的關鍵詞,當你發現態數設少了,想補算能量更高激發態的時候有一定用處,見《在Gaussian中對不同能量區間分批計算激發態》(http://www.shanxitv.org/348)。
3.3 TDDFT的頻率計算、過渡態和IRC計算
Gaussian09支持TDDFT的解析梯度,所以用TDDFT優化激發態速度不錯,但是不支持解析的Hessian,必須基于解析梯度做一階有限差分獲得,因此做振動分析耗巨耗時,相當于6N步(N=原子數)幾何優化的耗時,對于大點的體系很難算得動,因此應盡量避免用TDDFT做振動分析。但Gaussian09支持CIS的解析Hessian,因此如果對激發態頻率、振動耦合的電子光譜精度要求不太高,在Gaussian中用CIS做振動分析足矣。找過渡態、走IRC眾所周知需要提供初始的Hessian。然而,TDDFT下寫calcfc計算初始的Hessian過于昂貴,因此在Gaussian09中找激發態的過渡態時建議用opt=(TS,modRedundant,noeigen)或者opt=(TS,gediis,noeigen),走IRC建議用建議用IRC(gradientonly,euler),這樣就不需要用TDDFT通過有限差分計算昂貴的Hessian了,可惜這種方式走IRC的成功幾率較低。
在Gaussian16中,已經支持TDDFT的解析Hessian了,因此對激發態找過渡態、走IRC都不需要什么特殊考慮,直接像對待基態一樣寫關鍵詞就行了,只不過需要多寫上TD而已,比如可以用# B3LYP/6-31G* TD(root=2,nstates=5) opt(TS,calcfc,noeigen)以當前結構為初猜結構找第2激發態上的過渡態。
3.4 激發態的波函數
默認情況下,做TDDFT計算的時候Gaussian產生的是基態的密度,即末尾輸出的多極矩是基態的,波函數分析模塊(L601)、NBO模塊(L607)分析的也是基態波函數。TDDFT計算時如果同時寫上density關鍵詞,說明產生當前級別的密度,即root指定的激發態密度。這樣多極矩、NBO分析、原子電荷等等都是對root指定的激發態計算的,輸出的.wfn/.wfx文件里記錄的也將是第root激發態的自然軌道。但此時chk里記錄的軌道依然是基態的DFT軌道,如果要把激發態自然軌道轉存到chk里需要多做一步,見Multiwfn手冊第四章開頭的說明或《詳談Multiwfn支持的輸入文件類型、產生方法以及相互轉換》(http://www.shanxitv.org/379)。更簡單的做法是直接通過Multiwfn基于fch里的激發態密度矩陣產生激發態自然軌道,見《在Multiwfn中基于fch產生自然軌道的方法與激發態波函數、自旋自然軌道分析實例》(http://www.shanxitv.org/403)。
3.5 T1的優化
優化T1值得專門一說。有兩種做法,一種是用UDFT(也叫UKS),即自旋多重度設3直接優化,不寫TD關鍵詞,比如0 3結合# PBE1PBE/6-31G* opt,這樣是通過SCF方式直接算T1態并優化它,不牽扯TDDFT計算;另一種是自旋多重度設1同時用TD(triplet),比如0 1結合# PBE1PBE/6-31G* TD(triplet) opt,這樣每一步就是先計算單重態基態作為參考態,再通過TDDFT計算T1態并優化它。這兩種方式原理上都合理,結果通常相近,但強烈建議用UDFT方式,因為其耗時只比優化單重態基態增加一點,而用TDDFT優化T1的話計算量則要增加約一個數量級。
不過UDFT方法缺點是只能優化T1,而TDDFT則可以優化任意三重態激發態,而且能夠同時給出軌道躍遷信息。
經常有初學者寫成0 3結合TD(triplet) opt,這樣算出來什么都不是,根本就不是優化T1。
3.6 溶劑模型
Gaussian里TDDFT計算時寫上諸如SCRF=solvent=xxx就能在TDDFT計算時使用表現xxx溶劑的PCM溶劑模型。前面提到的線性響應(LR)和態特定(SS)方式都能支持,詳情如下
線性響應模型(LR):是SCRF關鍵詞默認的。對耗時增加不多,而且TDDFT依然有解析梯度,因此最適合激發態幾何優化。但如前所述,對溶劑效應表現得相對粗略。
態特定(SS):需要在SCRF里面寫ExternalIteration(等價于G09老版本中的statespecific),溶劑場會與root指定的激發態的密度迭代到自洽,雖然比LR更準但很耗時間,而且一次只能對一個激發態來做。SS還使得TDDFT沒有解析梯度,因此SS基本沒法用在激發態優化上。
考慮非平衡溶劑(neq)效應只是對垂直吸收、發射過程而言的,優化過程中應當使用平衡溶劑(eq),因為隨著溶質結構的變化溶劑的朝向會同步變化。
Gaussian中使用LR的時候,在特定結構下計算激發態時,對激發態來說是非平衡溶劑(LR-neq),即溶劑慢部分對于基態是平衡的,只有快部分響應了激發態;在激發態幾何優化時對激發態用的是平衡溶劑(LR-eq),即溶劑快慢部分都是對于激發態平衡的。計算中輸出的基態能量總是在基態的平衡溶劑下的。
Gaussian中使用SS的時候,對激發態默認使用平衡溶劑(SS-eq)。基于SS計算激發態時,當默認的平衡/非平衡溶劑設定和實際情況不符時,需要在SCRF里寫read關鍵詞讀取額外的溶劑設定,這些設定在輸入文件末尾空一行后書寫。在初態計算時用noneq=write來寫入初態時溶劑慢部分的信息,然后在末態計算時再用noneq=read來讀取初態溶劑慢部分的信息。
如果對溶劑模型還糊涂沒關系,直接仔細follow后文的例子就行了,不用想太多。
4. 實例
下面的例子不拿具體體系為例,就只是把用的步驟和關鍵詞介紹一下,一定要舉一反三。用的泛函和基組是隨便選的,示意一下而已。假設體系的基態是單重態。
4.1 氣相下計算最低5個垂直激發能
1 基態幾何優化:# PBE1PBE/6-31G* opt
2 取上一步優化得到的結構,#p PBE1PBE/TZVP TD(nstates=5)
輸出文件中會看到以下輸出,這些是基態到各個激發態的躍遷電、速度、磁偶極矩矢量,以及振子強度(Osc.)。Dip. S.是躍遷偶極矩各個分量平方和。
Ground to excited state transition electric dipole moments (Au):
state X Y Z Dip. S. Osc.
1 -0.1494 0.1449 -0.0654 0.0476 0.0062
2 -0.0829 0.0804 0.0707 0.0183 0.0030
3 0.3706 -0.0971 0.0232 0.1473 0.0252
4 0.3399 0.0539 -0.2128 0.1637 0.0305
5 -0.0200 -0.0149 -0.2283 0.0527 0.0101
Ground to excited state transition velocity dipole moments (Au):
state X Y Z Dip. S. Osc.
1 0.0368 -0.0331 0.0186 0.0028 0.0095
2 0.0436 -0.0233 -0.0285 0.0033 0.0089
3 -0.0787 0.0272 -0.0027 0.0069 0.0180
4 -0.1240 -0.0026 0.0725 0.0206 0.0492
5 -0.0070 -0.0017 0.0706 0.0050 0.0117
Ground to excited state transition magnetic dipole moments (Au):
state X Y Z
1 -0.3277 0.4784 0.1051
2 -0.0701 -0.7474 -0.6712
3 -0.2194 0.0764 -0.5296
4 0.2326 0.2713 0.0772
5 0.0186 0.0527 -0.0944
然后會輸出速度表象下的轉子強度R(velocity)以及長度表象下的轉子強度R(length),繪制ECD才需要。
<0|del|b> * <b|rxdel|0> + <0|del|b> * <b|delr+rdel|0>
Rotatory Strengths (R) in cgs (10**-40 erg-esu-cm/Gauss)
state XX YY ZZ R(velocity) E-M Angle
1 -20.9390 -24.0915 -48.3977 -31.1427 146.38
2 42.4101 28.6271 25.4666 32.1679 54.36
3 3.9375 32.5287 20.8226 19.0963 64.44
4 -7.0182 -38.0949 -15.4724 -20.1952 117.12
5 -17.5879 0.1833 0.3918 -5.6709 152.16
1/2[<0|r|b>*<b|rxdel|0> + (<0|rxdel|b>*<b|r|0>)*]
Rotatory Strengths (R) in cgs (10**-40 erg-esu-cm/Gauss)
state XX YY ZZ R(length)
1 -34.6361 -49.0075 4.8628 -26.2603
2 -4.1120 42.4792 33.5736 23.9803
3 57.4977 5.2465 8.6921 23.8121
4 -55.9240 -10.3458 11.6247 -18.2150
5 0.2632 0.5565 -15.2443 -4.8082
然后輸出各個激發態的信息,如下所示。從輸出的第一激發態的信息中可知它是單重態,不可約表示是A,激發能是5.3393eV (232.21nm),振子強度是0.0062。E(TD-HF/TD-KS)后面的值是這個激發態的能量,即前面輸出的SCF Done對應的基態能量加上這里輸出的激發能。root設多少、設不設都不會影響激發能的輸出,但是只有root選的激發態(此例沒設,默認為1),程序才會直接將它的總能量輸出出來。諸如23 -> 25 -0.20249這樣的輸出的含義看前面提到的《電子激發任務中軌道躍遷貢獻的計算》就行了,這里不提了。
Excited State 1: Singlet-A 5.3393 eV 232.21 nm f=0.0062 <S**2>=0.000
23 -> 25 -0.20249
24 -> 25 0.67156
This state for optimization and/or second-order correction.
Total Energy, E(TD-HF/TD-KS) = -323.278949720
Copying the excited state density for this state as the 1-particle RhoCI density.
Excited State 2: Singlet-A 6.6806 eV 185.59 nm f=0.0030 <S**2>=0.000
23 -> 25 0.47726
24 -> 25 0.18113
24 -> 26 0.44412
24 -> 27 -0.16152
...略
值得一提的是,雖然nstates設了5,此例得到了5個態的激發能,但是如前所述,越高的態激發能越不準。因此如果確實要得到準確的5個最低的激發能,建議將nstates設為10。
之后,利用這個Gaussian輸出文件,通過Multiwfn就可以繪制UV-Vis和ECD光譜了,詳見前述的《使用Multiwfn繪制紅外、拉曼、UV-Vis、ECD和VCD光譜圖》。用gview也可以繪制,但是可調選項沒有Multiwfn靈活,功能沒有Multiwfn強大,還是收費的。
假設你要考察第i激發態的波函數信息(如偶極矩),并同時得到相應的wfn文件用于Multiwfn的分析,這樣寫關鍵詞:#p PBE1PBE/TZVP TD(nstates=5,root=i) density out=wfn,然后末尾空一行寫上wfn文件的輸出路徑。這個任務算完后輸出文件末尾顯示的偶極矩、四極矩,還有Mulliken電荷等等都是第i激發態的。如果比如還要對這個態做NBO分析,再寫個pop=NBO就完了。
4.2 氣相下計算某分子S1結構和熒光發射能
1 基態幾何優化:# PBE1PBE/6-311g(d) opt (如果你覺得初始結構已經比較合理的話,這一步可以省)
2 取上一步優化得到的結構,做S1態優化:# PBE1PBE/6-311g(d) TD opt (TD默認是root=1,默認的nstates=3對于優化S1一般夠了,所以這里只寫個TD)
第二步最后的結構就是S1結構了。每一步優化都會做一次電子激發計算,所以會看到很多上一節那樣的輸出。最后一次輸出的第一激發態的激發能就是熒光發射能了。
利用第二步的輸出文件就可以繪制熒光光譜了。這里假定是符合kasha規則從S1發射的,所以繪圖時要把一同輸出的S2、S3的信息抹掉。如果用gview繪制,手動將S2、S3態的振子強度設為0,再按照繪制UV-Vis光譜操作即得到熒光光譜。若用Multiwfn繪制則不需要手動修改輸出文件,將輸出文件載入Multiwfn,依次輸入以下命令即可
11 //繪制光譜圖
3 //UV-Vis
20 //修改振子強度
2,3 //選擇第2、3激發態
0 //振子強度設為0
0 //繪制光譜
如果你還有不明白的地方,看Multiwfn手冊4.11.11節,里面有個完整的繪制BODIPY熒光光譜的例子。
順帶一提,如果你要優化S4,可以寫比如# PBE1PBE/6-311g(d) TD(nstates=7,root=4) opt。
4.3 氣相下計算T1結構和磷光發射能
有兩種方法做這個事情。
方法一:
1 優化T1結構:# B3LYP/6-31G* opt,電荷和自旋多重度為0 3。
2 取上一步優化得到的結構,用# B3LYP/6-31G* TD(triplet),電荷和自旋多重度為0 1。第一激發態的激發能就是磷光發射能了。
方法二:
1 優化T1結構:# B3LYP/6-31G* opt,電荷和自旋多重度為0 3。取最后一步的能量,作為T1結構下T1能量。
2 取上一步優化得到的結構,0 1下用# B3LYP/6-31G*計算單點能,得到T1結構下S0能量。
3 將第1步的能量減去第2步的能量即是磷光發射能
雖然兩個方法結果有定量差異,但原理上都對,由于方法二還考慮了軌道弛豫,所以原則上會更精確一些,而且由于不用做TDDFT計算所以耗時短,缺點是沒法像方法一那樣得到軌道躍遷信息。另外,如果你還用TDDFT算了熒光,那么建議用方法一算磷光,這樣都在TDDFT下計算結果更有可比性。
由于Gaussian不支持TDDFT級別下的旋軌耦合計算,所以得不到磷光發射對應的振子強度(輸出的數值為0),因此沒法繪制磷光光譜、計算磷光速率。支持MCSCF、MRCI等級別下做旋軌耦合的程序不少,不過這類方法很難用于大體系。目前支持TDDFT下考慮旋軌耦合得到振子強度的程序不算多,只有ADF(巨貴而且坑爹,別買)、Dalton(免費,推薦使用,計算例子見http://bbs.keinsci.com/thread-2455-1-1.html,以及計算化學公社量子化學版里面Dalton分類的帖子),以及Dirac等支持二/四分量相對論TDDFT的程序(使用門檻略高)。如果你只需要計算單-三重態間的旋軌耦合矩陣元的話,可以將Gaussian與PySOC結合使用,見《使用Gaussian+PySOC在TDDFT下計算旋軌耦合矩陣元》(http://www.shanxitv.org/411)。更好、更方便的選擇是用ORCA,見《使用ORCA在TDDFT下計算旋軌耦合矩陣元和繪制旋軌耦合校正的UV-Vis光譜》(http://www.shanxitv.org/462)。
4.4 基于線性響應溶劑模型做TDDFT計算
這個例子包含4個任務,你需要研究什么問題就follow哪個,沒有順序關系。此例是通過線性響應方法,通過PCM溶劑模型表現乙醇環境。
(1)溶劑下基態幾何結構優化:# PBE1PBE/6-311G* opt scrf=solvent=ethanol
(2)計算溶液中的最低20個態的激發能。取(1)得到的結構,用這些關鍵詞:# PBE1PBE/6-311G* TD(nstates=20) scrf=solvent=ethanol。然后如4.1節所說用Multiwfn或gview繪圖可以得到溶劑下的吸收譜。
(3)優化溶液中的S1結構:取(1)得到的結構,用這些關鍵詞:# PBE1PBE/6-311G* TD scrf=solvent=ethanol opt
(4)計算溶液中的熒光發射能:直接取(3)輸出文件當中最后一次的S0->S1激發能即可。可以如4.2節所說用Multiwfn或gview繪制溶劑下的熒光譜(這里強調一點,獲得熒光發射能不要取(3)的結構再單獨做一次電子激發計算得到S0->S1的能量,因為此時激發態處于非平衡溶劑,和發射光譜的溶劑實際特征恰相反。如果你就是要取(3)的結構單獨做一次電子激發計算,比如想使用比優化激發態更大的基組算發射能,那么可以在TD里加上Eqsolv,此時溶劑對激發態是平衡的,和實際發射的情況一致)。
如果要算磷光發射,把(3)改為用UDFT方式優化T1,然后在(4)中把TD里寫上triplet即可。
可見,基于線性響應模型做TDDFT計算和氣相下區別僅僅在于加上SCRF關鍵詞而已,什么其它區別都沒有,計算耗時也增加不多,十分省事。但如果非要用原理上更準確的特定態模型,那么如下一節所示,就繁瑣多也慢多了。
4.5 基于特定態溶劑模型做TDDFT計算
這個例子包含4個任務,你需要研究什么問題就follow哪個,沒有順序關系。此例是通過特定態方法,靠PCM溶劑模型表現乙醇環境。
(1)溶劑下基態幾何結構優化:和4.4節的(1)等同。
(2)計算溶液中基態到S1態的激發能:
特定態方法計算激發能時,默認情況下激發態處于平衡溶劑下,但實際中應當處于非平衡溶劑下(慢部分對基態是平衡的),因此需要做兩步才能正確表現非平衡溶劑效應
第一步:在(1)得到的結構下,先在溶劑下做基態單點計算,把基態的溶劑信息寫到chk里,同時得到基態在平衡溶劑下的能量(SCF Done后面的值):
# PBE1PBE/6-311G* scrf(read,solvent=ethanol),末尾空一行寫noneq=write
第二步:做電子激發計算,并讀取上一步在chk里寫入的基態溶劑信息,由此得到激發態在非平衡溶劑下的能量:
# PBE1PBE/6-311G* TD scrf(externaliteration,read,solvent=ethanol) geom=check guess=read,末尾空一行寫noneq=read
這一步計算耗時會很長,會反復計算許多次激發態,這是為了讓溶劑場與S1態電子密度相互自洽(即外迭代過程)。輸出文件末尾的下面這條信息告訴了你特定態方法+非平衡溶劑下的激發態的能量:
After PCM corrections, the energy is -153.527526510 a.u.
將這個能量減去第一步的能量就是所需的激發能了。
(3)優化溶液中的S1結構:和4.4節的(3)等同。特定態方法下雖然也能做幾何優化,但巨慢,所以優化激發態還是應當用線性響應模型。
(4)計算溶液中的熒光發射能:
第一步:在(3)得到的結構下,獲得平衡溶劑下的激發態能量,并將激發態溶劑信息寫入chk:
# PBE1PBE/6-311G* TD scrf(externaliteration,read,solvent=ethanol),末尾空一行寫noneq=write
激發態能量應當取末尾輸出的After PCM corrections后面的能量值。
第二步:做基態計算,讀取第上一步在chk里寫入的激發態溶劑信息,得到非平衡溶劑下的基態能量(SCF Done后面的值):
#P PBE1PBE/6-311G* scrf(read,solvent=ethanol) geom=check guess=read,末尾空一行寫noneq=read
然后將第一步的能量減去第二步的能量就是熒光發射能。
可見,用特定態方法比用線性響應方法繁瑣得多也耗時得多。這一節涉及的激發態是S1態,如果考察第i激發態就把TD里面寫上root=i即可。如果要算磷光發射,把(3)改為用UDFT方式優化T1,然后在(4)中把TD里寫上triplet即可。
5 其它問題
有幾個問題值得在這里說一下。5.1 注意優化激發態時破壞對稱性
就是激發態和基態的對稱性往往是不同的,優化小分子激發態的時候不注意這一點往往得不到真正的激發態極小點結構。例如乙醛,基態是Cs對稱性,但是S1的極小點結構并不具有Cs對稱性。因此,直接拿它的基態結構作為初始結構來優化S1是萬萬不可的,否則得到的S1結構會依然保持Cs對稱性,做振動分析會發現虛頻。為得到真正極小點結構,應當在優化S1前先把基態的結構人為地扭曲一下使得Cs對稱性被破壞,比如隨意微微扭動一個二面角。激發態對稱性往往低于基態,所以如果基態有對稱性,建議激發態優化前都先人為地稍微破壞一下對稱性。5.2 注意構象問題
柔性分子會有諸多構象,不同構象對應的吸收光譜也不一樣。計算吸收光譜時,如果用的構象不是能量最低的構象,那么會和實驗譜差很多。如果多個構象能量相差不太大,而且彼此間在實驗溫度下足矣越過勢壘能夠相互轉化,那么必須計算各自的波爾茲曼分布對光譜做權重平均,權重的計算方法見《根據Boltzmann分布計算分子不同構象所占比例》(http://www.shanxitv.org/165),權重平均的光譜繪制見《使用Multiwfn繪制構象權重平均的光譜》(http://www.shanxitv.org/383)。但對于發射光譜則不考慮構象分布這個問題,因為激發態壽命短暫,激發態分子來不及在各個激發態極小點之間達到熱力學平衡狀態,所以就在基態極小點作為初始結構優化出的激發態結構下計算發射光譜就行了。
5.3 激發能算出來是負值怎么辦?
有的時候算出來的一個或多個激發能為負值,這說明SCF部分產生的參考波函數(即HF或DFT波函數)不是當前結構下真正的基態波函數,此時得到的激發能也沒有意義。此時若用stable關鍵詞進行波函數穩定性檢測,一定會也提示波函數不穩定,解決方法:
先檢查自旋多重度設定的合理性,看是否自旋多重度設定的和當前結構下實際基態波函數不符(比如當前結構下基態是單重態但卻誤設了三重態)。
若確信自旋多重度沒設錯,則做一次單點計算,同時寫stable=opt關鍵詞讓Gaussian自動找出真正的基態波函數,然后做激發態計算的時候寫guess=read讀取其作為初猜波函數。
按以上方式處理后,若激發能都為正說明沒問題了。
5.4 怎么分析電子激發?
光是算算激發能、光譜,經常會顯得研究很膚淺,完全沒有在電子激發本質、特征層面上進行討論。Multiwfn是電子激發分析最強大同時也是最好用的工具,電子激發的類型是什么、牽扯到了哪些片段/原子/原子軌道、電子激發時哪個片段向哪個片段轉移了多少電子、電子激發導致的正負電荷分離程度如何、哪些分子片段或分子軌道對振子強度有關鍵性影響、電子激發導致成鍵特征發生了什么變化等等全部細節都可以用Multiwfn非常容易地分析得特別透徹。以后經常做電子激發計算的讀者務必要仔細看《Multiwfn支持的電子激發分析方法一覽》(http://www.shanxitv.org/437),此文有對電子激發分析手段十分全面的論述,熟練掌握后你會發現Multiwfn是電子激發研究完全離不開的工具。
5.5 注意勢能面交叉對激發態優化的影響
激發態的能級分布往往很密集,彼此之間在很多結構下存在交叉,再加上激發態勢能面有時候還有分叉之類特征,導致激發態的優化往往比基態的優化的情況復雜不少,比如會出現對不同激發態優化最終都優化到了相同結構,或者一開始優化的是比如S4態,但是發現在最終結構下這個態的能量卻并不是第四低的。強烈建議讀者在徹底掌握本文所述內容后閱讀這篇文章:《談談勢能面交叉對激發態優化的影響》(http://www.shanxitv.org/468)。把這篇文章內容徹底搞明白了,對激發態優化時遇到的一些看似奇怪的現象就能理解到底是怎么回事了。