基于背景電荷計算分子在晶體環境中的吸收光譜
基于背景電荷計算分子在晶體環境中的吸收光譜
文/Sobereva@北京科音 2020-Dec-23
1 前言
用Gaussian、ORCA等主流量子化學程序計算分子在真空中、在溶劑環境中的激發態和吸收光譜是非常簡單的事,參考比如《Gaussian中用TDDFT計算激發態和吸收、熒光、磷光光譜的方法》(http://www.shanxitv.org/314)、《Simulating UV-Vis and ECD spectra using ORCA and Multiwfn》(http://www.shanxitv.org/485)。也有不少人需要計算分子在其晶體環境中的吸收光譜,這需要表現出周圍分子產生的影響。筆者在網上經常看到有人對ONIOM一知半解,亂用ONIOM來試圖實現這個目的,普遍做法嚴重不當,比如居然用對靜電勢重現性巨差的QEq電荷、居然不知道應當使用電子嵌入,導致結果根本不靠譜卻渾然不知。在本文,筆者就專門介紹和演示如何正確地借助背景電荷來計算分子在晶體環境中的吸收光譜。本文的這種做法是被廣為接受的。
2 原理
在講具體例子前,先說一些最基本常識。背景電荷(background charge)在多數主流量子化學程序里都支持,它是指位于特定坐標的點電荷,其坐標和電荷值都是用戶在輸入文件里設置的。背景電荷對體系產生的靜電作用,影響體系的電子結構,也因此影響體系的各方面特征,顯然也包括電子激發相關性質,如激發能、振子強度、電子光譜。
在分子晶體中,分子是緊密排布的。下文管被計算激發態的那個分子叫中心分子,在晶體環境中它被周圍一層分子圍繞,這些分子在下文稱環境分子。顯然,要考察分子在晶體中的電子激發問題,應當把環境分子對它的影響恰當地考慮。環境分子與中心分子之間主要有范德華作用和靜電作用。如果涉及到結構優化的話,這兩種作用都需要表現出來。但在本文中,我們只計算晶體結構下的吸收光譜,不僅不涉及結構優化問題,而且環境分子與中心分子的范德華作用不會對其電子結構產生“直接”的影響,所以我們并不需要考慮范德華作用。環境分子對中心分子的靜電作用有不同方式描述,比較高級的可以考慮用分布多級展開等,但通常情況下通過背景電荷以這種最粗糙、最省事的方式來表現就夠了,這可以在一般量子化學程序計算中使用,而且對耗時的影響可忽略不計。具體來說,就是在環境分子各個原子核位置放置一個背景電荷,令電荷數值等于原子電荷。計算原子電荷的方法非常多,由于要求背景電荷必須能盡可能好地體現環境分子對中心分子的靜電作用,因此當前所用的原子電荷必須對分子范德華表面附近的靜電勢有較好的重現性,毫無疑問首選是擬合靜電勢電荷。擬合靜電勢電荷是一類方法,有不同的具體實現,一般就用比較流行的CHELPG或者MK電荷即可,后文都用CHELPG。用知名的RESP電荷也可以,但對于當前問題不比CHELPG有任何優勢。如果你對原子電荷了解甚少的話,務必閱讀《原子電荷計算方法的對比》(http://www.whxb.pku.edu.cn/CN/abstract/abstract27818.shtml)和《RESP擬合靜電勢電荷的原理以及在Multiwfn中的計算》(http://www.shanxitv.org/441)了解相關知識。看過之后自然就知道,用諸如QEq、NPA、AIM等對靜電勢重現性極差的原子電荷當背景電荷表現環境分子產生的靜電作用是完全不能接受的。
X光衍射測得到的晶體結構中氫的位置一般是明顯不準確的,見《實驗測定分子結構的方法以及將實驗結構用于量子化學計算需要注意的問題》(http://www.shanxitv.org/569)。因此在計算中心分子之前,中心分子的氫的位置是必須優化的。這相當于限制性優化,即在優化過程中將重原子(非氫原子)位置凍結在X光衍射測的坐標上,見《在Gaussian中做限制性優化的方法》(http://www.shanxitv.org/404)。在計算環境分子的擬合靜電勢電荷之前,也最好對環境分子的氫進行優化。顯然,優化時不能對所有原子都做,因為真空環境和晶體環境明顯不同,不凍結重原子的話體系結構就可能變得和晶體狀態明顯不符。
有人可能搞不明白用背景電荷和用ONIOM來實現環境分子對中心分子的影響有什么區別。實際上,用ONIOM可以實現和使用背景電荷相同的效果。具體來說,是從晶體中摳出一個團簇,將中心分子設為高層,用量子力學(QM)方式描述,而將環境分子設為低層,用分子力學(MM)描述,并將每個低層原子的電荷值設成擬合靜電勢電荷,并且要求程序用電子嵌入(electronic embedding),從而使得MM部分的原子電荷可以極化QM部分的電子結構。ONIOM的基本知識和應用我在北京科音基礎量子化學培訓班(http://www.keinsci.com/workshop/KBQC_content.html)里講得非常詳細,這里就不再多說了。對于單純計算吸收光譜的目的,我非常不建議通過ONIOM來實現,因為這不僅把事情搞復雜、輸出文件形式特殊,而且Gaussian做ONIOM得到的chk文件里不包含高層部分的波函數,因此之后無法用Multiwfn(http://www.shanxitv.org/multiwfn)對中心分子做各種波函數分析,無法做很流行的空穴-電子、NTO、躍遷密度矩陣圖等電子激發分析(見《使用Multiwfn做空穴-電子分析全面考察電子激發特征》http://www.shanxitv.org/434和《Multiwfn支持的電子激發分析方法一覽》http://www.shanxitv.org/437),因此局限性特別大,純粹是給自己找麻煩。只有當涉及到幾何優化的問題,比如在晶體環境中優化分子的激發態,那才真的有必要用ONIOM,因為這樣才能在優化時表現必不可少的環境分子和中心分子間的范德華作用,而這不屬于本文的范疇。
3 實例:桂皮酰胺(cinnamide)晶體的UV-Vis光譜
通過這個例子筆者演示如何實際進行計算。主要過程包含以下步驟
(1)構建晶體的超胞
(2)摳出分子團簇
(3)優化氫原子位置
(4)分離中心分子和環境分子
(5)對各個環境分子計算CHELPG電荷
(6)構建包含背景電荷的中心分子的TDDFT輸入文件并計算光譜
下文提到的每一個文件都可以在這個文件包里找到:http://www.shanxitv.org/attach/579/file.zip。本文涉及的程序有VESTA 3.3.8、Multiwfn 3.8(dev)、VMD 1.9.3、Gaussian 16 A.03、GaussView 6.0.16(本文里許多GaussView的特征在更老的版本里沒有)。Multiwfn可在http://www.shanxitv.org/multiwfn免費下載,VMD可在http://www.ks.uiuc.edu/Research/vmd/免費下載,VESTA可在http://jp-minerals.org/vesta/en/免費下載。
3.1 構建晶體的超胞
桂皮酰胺晶體的cif文件是本文文件包里的cinnamide.cif。為了能摳分子團簇,我們先把原胞擴展成足夠大的超胞。使用GaussView獲得超胞的做法在《基于分子晶體cif文件摳出分子團簇結構》(https://www.bilibili.com/video/av35864488/)里我已經詳細演示了,用GaussView的缺點是當產生的超胞原子數很多的話,GaussView可能要卡很久,而且GaussView也是收費的。這里筆者演示使用VESTA程序來實現這個目的。
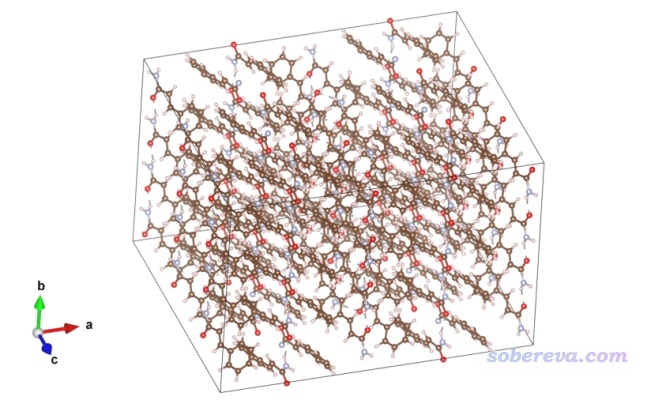
啟動VESTA,把cinnamide.cif拖入,選Edit - Edit Data - Unit Cell,再選Transform,將旋轉矩陣的三個對角元分別設為2、4、3,代表在第1、2、3個方向延展復制成原先的2、4、3倍,然后點OK,然后選“是”,再點一次OK。此時的結構如下所示,明顯超胞已經足夠大了,肯定能挖出一個中心分子+最近一層環境分子的團簇。
然后選File - Export Data,格式選xyz,保存為supercell.xyz。問是否save hidden atoms too時選“是”。
3.2 摳出分子團簇
這部分的操作我在《基于分子晶體cif文件摳出分子團簇結構》(https://www.bilibili.com/video/av35864488/)中已經詳細演示了。將Multiwfn文件包里的examples\getcluster.vmd拷到VMD目錄下。然后啟動VMD,將supercell.xyz載入VMD,然后在VMD的命令行窗口里輸入source getcluster.vmd運行此腳本,文本窗口最后會提示same fragment as {within 3.5 of fragment 31},說明用這個選擇語句可以選中我們需要的團簇部分。如果對VMD選擇語句不懂的話,看《VMD里原子選擇語句的語法和例子》(http://www.shanxitv.org/504)。
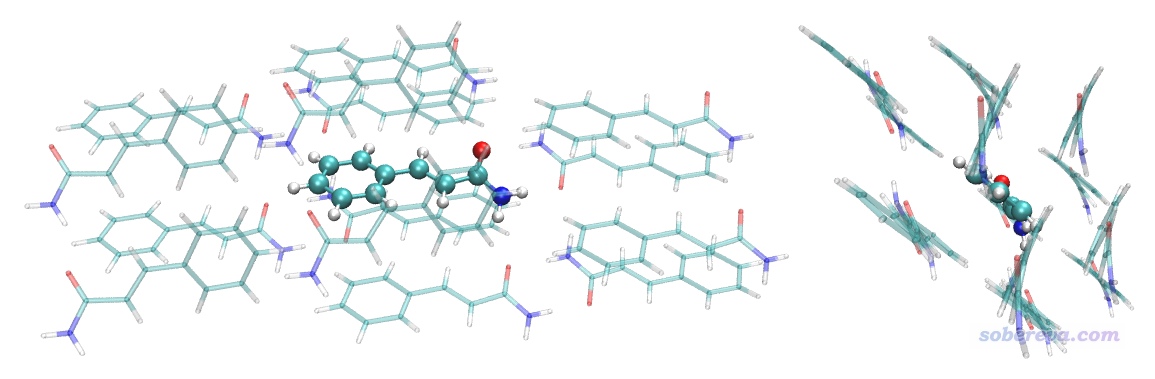
然后我們看看這個選擇語句選中的區域到底是不是我們實際想要的,于是在VMD中選擇Graphics - Representation,將same fragment as {within 3.5 of fragment 31}復制到Selected Atoms文本框里,然后按回車,就把團簇部分顯示出來了。之后若恰當修改顯示方式,讓位于中間的fragment 31部分CPK方式顯示,可看到下圖,兩個視角都給出了。可見確實中心分子周圍緊緊包裹了一層環境分子,沒有缺的也沒有多余的,故是很理想的模型。
然后在VMD main窗口里點擊當前體系,點右鍵選Save Coordinate,File type選mol2,Selected atoms輸入same fragment as {within 3.5 of fragment 31},然后點Save保存為cluster.mol2。
3.3 優化氫原子位置
當前團簇一共300個原子,包括1個中心分子和14個環境分子。對于Gaussian用戶,優化氫原子的位置可以用B3LYP-D3(BJ)/6-31G*,對于目前主流的雙路服務器來說做這個任務沒明顯壓力。這里為了省時間,就用Gaussian 16里的PM7半經驗方法優化,雖然優化的精度差一些,但比起不做優化強得多,結果也算定性合理了。將cluster.mol2載入GaussView,然后保存為cluster_optH.gjf,將內容修改為下面這樣,代表只優化氫原子,詳見《在Gaussian中做限制性優化的方法》(http://www.shanxitv.org/404)。
# PM7 opt=ReadOpt
<---空行
generated by VMD
<---空行
0 1
[坐標部分]
<---空行
noatoms atoms=H
<---空行
<---空行
這個優化任務耗時不高,在筆者的普通4核筆記本上花了20分鐘,共優化17步收斂了。你會發現優化后氫涉及的化學鍵比初始結構長了不少,也合理了很多。比如原先的氨基的兩個N-H鍵一個是0.909埃,一個是0.938,明顯偏短。而優化后,沒形成氫鍵的N-H是1.0埃左右,形成了氫鍵的稍微長一點,是1.02~1.05埃。
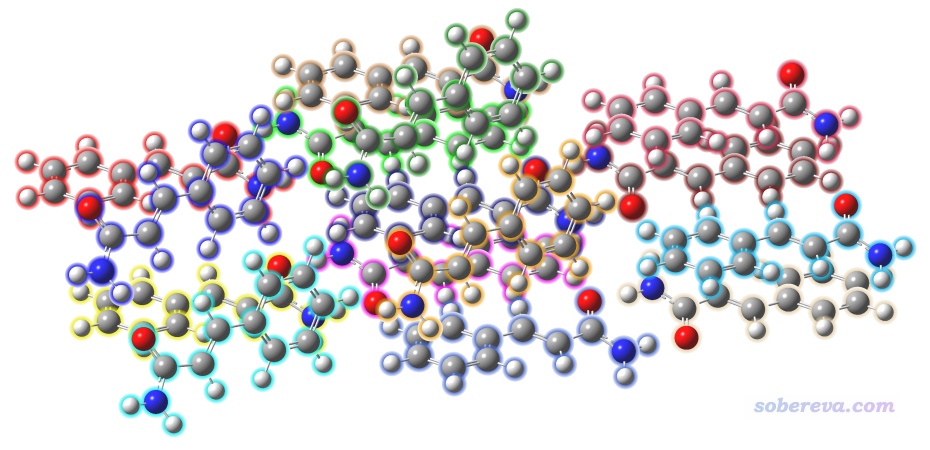
目前還有一個問題,是團簇中各個分子里面的原子序號不連續,這導致沒法用下一節介紹的工具便利地自動產生各個環境分子的輸入文件。為解決此問題,用GaussView打開上面算完的cluster_optH.out,然后點擊Edit - Standardize Z-matrix,此時每個分子里面的原子序號就變得連續了。下圖將1-20、21-40、41-60...281-300原子序號的部分用不同顏色分別顯示,可見確實是每個分子里面的序號都是連續的。
3.4 分離中心分子和環境分子
在GaussView中,在中心分子的任意一個原子上點右鍵,然后選擇Select fragment of Atom x,使整個分子成為黃色,然后點Ctrl+X挪到剪切板里,再新建一個窗口,點Ctrl+V將中心分子粘進來(此時笛卡爾坐標和在團簇中相同),保存為center.gjf。之前的窗口中剩下的都是環境分子,保存為environ.gjf。
3.5 對各個環境分子計算CHELPG電荷
下面來計算環境分子的CHELPG電荷。我不建議將所有環境分子當做一個整體來一次性計算它們的CHELPG電荷,而應當對每個分子分別單獨計算CHELPG原子電荷,原因有二:
(1)計算耗時和原子數絕對不是呈線性正比關系,而一般是呈三次方左右的正比關系,因此所有環境分子一起算的話耗時遠高于每個分子單獨算之和,尤其是對于環境分子的總原子數很多的情況。
(2)CHELPG電荷是通過擬合靜電勢得到的。根據此方法的擬合點的分布規則,若將所有環境分子視為整體確定擬合點的位置,有些情況下可能在中心分子區域分布的點數太少,導致擬合出的環境分子的CHELPG電荷對中心分子區域的靜電勢重現性不理想,也因此相應的背景電荷沒法表現好環境分子對中心分子的靜電作用。
不過,將環境分子視為整體來算CHELPG電荷在原理上可以體現出環境分子間的耦合效應對環境分子的電子結構的影響,不過這點對于當前這種中性體系來說可以忽視。
可能有人覺得沒必要對每個環境分子都挨個算電荷,認為只需算一個分子的CHELPG電荷,然后直接簡單復制N次就能得到所有環境分子的原子電荷。這種想法在實際中是有問題的:
(1)雖然我們已經讓每個環境分子內部的原子序號是連續的,但原子順序卻往往是不同的。比如第一個環境分子可能是C1,C6,N2...,第二個環境分子可能是N2,C6,C1...
(2)晶體中分子可能有不止一個構象,而擬合靜電勢電荷受構象影響很大
(3)有的時候考察的是共晶,甚至環境分子都不止一種
為了使分別計算各個環境分子的過程盡可能省事,我寫了一個名為splitwhole的程序,可以在http://www.shanxitv.org/soft/splitwhole_1.0.zip下載。其中帶后綴的是Windows下可執行文件,無后綴的是Linux下可執行文件。可以將含有一批原子的gjf文件提供給此程序,然后由用戶輸入各個片段里的原子序號,splitwhole就會基于當前目錄下的模板文件template.gjf產生各個片段的gjf文件,之后就可以批量計算了。下面我們就借助這個程序實現對每個環境分子算CHELPG電荷。
創建一個名為template.gjf的模板文件,放在當前目錄下,內容如下所示,任務是計算CHELPG電荷,并且用了nosymm關鍵詞確保Gaussian在計算時不自動平移、旋轉體系。nosymm的具體解釋看《談談Gaussian中的對稱性與nosymm關鍵詞的使用》(http://www.shanxitv.org/297)。CPU核數、內存上限請酌情設置。用此例的B3LYP/def-TZVP計算級別就足夠得到合理的CHELPG電荷,如果想要更好可以用def2-TZVP。[GEOMETRY]這行代表此處會被替換為用戶選擇的片段中的原子坐標。如果是在Linux下運行。
%mem=5000MB
%nprocshared=36
# B3LYP/TZVP pop=CHELPG nosymm
<---空行
Title Card Required
<---空行
0 1
[GEOMETRY]
<---空行
<---空行
啟動splitwhole,然后依次輸入
environ.gjf //上一節得到的包含了所有環境分子坐標的gjf文件
14 //指定14個片段,對應14個環境分子(environ.gjf一共280個原子,除以每個分子的原子數20便知)
a 20 //代表將1-20、21-40、...、261-280依次定義為這14個片段
然后當前目錄下就出現了frag_0001.gjf、frag_0002.gjf...frag_0014.gjf,分別對應14個環境分子的輸入文件,計算設定和template.gjf里的精確相同。
下面批量計算這14個gjf文件,可以用此文的腳本:《使用Gaussian時的幾個實用腳本和命令》(http://www.shanxitv.org/258)。在筆者雙路E5-2696v3共36核機子上3分鐘就都算完了,產生的out文件在本文的文件包里都給了。
在本文文件包里我提供了一個批量提取坐標和CHELPG電荷的Bash shell腳本getCHELPG.sh。注意里面開頭有一行natm=20,代表當前被處理的體系原子數是20,算其它體系的時候別忘了改。將此腳本拷到14個out文件所在目錄下,運行此腳本,就會依次從當前目錄下所有out文件中提取信息,并在當前目錄下產生bkchg.txt文件,前三列是提取出的原子的XYZ坐標(埃),最后一列是CHELPG電荷,如下所示:
-11.079484 3.713347 -0.838676 0.272660
-12.081139 4.631418 -1.104034 -0.210476
-11.445682 2.427925 -0.472243 -0.188136
-13.413720 4.251740 -1.034406 -0.034063
-11.829578 5.657487 -1.375829 0.110713
-9.685301 4.146687 -0.924873 -0.163191
-12.773778 2.065556 -0.387473 -0.061724
...略
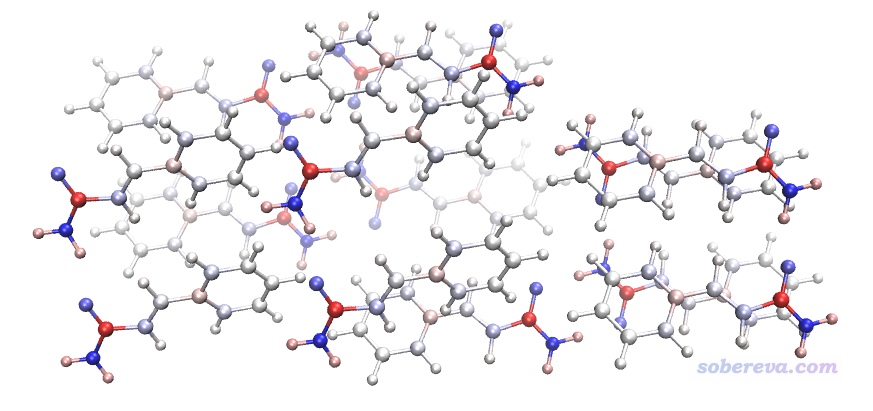
感興趣的讀者可以把environ.gjf里的原子名那里一列復制到bkchg.txt的第一列,然后另存為bkchg.chg。之后可以通過Multiwfn把這個文件轉化為bkchg.pqr文件,在VMD里顯示并根據charge屬性著色就可以非常直觀地顯示環境分子的原子位置和CHELPG電荷了。具體操作看《使用Multiwfn+VMD以原子著色方式表現原子電荷、自旋布居、電荷轉移、簡縮福井函數》(http://www.shanxitv.org/425)。下圖用的色彩刻度是-0.9~0.9,顏色根據藍-白-紅變化,因此越藍的原子電荷越負、越紅的越正,基本是白色的說明原子電荷接近0。可見跟我們期望的完全一致,比如苯環部分的原子電荷都不大,氨基氮的電荷很負、上面的氫的電荷比較正。
3.6 構建包含背景電荷的中心分子的TDDFT輸入文件并計算光譜
下面我們終于可以開始做最終的計算了。上一節的bkchg.txt里的內容滿足Gaussian的背景電荷的格式定義,因此只需要將其復制到3.4節創建的中心分子的輸入文件center.gjf的坐標末尾空一行的地方,并且加上charge關鍵詞代表當前任務要從輸入文件末尾讀取背景電荷設置。我們把計算任務設為PBE0/6-31G*下用TDDFT算20個激發態,不熟悉相關知識的話看《Gaussian中用TDDFT計算激發態和吸收、熒光、磷光光譜的方法》(http://www.shanxitv.org/314)。改好的輸入文件是本文文件包里的center_bkchg.gjf,內容如下
#P PBE1PBE/6-31G* TD(nstates=20) charge nosymm
<---空行
Title Card Required
<---空行
0 1
C -0.86593500 0.25621000 0.11508600
C -1.86773300 1.17440100 -0.15059900
C -1.23222000 -1.02932800 0.48142100
C 0.52813200 0.68962600 0.02868400
...略
<---空行
-11.079484 3.713347 -0.838676 0.272660
-12.081139 4.631418 -1.104034 -0.210476
-11.445682 2.427925 -0.472243 -0.188136
-13.413720 4.251740 -1.034406 -0.034063
-11.829578 5.657487 -1.375829 0.110713
...略
<---空行
<---空行
現在對這個文件進行計算,輸出文件是本文文件包里的center_bkchg.out,我們對它可以照常繪制光譜圖。我們也對比一下不帶背景電荷計算的情況,相應的輸出文件是本文文件包里的center_only.out。利用Multiwfn可以非常方便地將兩種情況的光譜放在一起顯示,見《使用Multiwfn繪制紅外、拉曼、UV-Vis、ECD、VCD和ROA光譜圖》(http://www.shanxitv.org/224)的第6節。UV-Vis對比圖如下
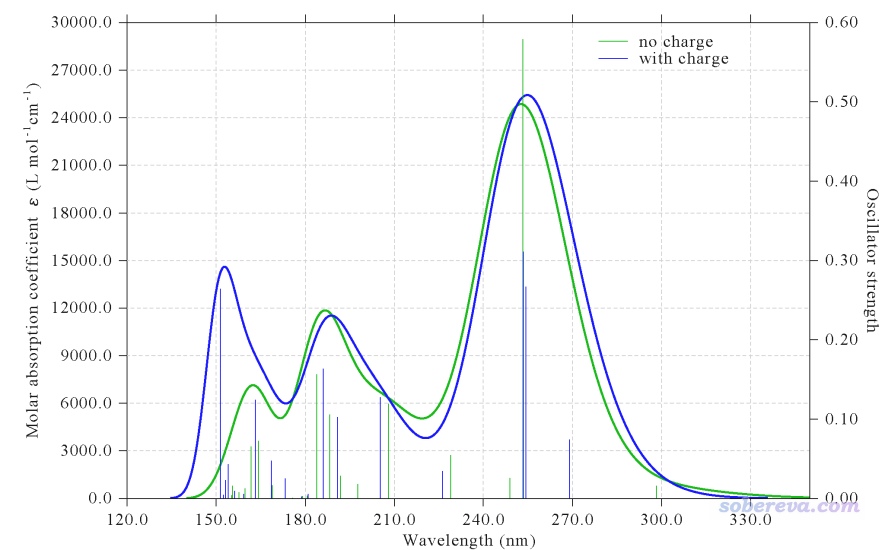
圖中綠線是沒背景電荷的情況,藍線是有背景電荷的情況。通過對比可見對于當前體系,在波長不是特別低的區域,背景電荷對UV-Vis光譜特征雖然有一些影響,但影響不算很顯著。不過你若仔細看輸出文件的話,會發現背景電荷對很多激發態的激發能和振子強度的影響其實是非常明顯的。比如S1態在沒加背景電荷的時候激發能是4.157 eV,加了背景電荷后是4.608 eV,振子強度也從原先的0.016增大到了0.074。對于其它一些分子,加不加背景電荷可能對光譜曲線形狀、較強的峰的位置都有顯著影響。
4 總結
本文非常詳細地介紹了如何通過背景電荷表現分子晶體中周圍的環境分子對被計算的分子的靜電作用,使得通過一般的量子化學程序也可以計算晶體中的分子激發態和吸收光譜。當然,也可以在帶著背景電荷計算的時候產生波函數文件,從而用Multiwfn考察分子環境中的電荷分布、成鍵、電子離域特征等問題。本文以Gaussian計算桂皮酰胺作為例子進行了演示,對于其它一般分子的情況也都是同樣的處理過程。在很多其它程序如ORCA里也同樣可以類似地設置背景電荷來計算,請讀者舉一反三來實現。
當前我們在計算時只考慮了距離中心分子最近的一層環境分子。雖然還可以再考慮更外層的環境分子,但不會有什么顯著的改進,因為當前分子是中性的,根據電多極相互作用公式,這樣的分子間的靜電作用隨距離衰減是很快的。但如果是陰陽離子構成的晶體,環境分子多考慮幾層有益。