實驗測定分子結構的方法以及將實驗結構用于量子化學計算需要注意的問題
實驗測定分子結構的方法以及將實驗結構用于量子化學計算需要注意的問題
文/Sobereva@北京科音
First release: 2020-Sep-10 Last update: 2021-Jan-4
筆者在網上答疑時,經常有人問類似于“實驗測定的分子結構,做量子化學計算時還需要優化么?”這種問題。我覺得值得寫一篇文章將實驗測定分子結構的原理科普一下,并順帶談談相關問題。搞懂了測定原理、測定時的環境和計算時的條件的差異,才能充分搞懂實驗測出的結構和理論計算的有沒有可比性、在對比時需要注意什么、是否能將實驗的結構直接用在理論計算中。下面對四種常見的測定分子結構的方法依次進行介紹和討論。
1 微波譜(microwave, MW)
微波譜是測量氣相小分子結構的常用方法,只能用于有永久偶極矩的分子,否則沒有信號。分子的轉動態能級差由轉動光譜得到,信號一般出現在微波、遠紅外區。轉動能級決定于轉動常數和轉動量子數(但還有離心畸變項等影響),轉動常數可以基于轉動慣量得到,而轉動慣量又是基于原子坐標和原子質量計算的(Multiwfn手冊3.100.21節有公式)。由于微波譜(微波范圍的轉動譜)可以測定轉動常數,因此給出了分子幾何結構信息。雙原子分子是可以通過微波譜直接給出具體結構(鍵長)的,而對于多原子分子來說微波譜測定的轉動常數只是體現了結構的一個側面,還需要結合其它手段才可能得到所有原子的坐標,比如可以將測定的轉動常數作為約束條件,用量子化學方法優化,或者基于紅外光譜轉化出的力場進行幾何優化,最終得到所有原子坐標。(對于非線型體系,轉動常數只有A、B、C這三個,通常小于內坐標數,因此原理上就不可能光靠測定轉動常數獲得完整的幾何結構。但如果將體系中的原子做同位素替換,測量不同情況下的轉動常數從而增加已知信息量,原理上倒也能獲得完整的幾何結構)
需要注意的是,實驗測定的是有效轉動常數,而勢能面極小點結構(也稱平衡結構)對應的是平衡轉動常數,二者之間的差異由各個振動模式的振動-轉動相互作用常數{α}以及振動量子數決定,這體現了振動運動對實際轉動常數的等效影響。利用振動基態和振動激發態的轉動光譜數據可以得到α,再結合微波譜給出的有效轉動常數,就能得到與極小點結構直接對應的平衡轉動常數。如果缺乏實驗的α,α也可以通過理論計算得到,比如在Gaussian里用freq=anharm關鍵詞就可以給出考慮了非諧振校正的α矩陣。
值得一提的是,一些文章里把量子化學程序計算的轉動常數與實驗微波譜測定的值相對照判斷幾何優化的結構的準確性。Grimme的ROT34測試集文章就是如此,包含了34種中等尺寸有機分子的實驗轉動常數,結合理論計算的α,測試了一系列理論方法的結構優化精度,見J. Comput. Chem., 35, 1509 (2014)。在J. Phys. Chem. A, 119, 2058 (2015)中,Barone等人根據實驗轉動常數結合量子化學算的α確定了47種有機分子的氣相精確結構,稱為B3se半實驗數據集,之后在J. Chem. Theory Comput., 12, 459 (2016)中Adamo等人還用這個數據集測試了主流泛函優化結構的精度。
2 氣相電子衍射(gas electron diffraction, GED)
電子衍射技術用于固體有掃描透射顯微鏡(STM)、電子背散射衍射技術(EBSD)等,也可以用于氣體,稱為氣相電子衍射。這是把氣體分子噴到衍射腔里,射入加速后的電子并測量衍射數據,由此可以獲得各原子間的距離信息(例如可給出徑向分布函數曲線),相當于測定了距離矩陣,也等同于得到了分子的幾何結構信息。但單靠這種做法只能測定很簡單的分子的結構,即不同原子間距離差異較大,因而容易通過峰位置指認不同原子間距離的情況。如果與轉動譜等其它實驗的信息相恰當結合,則可以更準確、測定更多的分子結構。對于分子略復雜而導致難以直接靠GED曲線確定所有原子位置的情況,也有不少研究將量子化學優化出的結構在GED解析時作為限制,比如J. Phys. Chem. A, 124, 5204 (2020)。
氣相電子衍射測的是各原子間的熱平衡距離,即對應于熱可及振動態的權重振動平均距離,顯然這樣的原子間距離和精確勢能面極小點結構對應的原子間距離是有不可忽略的差異的。即便只是在振動基態,由于勢能面的非簡諧性,振動平均結構也和極小點結構是存在一定差異的,在此文里專門說過:《談談溫度、壓力、同位素設定對量子化學計算結果產生的影響》(http://www.shanxitv.org/423)。量子化學優化的坐標若要和GED給出的相對比,GED實驗對應的溫度應當較低,以盡量減少熱平均距離和精確勢能面極小點的差異。例如J. Chem. Theory Comput., 2, 1282 (2006)中構建了衡量DFT泛函優化過渡金屬配合物精度的測試集,其中GED的數據都是刻意挑室溫下或比之略高的,而那些常見的1000 K或更高溫度下的GED數據都沒納入測試集。關于GED的更多細節、優缺點等信息可以參看Structure of Free Polyatomic Molecules-Basic Data (Kuchitsu,1998)一書第一章的介紹和討論。這本書里收集了大量氣相小分子、復合物的坐標,很大一部分都是靠MW、GED或者多種方法聯用來測定的。順帶一提,免費的UNEX(http://unexprog.org)程序可以通過GED數據或者轉動常數,或者將二者相結合來確定分子結構。
以上兩種做法是最常見的獲得氣相分子結構的方法,盡管也有些文章還通過別的手段推斷出了氣相分子結構。有些文章在給出氣相分子結構的時候明確說了是振動平均結構還是極小點結構,如果你要判斷幾何優化得準不準,顯然應當參照后者來對比;如果你要直接用實驗的結構來做一些計算,也應當用后者,此時不需要再對結構優化(但如果你是之后要做振動分析,由于振動分析的級別必須和幾何優化完全一樣,因此還是要先優化)。
3 X光衍射
培養足夠大尺寸的單晶,然后做X光衍射是實驗測定化學體系幾何坐標最常用的做法。此方法本質上是獲得晶體中的電子密度格點數據,再根據特定方法轉化為原子核位置。這實質上對應的是熱平均結構。特別要注意X光衍射測定的分子結構是晶體環境下的,由于分子間相互作用、堆積效應,勢必會影響結構,顯然多多少少會與氣相結構存在差異。具體差異有多大,和分子的柔性、晶體環境中的相互作用非常密切。例如吡咯,屬于剛性很強的分子,所以在晶體環境中和在氣相中結構差異甚微。而對于柔性體系,晶體環境對構象的影響可能是相當大的,比如二面角能差好幾十度。例如下面聯苯這個例子,氣相中是有二面角的,而在晶體環境中則成了平面的了
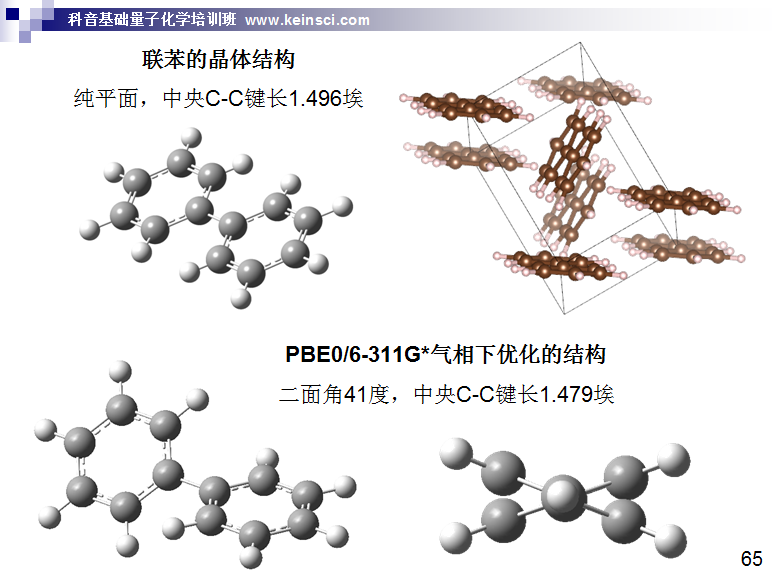
所以,對于柔性體系,不能用晶體結構和氣相中優化的結構相對比來判斷優化得是否準確。如果你的目的是考察真空狀態下某個柔性分子的特征,顯然也不能拿晶體結構未經優化直接用。
對于剛性體系,拿量子化學優化的結構與X光衍射的結構相對比來判斷優化得是否靠譜,通常來說還算說得過去,盡管原理上并不嚴格,畢竟這種實驗測的不是孤立狀態勢能面極小點結構,而且晶體中的分子間相互作用總是不可避免的。如果體系雖然是柔性,但只是將量子化學優化的某些剛性變量與實驗的對比,倒是沒明顯問題。比如一個過渡金屬配合物,其配體可能有很多柔性變量,但配位鍵的剛性通常都是很強的,所以如果只關心配位鍵鍵長的優化精度,一般可以與X光衍射的對比(但如果實驗的解析度太差也不行)。
關于X光衍射有一個非常需要注意的是氫原子的位置的測定問題。相關討論詳見J. Phys. Chem. Lett., 12, 463 (2021)。X光衍射實驗普遍用的是獨立原子模型(independent atom model, IAM)方法獲得原子位置,它將分子的電子密度近似視為孤立狀態下的球對稱的各個原子密度的疊加,但這忽略了原子的電子密度分布的各向異性,此問題對于帶電子很少的氫特別嚴重,會導致得到的與氫相關的化學鍵明顯偏短(對于其它元素倒沒什么問題)。后來有人提出了Hirshfeld原子精修(Hirshfeld atom refinement, HAR),需要迭代進行,原子坐標隨著迭代逐漸變化,每一輪迭代都要利用量化計算得到電子密度并涉及到做Hirshfeld劃分。即便X光衍射精度較差,利用HAR依然可以得到和中子衍射測定的相仿佛的氫原子坐標(中子衍射是最為準確的獲得氫的坐標的方法)。但HAR沒有考慮環境效應,對于形成氫鍵的情況,環境效應對相應氫原子的位置影響絕不可忽視,通常能對鍵長影響零點零幾埃。雖然用大團簇模型計算來考慮環境效應最理想,但是太昂貴。有人在使用HAR時,在量化計算過程中將環境分子的原子通過點電荷表現它對中心分子的影響,對結果有所改進,但還不算很理想。后來又有人提出了HAR-ELMO方法,將周圍一定距離內的分子的波函數用極度定域化分子軌道(ELMO)表現,相當于以嵌入方法對中心分子進行計算,因此以比較便宜的方式較真實地展現了環境效應,得到的參與氫鍵的氫的鍵長和中子衍射得到的非常接近了,誤差整體<=0.01埃。
雖然如上所述,目前已經有了能基于X光衍射較準確測定氫原子位置的方法,但由于已有的X光衍射的結構大多都是基于IAM方法確定的,因此千萬不要輕易把X光衍射實驗給出的cif文件里的氫的位置當做什么準確位置。對這種情況,如果你要把X光衍射得到的分子坐標用于量子化學計算,氫的位置是無論如何也要經過優化的。而重原子是否優化,看你的具體要求。如果是要研究分子在真空中或者溶劑環境中的狀態,那么所有原子都得優化;如果是想盡可能表現晶體環境中分子的狀態,那么就不要優化重原子了,只優化氫原子即可。具體來說,就是做凍結重原子的限制性優化,在此文說了做法《在Gaussian中做限制性優化的方法》(http://www.shanxitv.org/404)。注意這和前面說的HAR明顯不同,因為這種計算不涉及將X光實驗測定的電子密度當做確定坐標的參考依據。顯然,只優化氫的話,由于分子沒有完全優化,當做孤立體系做振動分析很容易出現虛頻,但這是不可避免的,也不要去在意這點。
在晶體環境中,周圍的分子不僅僅影響被考察的分子的幾何結構,對其電子結構也同樣有影響。如果分子的極性較強(怎么考察看《談談如何衡量分子的極性》http://www.shanxitv.org/518),計算當前分子時還應當恰當考慮周圍分子對當前分子的極化作用,這對于計算諸如UV-Vis譜等方面可能影響不小。其中實現起來最容易,也是在絕大多數量子化學程序中都能實現的辦法,是加上背景電荷,詳見《基于背景電荷計算分子在晶體環境中的吸收光譜》(http://www.shanxitv.org/579)中的非常詳細的介紹和實例。
4 中子衍射
對于單晶,中子衍射和X光衍射一樣可以測定晶體中的原子位置。X光衍射對應的是X光與電子云相互作用,而中子衍射對應的是中子與原子核相互作用,后者相對于前者的優點是可以直接測定氫原子核的位置(因為氫核會發生中子衍射),而且可以區分不同同位素。中子衍射除了用于晶體外,還可以用于氣體、液體、無定型固體、粉末,可以獲得徑向分布函數等數據。現有的中子衍射測定的分子結構數目遠少于X光衍射,因為需要核反應堆或散裂源作為中子源,這明顯不是一般實驗室可以擁有的,只有國家級的機構才有。而且用于中子衍射的單晶需要比X光衍射的有更大的尺寸,在樣品制備上也難得多。在劍橋晶體學數據庫CSD里既有X光衍射的也有中子衍射測定的結構。
最后再順帶說一下,網上有一些分子庫,比如pubchem、chemspider等等,里面能直接下載分子結構文件。大家千萬別隨便下下來就用、甚至以為那就是真實結構。網上的一些分子結構文件如果沒注明來源的話,很有可能只是根據分子的二維結構通過CORINA等等簡單、經驗的算法自動構建的,不僅不是什么真實結構,還非常粗糙,對于成鍵方式特殊的體系可能跟真實結構差異巨大。