在VMD中繪制Gaussian計算的分子振動矢量的方法
在VMD中繪制Gaussian計算的分子振動矢量的方法
文/Sobereva@北京科音
First release: 2020-Sep-8 Last update: 2022-May-19
Gaussian用戶觀看freq任務產生的振動矢量一般都是通過GaussView看(雖然也有ChemCraft等其它一些程序也可以看)。然而,起碼對于GaussView 6來說,GaussView顯示振動矢量的一個很大不足是箭頭太細,而且頭部不夠粗,導致有時候都看不清楚,放在文章里不夠美觀。另外,GaussView繪制分子結構的作圖選項不夠靈活,而且還收費。VMD是極其流行的化學體系可視化程序,免費、靈活、圖像效果好,本文介紹如何通過筆者寫的VMD作圖腳本非常方便地繪制Gaussian的振動分析任務產生的振動矢量。VMD可以在http://www.ks.uiuc.edu/Research/vmd/免費下載。
在這里下載筆者編寫的繪圖腳本和示例文件:http://www.shanxitv.org/attach/567/file.zip。此腳本至少對于目前撰文時的VMD正式版中最新的1.9.3、Gaussian 09和16是完全適用的。
這里以繪制多巴胺的振動矢量為例進行演示。把文件包里的dopamine.out放到VMD目錄下,這是多巴胺的Gaussian的freq任務的輸出文件。然后我們得把這個.out文件轉化成一個VMD可以認的結構文件的格式,比如可以把此文件載入GaussView,然后另存為.pdb或.mol2文件。也可以下載Multiwfn(http://www.shanxitv.org/multiwfn),啟動Multiwfn后載入此文件,然后選主功能100的子功能2,通過相應選項導出為.pdb或.xyz文件。
把文件包里的drawarrow.tcl和GauNorm.tcl都放到VMD目錄下,然后用文本編輯器打開GauNorm.tcl,把開頭的set filename后面的文件名改為dopamine.out。之后啟動VMD,把多巴胺的結構文件載入VMD,然后在文本窗口輸入source GauNorm.tcl執行此腳本,此時振動矢量信息就被讀入了,與此同時定義了名為norm的繪制振動矢量的命令。之后在VMD的文本窗口輸入比如norm 4,就可以把4號振動模式通過箭頭畫出來。
norm后面還可以接第2個參數,用來設置箭頭長度是正則矢量的幾倍,數值越大箭頭越長,默認是3。norm后面還可以接第3個參數,用來設置箭頭的半徑,默認為0.05。比如norm 5 6 0.07就代表用6倍長度、0.07的半徑繪制第5個振動矢量。默認是用黃色繪制箭頭,如果想用別的顏色,把GauNorm.tcl中的draw color后面的yellow改成其它顏色名,比如cyan。
此例輸入norm 17 5,然后令分子以CPK方式顯示(在Graphics - Representation里把Drawing method改為CPK,再把Sphere Scale設為0.6),效果如下,可見非常理想!和GaussView顯示的相對比,可見展現的信息是相同的,而GaussView畫的箭頭相比之下明顯太小氣了。
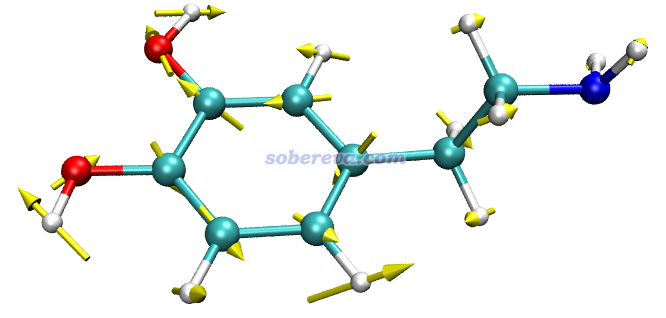
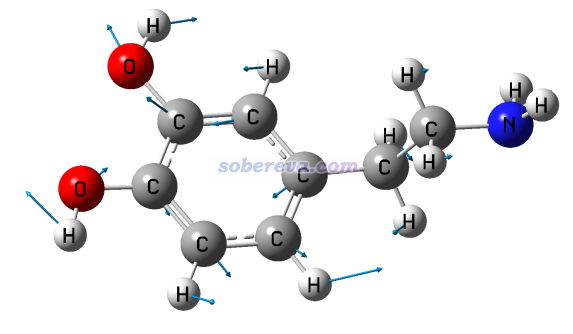
注意GauNorm.tcl開頭還有個set ilinear語句,如果當前體系是線型體系,必須把后面的值改為1。
如果你在Gaussian做freq或opt freq任務中按照《在Gaussian中做限制性優化的方法》(http://www.shanxitv.org/404)中的做法將N個原子的笛卡爾坐標凍結了,運行source GauNorm.tcl之前必須把里面set nfreeze后面的值設為N(默認為0,沒有原子被凍結)。