在VMD中顯示Gaussian計算的原子受力
在VMD中顯示Gaussian計算的原子受力
文/Sobereva@北京科音 2020-Sep-13
在量子化學研究中,原子受力往往是非常值得關注的量。Gaussian程序可以通過force關鍵詞計算原子的受力,然后在GaussView中可以根據原子受力大小進行著色,但是沒法繪制出受力矢量。本文介紹如何在免費、靈活、強大的VMD程序中,通過筆者自寫的tcl腳本,將Gaussian計算的原子受力非常直觀地通過箭頭展現出來,這對于一些量子化學問題的研究很有益處。VMD可以在http://www.ks.uiuc.edu/Research/vmd/免費下載,本文使用VMD 1.9.3,Gaussian使用G16 A.03。
在此處下載繪圖腳本和示例文件:http://www.shanxitv.org/attach/568/file.zip。將其中的forcevec.tcl和drawarrow.tcl復制到VMD目錄下。
用文本編輯器打開forcevec.tcl,可看到開頭有幾個設置:
set filename:載入的Gaussian輸出文件路徑
set sclfac:以a.u.為單位的原子受力矢量乘上這個系數就是繪制出的箭頭的長度。應根據實際圖像效果反復嘗試找到最合適的
set rad:箭頭的粗細
set color:箭頭的顏色名
set crit:如果某個原子的受力大小小于所有原子最大受力大小乘以這個系數,則這個原子的受力就不會用箭頭繪制出來,由此可避免在受力相對非常小的原子上出現非常短的、沒什么實際意義的箭頭。默認為0.1
例1:聯苯的基態極小點結構下在S1勢能面上的原子受力
本文文件包里的biphenyl_S1force.out是在優化過的聯苯的基態極小點結構下,用TDDFT算的S1激發態的原子受力的輸出文件,用到的關鍵詞在此文件里已經體現了。
把biphenyl_S1force.out載入GaussView,保存成pdb文件,然后把pdb文件載入到VMD里。然后將biphenyl_S1force.out挪到VMD目錄下,用文本編輯器打開forcevec.tcl,把set filename后面的內容改為biphenyl_S1force.out,把set sclfac后面的內容改為20,然后保存文件。之后在VMD的文本窗口里輸入source forcevec.tcl,腳本就從biphenyl_S1force.out里讀取原子受力,然后繪制出了箭頭。把背景改成白色,在Graphics - Representation里把顯示方式改為Licorice后看到下圖
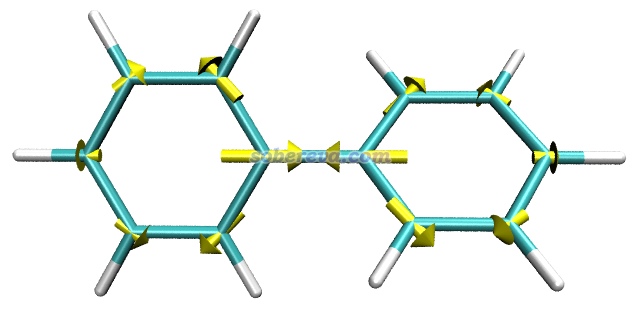
根據原子受力可見,在體系所處的這個Franck-condon點位置,聯苯中央的C-C鍵傾向于縮短。實際上在PBE0/6-311G*級別下,聯苯的基態極小點結構下這個C-C鍵鍵長為1.478埃,而同級別下用TDDFT優化的S1態極小點下這個鍵長為1.413埃,明顯短了很多。另外,從上圖可見其它原子也有顯著受力,這體現出苯環結構將要自發進行調整,確實S1態極小點下苯環的各個鍵長相對于S0態都有顯著變化。
例2:暈苯與富勒烯復合物
筆者之前在PM6-D3下對暈苯與富勒烯形成的復合物進行了優化。這里看一下如果在優化的復合物結構的基礎上,若把富勒烯與暈苯距離稍微拉遠一點,原子受力是什么樣的。本文文件包里complex_force.out就是在人為拉遠的結構下用PM6-D3做force任務的輸出文件,complex_force.pdb是相應的結構文件。將complex_force.pdb載入VMD,把complex_force.out復制到VMD目錄下,將forcevec.tcl里的set filename后面寫上complex_force.out。由于弱相互作用對應的原子間相互作用很弱,所以在set sclfac后面寫上一個比較大的值,這里用1500。在VMD文本窗口里運行source forcevec.tcl,進一步調節下作圖設置后看到下圖
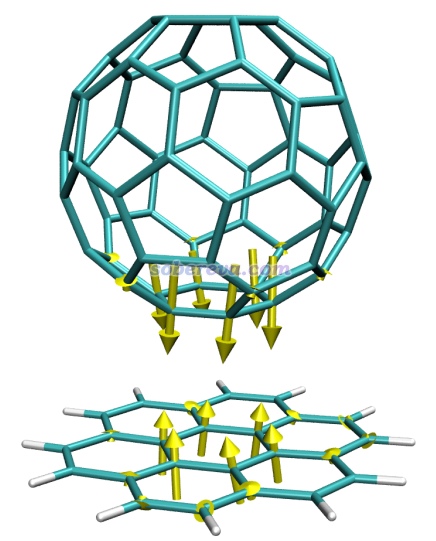
可見,由于富勒烯與暈苯之間的色散吸引作用,再加上當前結構下二者間距比平衡距離更遠,兩個分子彼此間挨得較近的原子上都出現了令二者距離進一步拉近的力。
例3:外電場下的18碳環的受力
筆者對18碳環及類似體系做過大量的理論研究,匯總見http://www.shanxitv.org/carbon_ring.html。其中,筆者在名為Ultrastrong Regulation Effect of the Electric Field on the All-Carboatomic Ring Cyclo[18]Carbon(https://chemistry-europe.onlinelibrary.wiley.com/doi/10.1002/cphc.202000903)的論文中全面、深入研究了外電場對18碳環的幾何結構、電子結構、電子吸收光譜等特征的影響,此文的介紹見《一篇文章深入揭示外電場對18碳環的超強調控作用》(http://www.shanxitv.org/570)。在此文中通過類似于上文的做法繪制了下面的圖(需要利用專門的tcl腳本)
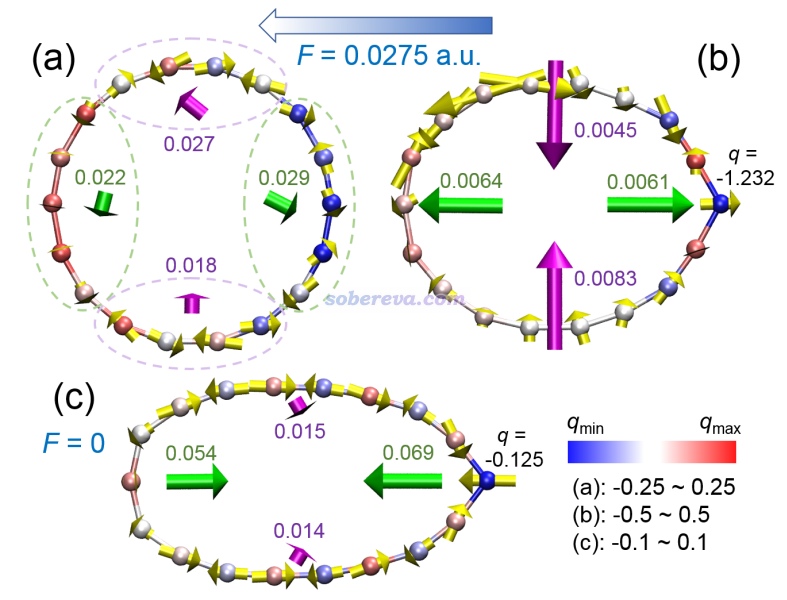
此圖體現的信息的詳細解釋請看上面提到的論文,這里只是簡單提一下。這個圖繪制了各個原子的受力,還用綠色箭頭展現了左、右兩側原子的總體受力,用紫色箭頭展現了上、下兩側原子的總體受力,劃分方式在圖(a)中通過虛線體現了。原子的顏色體現的是ADCH方法算的原子電荷,實現方法在《使用Multiwfn+VMD以原子著色方式表現原子電荷、自旋布居、電荷轉移、簡縮福井函數》(http://www.shanxitv.org/425)中介紹了。圖(a)體現出18碳環在原始結構下,如果剛加上外電場,此刻的受力并不會立刻造成碳環的整體變形,但會明顯造成鍵長發生改變的傾向。圖(b)體現出,在外電場下當碳環的結構已稍微經過弛豫、出現了一些變形后,此刻的原子受力就會明顯傾向于讓18碳環在順著電場方向拉長,而在垂直于電場方向壓扁,從而使碳環變得更加像橢圓。圖(c)是在電場下優化后的結構,但是此刻撤掉了外電場,由綠色箭頭可見此時18碳環有明顯的恢復成原本圓形的傾向,體現出彈性特征。此圖清晰地展現出電場下18碳環這個體系的獨特的力學特性,如果不把受力繪制成箭頭,很難予以這樣深入的探討。
若要繪制一批原子的總受力,可以在source forcevec.tcl之后,把下面的代碼拷到VMD文本窗口里。這部分代碼原本是筆者用來繪制上圖中左側5個原子總受力用的。4 5 6 7 8是被計算總受力的原子序號,總受力矢量及其模也會被輸出到屏幕上。在繪制箭頭時,為了讓位置比較舒服,箭頭中心位置取的是$atmleft 3 9 2 10的幾何中心,即4 5 6 7 8加上相鄰的3 9 2 10四個原子。
set atmleft "4 5 6 7 8"
draw color green
set fxleft 0
set fyleft 0
set fzleft 0
foreach i $atmleft {
set fxleft [expr $fxleft+$fx($i)]
set fyleft [expr $fyleft+$fy($i)]
set fzleft [expr $fzleft+$fz($i)]
}
puts "Left side force: $fxleft $fyleft $fzleft"
set normval [expr sqrt($fxleft**2+$fyleft**2+$fzleft**2)]
puts "Norm: $normval"
drawarrow "serial $atmleft 3 9 2 10" $fxleft $fyleft $fzleft $sclfac 0.2