使用Multiwfn做IGMH分析非常清晰直觀地展現化學體系中的相互作用
使用Multiwfn做IGMH分析非常清晰直觀地展現化學體系中的相互作用
文/Sobereva@北京科音
First releas: 2022-Mar-7 Last update: 2022-Oct-27
0 前言
目前已有不少將化學體系中的相互作用以圖形化方式展現的方法,如NCI(也稱RDG)、aNCI、IGM、aIGM、IRI、ELF、LOL、SCI、價層電子密度,等等,它們各具特點,其中一些已被廣為使用。本文將全面介紹筆者近期提出的一種重要的圖形化展現自定義片段間和片段內相互作用的方法,稱為independent gradient model based on Hirshfeld partition (IGMH),這是對2017年提出的IGM方法的重要改良。此方法已經實現在了知名的波函數分析程序Multiwfn(http://www.shanxitv.org/multiwfn)的最新版中。從本文給出的應用例子讀者將會看到,IGMH在展現化學體系中的相互作用方面極其靈活和方便,圖像效果非常理想,普適性特別強,有極高的實用價值。
IGMH方法的非常詳細的介紹文章已經發表如下,非常歡迎讀者們閱讀
Tian Lu, Qinxue Chen, Independent gradient model based on Hirshfeld partition: A new method for visual study of interactions in chemical systems, J. Comput. Chem., 43, 539-555 (2022) https://doi.org/10.1002/jcc.26812
此文可以通過此鏈接直接免費閱覽:
https://onlinelibrary.wiley.com/share/author/H659SPFCDKPCSJUVG5YT?target=10.1002/jcc.26812
使用Multiwfn做IGMH分析發表文章時,除了按照程序包內的How to cite Multiwfn.pdf文檔的說明恰當引用Multiwfn程序外,也勿忘引用上面這篇IGMH原文(另:之前已經有大量Multiwfn用戶使用IGMH方法的前身IGM發表了共約200篇研究文章。以后如果讀者仍使用IGM功能發表文章的話,除IGM和Multiwfn原文外也請引用以上文章,其中對IGM方法以及筆者對IGM的擴展做了系統性的介紹,文中還給出了IGM在Multiwfn中的實現細節)。
2022-Oct-27補充:后來筆者寫了一篇對IGMH原文里一個實現細節的勘誤文章,并做了相關理論分析,見ChemRxiv (2022) DOI: 10.26434/chemrxiv-2022-g1m34(https://doi.org/10.26434/chemrxiv-2022-g1m34)。建議此文和上述IGMH原文一并引用。
2023-May-31補充:《一篇最全面介紹各種弱相互作用可視化分析方法的文章已發表!》(http://www.shanxitv.org/667)介紹的筆者綜述文章中對IGMH有極其充分的介紹并給了諸多應用例子,歡迎閱讀和引用!
下文將對IGMH的各個方面進行介紹,包括以下內容
第1節:對IGMH的基本原理做簡要說明,使讀者快速了解此方法的思想
第2節:給出IGMH用于各類體系的效果展示,使讀者明白IGMH分析都能以什么方式研究什么問題
第3節:將IGMH和IRI、NCI、IGM方法進行對比,便于讀者了解什么時候適合用什么圖形化分析方法
第4節:對Multiwfn中的IGMH功能和所需的輸入文件等方面進行說明
第5節:給出使用Multiwfn做IGMH分析的具體例子
第6節:對本文內容做總結
Multiwfn在化學鍵和弱相互作用分析上極為強大,有眾多的功能,IGMH只是其中之一,各種方法匯總介紹見《Multiwfn支持的弱相互作用的分析方法概覽》(http://www.shanxitv.org/252)和《Multiwfn支持的分析化學鍵的方法一覽》(http://www.shanxitv.org/471)。在Multiwfn與波函數分析培訓班(http://www.keinsci.com/workshop/WFN_content.html)里筆者也會做非常全面、系統性的講解。很值得一提的是IRI是筆者提出的另一種相互作用圖形化分析方法,已被很多文章使用,介紹見《使用IRI方法圖形化考察化學體系中的化學鍵和弱相互作用》(http://www.shanxitv.org/598),它和IGMH的用處既有共性又有互補性,這在后文將會看到。
1 IGMH的原理簡介
IGMH的詳細介紹請讀者仔細閱讀前述的IGMH方法的原文,本節只是把關鍵信息做簡要說明。
1.1 IGMH和IGM方法對相互作用區域的展現
IGMH的基本思想和其前身IGM是共通的,都是使用三維函數δg來把原子間相互作用區域直觀展現出來。為了便于說明,我們首先看一個最簡單的情況,氫氣分子H2。在H-H鍵軸上,兩個氫的電子密度曲線圖如下圖(a)所示
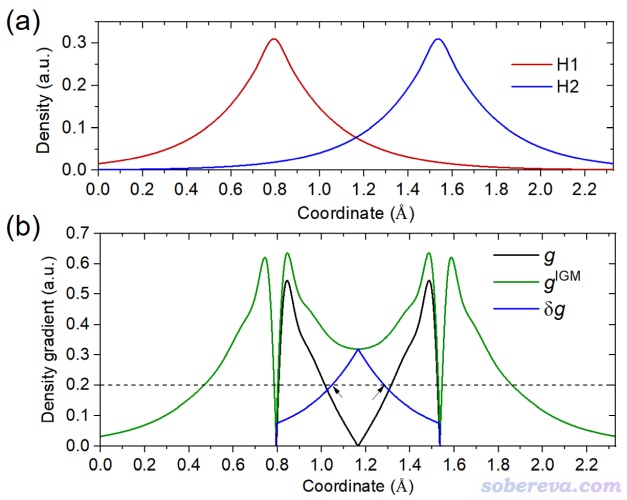
上圖(b)中的黑色曲線對應的g代表gradient,是兩個H原子電子密度梯度之和。在兩個H正中央g精確為0,這是因為在此處兩個H的電子密度梯度大小相同但符號相反,正好抵消了。上圖(b)中的綠色曲線對應的g_IGM是IGM(independent gradient model,獨立梯度模型)型電子密度梯度,相當于兩個H原子的電子密度梯度的絕對值加和,因此不會因為符號差異而出現相互抵消,仿佛兩個原子是彼此“獨立”的。顯然g_IGM是g的上限。上圖(b)中的藍色曲線是δg,對應于g_IGM減去g。由圖可見在差不多X=1.2埃處,δg達到了極大點。δg在原子間相互作用區域能有這么個峰出現,來自于兩個原子的密度存在重疊,也暗示原子間相互作用的存在。在分子的平衡結構下,原子間密度重疊越大,這個峰往往越高,也暗示出原子間相互作用越強因而使得原子間距離越近。如果倆原子間都沒有可查覺的相互作用,g和g_IGM就不會有差異,δg也就不會出現峰。
上圖(b)中畫了一個Y=0.2的虛線,和δg的交點用黑色箭頭標注了。這體現出,對這個體系,若把δg繪制成等值面圖,在0.2等值面數值(isovalue)的情況下就會出現一個等值面,包裹的區域就是上圖中兩個箭頭之間的區域。可見δg函數的等值面可以用來把原子間存在明顯相互作用的區域圖形化展示出來,而且等值面數值取得越小,越弱的相互作用區域就越可能也出現相應等值面。而等值面數值取得較大時,則只會有較強的相互作用區域被展現出來。
上面給出的情況是一維情形,實際研究的當然都是三維情況。對于三維情況以如下方式定義g、g_IGM和δg。粗體r是坐標矢量,i循環所有原子,| |符號代表取里面矢量的模,▽是矢量微分算符,ρ_i代表i原子的電子密度。
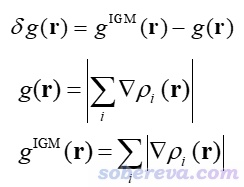
δg可以展現當前體系中所有原子間的相互作用。為了能專注于展現特定片段間(interfragment)和片段內(intrafragment)的相互作用,可分別定義δg_inter和δg_intra,如下所示。其中A循環用戶定義的各個片段
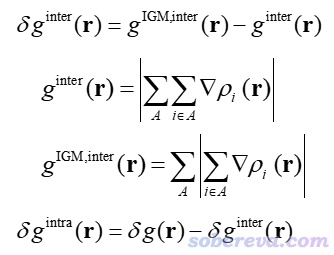
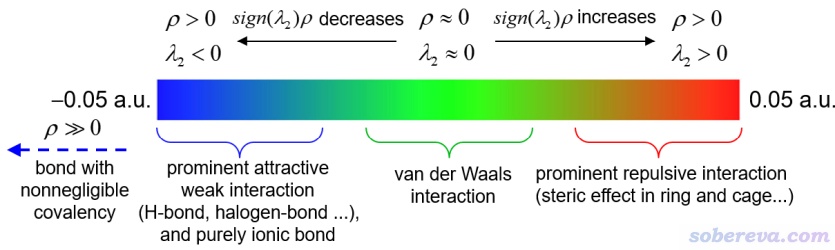
IGM和IGMH方法不僅能展現相互作用區域,還能將sign(λ2)ρ函數通過不同顏色投影到δg、δg_inter、δg_intra的等值面上展現相互作用類型和強度。λ2是電子密度Hessian矩陣第二大本征值,sign()是取符號的意思。相互作用區域里某個位置的λ2若小于0體現出在此處原子間有吸引作用,大于0則體現互斥作用(但這只是個對多數相互作用區域適用的粗糙的、經驗性的判據,切勿教條化理解)。ρ是當前體系實際電子密度,在相互作用區域此值越大暗示相互作用越強。sign(λ2)和ρ相乘得到的sign(λ2)ρ顯然有能力對相互作用類型和強度進行區分。在IGMH原文里筆者給出了下圖,可以作為IGMH圖中sign(λ2)ρ的標準著色方式,不同顏色對應的常規解釋也都給出了
1.2 IGMH與IGM的差異
以上內容對于IGM和IGMH來說都是完全相同的,這兩種方法關鍵的不同在于原子的電子密度ρ_i怎么定義。IGM方法我之前在《通過獨立梯度模型(IGM)考察分子間弱相互作用》(http://www.shanxitv.org/407)和《使用Multiwfn結合CP2K通過NCI和IGM方法圖形化考察固體和表面的弱相互作用》(http://www.shanxitv.org/588)專門介紹過。IGM直接用原子在孤立狀態下的球對稱化的電子密度當做式中的ρ_i,分布僅由元素決定而不體現實際化學環境影響。這么做的好處是計算起來很快,在高效的Multiwfn中算幾千個原子都能做到,而且只需要提供體系的坐標和元素信息就能做IGM分析,因為各種元素原子的球對稱化的電子密度分布在Multiwfn程序中是直接內置的。但代價是,IGM的物理意義不嚴格,原子密度分布沒有體現體系的實際電子結構,而且最關鍵的是,往往圖像效果爛。如后文示例可見,IGM圖中δg和δg_inter函數的等值面往往特別肥厚,不僅丑陋,還經常延伸到距離原子核較近因而電子密度較大的位置,導致在根據弱相互作用區域sign(λ2)ρ的顏色判斷時往往會高估作用強度。
為了解決IGM的這些問題,IGMH使用Hirshfeld原子空間劃分的方式從體系實際的電子密度中劃分出各個原子的電子密度,由于考慮了實際電子結構和化學環境,從物理意義上就比IGM強。這個改進還同時使得IGM等值面太肥厚的問題被理想地解決,IGMH方法的δg/inter/intra等值面總是比較薄且美觀,能夠純粹地展現出相互作用區域的特征。IGMH的實現細節在IGMH原文2.2節有具體說明,本文就不再過多敘述了。IGMH和IGM還有個差異在于,IGMH分析中用的sign(λ2)ρ是基于實際電子密度計算的,而IGM分析中的sign(λ2)ρ是基于粗糙的準分子(promolecular)密度算的,準分子密度只是各個原子孤立狀態電子密度的簡單疊加,沒有體現原子間相互作用對電子密度的改變。
由于IGMH要基于實際電子密度做分析,因此輸入文件必須包含波函數信息,這需要做量子化學或第一性原理計算得到。另外,IGMH計算形式比IGM更為復雜,計算量更大。顯然,做IGMH分析的總耗時是明顯高于IGM的。不過以如今主流的幾十核雙路服務器的計算能力來說,IGMH分析用于幾百個原子體系完全沒問題。所以,除非真是碰到巨大體系用IGMH確實算不動,否則*強烈*不建議用IGM,而總應當用IGMH。實際上,整個IGM/IGMH分析流程中真正最耗時的是體系的幾何優化(一般都是用DFT方法),IGMH和IGM分析本身的耗時通常只占幾何優化的零頭。從這個角度來說,IGM和IGMH分析能處理的體系尺度并沒有實質性區別(除非你用很便宜的分子力場或半經驗層面的方法做幾何優化,此時的IGM、IGMH分析的準確性無疑會打一定折扣)。
1.3 定量化分析原子和原子對對相互作用的貢獻
為了能讓IGMH在分析片段間相互作用時也能做一些定量分析討論,筆者定義了一些指數,它們對于IGM也是同樣適用的。
原子對δg指數(atomic pair δg index)記為δG_pair,定義如下。其中A和B是要被考察相互作用的兩個片段,i和j分別是兩個片段中的原子
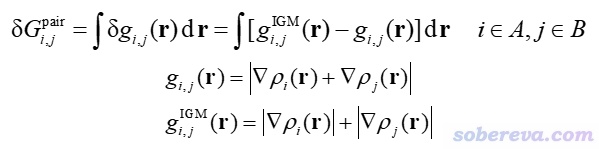
原子對的δG_pair(%)定義為它的δG_pair占兩個片段間所有δG_pair總和的百分比,可以粗略體現原子對對片段間相互作用的貢獻程度。
原子δg指數記為δG_atom,它可以衡量各個原子對片段間作用的貢獻程度,其定義如下,其中i是一個片段里的原子,j循環另一個片段里的所有原子。相應地還可以定義δG_atom(%)用于衡量某原子對片段間作用貢獻的百分比。
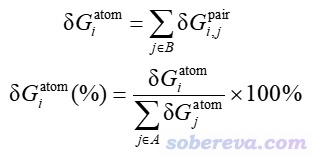
上面這些指數很有實用性,可以使研究者快速判斷哪些原子或原子對對片段間結合起到主要作用。但畢竟這些指數只是以較為簡單方式定義的,并沒有充分考慮物理層面的因素,因此不要指望能和片段間相互作用能會有太密切聯系。它們只適合粗略判斷哪些原子和原子對較為重要、比較值得關注。如果要從相互作用能角度考察原子間相互作用的話,可以用《使用Multiwfn做基于分子力場的能量分解分析》(http://www.shanxitv.org/442)里介紹的EDA-FF方法,在《18碳環(cyclo[18]carbon)與石墨烯的相互作用:基于簇模型的研究一例》(http://www.shanxitv.org/615)一文中也有其應用實例。
2 一些典型的IGMH分析結果展示
這一節展示一些IGMH圖,以令讀者對IGMH有直觀的認識、充分了解其重要實用價值。圖片都來自IGMH原文,原文里面有詳細的說明和討論,這一節里筆者對每個例子只是簡單地提幾句。這些圖用的sign(λ2)ρ的色彩刻度基本都和前文1.1節給出的一致,有些圖為了顏色更便于觀察,將色彩刻度下限和上限做了適當的調節。為了圖像效果盡可能好,這些圖的等值面數值都經過反復調節,選取了最能充分、清楚展現感興趣的相互作用的值。所有結構都用恰當的級別做過幾何優化。
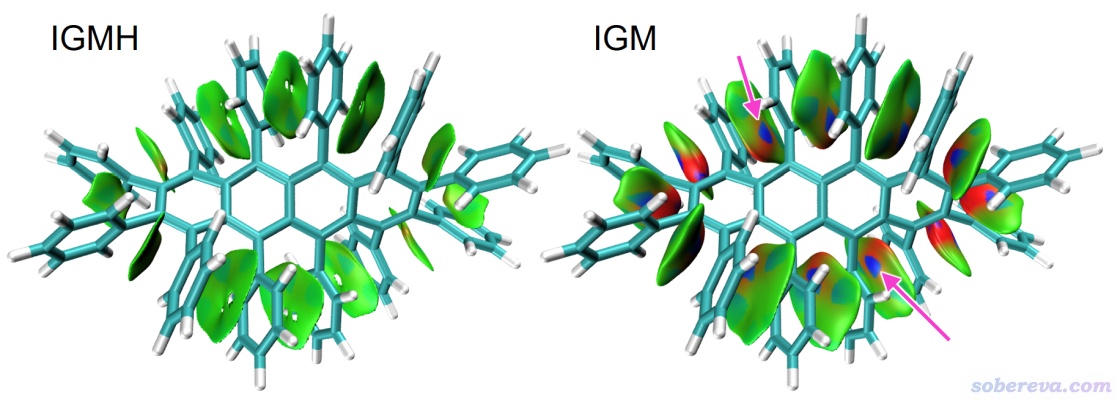
我們先看一系列片段間弱相互作用分析例子,這也是IGMH分析最常用的情況。下圖是dodecaphenyltetracene體系的IGMH和IGM的sign(λ2)ρ著色的δg_inter等值面圖,每個苯基片段定義為一個片段。在IGMH圖中相鄰苯基間有綠色大面積扁片,基本平行于苯環,這是典型的pi-pi堆積特征。pi-pi堆積作用特征都是作用面積大,而作用區域電子密度很低。關于pi-pi堆積更多知識看《全面探究18碳環獨特的分子間相互作用與pi-pi堆積特征》(http://www.shanxitv.org/572)。IGMH圖將當前這個側鏈很擁擠的分子中的重要的弱相互作用十分清楚地展現了出來,而下圖中的IGM圖的效果就遠不如IGMH了,如粉色箭頭所指,個別地方出現了很詭異的顏色。這就是因為前面說的IGM圖經常存在的問題,由于等值面太肥厚,導致它延展到了距離原子核較近因而電子密度較高的區域,這顯然會對弱相互作用特征造成嚴重的誤判。
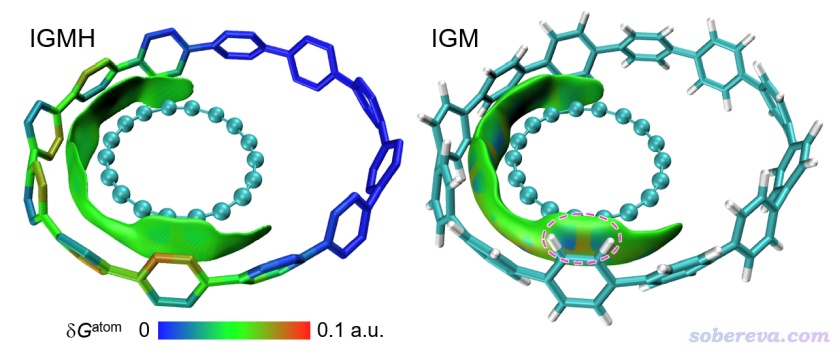
18碳環是幾何和電子結構都很奇特的體系,近些年有大量理論研究文章出現,筆者也做了大量理論研究,匯總見http://www.shanxitv.org/carbon_ring.html。有一篇他人的研究工作涉及的體系是將18碳環置入一個[12]CPP大環分子里,當時其作者還用Multiwfn做了IGM分析。將這個體系里18碳環和[12]CPP分別定義為兩個片段繪制的IGMH和IGM圖如下所示。其中IGMH圖里為了令原子對片段間結合的貢獻能夠直觀展現,令原子按照δG_atom值進行了著色,色彩刻度條也在圖中給出了。由此圖可見,IGMH圖非常好地展現出18碳環與[12]CPP之間大面積的色散作用主導的主要相互作用區域,而且圖中綠色和棕色把[12]CPP與18碳環相互作用最顯著的原子清晰地高亮了出來。IGM圖就差遠了,由于其過于肥厚的等值面,導致圖中虛線標注的區域出現了錯誤的著色,令人誤以為這地方有特殊的相互作用(這個體系研究文章原作者根據IGM圖做的結論是The regions colored in blue, on the other hand, reflect a rather different type of interaction,明顯是被IGM方法誤導了!)。
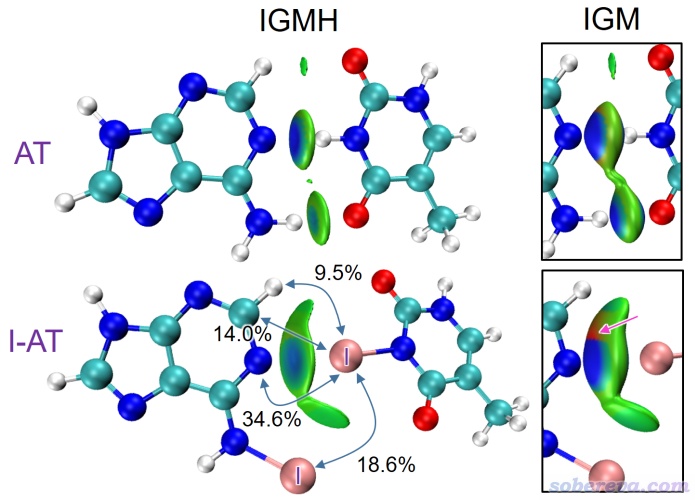
下圖是AT堿基對和碘代AT堿基對的IGMH和IGM圖,兩個單體各自定義為一個片段。可見IGMH圖很好地將AT堿基對之間兩個氫鍵作用區域展現了出來。對于AT,N...H-N和N-H...O的等值面中心都呈藍色,因此都是比范德華作用明顯更強的吸引作用,結合體系結構可判斷這顯然是典型的氫鍵,而由于前者的藍色要比后者深得多,可以認為N...H-N氫鍵明顯比N-H...O氫鍵更強。讀者若根據筆者在《透徹認識氫鍵本質、簡單可靠地估計氫鍵強度:一篇2019年JCC上的重要研究文章介紹》(http://www.shanxitv.org/513)中介紹的文章里提出的基于atoms-in-molecules (AIM)理論的判斷氫鍵強度的方法來考察,也會得到同樣的結論。AT的圖中C-H...O之間有很小的綠色等值面,說明其作用區域電子密度很低,明顯這只能算是色散吸引為主的很弱的作用,遠達不到一般氫鍵的強度。在碘代AT堿基對的圖中可看到N和I之間的等值面呈現藍色,這展現出明顯的靜電作用主導的鹵鍵作用。其它的色散作用主導的相互作用區域在此圖中也能清清楚楚地看出來。IGMH圖中還標注了幾個原子對的δG_pair(%)值,以此體現它們對堿基之間結合的貢獻比例。可見給出的數值很能說明問題,其中最大的正是N...I原子對,貢獻34.6%,而其它幾個原子對的貢獻也不容忽視(還是要強調,這個百分比只能粗略體現重要性的高低,切勿當成對作用能的貢獻來分析討論一番)。下圖還給了IGM圖,可見遠不如IGMH圖好看,等值面不僅太鼓,還有著色不合理的區域出現(如粉色箭頭所指)。此外,IGM圖上兩個氫鍵和一個鹵鍵的作用區域顏色都是深藍色,作用強度差異都難以靠顏色區分。
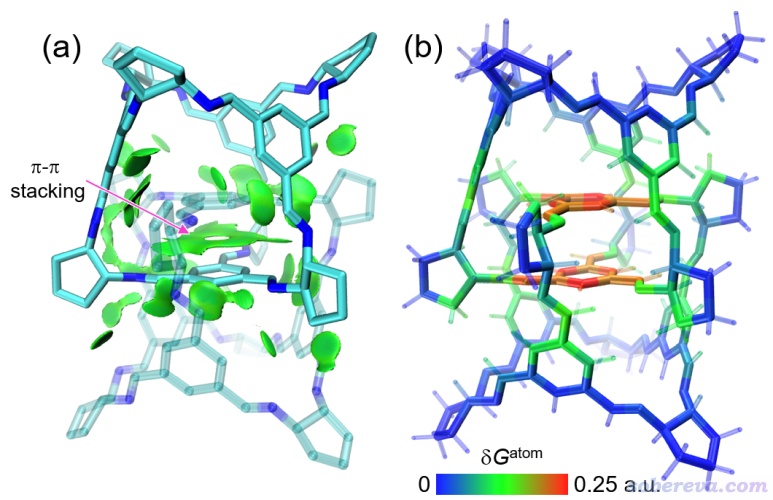
下圖是一個三重聯鎖共價有機籠體系,由兩個四面體籠嵌套構成,要想分離兩個單體需要破壞共價鍵才行。IGMH分析中將兩個單體各做為一個片段。下面(a)圖是IGMH著色等值面圖,(b)圖是將原子按照δG_atom進行著色的圖。可見IGMH將兩個單體之間色散主導的作用區域,特別是兩個分子中央的pi-pi堆積區域充分展現了出來,這也驗證了這個體系的合成者在其原文里基于幾何結構對pi-pi堆積存在性所作的推斷。顯然,光從幾何結構上憑經驗和感覺去分析分子間相互作用,遠不如做個IGMH分析直觀、嚴格、有說服力。從δG_atom著色圖上可以進一步看出,對兩個單體相互作用貢獻最大的就是pi-pi堆積涉及的那些原子,其次是周圍的原子,而整個體系頂端和底端藍色區域原子的δG_atom幾乎為0,因此對結合的重要性相對很低,這也是因為它們距離另一個單體原子相對較遠。
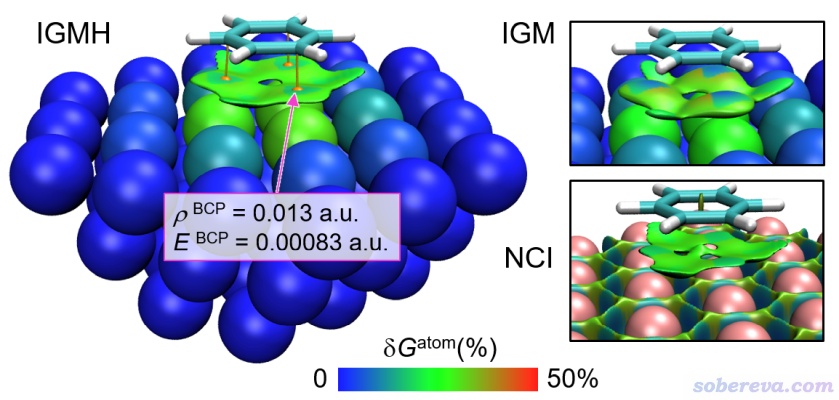
筆者之前寫過《使用量子化學程序基于簇模型計算金屬表面吸附問題》(http://www.shanxitv.org/540),專門講怎么用量子化學程序基于金屬的簇模型考察金屬表面吸附。這類表面吸附體系也非常適合用IGMH考察吸附作用。下圖是Ag(111)表面吸附苯分子的IGMH、IGM和NCI圖,在IGMH和IGM分析中苯分子和Ag簇分別定義為兩個片段。在IGMH圖中,還按照《使用Multiwfn+VMD快速地繪制高質量AIM拓撲分析圖(含視頻演示)》(http://www.shanxitv.org/445)的做法把金屬和苯之間的鍵臨界點(BCP,圖中的桔黃色圓球)和相應的鍵徑(圖中棕色細線)也同時畫了出來,并把BCP位置的電子密度和能量密度標了出來。此外,還對金屬原子根據δG_atom(%)進行了著色。從IGMH圖上可明顯看出苯和Ag之間有色散主導的大面積物理吸附作用區域,對吸附貢獻最大的Ag原子就是苯下面正對著的三個。對于大面積的相互作用,AIM理論中的BCP沒法將作用區域像IGMH或IRI等方法那樣全面充分展現,但BCP出現的位置普遍被認為是相互作用區域中最具代表性的位置,因此將BCP的屬性和IGMH或IRI等圖形化分析方法相結合還是很有益的,可以將作用區域的特征定量展現,而鍵徑則可以把原子間有代表性的相互作用路徑給直觀表現出來。不了解BCP、鍵徑這些AIM相關概念的話看《AIM學習資料和重要文獻合集》(http://bbs.keinsci.com/thread-362-1-1.html)。從下圖可看到苯正好和下面與之作用最強的三個Ag之間有BCP和相應的鍵徑,ρ_BCP的信息定量體現出作用區域的電子密度數值很低,而BCP處的能量密度(E)為正則體現出這個吸附作用的本質是非共價作用,這個判據在《Multiwfn支持的分析化學鍵的方法一覽》(http://www.shanxitv.org/471)中我專門介紹過。下圖中的IGM圖和之前的例子一樣也是等值面太肥,不僅不好看,上面的藍色區域還令人誤以為作用區域電子密度其實不低。下圖也給出了這個體系的NCI分析圖,NCI方法在《使用Multiwfn結合CP2K通過NCI和IGM方法圖形化考察固體和表面的弱相互作用》(http://www.shanxitv.org/588)和《使用Multiwfn圖形化研究弱相互作用》(http://www.shanxitv.org/68)中我專門介紹過。下圖的NCI圖在吸附作用區域和IGMH圖很類似,但是在Ag原子間出現了大量等值面,這些等值面雖然無害,但在視覺上妨礙我們考察真正感興趣的吸附作用區域。
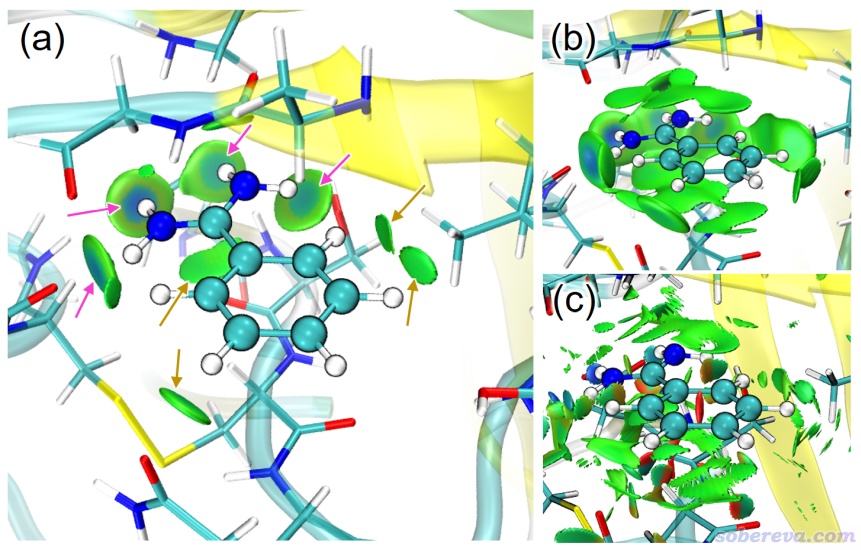
下圖展示的是芐脒陽離子配體與胰蛋白酶之間的相互作用。實際計算時把配體和周圍與之作用的殘基挖了出來作為簇模型來計算,做法可參考《要善用簇模型,不要盲目用ONIOM算蛋白質-小分子相互作用問題》(http://www.shanxitv.org/597)中的討論。實際作圖時,我還把整個蛋白質用New Cartoon方式顯示出來便于觀看骨架位置,涉及到的一些操作細節可參照《使用Multiwfn做aNCI分析圖形化考察動態過程中的蛋白-配體間的相互作用》(http://www.shanxitv.org/591)。下圖(a)和(b)分別是等值面取0.006和0.002 a.u.時候的IGMH等值面圖。可見(a)圖清楚地把配體和周圍殘基間的氫鍵作用(粉色箭頭所指)和明顯的范德華作用(棕色箭頭所指)都展現了出來。(b)圖由于用了更小的等值面數值,使得更多區域更弱的相互作用也同時被展現了出來。可見通過等值面數值的不同選取,可以決定是否展現較為次要的相互作用。下圖的(c)是NCI分析的圖,可見雖然此方法也能展現出蛋白-配體的相互作用,但由于沒法像IGMH那樣定義片段,導致各種相互作用全都出現,包括配體內的和蛋白質殘基之間的,因此圖像看著很亂,有些我們感興趣的等值面還被不感興趣的等值面所遮擋,給討論帶來了很大麻煩。另外,NCI圖對格點質量的敏感性遠高于IGMH,當前NCI圖和IGMH的格點間距是一樣的,IGMH圖里的等值面很平滑,而NCI等值面卻有很多難看的鋸齒。實際上此例用的格點質量已經挺高的了(格點間距為0.15 Bohr,已經很小了),雖然進一步減小格點間距會使得NCI圖更光滑,但耗時也會劇增,因為計算量和格點間距的三次方呈反比。
IGMH分析不僅可以用于分子,也可以用于周期性體系。例如下圖是Multiwfn基于CP2K第一性原理程序對冰計算的周期性波函數得到的IGMH圖,任意選取的一個水和其它的水分別被定義為兩個片段。可見中間的水與周圍的水之間的四個氫鍵被藍色的等值面展現得非常清楚。
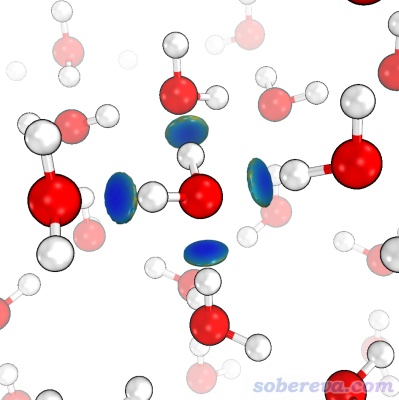
下圖通過IGMH展現了沸石與其中吸附的甲苯之間的相互作用,沸石和甲苯各定義為一個片段,結構使用CP2K做優化得到。由圖可見甲苯與周圍的沸石原子有大面積的相互作用。由于等值面都是綠色,說明吸附是色散作用主導的物理吸附。
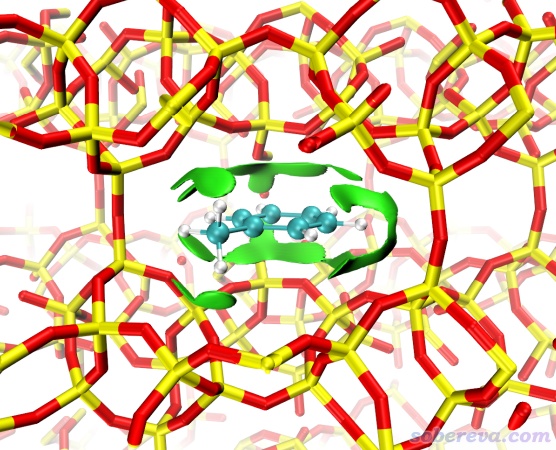
下圖通過IGMH展現了一種多孔共價有機框架化合物(COF)的層間相互作用。可見兩層之間有無限延展的大面積的綠色等值面,無疑說明這種COF層間靠廣闊的pi-pi堆積作用相結合。由圖也可以看到等值面在晶胞邊緣處被合理地截斷,也體現出Multiwfn中的IGMH可以完美地考慮周期邊界條件。
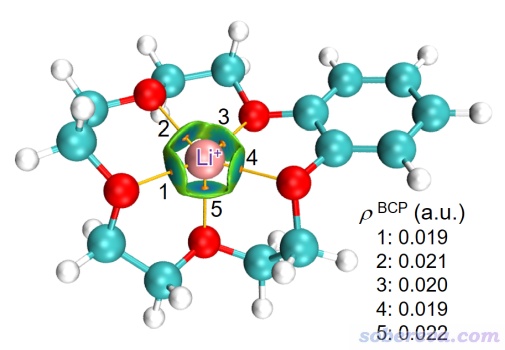
前面的例子主要是通過IGMH展現弱相互作用,而實際上IGMH對于很強的,甚至化學鍵程度的作用也同樣可以很好地展現。例如下圖是冠醚衍生物結合鋰離子形成的體系,IGMH分析時將鋰離子和冠醚定義為兩個片段,由圖可見確實IGMH圖將鋰離子與周圍帶負電的氧之間的高度離子性相互作用區域很好地展現了出來。另外,此圖里還把鍵臨界點和鍵徑一起畫了出來,每個臨界點的電子密度都在圖上給出了,通過這些數據便于定量討論不同的氧與鋰離子作用強度的差異。這有明顯益處,畢竟光是從等值面顏色上判斷容易有視覺誤差、顏色差異較小時難以從視覺上區分,而且看到的顏色還多多少少受可視化程序里光源設置的影響。
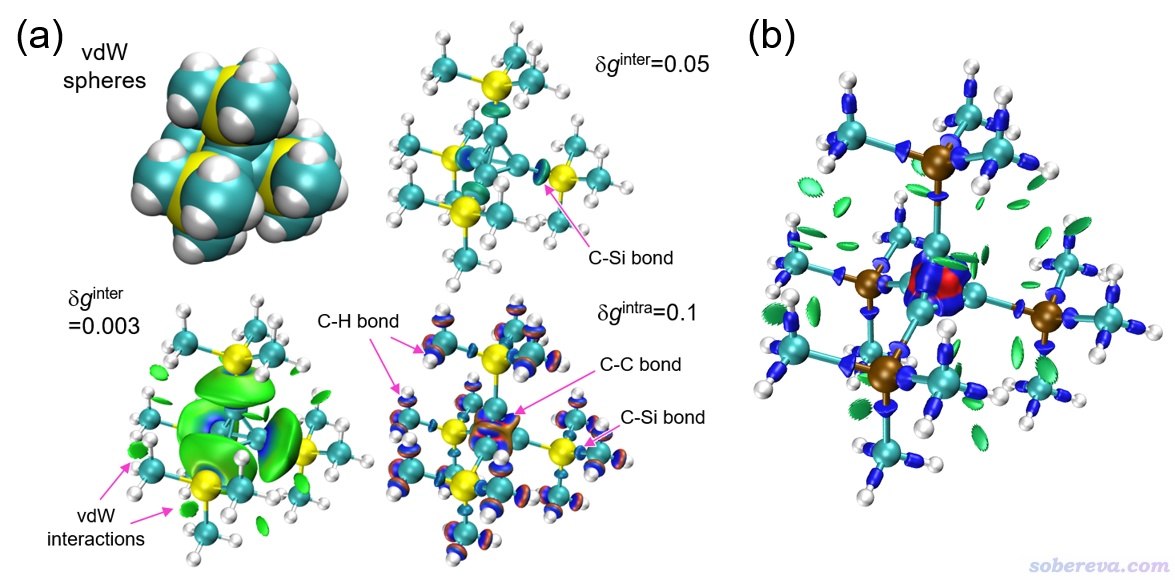
下面的例子是四(三甲基甲硅烷基)四面體體系。下圖左上角是此分子的范德華球疊加圖,可見這個體系相當擁擠,甲基之間距離很近,理應有明顯的范德華作用。對這個體系做IGMH分析時將體系中央的四面體C4部分作為一個片段,上面連接的所有基團作為另一個片段。下圖(a)里面給出了IGMH的δg_inter為0.05時的填色等值面圖,由于當前等值面數值取得比較大,因此只有兩個片段間比較強的相互作用被展現了出來,即C-Si共價鍵作用區域。當δg_inter等值面數值設小到0.003時,如下圖左下角的圖所示,甲基之間的范德華作用區域也被展現了出來,但這個時候C-Si鍵對應的等值面就顯得很肥胖,不好看了。前面例子里主要都是分析片段間作用,而下圖里還給出了δg_intra為0.1的填色等值面圖,用以展現片段內相互作用。可見兩個片段內的C-C鍵、C-Si鍵、C-H鍵作用都被藍色的等值面所展現出來了,藍色也體現出成鍵區域電子密度是很大的,這是共價鍵作用區域的共性(此圖的sign(λ2)ρ色彩刻度范圍設的是-0.2到0.2 a.u.,藍色體現出那部分區域的電子密度肯定>=0.2 a.u.)。值得一提的是,對應化學鍵的等值面周圍有一圈紅色,這沒有明確的物理意義,沒必要關注和予以解釋。下圖的(b)圖給出的是sign(λ2)ρ著色的IRI等值面圖,可見一張圖就把弱相互作用和化學鍵作用區域同時清楚地展現了出來,C4中的三元環里的強烈的位阻區域也被圖中央的紅色等值面部分所展現。可見IRI和IGMH各有各的價值,IRI分析雖然沒法分片段分析,但能夠在一張圖里把各種強度的相互作用同時充分展現出來,這是IGMH分析做不到的。
下圖是P4和Li6兩種原子團簇結構。這回我們不劃分片段,直接用sign(λ2)ρ著色的δg函數來展現體系中的所有相互作用。從下圖左上角的IGMH方法的δg圖上可以看到P-P化學鍵區域(等值面為藍色的部分),以及三個P之間的環形位阻區域和四個P之間的籠形位阻區域(等值面為紅色的部分),都被正確地展現了出來。IGM方法的δg函數的圖在下圖也給出了,可見效果較爛,位阻作用區域描述得亂七八糟,而且本來四面體籠狀結構有很強的籠張力,成鍵區域的電子理應往外凸才對,結果圖中描述化學鍵的藍色等值面主體出現區域卻反倒相對于P-P鍵軸往里收縮,可見IGM方法在此例表現得很差。下圖右上角是P4的IRI圖,可見它雖然和IGMH的δg圖有異,但也把各種特征作用區域理想地展現了出來,而且相對來說IRI的圖還顯得更簡單干凈一些。
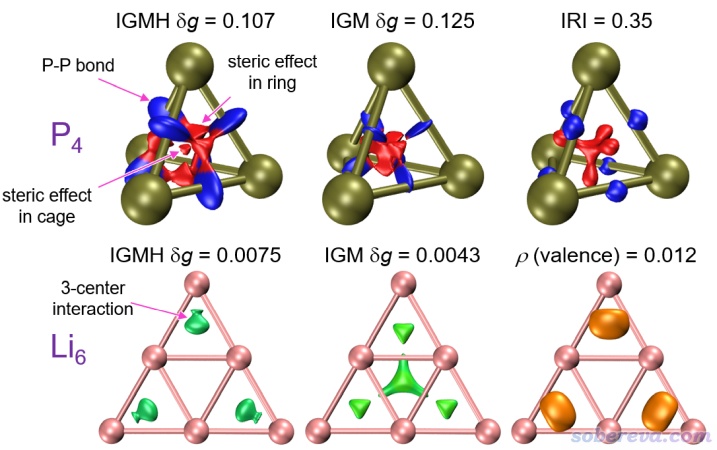
上圖的Li6體系邊緣的三個Li之間存在明顯的三中心鍵作用,這通過Multiwfn中的大量分析手段都能很好地展現出來,包括電子定域化函數(參看《ELF綜述和重要文獻小合集》http://bbs.keinsci.com/thread-2100-1-1.html里的相關文章和我寫過的大量相關博文)、定域化軌道(見《Multiwfn的軌道定域化功能的使用以及與NBO、AdNDP分析的對比》http://www.shanxitv.org/380)、變形密度圖(見《使用Multiwfn作電子密度差圖》http://www.shanxitv.org/113)、多中心鍵級(見《衡量芳香性的方法以及在Multiwfn中的計算》http://www.shanxitv.org/176)、AdNDP(見《使用AdNDP方法以及ELF/LOL、多中心鍵級研究多中心鍵》http://www.shanxitv.org/138)等等等等,以及筆者在DOI: 10.3866/PKU.WHXB201709252一文中提出的價層電子密度分析方法。上圖右下角給出了價層電子密度圖,可見邊緣三個Li之間的價層電子密度顯著高于中間三個Li之間的,因此有對應于三中心鍵的等值面出現。IGMH的δg圖也完全正確地反映出了三中心鍵的存在。而上圖中的IGM的δg圖則對成鍵方式給出了完全錯誤的描述,在中心的三個Li之間也給出了明顯的等值面,這明顯會誤令人以為這個地方也同樣有三中心鍵出現。IGM之所以出現這么嚴重的錯誤正是在于IGM方法只是基于幾何結構做的分析,沒有像IGMH那樣考慮實際電子結構。
3 該選擇什么方法圖形化展現相互作用?
肯定有不少讀者之前也都用過Multiwfn做過NCI、IGM、IRI分析,他們都是重要的圖形化分析方法。我覺得很有必要專門說一下怎么選用。
? IGMH:如果你的目就是分析特定片段間或片段內的相互作用,很明確地知道片段該怎么定義,那么IGMH絕對是最理想的選擇。從上一節的大量例子可看出,IGMH對各類體系表現得都很理想,圖像很美觀。
? IRI:如果你想將體系中所有相互作用(不分類型和強弱)在一張圖里同時展現出來,肯定要用IRI,這在上文的幾個例子里已經有所體現。更多的例子和介紹見《使用IRI方法圖形化考察化學體系中的化學鍵和弱相互作用》(http://www.shanxitv.org/598)以及IRI原文Chemistry—Methods, 1, 231 (2021) DOI: 10.1002/cmtd.202100007。另外,IRI還有個重要優點是可以很好地展現出化學反應過程中化學鍵和弱相互作用的變化和過渡,還可以做成生動的動畫予以動態展現,這在IRI介紹文章里都給出了,一定記得看。此外,IRI還有個變體IRI-pi,對于專門研究pi電子作用極有用處,在上文以及《在Multiwfn中單獨考察pi電子結構特征》(http://www.shanxitv.org/432)中都提到了。
? IGM:有了圖像效果好得多且物理上更嚴格的IGMH后,在《通過獨立梯度模型(IGM)考察分子間弱相互作用》(http://www.shanxitv.org/407)中介紹的IGM基本就不用再考慮了。IGM仍有點用處的地方也就是這幾個:(1)分析巨大體系時出于耗時考慮不得不用IGM (2)僅僅是想很粗略、快速地預覽一下片段間弱相互作用出現的區域 (3)有坐標文件,但由于特殊情況不方便得到用于IGMH分析的波函數文件。
? NCI:2010年提出的NCI方法專門用來圖形化展現弱相互作用,流行程度很高,Multiwfn用戶做NCI分析發表的文章到目前為止大概得有2000篇了。而有了IGMH和IRI后,NCI就基本沒用了。IRI比NCI能展現更豐富的信息,尤其是能同時展現出強相互作用。而IGMH由于能自定義片段,比NCI方便靈活太多了,避免了無關區域的等值面妨礙分析,而且IGMH不需要特別精細的格點就能得到較平滑的等值面,從這個角度來說IGMH比NCI耗時還更低,這些在上文的蛋白-配體作用分析例子上都體現了。除此之外,IGMH還能額外給出原子和原子對的定量指標體現它們對相互作用的貢獻。所以,若無特殊理由就沒必要再用NCI了。
再順帶提一下,IRI、IGMH這樣的圖形化分析方法和極其流行的AIM理論基于臨界點方式的拓撲分析不僅不沖突,還有很強互補性。IRI和IGMH長處是直觀、定性分析,而AIM拓撲分析的長處是在定量層面上討論,可以給你相互作用區域最有代表性的點的各種屬性,根據這些屬性能做大量分析討論,在《Multiwfn支持的分析化學鍵的方法一覽》(http://www.shanxitv.org/471)里有很多介紹。此外,AIM理論里的鍵徑可以把原子間最有代表性的相互作用路徑描繪出來,光從圖形展示的層面上來講也是有益的。還值得一提的是,有些相互作用只能靠IRI、IGMH等圖形分析方法來展現,例如很多體系中明顯的內氫鍵找不到對應的鍵臨界點,因此無法靠AIM拓撲分析來研究,這個問題在IRI原文2.4節里有深入討論。
4 Multiwfn中的IGMH分析功能
如果你不了解Multiwfn的話,強烈建議看《Multiwfn入門tips》(http://www.shanxitv.org/167)和《Multiwfn FAQ》(http://www.shanxitv.org/452)了解相關常識性知識。做IGMH分析一定要用2022年2月及之后更新的Multiwfn版本。繪制IGMH填色等值面圖需要結合免費的VMD程序,可以在http://www.ks.uiuc.edu/Research/vmd/下載,目前強烈建議用VMD 1.9.3版(切勿用bug奇多的1.9.4非正式版)。
在Multiwfn中做IGMH分析非常簡單,通常就是以下步驟,這里簡要提一下,具體細節看后文的例子
(1)啟動Multiwfn,載入波函數文件,進入主功能20的子功能11
(2)輸入片段總數,以及每個片段包含的原子序號
(3)設置格點。格點涵蓋的范圍要至少包含感興趣的等值面出現的區域。格點間距越小,等值面會越平滑、圖像效果越好,但計算耗時越高。關于設定格點的常識性知識看《Multiwfn FAQ》(http://www.shanxitv.org/452)里的Q39。
(4)Multiwfn開始對每個格點計算δg、δg_inter、δg_intra、sign(λ2)ρ這些函數。耗時和體系大小、基組大小、格點數、CPU性能有關。Multiwfn的這部分功能做了充分的并行化,計算量大的情況應當在較好服務器CPU上算,并且記得把settings.ini里的nthreads參數設為CPU實際物理核心數。
(5)計算完畢后出現了后處理菜單,之后:
? 如果要計算δG_pair、δG_atom等前面提到的指數,就選選項6,然后這些指數就會輸出到屏幕上提示的文本文件里。還同時可以輸出atmdg.pdb,在VMD里基于它就可以對原子著色來展示原子的δG_atom或δG_atom(%)值
? 如果要繪制IGMH等值面圖,就選擇選項3,將δg、δg_inter、δg_intra、sign(λ2)ρ函數的格點數據導出為各自的cub文件。之后結合Multiwfn文件包里提供的VMD作圖腳本IGM_inter.vmd和IGM_intra.vmd就可以分別得到sign(λ2)ρ著色的δg_inter和δg_intra等值面圖
如果你之前已經會用Multiwfn做IGM分析,那么IGMH都不用學,因為IGMH和IGM分析操作上的差異僅僅在于進入主功能20之后選主功能11還是10,以及輸入文件是否含有波函數信息,其它地方都是完全相同的。
只要是Multiwfn可識別的含有波函數信息的文件都可以當IGMH分析的輸入文件。比如wfn、wfx、fch、mwfn、molden等等格式的文件都可以用,主流的量子化學程序如Gaussian、ORCA、GAMESS-US、xtb等至少可以產生其中的一種,讀者可參看《詳談Multiwfn支持的輸入文件類型、產生方法以及相互轉換》(http://www.shanxitv.org/379)里的詳細說明,怎么用主流程序產生這些文件在文中也都寫明了。如果是研究周期性體系,應當用CP2K產生molden格式的波函數文件,并且需要自行把晶胞信息加入到此文件里,詳見《使用Multiwfn結合CP2K通過NCI和IGM方法圖形化考察固體和表面的弱相互作用》(http://www.shanxitv.org/588)里的相關說明。
做IGMH分析用的波函數不需要用昂貴的級別產生,用一個對當前體系適合的DFT泛函,結合一個不低于2-zeta檔次帶極化函數的基組就夠了。可見這對計算級別的要求和幾何優化相仿佛,因此通常就用幾何優化最后順帶產生的波函數做IGMH分析就可以了。對于Gaussian用戶,一般用B3LYP-D3(BJ)/6-311G*就可以。對于ORCA用戶也可以用這個級別,但用B97-3c或r2SCAN-3c方法明顯更劃算,不了解這倆方法的話看《詳談Multiwfn產生ORCA量子化學程序的輸入文件的功能》(http://www.shanxitv.org/490)中的敘述。對于CP2K用戶,一般用PBE-D3(BJ)結合pob-TZVP基組或更便宜些的6-311G*就可以。
如果體系很大,用DFT優化結構太耗時,用xtb等程序通過很廉價的半經驗層面的GFN-xTB方法做優化也可以。xtb產生的molden文件也可以用于IGMH分析,結果大多數情況可以接受,但是由于GFN-xTB級別的波函數質量還是明顯比DFT要差,基于它做的IGMH圖和基于DFT波函數得到的圖還是有可查覺的差距。所以最好還是基于像樣的半經驗方法或GFN-xTB方法優化的結構,用DFT算個單點任務得到用于IGMH分析的波函數文件。
5 用Multiwfn做IGMH分析實例
5.1 對分子和周期性體系做IGMH分析的例子
極度詳細、完整、圖文并茂的使用Multiwfn對分子體系和周期性體系做IGMH分析的教程可以直接在http://www.shanxitv.org/multiwfn/res/IGMH_tutorial.zip下載(此教程在IGMH原文里提到了,算是官方教程),所有涉及的文件在里面也都一并提供了。此教程多達40頁,對IGMH分析的所有操作和要點都描述得特別細致,之前完全沒有用過Multiwfn和VMD的讀者肯定也能照著此教程順利作出前文給出的各種各樣體系的IGMH圖。而且此文檔里面還給出了前文沒有舉例的曲線圖、平面填色圖、sign(λ2)ρ與δg之間的著色散點圖的繪制方法。
另外,《通過獨立梯度模型(IGM)考察分子間弱相互作用》(http://www.shanxitv.org/407)和《使用Multiwfn結合CP2K通過NCI和IGM方法圖形化考察固體和表面的弱相互作用》(http://www.shanxitv.org/588)里分別給了對分子體系和周期性體系做IGM分析的例子。讀者只要把文中在主功能20里面選子功能10的地方替換成子功能11,并且改用含有波函數信息的文件作為輸入,得到的就是IGMH分析結果了。因此如果看前面給的IGMH教程中有不理解的地方也可以同時參照這倆IGM教程。
VMD中的一些操作、設置對于IGMH分析和NCI分析是互通的,如果你按照上面的IGMH教程操作的時候在一些地方由于缺乏計算機基本常識、對描述的理解存在偏差等原因卡殼,那么也不妨看一下《使用Multiwfn做NCI分析展現分子內和分子間弱相互作用》(https://www.bilibili.com/video/av71561024)這個用Multiwfn結合VMD做NCI分析全流程的視頻演示,仔細看過后之前做IGMH分析卡殼的地方可能也就明白了。
之前筆者寫過《用Multiwfn+VMD做RDG分析時的一些要點和常見問題》(http://www.shanxitv.org/291),里面有很多關于改進VMD里作圖效果的討論,對于在VMD中繪制IGMH填色等值面圖也是同樣適用的,建議讀者看完前述教程后參考。
常有人問怎么IGMH圖作完了發現上面沒有等值面,這大概率是因為片段間相互作用很弱,而默認的等值面數值不夠小所致的。把等值面的數值手動改小到比如0.005甚至0.003,肯定能看見。如果此時還看不見等值面,那就說明片段間的相互作用可以完全忽略了,片段間的距離應該很遠。
5.2 基于簇模型利用IGMH分析考察分子晶體中的相互作用
這一節的目的是介紹如何便利地通過簇模型做IGMH分析考察分子晶體中的弱相互作用,這個例子在前述教程和相關博文里都沒有,因此是對IGMH官方教程的一個補充。請讀者務必先把前述教程看了再看本節的這個補充教程。做這一節的例子時,讀者務必用2022年3月及以后更新的Multiwfn,否則例子中涉及的功能沒有。
這里說的簇模型是指把晶體中的一個分子和與之有直接接觸的一圈分子提取出來作為一個簇,中心分子和周圍分子分別定義為IGMH分析的兩個片段,這樣分析結果就能完整展現晶體中的分子與周圍環境分子的相互作用了。雖然基于CP2K計算產生的周期性波函數也能做分子晶體的IGMH分析,但使用簇模型來做有幾個優點:
(1)計算只考慮中心分子和鄰近的一圈分子,可避免計算更外圍、和中心分子沒直接作用的分子導致浪費計算量。而且由于此時的IGMH分析過程中不需要考慮周期性,計算耗時比考慮周期性時低得多
(2)避免更外圍分子的結構顯示在圖中令圖像混亂(雖然作為周期性體系分析時也可以在VMD中恰當設置來避免顯示無關分子,但要多花費點步驟)
(3)不需要用第一性原理程序做周期性計算,而只需要用量子化學程序就可以做,因此對量子化學研究者們更方便
本節涉及到的各種文件都可以在http://www.shanxitv.org/attach/621/file.rar里下載。這一節用的例子是尿素晶體,其cif文件是里面的urea.cif。我們將用Gaussian程序做量子化學計算。
首先要做的是基于晶體結構構造尿素團簇。這不需要自己先手動構造超胞,然后再從里面費勁地摳團簇,因為Multiwfn直接就提供了超級便利的一鍵產生“中心分子+臨近一圈分子”的團簇結構的功能。啟動Multiwfn,然后輸入
urea.cif //輸入cif文件的實際路徑
300 //主功能300
7 //幾何相關操作。這部分的功能在《Multiwfn中非常實用的幾何操作和坐標變換功能介紹》(http://www.shanxitv.org/610)中有全面介紹和示例
25 //構造“中心分子+臨近一圈分子”的團簇
1 //當前晶胞里1號原子所在的分子將被作為中心分子(由于尿素晶體里所有分子都是等價的,所以當前隨便輸入一個原子序號即可)
[回車] //這代表若一個周圍分子與中心分子間最近原子對距離小于這倆原子的范德華半徑和的1.2倍,則這個周圍分子就被整個納入團簇
Multiwfn瞬間就構造出了團簇,從屏幕上的提示可看到這個簇有88個原子,并且屏幕上還巨貼心地把中心分子中的原子序號給了出來,當前為1-3,25,32-34,69。把這個序號記下來,之后IGMH分析時要用到。
現在可以選菜單中的選項0看一眼新構造出的團簇是什么樣,如下所示,可見非常理想,確實是中心分子被周圍一圈分子所圍繞
點圖形窗口右上角的RETURN按鈕關閉窗口,然后選選項“-3 Output system to .gjf file”,再輸入cluster.gjf,當前團簇結構就保存為了當前目錄下的cluster.gjf,這是Gaussian的輸入文件格式。Multiwfn現在可以關了。
如《實驗測定分子結構的方法以及將實驗結構用于量子化學計算需要注意的問題》(http://www.shanxitv.org/569)所提到的,X光衍射得到的晶體結構中氫的位置大多是不準的,所以我們要對這個簇里的氫原子位置做優化,而重原子的位置則應固定住,否則在真空中優化后,團簇結構會和晶體狀態出現偏離。怎么在Gaussian里只優化氫在《在Gaussian中做限制性優化的方法》(http://www.shanxitv.org/404)中專門說過。把cluster.gjf內容改成下面這樣,坐標部分不動。由于當前牽扯氫鍵,所以特意給氫加了極化函數。
%chk=cluster.chk
#P B3LYP/6-31G** em=GD3BJ opt=readopt
[空行]
Generated by Multiwfn
[空行]
0 1
[坐標部分略...]
[空行]
noatoms atoms=H
[空行]
[空行]
然后用Gaussian運行cluster.gjf,算完之后當前目錄下就出現了cluster.chk,里面記錄了優化后的結構的波函數信息。按照《詳談Multiwfn支持的輸入文件類型、產生方法以及相互轉換》(http://www.shanxitv.org/379)所述,輸入formchk cluster.chk命令將之轉化為Multiwfn可以直接載入的cluster.fch文件。
下面就是做IGMH計算了。啟動Multiwfn,然后輸入
cluster.fch
20 //弱相互作用可視化分析
11 //IGMH
2 //定義兩個片段
1-3,25,32-34,69 //第一個片段里的序號,此例對應中心分子序號
c //所有其余原子作為第二個片段,等同于輸入4-24,26-31,35-68,70-88
11 //格點設置方式為:選中一批原子,在其四周擴展一定距離定義盒子(即格點分布區域),并指定格點間距。這種模式設置格點最適合當前情況
1-3,25,32-34,69 //盒子所圍繞的中心分子中的原子序號
2 A //在中心分子四周擴展2埃定義盒子,通常這足夠大了(如果最終從IGMH圖中發現有些等值面被截斷,可以再加大)
0.15 //格點間距(Bohr)。越小等值面越光滑,而計算耗時越高。鑒于當前體系較小,耗時必定不高,所以用了這樣很精細的格點以得到盡可能理想的圖像
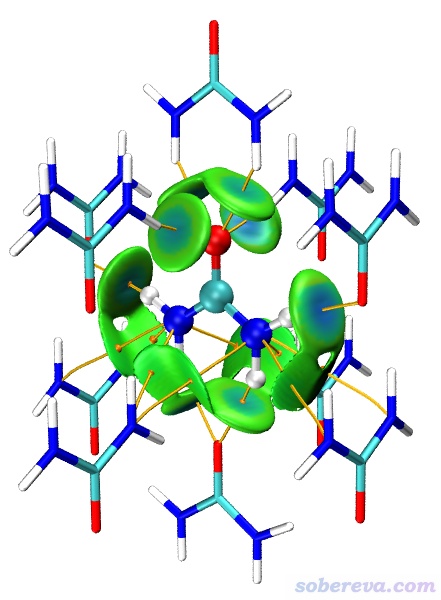
算完后在后處理菜單選擇3將格點數據導出成當前目錄下的cub文件,然后將其中的dg_inter.cub和sl2r.cub挪到VMD目錄下,也把Multiwfn文件包里的examples目錄下的IGM_inter.vmd腳本挪到VMD目錄下。啟動VMD,然后輸入source IGM_inter.vmd,即看到下圖中δg_inter為0.01的圖,此圖中等值面上的藍色部分將所有的氫鍵作用區域都很好地展現了出來。若進入Graphics - Representation,點擊其中第二項,把Isovalue從0.1改小為0.004,看到的就是下圖右側的圖,可見此圖中綠色扁片部分把相對較弱的范德華作用區域也給展現了出來。
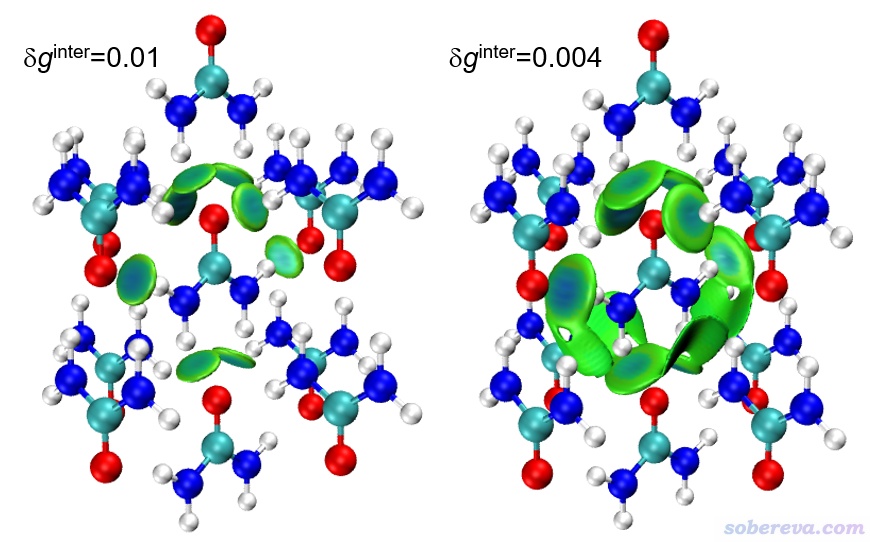
由此例可見,依靠Multiwfn以簇模型方式考察分子晶體中的弱相互作用特別便利。之后大家還可以進一步調整圖像效果,比如把中心分子和周圍分子用不同顯示方式進行區分以凸顯中間分子,還可以把鍵臨界點和鍵徑也畫上去,如下所示
下面順帶說一下怎么把上圖的中心分子和周圍分子間的鍵臨界點(BCP)和鍵徑顯示出來,步驟較多,不感興趣的讀者可直接略過。如果你對Multiwfn里拓撲分析功能一無所知的話,先看《使用Multiwfn做拓撲分析以及計算孤對電子角度》(http://www.shanxitv.org/108)和Multiwfn手冊4.2節的一堆例子了解基本操作。
按照《使用Multiwfn+VMD快速地繪制高質量AIM拓撲分析圖(含視頻演示)》(http://www.shanxitv.org/445)的做法在VMD里結合我提供的腳本通過aim命令可以顯示出臨界點和鍵徑,再運行source IGM_inter.vmd命令,則所有臨界點、鍵徑以及IGMH等值面就會一起顯示出來,但此時周圍尿素分子間的臨界點和鍵徑也會出現,令圖像較亂。若想只保留中心分子和周圍分子間的BCP和鍵徑應當這么做:啟動Multiwfn,載入cluster.fch,然后依次輸入
2 //拓撲分析
2 //以每個原子核為初猜搜索臨界點
3 //以每一對原子中點為初猜搜索臨界點
8 //產生鍵徑
-5 //處理鍵徑
8 //只保留連接兩個片段間的鍵徑和對應的BCP而刪除其它的
1-3,25,32-34,69 //中心分子的原子序號
4-24,26-31,35-68,70-88 //其余分子的原子序號
y //連接兩個片段以外的鍵徑已被刪除,這里輸入y也刪除它們對應的BCP
6 //把當前鍵徑導出為paths.pdb
0 //返回
-4 //修改臨界點
2 //刪除某些臨界點
5 //刪除對當前沒意義的(3,+1)型臨界點
6 //刪除對當前沒意義的(3,+3)型臨界點
0 //返回
6 //把臨界點導出為CPs.pdb
現在關閉Multiwfn,把paths.pdb和CPs.pdb挪到VMD目錄下,啟動VMD,運行aim命令,就會看到只有核臨界點、中心分子和周圍分子間的BCP和鍵徑被顯示了出來。再執行source IGM_inter.vmd,看到的圖像就和上面的差不多了。順帶提示一下,在VMD里用選擇語句選擇中心分子可以用serial 1 to 3 25 32 to 34 69,選擇周圍分子可以用not {serial 1 to 3 25 32 to 34 69}。上圖設了兩個representation,中心分子部分Drawing Method是CPK,周圍部分是Licorice。
6 總結
本文對筆者提出的IGMH方法的原理和不少細節做了介紹,給出了大量應用范例,充分體現出了IGMH的圖像效果十分理想,在分析化學體系中的相互作用方面具有極高的實用性和很好的普適性。IGMH和IRI、NCI、IGM等圖形化相互作用分析方法之間的差異和選用也在文中做了討論。本文還介紹了強大的Multiwfn波函數分析程序中的IGMH分析的功能,并給出了具體的做IGMH分析的操作示例。可以看到IGMH分析在Multiwfn中使用特別方便,步驟簡單,很容易上手。由于IGMH擁有諸多優點,又有不可替代的重要價值,并且有高效、易用的Multiwfn可以對各類體系實現IGMH分析的程序,IGMH在未來一定會非常流行。