使用量子化學程序基于簇模型計算金屬表面吸附問題
使用量子化學程序基于簇模型計算金屬表面吸附問題
文/Sobereva@北京科音
First release: 2020-Mar-8 Last update: 2020-Mar-10
簡介:本文首先對量子化學程序基于簇模型計算表面反應或吸附問題做簡單介紹,然后基于ORCA量子化學程序,簡單演示如何計算苯分子在Ag(111)表面上的吸附能。本文目的是令讀者認識到量子化學程序研究表面問題的可行性,并了解實際計算中需要值得注意的地方。
筆者還另有一篇博文章《18碳環(cyclo[18]carbon)與石墨烯的相互作用:基于簇模型的研究一例》(http://www.shanxitv.org/615),也和本文主題密切相關,建議讀者一看。
1 關于簇模型
一說到基于量子力學方法的固體表面反應/吸附的計算,很多人就首先想到用第一性原理程序諸如Quantum Espresso、CP2K去做,甚至以為以計算孤立體系為主的Gaussian、ORCA等量子化學程序完全不適合,實際上這是不對的。很多固體表面問題的計算都可以通過構建簇模型(cluster model)來使用量子化學程序計算。所謂簇模型就是把反應/吸附位點附近區域的原子挖出來當做孤立體系計算。
用量子化學程序通過簇模型來算和使用第一性程序通過周期性方式來算這類問題各有利弊。用簇模型的優點在于:
? 整體來說,量子化學程序支持的理論方法遠比第一性原理程序豐富得多得多,選擇余地明顯更大,還可以做到更高精度(如雙雜化、DLPNO-CCSD(T)等)。用雜化泛函相對于純泛函的耗時增加程度遠低于基于平面波的第一性原理程序。
? 在計算設置上更方便,也不需要考慮k點、真空層、分子與相鄰鏡像作用、偶極校正之類亂七八糟的事。
? 支持的任務類型更豐富,關鍵詞寫著省事,可視化程序也多。例如Gaussian vs. VASP
? 結合Multiwfn可以做非常豐富的波函數分析,使文章的分析討論部分明顯更充實、更上檔次。參考比如《Multiwfn支持的分析化學鍵的方法一覽》(http://www.shanxitv.org/471)、《Multiwfn支持的弱相互作用的分析方法概覽》(http://www.shanxitv.org/252)、《Multiwfn支持的電子激發分析方法一覽》(http://www.shanxitv.org/437)等大量文章,在《Multiwfn入門tips》(http://www.shanxitv.org/167)里有所有Multiwfn做分析的博文匯總。而使用第一性原理程序的話,能做的分析少得多,不過《使用Multiwfn結合CP2K通過NCI和IGM方法圖形化考察固體和表面的弱相互作用》(http://www.shanxitv.org/588)
用簇模型的弊端在于:
? 需要合理考慮邊界效應。邊界效應此處指的是使用有限的簇模型作為周期性體系的近似導致的對被研究問題的計算精度的影響。邊界效應取決于兩點:(1)簇的尺寸 (2)對邊界的處理。簇的尺寸越大、讓邊界原子所處的環境越像體相,則邊界效應越小。
簇的大小選取有很大任意性。固體部分截的原子太少的話邊界效應太強,結果不準;而截得大的話,則計算耗時太高。除非團簇截得極大,否則都要考慮怎么恰當處理邊界。比如有機體系(如石墨烯),邊界往往用氫飽和,見比如《18碳環(cyclo[18]carbon)與石墨烯的相互作用:基于簇模型的研究一例》(http://www.shanxitv.org/615);如果是無機晶體,有用贗氫、capped ECP、用大量背景電荷表現更長程區域原子的靜電勢等做法;如果是純金屬的話倒是不需要做特別的考慮。
? 對于原子致密排布的塊狀固體材料(不是那種稀疏的、有孔洞的諸如MOF等,或者單層材料),用簇模型的話耗時比起用第一性原理程序當周期性算往往更高。但這點具體看用什么程序、什么基組、數值方面的設定等等,不能一概而論。
? 用簇模型算含有過渡金屬的無機晶體有時會在SCF收斂上遇到麻煩,有時需要大量折騰。但也不是絕對解決不了,可結合《解決SCF不收斂問題的方法》(http://www.shanxitv.org/61)里的說明恰當嘗試。
總的來說,用第一性原理程序做周期性計算研究表面問題是比較主流的做法,文章非常多,但簇模型計算這類問題也絕非很稀罕,相關研究文章也不少。還有的研究將兩種方式相結合,取二者長處,比如ACS Catal., 8, 3825 (2018)研究石墨烯上催化二氧化硫與氧氣反應形成硫酸鹽,就先用了VASP做了周期性計算優化了極小點和過渡態,再用Gausisan基于VASP優化的結構用簇模型做了單點計算,將得到的波函數用Multiwfn分析了催化反應機理以及石墨烯表面與被催化物質的弱相互作用。
2 簇模型計算表面問題的實際例子:苯分子在Ag(111)表面物理吸附的結合能的計算
下面就通過一個簡單例子,說明如何基于簇模型計算物理吸附的結合能。
2.1 相關說明
本例涉及的各種文件都可以在這里下載:http://www.shanxitv.org/attach/540/file.rar。
這個體系在DFT-D3原文J. Chem. Phys., 132, 154104 (2010)中被研究過,苯與銀表面間距的剛性掃描圖如下所示
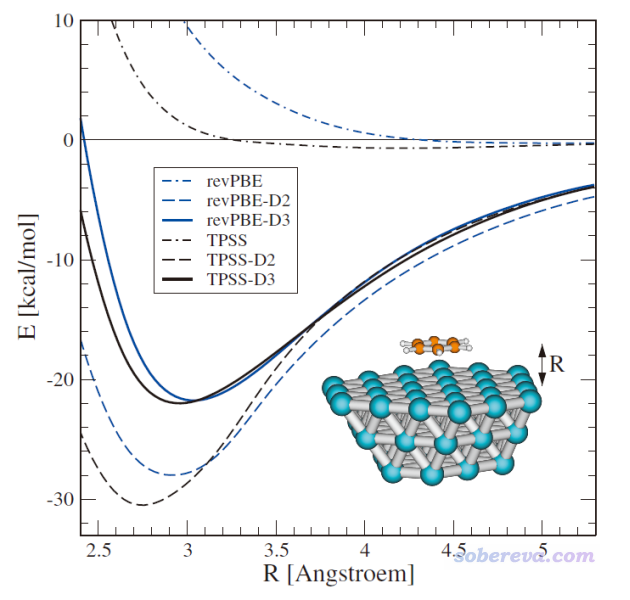
文中也說了,實驗測定的苯吸附到Ag(111)上的焓變是-13 kcal/mol。
下面我們來算苯分子在Ag(111)表面物理吸附對應的結合能。這里說的結合能是習俗上定義的,即只考慮電子能量的變化,而且片段能量用的是復合物中的結構算的。算這個問題分為以下幾步
(1)構建金屬簇
(2)把苯分子放進去得到復合物初猜結構
(3)優化復合物
(4)計算各個片段的單點能
(5)求差得到結合能
之后還可以再用Multiwfn做一些波函數分析。
嚴格來說,就算哪怕不計算熱力學量,優化完復合物也應當做一下振動分析來看看虛頻情況,如果苯分子有沿表面運動的虛頻模式,應當恰當消掉,基本策略參考《Gaussian中幾何優化收斂后Freq時出現NO或虛頻的原因和解決方法》(http://www.shanxitv.org/278)。但由于當前體系不小,做振動分析耗時很高,本文就忽略這點了。
本例使用ORCA程序,一方面是因為免費,另一方面是支持RIJCOSX,可以令這種大團簇體系在單點計算和優化時耗時降低極多,明顯快過Gaussian。不過ORCA做振動分析慢,也是本文沒做振動分析的原因。ORCA的基本常識看《量子化學程序ORCA的安裝方法》(http://www.shanxitv.org/451)和《基于ORCA量子化學程序對分子做優化、振動分析、觀看紅外光譜、觀看軌道的簡單演示》(https://www.bilibili.com/video/av59599938)。本文用的是ORCA 4.2.1。
2.2 建模
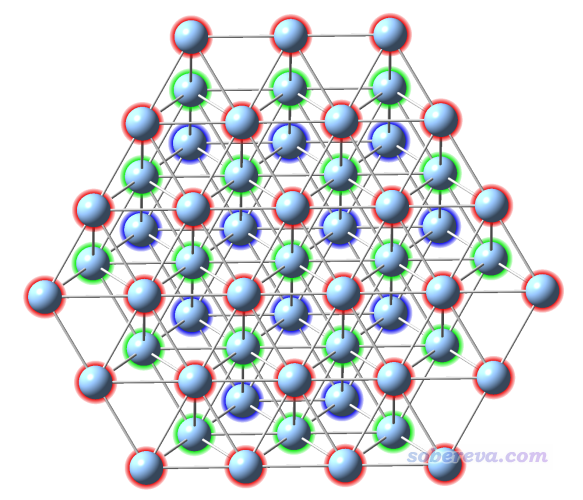
本文用的對應Ag(111)表面的團簇結構就按照上一節文獻里的圖來構建就行了,用什么程序隨意。比如可以去ICSD找到Ag的晶體結構,用VESTA、GaussView、Avogadro之類復制延展成足夠大的復晶胞,然后一點點刪原子就可以得到。你也可以用某貴得離譜的有切表面功能的可視化程序先載入自帶的庫里的Ag原胞,用切表面的選項切出(111)表面,然后再復制延展成面積足夠大、至少有三層原子厚度的表面,之后再一點點刪多余的原子。為了不讓邊界效應太顯著,對于簇模型研究表面問題,一般建議至少三層原子,而且固體團簇要比被吸附的分子明顯大一圈。像上面圖片里構造的Ag58團簇對于研究苯的吸附就是很得當的,不大不小。再大一圈的話耗時就忒高了,再小一圈的話邊界效應就較為顯著了。而且為了節約原子來降低耗時,可見上圖中的團簇是上面寬下面窄,比起上下一樣寬時原子數明顯更少。由于苯分子豎著看起來是近乎圓形的,因此團簇的表面最好也接近圓形。顯然,分子如果是長條形的,簇的表面也應當是長條形。
筆者建立好的簇如下所示,為了看得清楚每層用了不同顏色。在gview里可能默認沒有把Ag-Ag之間判斷為成鍵,導致構建簇模型的時候不好判斷原子位置,可以根據《談談原子間是否成鍵的判斷問題》(http://www.shanxitv.org/414)文中所述自定義成鍵判斷閾值,筆者設的是小于3埃就判斷成成鍵。
然后用把苯分子放在金屬簇的上方正中央位置。距離不要太遠,要不然優化到平衡位置需要多花很多步;也不要太近,本來過渡金屬團簇就是SCF難收斂的典型,如果被吸附分子放得太近導致電子結構變得更復雜的話會雪上加霜。本文把苯分子放在團簇表面上方3埃的位置。從DFT-D3原文里那張勢能面掃描圖來看,TPSS-D3的極小點位置也差不多就是3埃。
當前的結構不能直接就拿去優化,因為簇模型里邊界的原子沒有感受到真實體相環境的束縛,如果不把邊界的原子進行固定就優化的話,可能導致邊界的原子位置大幅改變,和真實情況明顯不符。因此我們把最靠邊的Ag都凍結,而中間的,即與苯分子密切接觸的Ag原子不設凍結,這是為了讓它們的位置自發地調整來響應相互作用造成的實際的結構變化。
下圖就是筆者在GaussView里最終構建的復合物初始結構,為了看得清楚給了兩個視角,黃色是被凍結的原子。在GaussView的Tools - Atom Selection界面里可以直接查詢到這些被選成黃色的原子的序號,即1-11,14,17,19-23,26,29,32,34-38,41,44,46-50,52,54-58。
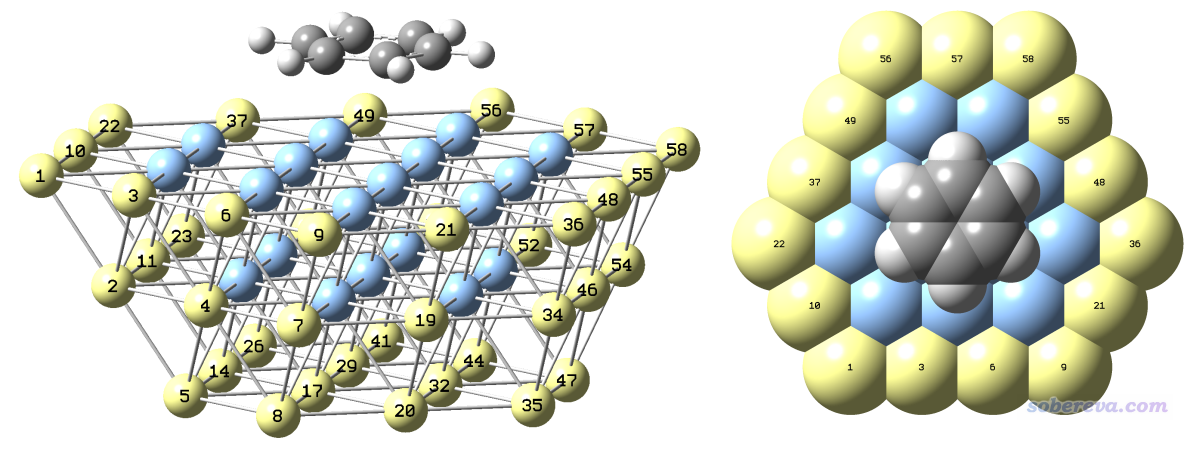
這個初始結構用GaussView保存出來的gjf文件是本文文件包里的Ag58_benzene_freeze.gjf。
值得一提的是,但凡若有可能,都最好讓簇模型處于閉殼層狀態,這樣算起來比開殼層的情況快得多。對于當前的體系,苯分子是閉殼層的,Ag是奇數個電子而且靠s電子成鍵(成鍵情況比較簡單),故只要Ag原子是偶數個,就可以讓整個體系是閉殼層。當前Ag是58個,所以整個體系是閉殼層。如果你用的銀團簇是比如Ag59或者Ag57,那么整體就是開殼層了,算起來就費勁多了。所以在設計金屬團簇的時候應當注意考慮這點。
2.3 幾何優化
下面用Multiwfn產生ORCA程序對上面的體系做限制性優化的輸入文件。相關信息看《詳談Multiwfn產生ORCA量子化學程序的輸入文件的功能》(http://www.shanxitv.org/490)。本文用的是2020-Mar-5更新的Multiwfn,可在官網http://www.shanxitv.org/multiwfn下載。啟動Multiwfn,然后輸入
Ag58_benzene_freeze.gjf //gjf文件的實際路徑,Multiwfn會從中讀取結構
oi //產生ORCA輸入文件
complex_opt.inp //產生的文件的名字
0 //設置任務類型
2 //優化
-3 //設置原子凍結
1-11,14,17,19-23,26,29,32,34-38,41,44,46-50,52,54-58 //被凍結的原子序號
3 //用RI-B3LYP-D3(BJ)/def2-TZVP(-f)
然后我們就在當前目錄下得到了complex_opt.inp文件。把其中的B3LYP改成PBE0,把def2-TZVP(-f)改為def2-SV(P),把CPU核數和內存分配量設為實際情況,其它內容不變。此時這個文件對應于PBE0-D3(BJ)/def2-SV(P)下進行限制性優化,是我們實際要用的。
這里解釋下為什么用這個級別做優化。前面我的博文里提到過,在ORCA里開RI時,純泛函的耗時遠低于雜化泛函,而且純泛函PBE對純金屬的描述也不錯,而之所以這里非要用雜化泛函PBE0,是因為用純泛函時SCF收斂難度比雜化泛函通常明顯更大,尤其是當前這種過渡金屬團簇類型的體系本來就是SCF難收斂的體系,嘗試過PBE就會發現收斂情況就像噩夢,所以用了雜化泛函。之所以把原本輸入文件里的B3LYP關鍵詞給改成了PBE0,這是因為J. Chem. Phys., 127, 024103 (2007)中專門說了B3LYP算金屬很爛,而量子化學領域里另一個很常用的雜化泛函PBE0則在這方面可靠得多,也有不少文章用PBE0算過Ag表面,如J. Phys. Chem. C, 117, 5075 (2013)。對于PBE0這種描述色散作用垃圾的泛函,在計算物理吸附時DFT-D校正顯然是需要的,見《談談“計算時是否需要加DFT-D3色散校正?”》(http://www.shanxitv.org/413)、《亂談DFT-D》(http://www.shanxitv.org/83),要不然結果就搞笑了。雖然目前ORCA也支持DFT-D4了,見《DFT-D4色散校正的簡介與使用》(http://www.shanxitv.org/464),但這里還是用D3,一方面是D3目前已經被極為廣泛地使用,比D4更放心一些,另一方面是D4比D3強的地方關鍵在于能考慮原子帶電狀態對色散校正的影響,而當前體系里金屬基本不帶電荷,因此用D4也不會帶來什么明顯好處(改進較大的是離子、過渡金屬配合物類型的體系)。
至于優化此體系用的基組的選擇,考慮到當前體系不僅原子數多,而且絕大多數都是重原子,本來耗時就肯定比較高,再加上幾何優化對基組不敏感(見《淺談為什么優化和振動分析不需要用大基組》http://www.shanxitv.org/387),所以基組用個便宜的2-zeta檔次的就夠了。lanl2DZ雖然便宜且也能基本滿足幾何優化目的,但沒有標配的輔助基組,如果用autoaux關鍵詞自動產生輔助基組的話不劃算。def2-SVP是ORCA用戶非常常用的中等偏小的基組,有標配的輔助基組,且對Ag是贗勢基組,耗時不會很高,用于優化比較合適。由于此基組對Ag有f極化函數,它會造成耗時增加不少,而對優化來說影響不大,因此我們實際用的是def2-SV(P),它把f極化函數給砍掉了,同時也砍掉了對氫的p極化,這對優化影響也甚微。
復合物優化任務的輸入文件是本文文件包里的complex_opt.inp,在雙路XEON E5-2696v3(共36核)服務器上花了10小時算完。產生的最終結構文件是本文文件包里的complex_opt.xyz。為了觀看優化過程的收斂趨勢,筆者用《OfakeG:使GaussView能夠可視化ORCA輸出文件的工具》(http://www.shanxitv.org/498)里的OfakeG工具將輸出文件轉成了贗Gaussian輸出文件,即本文文件包里的complex_opt_fake.out。用GaussView打開,收斂曲線如下所示,可見收斂還蠻順利的
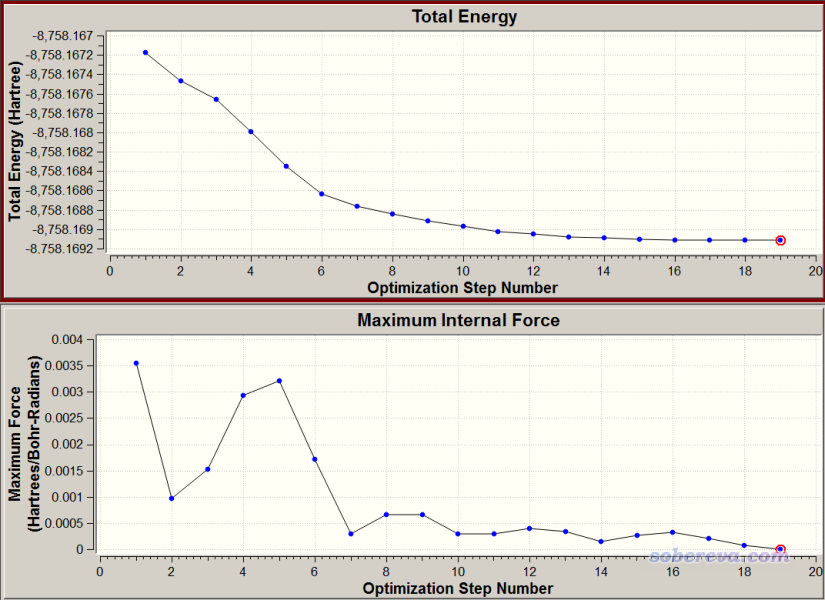
大家通過gview看結構變化也可以發現,由于復合物初始結構本來擺得就比較理想,所以優化過程中只是苯分子位置稍微調節了一下而已,整體沒有明顯變化,和苯接觸的銀原子也沒怎么動。
2.4 單點計算
現在產生單點任務的輸入文件。啟動Multiwfn,載入complex_opt.xyz,然后輸入
oi //產生ORCA輸入文件
complex_SP.inp //產生的文件的名字
4 //選擇計算級別
如果你對結合能計算精度要求不是特別高,或者計算條件不是很好,將產生的complex_SP.inp里的B3LYP改成PBE0就可以開始算了,此時對應于PBE0-D3(BJ)/def2-TZVP級別。
不過,有常識的人都知道,精確計算弱相互作用能需要考慮BSSE問題,見《談談BSSE校正與Gaussian對它的處理》(http://www.shanxitv.org/46)。在def2-TZVP下雖然BSSE問題已經不太大了,但對于弱相互作用能計算的影響還是不可忽視的,有四個解決辦法解決BSSE問題:
(1)做counterpoise校正。但這需要多花兩倍左右時間。若想要用這種做法,參看《在ORCA中做counterpoise校正并計算分子間結合能的例子》(http://www.shanxitv.org/542)。
(2)用gCP方法以免費方式近似解決BSSE問題。gCP校正的簡介見《大體系弱相互作用計算的解決之道》(http://www.shanxitv.org/214)。gCP對def2-TZVP有現成的參數,在ORCA里要用這種校正就寫上gCP(DFT/TZ)即可。而且gCP校正有解析梯度,完全可以用于前面的幾何優化過程中,在其輸入文件里寫上gCP(DFT/SV(P))即可。然而,為什么我們對此例不用gCP?這是因為gCP原本只對前四周期元素擬合了參數,對于之后的元素,會用同族的第四周期元素來湊合,比如Ag的gCP參數會用Cu的參數來湊合。這種湊合的做法,對于第四周期之后元素的原子數較少(比如過渡金屬配合物)的時候是可以接受的,但當前體系有一大堆Ag,其參數是影響結合能計算的重點,顯然這么湊合是不靠譜的近似。
(3)給基組加彌散函數,即改成ma-def2-TZVP,這樣可有效改進弱相互作用計算精度并減小BSSE問題。但這種做法我不推薦用于當前計算,因為此時沒有標配的輔助基組,用autoaux自動產生的不是特別劃算。而且本來當前體系SCF就比較難收斂,而眾所周知加彌散函數會導致SCF收斂難度暴增,對于當前這種原子堆得緊密的情況更是會導致線性依賴問題極度顯著,所以加彌散是餿主意。
(4)用更大的def2-QZVP基組。這個基組對于普通泛函的DFT計算而言已經基本達到了完備基組極限了。此時BSSE問題已可以忽略,而且比def2-TZVP時對電子結構描述又有少量改進,而且DFT-D3參數原本又是在def2-QZVP下搞的,因此在這個層面上顯得很完美。def2-QZVP的確相當貴,但在ORCA中利用RIJCOSX技術,用這么大的基組對當前體系進行單點計算,耗時依然是完全可接受的。因此,本例使用PBE0-D3(BJ)/def2-QZVP來做單點計算。
還有需要注意的是,通常基組越大SCF越不容易收斂。筆者嘗試過,在優化后的復合物結構下用def2-TZVP可以直接收斂(雖然SCF花了多達46圈),而用def2-QZVP則迭代幾十次之后依然沒有收斂的跡象,即便讀def2-TZVP收斂的波函數當初猜也不行。經嘗試,我發現把incremental Fock加速技術關閉后可以直接解決,即在輸入文件里加上%scf DirectResetFreq=1 end。
(補充:如果你是用ORCA 5.0及以后版本,請忽略下面grid4 gridx4部分的說明,輸入文件里也不要加上grid4 gridx4,否則會報錯)
另外,建議在關鍵詞里加上grid4 gridx4關鍵詞,使用比默認更好的交換相關泛函積分格點和COSX格點。之所以這樣,是因為當前我們希望算的結果比較準,故提升積分格點精度有好處。而且,當前體系大量原子致密堆積,默認積分格點下的積分精度差點意思。比如在def2-TZVP下用默認格點,在SCF收斂后自動做Final grid步驟(提升積分格點精度算最終能量)的時候提示如下
Change in XC energy ... -0.003667837
Integrated number of electrons ... 1144.003054396
Previous integrated no of electrons ... 1143.933959260
可見迭代過程用的積分格點對電子數的積分值1143.9339和整數相差不小。即便在final grid時,積分值1144.0030偏離整數仍不可忽略。反之,如果用grid4 gridx4,則輸出變為
Change in XC energy ... 0.001404285
Integrated number of electrons ... 1144.000217645
Previous integrated no of electrons ... 1144.003114364
可見積分精度改進不小,明顯更接近于整數。提升積分格點精度還有一個好處是對于某些體系可能令SCF收斂更容易。當前這種特征的體系結合大基組本來SCF就很難收斂,顯然多花點耗時來提升SCF收斂的可能性是劃算的。總之,對本文這種類型的體系在算單點的時候,都建議用grid4 gridx4,一舉多得,而對耗時的增加完全可以接受,而且還可能因為達到收斂限所需步數變少而反倒令耗時更低(在def2-TZVP下確實如此,默認格點下用了46輪收斂,grid4 gridx4下用了37輪,導致后者反倒耗時比前者還低了幾分鐘)。
考慮到上面提到的問題,最終我們對復合物用的單點任務的輸入文件的關鍵詞部分如下
! PBE0 D3 grid4 gridx4 def2-QZVP def2/J RIJCOSX noautostart miniprint nopop
%scf DirectResetFreq=1 end
計算用了40輪收斂,在2*E5-2696 v3 36核機子上花了5個小時。注意這類體系本身就是比較難收斂的,所以切勿看到跑了比如30輪左右還沒收斂,而且收斂情況還有些波動就把任務給斷了,一定要沉得住氣。
復合物算完后,我們把complex_SP.inp復制為benene_SP.inp并把其中銀原子都去掉,就成了苯分子的單點任務文件,計算之。再把complex_SP.inp復制為Ag58_SP.inp并把其中苯分子部分去掉,就成了銀團簇的單點任務文件,計算之。算完的同名out文件在本文文件包里都提供了。
2.5 計算結合能
從單點任務輸出文件中提取單點能,按照E_bind = E(Ag58_Benzene) - E(benzene) - E(Ag58)即可計算出結合能,即627.51*(-8760.730415916696+232.068063781533+8528.63
2371853557)=-18.8 kcal/mol。
前面提到,苯吸附過程的實驗焓變是-13 kcal/mol,我們算的和實驗定性吻合(比DFT-D3原文里勢能面掃描圖體現的結果還更好)。由于這個結合能已經算是很大了(苯二聚體結合能才-2.8 kcal/mol),而且這又不是容易算的體系,能算到這種精度就已經不容易了。我們的計算結果和實驗值的差異來自于幾個方面:
(1)邊界效應仍不可忽略,用更大的簇可能能改進結果
(2)PBE0-D3(BJ)理論方法的誤差。可嘗試其它泛函或結合其它的色散校正方式
(3)實驗數據對應焓變,本身和我們算的結合能就缺乏可比性。尤其是,形成復合物后,被吸附物被基底束縛,從而會誕生新的振動模式,這會對復合物的焓產生明顯貢獻(使之變得更正),忽略了這點會導致算出來的結合能偏大(即overbinding、結合能過負)
(4)忽略了從無限遠離到被吸附狀態過程中的分子和金屬表面結構變化對能量的影響。但由于當前的體系比較剛,這基本可以忽略
(5)實驗數據本身就有誤差
有一點非常值得一提,也是絕大多數人都忽略的,也就是DFT-D3的三體校正項,這在《DFT-D色散校正的使用》(http://www.shanxitv.org/210)中專門說過。平時我們用Gaussian、ORCA做計算的時候加D3校正都沒考慮三體校正項,因為這項對結合能的影響通常很小。然而,對于原子數很多,尤其是當前體系這樣原子還是緊密堆積的情況,三體校正項對結合能的影響可能是不容忽視的。
如上面的博文所述,ORCA里寫上ABC關鍵詞就可以在計算D3校正時也考慮三體校正項,但我們前面計算的時候沒寫這個。為了快速得到考慮三體校正項之后的結果,我們用Grimme獨立的dftd3程序來算。把分子坐標弄成xyz格式,運行dftd3 mol.xyz -func pbe0 -bj -abc,輸出的就是mol.xyz里的體系在PBE0泛函下的帶三體校正項的DFT-D3(BJ)色散修正能了。下面的表格里把ORCA輸出文件里的PBE0能量、dftd3給出的帶和不帶三體校正項(ABC)的DFT-D3(BJ)色散校正能都匯總到了一起
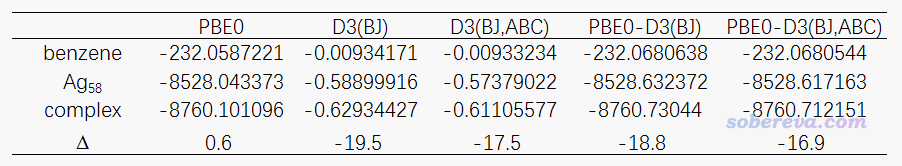
由上表可見,如果不考慮色散校正,結合能是正值,這是因為PBE0幾乎完全不能描述色散作用(這是苯與Ag表面吸引作用的最主要來源),考慮DFT-D3(BJ)校正后才定性正確。如果再把三體校正項考慮進去,算出來的結合能-16.9 kcal/mol比不考慮三體校正項時明顯更接近實驗的吸附過程的焓變,改進了約2 kcal/mol。如果再考慮到把焓的熱校正考慮進去還能進一步改進結果,且實驗數據本來就有誤差,可以認為當前計算模型的精度已經相當不錯了。
我建議大家用ORCA基于簇模型計算表面問題時都帶著ABC關鍵詞。幾何優化、振動分析帶不帶無所謂(PS:三體校正項沒有解析Hessian),關鍵是單點任務時應當帶上,反正對耗時沒有增加,還有可能對結果有不可忽視的改進。
2.6 弱相互作用分析
順帶一提,用簇模型算完之后可以結合Multiwfn做各種各樣的波函數分析。比如《使用Multiwfn圖形化研究弱相互作用》(http://www.shanxitv.org/68)和《使用Multiwfn做NCI分析展現分子內和分子間弱相互作用》(https://www.bilibili.com/video/av71561024)中介紹的NCI分析是非常流行的圖形化考察弱相互作用的方法,這里我們也將之用于苯與銀表面的物理吸附問題上。
是使用單點計算產生的波函數還是幾何優化產生的波函數來做NCI分析?我建議用后者,因為后者是在def2-SV(P)下產生的,前者是在def2-QZVP下產生的,用后者耗時顯然低得多得多,而且NCI分析對于波函數質量并不敏感,基組基本像樣即可。故基于優化任務產生的gbw文件,我們就可以做分析了。如《詳談Multiwfn支持的輸入文件類型、產生方法以及相互轉換》(http://www.shanxitv.org/379)所述,要么用orca_2mkl將gbw轉成molden輸入文件再載入Multiwfn,要么通過設置Multiwfn的settings.ini里的orca_2mklpath使得multiwfn能直接載入gbw文件了。對當前體系做NCI分析值得注意的一點是,為了節約時間,在設置格點時,應當讓空間范圍只涵蓋苯與金屬作用的區域,而不要把整個體系都納入。具體來說,在NCI分析功能設置格點的步驟中應選擇選項10 Set box of grid data visually using a GUI window,然后把盒子范圍調整成下面這樣
雖然當前體系很大,但由于盒子小,在基本能保證圖像精細的0.25 Bohr格點間距下,哪怕用4核機子也能很快就算出來結果。如果想要讓圖像更光滑,可在上圖的界面的右下角把格點間距再改小點,比如0.15 Bohr。下圖是0.19 Bohr時用VMD繪制出來的結果,色彩刻度用的-0.04~0.03 a.u.
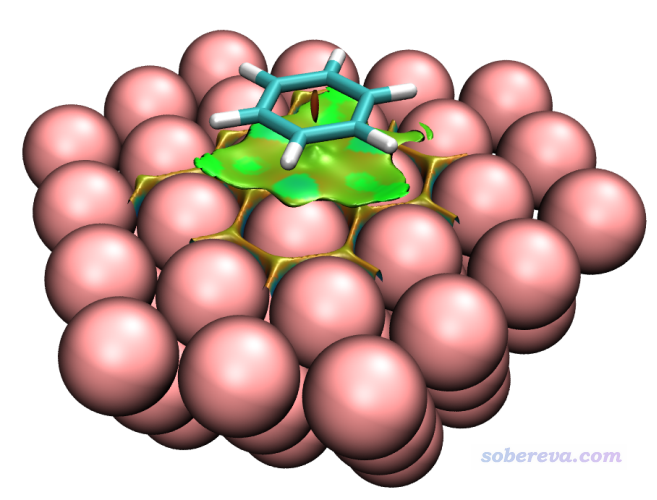
可見苯和銀表面之間有大面積的綠色區域,直觀展現出了苯的pi電子與銀表面電子海洋的大范圍色散相互作用,這有點類似于pi-pi堆積的景象(當然,銀沒有pi電子)。另外,在銀與銀之間也有等值面出現,我們不用管它,這體現的是Ag-Ag之間的作用,如果想屏蔽掉也可以,做法參看《用Multiwfn+VMD做RDG分析時的一些要點和常見問題》(http://www.shanxitv.org/291)。
3 總結&其它
本文通過苯在Ag(111)表面吸附的一個簡單例子,示例了怎么用簇模型研究表面問題,并且針對相關知識、涉及到的一些細節和要點做了討論,希望讀者舉一反三。本文的情況其實還算比較簡單的,不牽扯對邊界進行特殊處理的問題。
像當前這種不易SCF收斂的體系,可能光是在優化階段一開始就已經出現SCF不收斂了(這和復合物初始結構也有關。比如把苯放到距離銀表面2.7埃的距離,SCF就比3.0埃的時候難收斂,因為其更偏離平衡距離)。這種情況可以適度降低SCF收斂限。ORCA對優化默認用tightSCF收斂限比較嚴,如果發現SCF差一點就能收斂,但死活就是達不到收斂限,不妨用scfconv7關鍵詞,即把SCF收斂閾值設成單點任務默認閾值與tightSCF之間,這樣優化出來的結果一般也足夠精確。如果scfconv7都收斂不了,但是用scfconv6能收斂,在優化完了之后建議再用默認的tightSCF進一步優化確保結構準確。如果是初始結構太離譜導致SCF難收斂,自己又不知道大概怎么擺合適,用xtb程序做個預優化也可以,相關信息見《將Gaussian與Grimme的xtb程序聯用搜索過渡態、產生IRC、做振動分析》(http://www.shanxitv.org/421)。
對于單元素團簇類型體系,SCF收斂難度通常是:稀土>d族>ds族>主族。本文的Ag團簇的情況雖然已經不是很容易SCF收斂了,但還不是真正難搞的。諸如Fe團簇等,收斂難度遠高于Ag團簇,而且還有較高可能性出現波函數不穩定等情況,如果是初學者沒經驗的話不建議輕易去用簇模型做這些金屬的表面問題的計算。
值得一提的是,當前體系是C3v對稱性的。如果你用Gaussian來算的話,且在計算之前用gview對稱化為C3v,計算會快非常多,收斂也會比較容易。
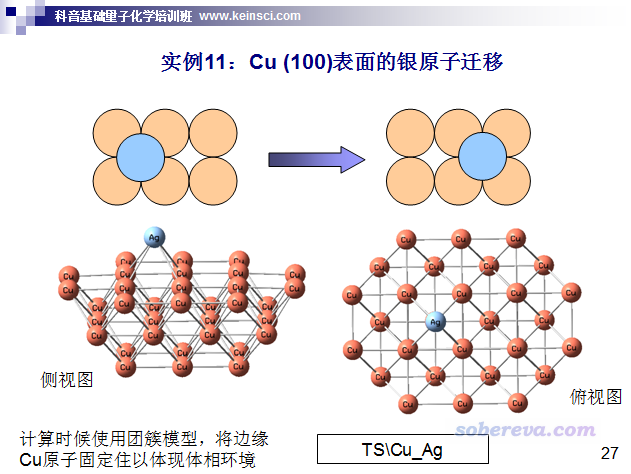
筆者講授的北京科音基礎量子化學培訓班(http://www.keinsci.com/workshop/KBQC_content.html)里的過渡態搜索部分有個實例也是基于簇模型的,如下,大家作為練習可以自行做一下。除了要凍結邊緣原子外,搜索過渡態和走IRC的過程和普通分子體系無異。
簇模型不僅限于研究表面問題。比如計算酶催化反應,整個蛋白酶動輒幾千原子,直接用量子化學算算不動,此時可以把反應位點區域挖出來,即保留底物以及周圍一圈跟它發生反應或者有顯著弱相互作用的殘基的側鏈和alpha碳,并且把alpha碳凍結并加氫飽和。這種研究方法可以參看這篇綜述:J. Am. Chem. Soc., 139, 6780 (2017)。筆者在北京科音高級量子化學培訓班里會講這種計算的具體細節。雖然也有很多人拿ONIOM(QM:MM)算這種問題,但補力場參數相當麻煩,好多任務和分析都做不了、容易出現亂七八糟問題,非常折騰,從結果上看一般也不比用簇模型有多大好處,所以沒有特殊情況的話我很不建議用ONIOM(QM:MM)。筆者專門有一篇文章說這事:《要善用簇模型,不要盲目用ONIOM算蛋白質-小分子相互作用問題》(http://www.shanxitv.org/597)。