要善用簇模型,不要盲目用ONIOM算蛋白質-小分子相互作用問題
要善用簇模型,不要盲目用ONIOM算蛋白質-小分子相互作用問題
文/Sobereva@北京科音 2021-May-22
初學者亂用、濫用Morokuma當年提出的ONIOM方法是一個長久以來就有的問題,我在網上答疑時經常強調初學者不要老想著用ONIOM。我主張ONIOM能不用就不用,因為麻煩事和細節問題太多,尤其是許多初學者連基本的計算都做不利落,還用ONIOM純粹是自討苦吃、自找麻煩。這兩天在思想家公社QQ群答疑時連著碰到兩個對量子化學計算一知半解的人問ONIOM的事,令我再次感到有的初學者但凡聽說過點ONIOM的東西,若他們的研究涉及到酶催化、蛋白質-配體相互作用之類的問題,就會言必曰ONIOM。他們對ONIOM的實用價值和使用的必要性的認識真是有著極大的誤區。在沒必要用ONIOM的時候非要用,往往會導致初學者窮折騰、白浪費大量時間精力,最終還可能結果不理想,甚至被誤導。筆者在此實在忍不住寫個小文發表一下我對于初學者用ONIOM算蛋白-小分子體系的個人觀點。此文主要是對初學者來說的,如果你用ONIOM都已經經驗豐富,那就沒必要看此文了。本文并不去詳細具體講ONIOM的原理和在Gaussian中的使用,這部分內容我在北京科音基礎量子化學培訓班(http://www.keinsci.com/workshop/KBQC_content.html)里會用100多頁ppt專門做全面、系統的講解并配以實例。
簡單來說,ONIOM是把兩個,甚至三個不同的計算級別組合到一起的一種普適的方法,此文主要只涉及前種情況。比如ONIOM(QM:MM)就是把量子化學方法(QM)和分子力學(MM)結合起來,這也被叫做IMOMM。這種計算常被用于研究蛋白質與小分子的相互作用上,其中把需要較好精度描述的部分,或者普通分子力場描述不了的涉及成鍵斷鍵的區域,用相對昂貴的量子化學方法描述。這部分叫做high layer (HL),一般包括小分子以及與之近鄰的氨基酸殘基。而與小分子距離更遠的相對來說不重要的區域,則使用耗時比QM低幾個數量級的MM來描述,這部分叫做low layer (LL)。HL和LL的并集就是整個體系。注意ONIOM(QM:MM)和常說的QM/MM并不相同,一個關鍵區別在于QM/MM相當于把QM和MM分別描述的兩塊區域以恰當的方式拼接起來,因此MM計算的時候不會涉及到含有小分子的high layer部分。而ONIOM(QM:MM)能量的計算方式是E(QM,HL)+E(MM,整體)-E(MM,HL),因此需要算三次能量,可見也涉及到通過MM算配體部分(因為配體在HL里)。
ONIOM的關鍵意義在于可以讓算不動的體系算得動。比如整個蛋白酶可能含有幾千個原子,用當下極好的機子,靠一般的量子化學程序以常規方式(不牽扯諸如分塊求解),在常用的DFT方法結合像樣基組的級別下算單點都算不動。而用ONIOM的話,可以把尺寸有限的最關鍵區域取為high layer用DFT描述,而其余部分靠極度廉價的MM描述,顯然這樣就算得動了。
看似ONIOM很美好,似乎不用就虧了似的,而我卻對初學者主張能不用ONIOM就不用ONIOM,自然是因為其弊端太多了,舉幾個相對來說明顯的問題:
(1)需要補力場參數:算蛋白-小分子相互作用問題時,ONIOM用得較多的是ONIOM(QM:MM)形式,由于牽扯到力場計算,往往就必須補力場參數,這是特別麻煩、勞神的事情。由于涉及到蛋白質,通常力場用的是Gaussian內置的AMBER力場,然而AMBER力場局限性很大,只適合生物分子以及非常普通的有機分子,且支持的元素很有限,如果體系里有個過渡金屬、硼之類不支持的元素,補參數那可就極為折騰、痛苦了。而且AMBER力場直接包含的成鍵項參數也只有成鍵方式很常見的情況,而算雜七雜八有機配體小分子的時候經常會提示缺bond、angle、dihedral等參數沒法跑,于是你還得借助比如AmberTools里的Antechamber或者其它什么工具試圖從GAFF等力場里弄來參數,有的時候還得靠Google搜參數、想辦法借用參數等,甚至有時必須自己擬合參數,真是又花時間,又需要經驗。初學者往往缺乏分子模擬方面與力場有關的知識,很可能補參數的時候把函數形式弄錯了、把單位弄錯了之類犯一些低級錯誤,導致計算用了錯誤的參數得到不合理的結果。初學者還很容易在輸入文件里自定義力場參數的地方格式寫不對,反反復復來來回回跟報錯作斗爭又耽誤大量時間。Gaussian官方的exploring那本書第3版里有ONIOM(QM:MM)的一個例子,就那么區區一個例子,在準備工作方面就花了多達好幾十頁講,正體現出ONIOM的麻煩程度有多高。初學者要是早知道同樣的問題其實靠簇模型就能容易地可靠地研究,估計早沒耐性折騰ONIOM了。
(2)需要設置原子電荷:Gaussian里的分子力場通過原子電荷來計算原子間靜電相互作用,所以做ONIOM(QM:MM)計算必須給原子設置原子電荷,而這也是麻煩事。對于蛋白質中的標準殘基,GaussView保存ONIOM輸入文件的時候直接就會賦予AMBER力場分子庫里預置的原子電荷,但是碰上非標準殘基,就得自己額外算原子電荷才行(比如構造模型體系,然后在Multiwfn里恰當施加約束算RESP電荷,見http://www.shanxitv.org/441)。順帶一提,有人以為在力場名后頭寫個=QEq讓Gaussian自動算QEq原子電荷就完事了,并不麻煩,這其實是大錯特錯,因為這種電荷對靜電相互作用描述極其垃圾,看《原子電荷計算方法的對比》(http://www.whxb.pku.edu.cn/CN/abstract/abstract27818.shtml)里面關于靜電勢重現性的討論,另外在此文也著重說了:《基于背景電荷計算分子在晶體環境中的吸收光譜》(http://www.shanxitv.org/579)。
(3)能用的理論方法有限:支持ONIOM的程序很少,截止到撰文時只有Gaussian、Q-Chem(僅支持力學嵌入)、MRCC(只支持QM和QM組合、力學嵌入)和ADF支持。因此諸如想在ORCA程序里開RIJCOSX做ωB97M-V泛函的ONIOM計算都無法直接實現,而這是非常有用的計算級別。
(4)計算控制不靈活:Gaussian里用ONIOM(QM:MM)的時候一些常規關鍵詞都不再兼容,比如opt里的柔性掃描設置、幫助收斂的許多設置等等,這是因為MM部分的優化默認用專門的代碼。雖然opt里加上nomicro后可以用常規的opt選項來控制優化和柔性掃描,但是速度比默認情況慢得多。
(5)幾何優化收斂問題:用ONIOM(QM:MM)的時候比起常規的計算幾何優化往往更難收斂。
(6)用隱式溶劑模型時慢:Gaussian里用ONIOM(QM:MM)的時候一旦開啟了隱式溶劑模型,速度會非常慢,因為原子數非常多的MM部分也要在溶劑模型計算時予以考慮,所以這部分耗時就不再是普通力場計算的耗時級別了。
由于上述問題,ONIOM遠沒有缺乏實踐經驗的一些初學者們想象的好用。
那么,不用ONIOM的話怎么算蛋白質-小分子相互作用類型的問題呢?答案是:簇模型(cluster model)。簇模型簡單來說就是把原本很大或者周期性的體系的關鍵部分挖出來當做一個孤立體系進行計算。之前筆者還寫過《使用量子化學程序基于簇模型計算金屬表面吸附問題》(http://www.shanxitv.org/540),也是簇模型的典型應用。其實簇模型算蛋白質-小分子作用差不多就等于把ONIOM的high layer拿出來單獨做量化計算,但是需要自己手動對邊界進行處理,如截斷化學鍵的地方通常需加氫進行飽和避免存在懸鍵。并且計算的時候要把邊界的非氫原子凍結,避免幾何優化的時候截出來的簇嚴重坍塌、變形,失去在完整體系中的形態。
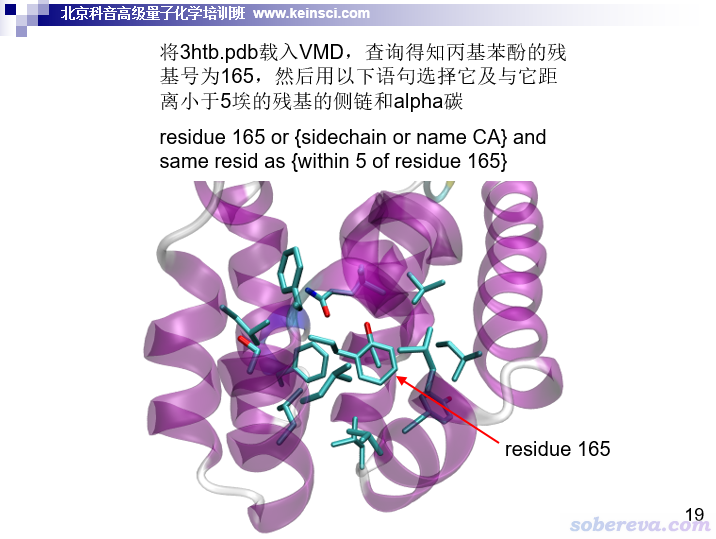
從已有的蛋白質-小分子復合物中挖團簇可以用VMD程序靠選擇語句把距離小分子空間距離較近的一大片都保存成結構文件,需要用到比如same resid as {within 8 of resname MOL}的選擇方式,參見《VMD里原子選擇語句的語法和例子》(http://www.shanxitv.org/504)。然后,再用GaussView打開VMD保存出來的粗略結構人工做進一步處理,比如恰當地加氫(注意恰當考慮質子化狀態),刪除除了alpha碳以外的蛋白質骨架原子(但并非總是要這樣做,應根據實際情況隨機應變,如骨架原子和小分子有明顯作用時就不能刪除),根據化學直覺恰當刪除一些不是必須保留的、和分子相互作用不密切的殘基來減少總原子數等等。如果你有個較好的雙路服務器,把原子數控制到三百個以內,用普通泛函結合中等大小基組做優化、找過渡態什么的就都能算得動了。模型構建好后,就可以用Gaussian等量子化學程序照常計算了,除了要凍結邊界原子外和計算普通體系無異,Gaussian里凍結原子的方式在此文說了:《在Gaussian中做限制性優化的方法》(http://www.shanxitv.org/404)。當然了,做耗時較高的DFT優化之前,可以先用比如Gaussian支持的PM6-D3,或者結合xtb程序在GFN-xTB級別做預優化,參見《將Gaussian與Grimme的xtb程序聯用搜索過渡態、產生IRC、做振動分析》(http://www.shanxitv.org/421)。
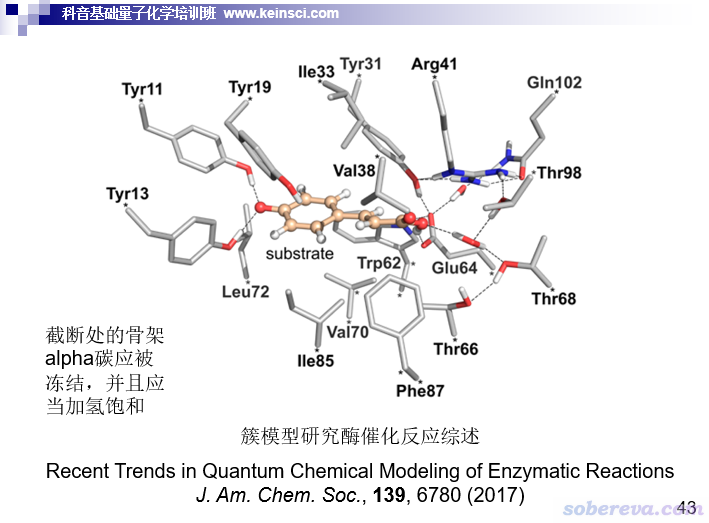
為了讓大家對簇模型有一個直觀的印象,這里放我培訓班里的一頁ppt
另一頁ppt,體現了VMD里如何恰當選取
另外,為了節約簇模型的計算時間,建議把距離小分子相對較遠的蛋白質原子用較小的基組。很不重要、與相互作用區域或反應位點很遠的原子甚至用3-21G這種垃圾基組都可以接受。這即是使用混合基組,見《詳解Gaussian中混合基組、自定義基組和贗勢基組的輸入》(http://www.shanxitv.org/60)。
簇模型計算用的結構和ONIOM的high layer的選取標準差不多。取多大是個重要問題,取大了算不動,取小了可能導致一些不可忽略的殘基沒考慮,這就必須結合實際計算條件、體系特點,根據理論化學直覺考量,沒經驗的話可以多參考一些文獻里的模型,像上面ppt里的簇就比較合適,明顯可見與小分子相互作用的殘基都被考慮了,非常穩妥。如果擔心考慮的殘基不夠充分,又不想讓計算量明顯更大而擴大體系,還是按前面說的,一定要善用混合基組來節約時間,恰當用混合基組就可以在同樣的計算量下使用更大的簇、令結果更穩妥、減少在模型構建時候的糾結。
順帶一提,以前看到有初學者居然用ONIOM來試圖實現不重要的區域用小基組,比如ONIOM(B3LYP/6-31G*:B3LYP/3-21G),這純粹就是胡搞瞎搞。這么做并不比用混合基組耗時低,而且會令結果精度更低(ONIOM分層的做法絕不是沒有代價的,這相當于在層間相互作用描述上引入了近似,尤其是對于需要截斷化學鍵的情況來說。好好了解一下ONIOM的原理、連接原子是怎么回事自然就清楚)。
算蛋白質-小分子作用問題,凡是用簇模型就很適合的情況,我總是建議用簇模型非ONIOM,因為簇模型沒有前述ONIOM存在的弊端,用著省心。相比ONIOM(QM:MM),簇模型的好處很明顯:不需要麻煩地補任何力場參數,不涉及計算原子電荷,計算時候和計算普通體系沒有任何差異(各種控制計算的關鍵詞都能照常用、各種任務都能照常做),任何量子化學程序都能算簇模型,用隱式溶劑模型也不至于令耗時暴增。
也要強調的是,ONIOM相比簇模型,絕對不是沒有任何用處。需要用ONIOM而不適合簇模型的情況有二
(1)與小分子有密切作用的殘基太多(比如小分子較大的情況就很常見),而且在保證結果合理的前提下把模型簡化到極限、也考慮恰當用混合基組來節約時間,但還是算不動,那這時候就得靠ONIOM了,可以把一些相對次要但又不能完全舍棄的殘基通過MM來描述。
(2)研究的過程(與小分子反應、結合等)可能引起蛋白質較大范圍區域結構的不可忽視的變化,這個時候顯然就不能單純用簇模型只考慮與小分子近鄰的殘基而且還把其骨架原子凍住了,而必須得通過力場把其它部分的蛋白質殘基也都明確表現出來。
隨著計算能力的提升,實際上ONIOM的用處是越來越小的。早年間,在DFT普遍也就算得動區區幾十個原子的“貧窮”的時代,顯然極難構造出能用的簇模型來合理描述蛋白質-小分子反應區域或結合位點,所以那個時代算這類問題ONIOM算是標準方法,不得不用。而如今,一萬多塊錢買個廉價服務器(如幾十核的雙路XEON E5 v3),拿DFT算二、三百個原子都沒太大壓力,大多數蛋白質-小分子作用的情況靠合理構建的簇模型都算得動,還非要屈辱地用ONIOM作甚,這不是自虐么?隨著計算能力進一步發展,以后DFT算>500個原子早晚也會沒有壓力,到時候由于上面(1)的情況而必須用ONIOM的情況基本就完全消失了。
再順帶一提,很多早年間的ONIOM計算在現在來看基本就屬于搞笑了。比如下面的ppt給了一個老文章里的例子
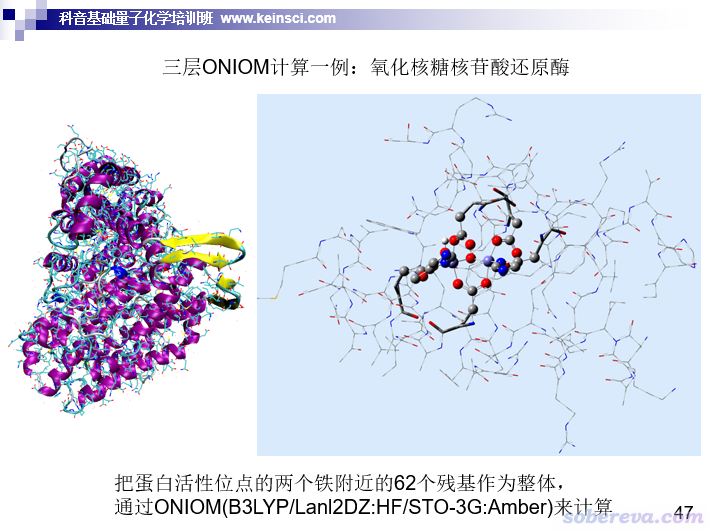
這個例子單純是為了展示3層ONIOM是怎么回事,如今再用這么low的量化級別做計算,文章鐵定發不出去。而且此例分成三層在如今來看沒有任何意義,屬于瞎折騰,因為現在直接用DFT結合中等基組別說把此例的high layer和medium layer放一起算都能算得飛快,甚至再把與medium layer相鄰的一圈lower layer的殘基都納入然后作為簇模型來算也沒有壓力。所以,現在做蛋白質-小分子計算的新人,不要凈參考一些老文章,那時候ONIOM的意義、ONIOM的使用方式對如今來說大多沒有什么參考意義,盲目效仿只會自取其辱。PS:上例也是我強烈不推薦用ONIOM的情況,因為里面有鐵,補參數可費勁了!