使用Multiwfn做aNCI分析圖形化考察動態過程中的蛋白-配體間的相互作用
使用Multiwfn做aNCI分析圖形化考察動態過程中的蛋白-配體間的相互作用
文/Sobereva@北京科音
First release: 2021-Mar-4 Last update: 2022-Feb-22
1 前言
《使用Multiwfn圖形化研究弱相互作用》(http://www.shanxitv.org/68)介紹的NCI方法是如今用得非常普遍、廣為流行的圖形化研究弱相互作用的方法,后來我又在《使用Multiwfn研究分子動力學中的弱相互作用》(http://www.shanxitv.org/186)中介紹了averaged NCI(aNCI)方法,可以把動態過程中的平均化的相互作用予以直觀展現。aNCI對于研究蛋白和配體間的相互作用非常有價值,但那篇文章里我沒給出具體例子,估計有用戶實現起來會遇到一些技術上的困難,不過也有不少用戶已經很好地用Multiwfn做了這種分析討論了蛋白-配體結合問題,比如PLoS ONE, 13, e0196651 (2018)。本文的目的是詳細演示如何在Multiwfn中做aNCI分析來清晰直觀地展現蛋白和配體之間的弱相互作用,讓所有用戶都可以順利地運用這種分析研究實際問題。aNCI的原理在本文就不再累述了,請先仔細閱讀《使用Multiwfn研究分子動力學中的弱相互作用》了解相關背景知識。并且非常推薦閱讀《一篇最全面介紹各種弱相互作用可視化分析方法的文章已發表!》(http://www.shanxitv.org/667)介紹的筆者的綜述文章,其中含有對aNCI的詳細介紹并給出了典型的應用例子。
PS:雖然ligplot和ProteinPlus中的PoseView等程序也能以二維圖的方式展現配體與蛋白各殘基的相互作用,但那種展現方式過于抽象,不能體現三維空間關系,而且也不能體現實際動態環境的情況,局限性極大。用aNCI則嚴格得多、給出的信息豐富得多。
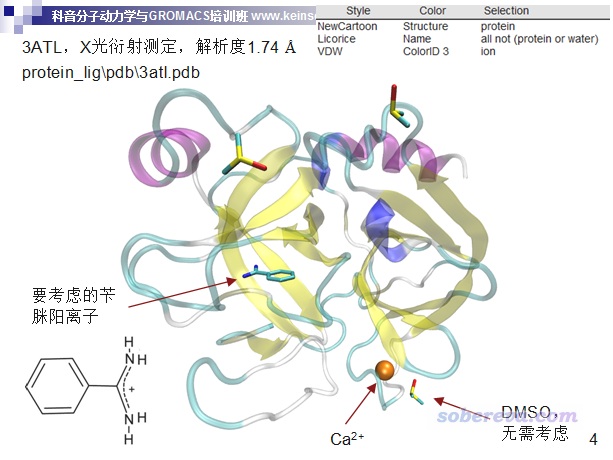
讀者請使用2021年3月4日及以后更新的Multiwfn版本,否則有的地方和本文所述不符。Multiwfn可以在http://www.shanxitv.org/multiwfn免費下載。本文用的VMD是1.9.3版,可以在http://www.ks.uiuc.edu/Research/vmd/免費下載。本文的例子是北京科音分子動力學與GROMACS培訓班(http://www.keinsci.com/workshop/KGMX_content.html)中我講蛋白質-配體復合物模擬的3ATL那個例子,此體系結構如下
對這類蛋白-配體復合物體系做aNCI分析總流程是:
(1)將配體位置凍結,做一段動力學模擬
(2)將配體連同與之作用的殘基保存成xyz軌跡
(3)用Multiwfn計算aNCI的格點數據
(4)對格點數據進行適當屏蔽以得到最佳的效果
下面依次介紹這些步驟怎么做。涉及到動力學程序的部分我用的是GROMACS,其它動力學程序的用戶也可以以類似的方式實現。本文用到的所有文件都可以在這里下載(cub文件太大就不提供了):http://www.shanxitv.org/attach/591/file.rar。
2 動力學模擬
對于aNCI分析蛋白-配體相互作用,我建議先做NPT模擬,使得體系達到充分平衡的狀態,然后再做NVT模擬,此時將配體的坐標凍結。凍結配體很重要,如果在動力學過程中允許配體亂動的話,aNCI圖可能會變得一團糟。注意如果在真實環境中,配體就是在多種構象之間反復切換,那么正確的考慮方式是對NPT軌跡的配體部分做簇分析,取容量較大的幾個簇各自的最有代表性的結構,然后凍結配體再做動力學和aNCI分析,也即aNCI分析是對配體處于不同典型的構象分別來做、分別討論的。本文的例子的配體是芐脒陽離子,并沒有多種構象的問題。
跑3ATL這個體系的凍結配體坐標的NVT動力學用的tpr和mdp文件,以及跑出來的xtc和gro文件都可以在本文的文件包里下載。為避免軌跡太大不好下載,這里給出的軌跡md_fix_nowat.xtc以及對應的最終結構的md_fix_nowat.gro是去水之后的。md_fix.mdp是這個過程用的mdp文件,可見里面有freezegrps = MOL和freezedim = Y Y Y,這是用來將配體(MOL殘基)固定用的。這個NVT動力學跑了1ns,每500步(1ps)保存一次軌跡,一共1001幀。通常來說有1000幀足夠得到能說明問題的aNCI圖了。注意aNCI分析耗時是和考慮的幀數成正比的。
3 獲得用于aNCI分析的xyz軌跡文件
注意絕對不要直接把整個蛋白+配體體系給Multiwfn用于aNCI分析,這樣會耗時極高,幾乎根本算不動。正確做法是只把配體以及與配體距離較近的殘基原子保存為xyz軌跡文件。
啟動VMD,載入md_fix_nowat.gro,刪除僅有的一幀,然后再載入md_fix_nowat.xtc。然后我們測試一下看看用什么樣的VMD選擇語句可以選中合適的區域,不了解語法的話看《VMD里原子選擇語句的語法和例子》(http://www.shanxitv.org/504)。把軌跡切換到第0幀,然后在Graphics - Representation里把選擇范圍設置為resname MOL or protein same resid as within 3.5 of resname MOL,這樣就把配體以及有任何一個原子在距離配體3.5埃范圍以內的殘基都選中。把顯示方式設為CPK,此時的圖像如下(我把配體加了個透明的表面以突出顯示)
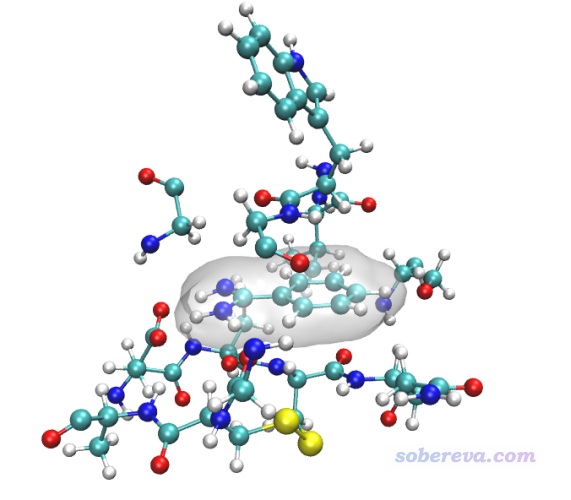
可見當前用的選擇語句很合適,如果發現不合適的話可以再調節within后面的值。現在選擇File - Save coordinate,selected atoms填resname MOL or protein same resid as within 3.5 of resname MOL,保存的文件類型選xyz,然后點Save按鈕,將這個局部區域保存為cluster.xyz。
有一個重點需要注意,aNCI分析必須知道各個原子對應什么元素,否則沒法構建準分子密度。因此,用于aNCI分析的xyz軌跡文件里應該包含各個原子的元素名(規范的xyz文件都應當如此),否則Multiwfn會根據原子名去猜,這樣很容易猜錯,比如CA就到底是鈣還是alpha碳就沒法區分。由于md_fix_nowat.gro里記錄的是GROMACS跑動力學時候的原子名,而此格式并不記錄元素信息,所以像上面這樣保存出的cluster.xyz里也是用的原子名。那么怎么讓Multiwfn能正確獲得元素信息呢?一個做法是手動把cluster.xyz里的原子名都替換成元素名,對于水盒子這種簡單的情況還好辦,原子名就三種因此就替換三次就可以,而對于當前體系顯然這么替換太費勁了。因此Multiwfn設計了一個特殊規則,也就是如果當前目錄下如果有和載入的xyz文件名相同但后綴是pdb的文件,就會優先從pdb文件記錄元素的那一列里讀取元素名。不過如果pdb里對某些原子沒有提供元素名,對這些原子Multiwfn仍會根據xyz文件里的原子名去猜,原子名里如果有數字會被事先自動去掉。所以,現在要做的是產生一個對應局部區域的含有元素信息的pdb文件,具體做法如下。
運行gmx editconf -f md_fix.tpr -o md_fix.pdb,將當前動力學的tpr文件轉化成pdb文件。因為tpr里有蛋白的元素信息,所以得到的pdb文件的蛋白部分也有元素信息,即新產生的md_fix.pdb的最后一列。但打開md_fix.pdb并找到MOL殘基的位置,會看到這樣得到的pdb中的配體部分是沒有元素信息的。不過對于此例這倒不是問題,因為cluster.xyz里配體部分的原子名都是比如C1、N8、H13這樣,除去數字后的字符和元素名直接相符,所以此例既不需要在md_fix.pdb里把配體部分的元素名再手動補上,也不需要把cluster.xyz里的配體的原子名都改為元素名(但對于其它體系的情況,二者最好改其一以求穩妥)。
注:順帶在這里說一下什么情況pdb是記錄元素信息的。在[ atomtypes ]里,如果原子類型名后面記錄了元素在周期表里的序號,那么tpr里使用這種原子類型的原子就是有元素信息的,因而轉成pdb后也有元素信息。GROMACS自帶的蛋白質力場的ffnonbonded.itp中的[ atomtypes ]里都定義了元素序號,因此轉出來的pdb里蛋白質部分總是有元素的。而對于小分子部分,就看你用的拓撲文件產生工具在拓撲文件里給的[ atomtypes ]里是否包含元素序號了,和程序有關,沒給的話也可以自己手動添加。
用VMD打開md_fix.pdb,然后選擇保存文件,選擇的范圍還是resname MOL or protein same resid as within 3.5 of resname MOL。這樣保存出的文件是本文文件包里的cluster.pdb,一共161個原子。
強調一下,前面保存cluster.xyz的時候我提了一句“把軌跡切換到第0幀”,千萬別漏了這個。因為不同結構下,用上述選擇語句選擇的原子序號、原子數可能是不同的。按照上述操作,cluster.xyz里記錄的原子是根據第0幀結構按照選擇語句判斷的那些原子,這些原子和cluster.pdb里是相同的,因為cluster.pdb的結構來自md_fix.tpr,這里面的坐標正是軌跡第0幀的。
4 計算aNCI格點數據
把cluster.xyz和cluster.pdb放到相同目錄下,然后啟動Multiwfn,輸入cluster.xyz的路徑,程序載入cluster.xyz里的坐標時也會把cluster.pdb里的元素信息載入。然后可以留意一下載入文件后Multiwfn在屏幕上輸出的化學組成信息,當前為Formula: H72 C53 N16 O18 S2,明顯是合理的,沒有不該出現的元素。也可以進入主功能0看一眼當前的結構有無異常的地方(此時顯示的結構是cluster.xyz里第一幀結構)。檢查沒問題后,就接著輸入
20 //圖形化分析弱相互作用
3 //aNCI分析
1,1001 //考慮的軌跡幀號范圍,從1到1001幀(注意Multiwfn的幀號從0開始計)
11 //設置格點。這里選的模式11專門適合aNCI分析,即選擇一批原子,然后往周圍擴展一定距離定義盒子范圍
144-161 //配體的原子序號(打開cluster.pdb,可見MOL殘基第一個原子和最后一個原子的原子序號分別是144和161)
3 A //往配體周圍擴展3埃定義盒子
0.15 //格點間距。數值越小要算的點數越多,耗時越高,而aNCI等值面會越光滑
然后程序就開始計算了。aNCI的耗時是不低的,嫌慢可以找個比較好的服務器算,這部分代碼做了充分并行化。在筆者的普通4核機子上經過不到半小時算完,而如果用36核服務器,3分鐘就能算完。
之后選6,aRDG(平均的約化密度梯度)格點數據會被導出到當前目錄下的avgRDG.cub,平均的sign(lambda2)rho會被導出到當前目錄下的avgsl2r.cub。如果還想繪制熱波動指數(Thermal fluctuation index, TFI)著色的aRDG等值面圖,這里再選7來再計算TFI,之后TFI格點數據會被輸出到當前目錄下的thermflu.cub。
5 繪制aNCI圖
把avgRDG.cub和avgsl2r.cub以及Multiwfn目錄下的examples\aNCI\avgRDG.vmd腳本挪到VMD目錄下。然后啟動VMD,在文本窗口里輸入source avgRDG.vmd執行此腳本繪制aNCI圖,當前圖像如下所示
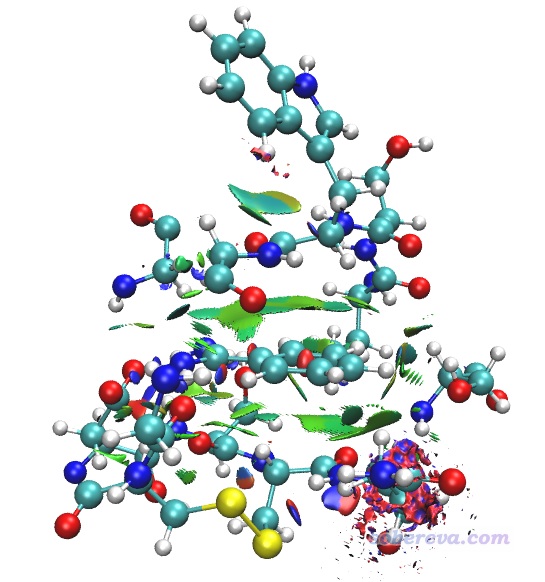
當前圖像已經不錯地展現了配體和周圍殘基的相互作用,但其它地方有很多零碎的多余的等值面,特別是上圖右下角那一坨很難看,所以需要對aRDG的格點數據進行屏蔽。在Multiwfn中,可以實現將距離某個片段較遠區域的aRDG格點數據的數值設為一個比較大的值(比如100),之后再繪制等aRDG值面圖的時候那部分區域就不會出現等值面了,下面就來這么處理一下。
這里先把原先avgRDG.cub改名為avgRDG_org.cub,然后啟動Multiwfn,載入avgRDG_org.cub,輸入
13 //處理格點數據
13 //設置遠離或接近某些原子的格點的數值
1.6 //設置原子范德華半徑乘的倍數,這個值可以反復試試直到效果滿意
100 //滿足條件的格點的數值設為100
2 //手動輸入原子序號
144-161 //配體的原子序號
0 //將當前的格點數據導出
avgRDG.cub //新的格點數據的文件名
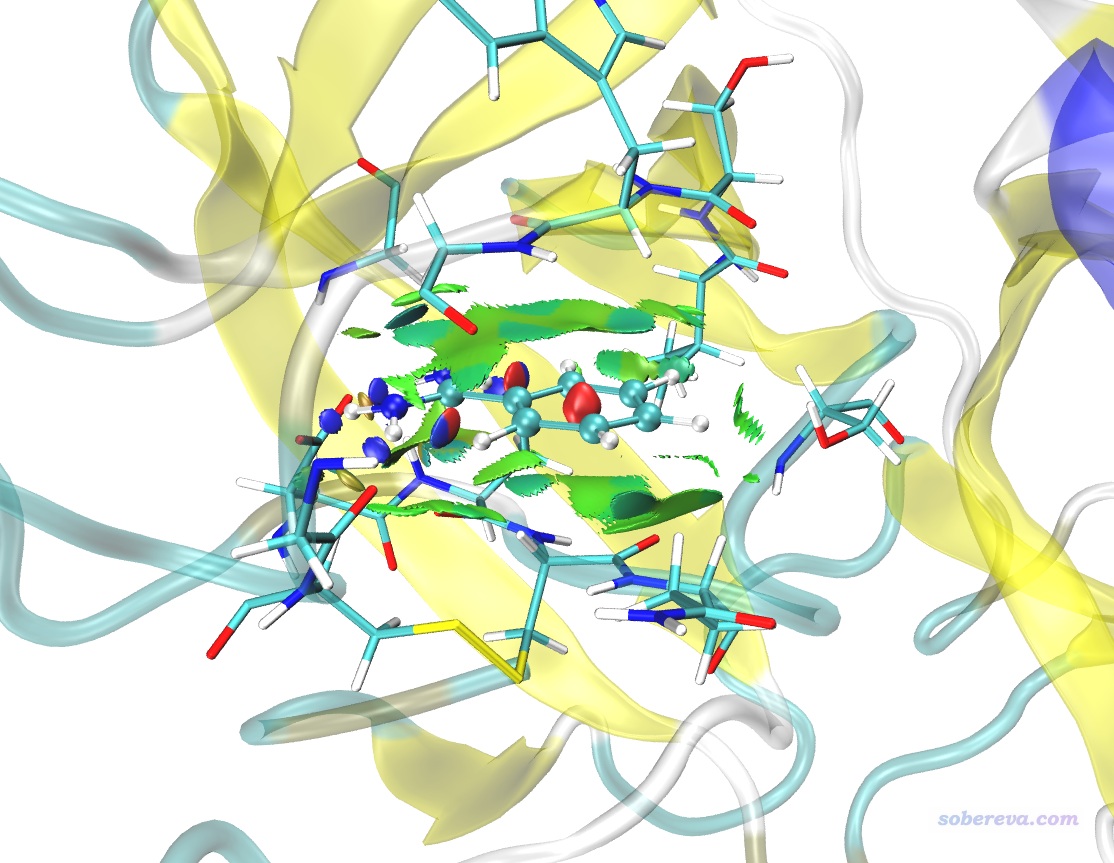
之后將avgRDG.cub挪到VMD目錄下替換掉原先的,再次用avgRDG.vmd腳本繪圖。之后再做一些適當調節,在Graphics - Representation里把顯示等值面的那個rep的isovalue稍微設大到0.3使得等值面更膨脹豐滿一些,以令邊緣的鋸齒減弱。然后把顯示當前幾何結構的rep的選擇范圍設為serial 144 to 161,把Sphere Scale設小為0.7。然后點Create Rep按鈕新建一個Rep,把選擇范圍設為not serial 144 to 161,Drawing Method設為licorice,bond radius設0.1,Material設Transparent。此時的圖像效果如下。
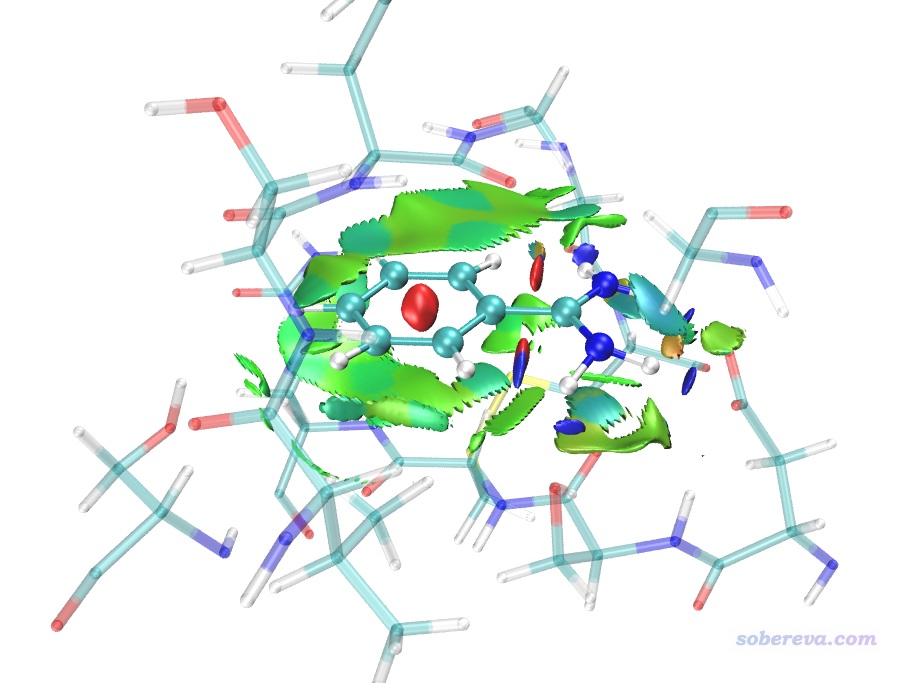
上圖的效果已經挺好了。大面積的綠色等值面展現出了苯環區域與周圍殘基形成的廣闊的范德華作用。配體的每個氨基氫與與之接觸的殘基間都有藍色等值面,體現出了與周圍殘基的較強的氫鍵作用。注:上圖中在配體的C-H和N-H之間有一塊藍色區域,切勿把這個解釋成分子內的強吸引作用。C-H和N-H之間并沒強烈的吸引作用,實際上這部分只是范德華作用而已,由于aNCI是基于準分子密度近似的,所以有些弱相互作用區域的電子密度不夠真實,可能偏高(沒體現Pauli互斥導致作用區域密度的下降)。
avgRDG.vmd腳本用的色彩刻度是-0.025到-0.025,顏色變化是藍-綠-紅。其默認的色彩刻度范圍比較窄,這令某些很弱作用的區域的等值面的顏色仍顯得發青。想避免的話可以自行把腳本里的-0.025和0.025分別改為-0.04和0.04,也可以在Graphics - Representation里選中顯示等值面的那個rep,然后點Trajectory標簽頁,手動輸入色彩刻度下限和上限為-0.04和0.04。發文章時圖中應附上的色彩刻度條,可以直接用Multiwfn自帶的examples\RGB_bar.png文件。
還可以把md_fix_nowat.gro一同載入VMD,用New Cartoon方式顯示其中蛋白質骨架結構,用Secondary structure著色,材質用Transparent,而之前的與配體作用的殘基材質改為AOEdgy。此時圖像如下,效果非常理想
下面我們再來繪制一下熱波動指數TFI著色的aRDG圖。啟動VMD,在文本窗口里輸入source avgRDG_TFI.vmd,然后對顯示方式稍作調節,即可看到下圖(這里對配體用了AOEdgy材質,可以令輪廓比較清楚)
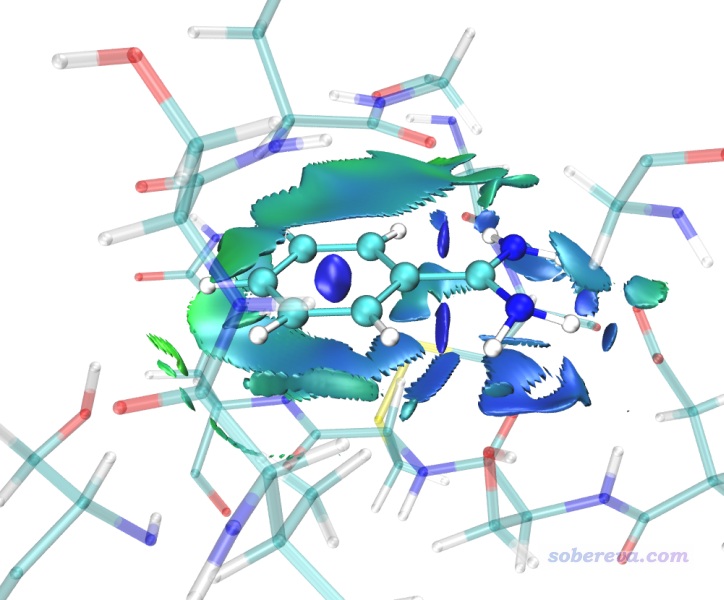
此圖中越藍的地方說明相互作用越穩定,在動力學過程中波動程度越低。由于芐脒陽離子在當前的蛋白環境中很穩定,所以大部分等值面都是藍色的,尤其是上圖右側兩個氨基附近的等值面都是藍色的,體現出氫鍵穩定程度非常高。在上圖左側等值面顏色發綠,體現出這部分作用的穩定性相對差一些。
6 總結&其它
本文演示了如何利用GROMACS+Multiwfn+VMD繪制蛋白-配體相互作用的aNCI圖。這種圖非常清楚直觀生動地展現了蛋白質和配體間的相互作用,能令這類問題分析的文章增色不少,鼓勵大家在實際中使用。這種做法也可以用于研究分子在其它受限環境中與外界的相互作用。
筆者之前在《通過獨立梯度模型(IGM)考察分子間弱相互作用》(http://www.shanxitv.org/407)里還介紹過IGM分析,比起NCI分析的好處是可以自定義片段,只讓片段間的相互作用顯示出來,就免去了做格點數據屏蔽的步驟,而且哪怕用比aNCI明顯更大的格點間距,等值面邊緣的鋸齒也很不明顯。筆者在aNCI的思想上提出了aIGM,即平均的IGM,專用于分析動態環境中的弱相互作用,此分析將另文說明。如果想現在就用的話,使用Multiwfn主功能20里的12 Averaged IGM analysis (aIGM)即可,介紹見Multiwfn手冊3.23.9節,使用此功能發文章時除了引用Multiwfn程序原文外也應引用的手冊這一節。