使用AdNDP方法以及ELF/LOL、多中心鍵級研究多中心鍵
后記:Multiwfn從3.4版開始支持了軌道定域化功能,如果沒有離域范圍很大的AdNDP軌道時可以直接代替AdNDP分析,結果基本相同,但用起來方便得多,詳見《Multiwfn的軌道定域化功能的使用以及與NBO、AdNDP分析的對比》(http://sobereva.com/380)。
使用AdNDP方法以及ELF/LOL、多中心鍵級研究多中心鍵
文/Sobereva @北京科音
First release: 2012-May-8 Last update: 2023-Apr-14
1 前言
Adaptive natural density partitioning(AdNDP,適應性自然密度劃分)是2008年由Zubarev和Boldyrev提出來的一種分析多中心鍵的方法,原文見PCCP,10,5207。AdNDP已經被用于不少體系,目前已有不少相關文章發表,比如原文中研究了硼團簇,J.Org.Chem.,73,9251研究了平面的芳香烴,JPCA,113,866研究了金團簇,Annu.Rep.Prog.Chem. C,107,124研究了很多多核過渡金屬化合物。我國李思殿等人也用AdNDP研究了不少團簇。
筆者開發的波函數分析程序Multiwfn中的主功能14可以實現AdNDP分析,操作非常方便,十分靈活,所得軌道還可以直接可視化、計算出軌道能量、分析軌道成份,比AdNDP原作者在其主頁上提供的AdNDP程序強大得多。如今已經有大量使用Multiwfn做AdNDP分析的文章發表。Multiwfn可以在其主頁http://sobereva.com/multiwfn上免費下載。希望通過本文,能令讀者了解到AdNDP的基本原理、分析流程和在Multiwfn中的使用方法。不了解Multiwfn的讀者強烈建議參看《Multiwfn FAQ》(http://sobereva.com/452)了解基礎知識。
由于ELF/LOL和多中心鍵級也都是重要的研究多中心鍵的方法,并且在Multiwfn里可以很方便地使用,所以本文還將它們通過實例與AdNDP做簡單的對比研究。一些關于ELF和LOL的基本知識可參看Multiwfn手冊的2.6節和《電子定域性的圖形分析》(http://sobereva.com/63)。
2 AdNDP方法的原理
NBO分析方法中,一般是通過體系的密度矩陣信息搜索出孤對電子(1c-2e)和雙中心雙電子鍵(2c-2e),將體系轉化為定域形式來描述,以期與Lewis式存在對應關系。而對于比如硼烷的情況,眾所周知存在三中心雙電子鍵(3c-2e),光靠Lewis式完全不能描述這種體系,因此NBO方法也被適當地擴展而能夠搜索出3c-2e鍵,在NBO程序中用了3cbond關鍵詞就可以實現。然而,對于離域性更強的含有三中心以上的多中心鍵的體系,NBO方法就很難處理了,會看到非Lewis成份非常大,它表示無法被單個Lewis式描述的成份。此時只得借助于自然共振理論(NRT),使強離域性體系通過多個由NBO方法給出的Lewis式互相共振來等效地描述。這實際上是個比較別扭、迫不得已而為之的描述形式,強行將多中心鍵靠雙中心鍵形式表現,使體系內在多中心鍵沒法清楚地展現出來。因此,很有必要將NBO方法進一步做擴展,使之不僅能搜索3c-2e,還能搜索4c-2e、5c-2e、6c-2e...AdNDP就是基于這種思路被提出來的,它是對NBO搜索方法的廣義化。原則上,AdNDP可以利用體系的密度矩陣信息搜索出所有Nc-2e鍵(N小于等于體系總原子數)。眾所周知,正則分子軌道,即我們一般說的分子軌道是高度離域化的,而NBO軌道是高度定域化的,可以說AdNDP是這兩種極端的描述形式之間的平滑的過渡,也許可以稱為半定域軌道。
NBO軌道的搜索過程是基于以自然原子軌道(NAO)為基的密度矩陣進行的,基本搜索過程是:先把對于成鍵沒用的內核軌道對密度矩陣的貢獻扣除,然后搜索1c-2e。也就是將密度矩陣的對應于每個原子的對角塊依次取出,對角化求本征值,如果某些本征值(即軌道占據數)大于某個預設閾值(通常設為一個很接近2.0的值),那么相應的本征向量就被認為是1c-2e而被取出保存,這即是孤對電子軌道。將這些孤對電子對密度矩陣的貢獻扣去之后,接下來搜索2c-2e。搜索方式是以窮舉方式進行的,比如體系有10個原子,那么總共要嘗試10!/(10-2)!/2!=45種組合。在嘗試比如A,C原子組合時,就會將密度矩陣的四個子塊A,A、A,C、C,A、C,C取出來拼成一個矩陣(假設A的基函數有M個,C有N個,那么拼合后的矩陣將是(M+N)*(M+N)個元素的矩陣)。然后將其對角化,如果有本征值大于閾值的本征向量,將會把它們取出保存作為A-C之間的NBO軌道,同時把它對密度矩陣的貢獻扣除。搜索三中心的時候也是類似,嘗試所有三種原子組合的情況,對每種組合取出相應的3*3=9個密度矩陣子塊拼成一個矩陣并對角化,之后取出占據數大于閾值的軌道作為3c-2e,再把它們對密度矩陣的貢獻扣除。NBO方法搜索軌道的過程到這一步就為止了,AdNDP方法中這個過程會繼續延續下去,在搜索完1c-2e、2c-2e、3c-2e后,還以完全相同的方法接著依次尋找4c-2e、5c-2e、6c-2e...
NBO軌道搜索過程中會涉及到一些細節問題,看似不起眼但實際上十分關鍵,比如軌道正交化以及搜索順序的確定,在這里并不打算談及。由于AdNDP方法要搜索更多中心的軌道,搜索過程細節的處理就變得更為關鍵,直接影響最終AdNDP的結果的合理性。用過AdNDP方法的用戶會發現,使用AdNDP作分析通常并不容易,搜索過程沒有絕對的規律可循,至少目前沒有一個完美的解決辦法,這是AdNDP方法最大的局限性,往往必須靠人工憑借經驗進行操作才能完成。在后文,我們將在研究四個實際體系的過程中了解到AdNDP分析的細節過程和一些要點。
注意,Multiwfn本身只是一個做AdNDP分析的靈活方便的工具,并不能克服AdNDP自身方法的含糊性,因此很有可能不同用戶由于操作過程(搜索和選取軌道的順序)的不同得到很不同的AdNDP圖樣(這里指AdNDP方法得到的所有雙電子軌道的集合,可視為廣義化的Lewis式),這就需要用戶自己在實踐過程中以及在閱讀AdNDP方法的相關文獻過程中逐漸尋找感覺、積累經驗。實際上,有時候對同一個分子會找出不少看似合理的AdNDP圖樣,很難判定哪種AdNDP圖樣正確哪種錯誤。例如,在J.Org.Chem.,73,9251當中對苯進行研究時,作者自己也說圖5的兩種AdNDP圖樣都是正確的。我想,讓體系由多個AdNDP圖樣相互共振表達,這或許可以視為是自然共振理論在多中心鍵層次上的廣義化。另外我想說的是,對復雜體系,AdNDP的分析結果有時很大程度上取決于使用者的直覺,所以不應認為文獻上給出的AdNDP圖樣就一定是最佳的,要有懷疑精神。包括AdNDP原文上分析B13+團簇得到的AdNDP圖樣我認為都不是最合理的,對照ELF圖,我認為其中圖7的IV和V的三中心鍵恐怕并不存在,而II和III的三中心鍵描述為四中心鍵應該更妥當。
雖然AdNDP一般都是分析閉殼層體系,但從形式上看,它也可以分析開殼層體系,這在Multiwfn中也是支持的,程序將會問你要分析Alpha密度還是Beta密度還是混合密度。如果分析的只是單一自旋的密度,那么就是尋找多中心單電子軌道了,相應地,搜索過程中占據數的閾值應該設為一個接近1.0的值。
3 在Multiwfn中做AdNDP分析的方法
注:讀者請務必使用2023-Apr-17及以后更新的Multiwfn版本,否則有些地方與下文所述情況不同。
3.1 分析Li5+團簇
在Multiwfn中做AdNDP分析需要NBO程序輸出的含有以NAO為基的密度矩陣。如果還想將軌道結果可視化或者輸出為cube文件,NBO程序輸出文件里還必須包含原始基函數與NAO間的變換矩陣,并同時提供Gaussian的.fch文件。這里所說的NBO程序可以是獨立運行的NBO程序(GENNBO),也可以是Gaussian的L607模塊(對應NBO 3.1),也可以是與量子化學程序掛接的NBO版本。本文假定用戶都是Gaussian用戶。
為了讓Gaussian輸出文件中包含以NAO為基的密度矩陣和原始基函數與NAO間的變換矩陣,必須使用pop=nboread關鍵詞,并在分子坐標末尾空一行寫上$NBO DMNAO AONAO $END。例如Li5+的輸入文件應為
%chk=C:\ltwd\Li5+.chk
# b3lyp/6-311+g(d) pop=nboread
[空行]
b3lyp/6-311+G* opted
[空行]
1 1
Li -0.00180800 1.40848000 -0.69649200
Li 0.00259400 -1.30839200 -0.87169100
Li -0.00056800 -0.10171300 1.56832400
Li -2.76113600 -0.00025300 -0.10041300
Li 2.76091800 0.00187800 -0.10041300
[空行]
$NBO DMNAO AONAO $END
這個輸入文件的結構已經在B3LYP/6-311+G*級別上優化過了。運行此文件得到Li5+.out,并且用formchk將chk文件轉成fch文件。注意,給Multiwfn用的Gaussian輸出文件必須是單點計算的輸出文件。
啟動Multiwfn,輸入Li5+.out的文件路徑,然后選主功能14就進入了AdNDP分析模塊。Multiwfn會先載入一些NAO信息和以NAO為基的密度矩陣,然后自動扣除內核NAO軌道對密度矩陣的貢獻,然后就會看到一堆選項。由于Li5+這個體系非常簡單,所以只需要用窮舉搜索的方法就行了。首先選2去窮舉搜索所有單中心軌道,但是沒有找到占據數高于閾值的軌道,這也是理所當然,從直覺上就知道Li5+不可能有孤對電子。當前的閾值就是選項4上顯示的值,目前版本中默認為1.7。接下來再按2,就開始窮舉搜索雙中心軌道,然后再按2,就窮舉搜索三中心軌道。但是也沒有發現可能的2c-2e和3c-2e軌道。再按2,經過窮舉搜索,從菜單前頭顯示的候選軌道列表中得知發現了兩個四中心軌道的占據數都高于閾值,它們的占據數都高達1.997,因此是理想的4c-2e軌道,此時選0,然后輸入2,就把這兩個軌道從候選列表中取了出來成為AdNDP軌道。
(注:當前的候選軌道列表總是自動顯示在菜單的前頭,為了避免其信息礙事,只有選擇5查看AdNDP軌道信息一覽表或者選13查看剩余電子分布時沒顯示出來。但任何時候都可以從中用選項0選取軌道,或者用選項8和10來分別查看它們和導出它們)
現在可以選7來觀看這兩個AdNDP軌道。Multiwfn首先會讀取Guassian輸出文件中的原始基函數與NAO間的變換矩陣,然后讓你輸入相應的fch文件的路徑,假設你已經把Li5+.fch放到了Li5+.out相同的目錄下,那么Multiwfn就會直接讀取。讀取完畢后,你會看到一個圖形界面,和Multiwfn主功能0提供的界面一模一樣,可以觀看分子結構,在右下角選相應的AdNDP軌道編號就能立刻顯示出等值面圖。對這個界面如果有不明白的地方可以看手冊3.2節的說明。我們得到的Li5+的兩個4c-2e如下圖所示(需要把bonding threshold調大一點,否則中間的三個Li不會與兩側的Li用棍棒接上。另外需要在菜單上Isosur#1 style里選擇Use solid face+mesh才會在等值面上顯示網格):
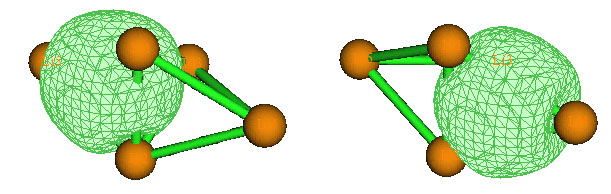
目前會看到菜單前頭的Residual valence electrons of all atoms in the search list后面顯示的數值為0.01989,這代表剩下的電子數,相應于NBO分析當中的非Lewis成份。由于剩下的電子已經遠小于2了,就不可能再去找到其它雙電子多中心鍵了,因此對Li5+的分析就到此結束了。
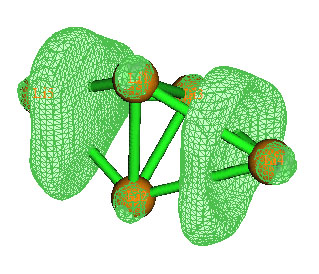
實際上,通過考察如下的Li5+的ELF=0.99等值面圖,也能了解到它有兩個四中心鍵,有兩個比較扁的定域性很高的區域出現。ELF等值面圖在Multiwfn中的做法可參見手冊4.5.1節。
3.2 分析Au20團簇
四面體型Au20團簇在JPCA,113,866文中被通過AdNDP方法研究過,本節我們也研究一下它內部的多中心鍵。下面的坐標是直接從此文獻中的補充信息中摘取的,是Td點群,請大家效仿上一節的輸入文件寫一個Au20的Gaussian單點輸入文件,計算完畢后轉換出對應的fch文件。此體系凈電荷為0,自旋多重度為1,理論級別最好和原文一致,使用B3PW91結合LANL2DZ贗勢基組計算。使用贗勢并不會妨礙價層密度的分析。如果大家懶得自己算,可以直接下載http://sobereva.com/attach/138/Au20.rar,里面包含了用于AdNDP分析目的的Gaussian輸出文件和fch文件。
Au 2.935949 -2.935949 2.935949
Au 2.935949 2.935949 -2.935949
Au -2.935949 -2.935949 -2.935949
Au -2.935949 2.935949 2.935949
Au -1.142441 -1.142441 1.142441
Au 1.142441 1.142441 1.142441
Au -1.142441 1.142441 -1.142441
Au 1.142441 -1.142441 -1.142441
Au 0.964601 0.964601 -3.140361
Au 3.140361 0.964601 -0.964601
Au 0.964601 3.140361 -0.964601
Au -0.964601 3.140361 0.964601
Au -0.964601 0.964601 3.140361
Au -3.140361 0.964601 0.964601
Au 0.964601 -3.140361 0.964601
Au 3.140361 -0.964601 0.964601
Au 0.964601 -0.964601 3.140361
Au -3.140361 -0.964601 -0.964601
Au -0.964601 -0.964601 -3.140361
Au -0.964601 -3.140361 -0.964601
將Gaussian輸出文件載入進Multiwfn并進入AdNDP分析界面后,還是先選2窮舉搜索單中心軌道。這次搜索出了占據數100個高于閾值的軌道,它們按照占據數由大到小編號顯示在候選列表中。選擇0并輸入100把它們全都取出作為AdNDP軌道。這些軌道對密度的貢獻被去掉后,屏幕上顯示目前還有22.3329個剩余電子。(提示:候選軌道有時會比較多,列表比較長,可以將命令行窗口豎向方向拉長一些以顯示更多信息)
接下來選2窮舉搜索二中心軌道,但是一無所獲,再選2窮舉搜索三中心軌道仍然一無所獲。再次選2窮舉搜索四中心軌道,這次發現了10個候選的(由于要做20!/(20-4)!/4!=4845次嘗試,所以略微耗時):
# 10 Occ: 1.7589 Atom: 5Au 7Au 14Au 18Au
# 9 Occ: 1.7589 Atom: 5Au 8Au 15Au 20Au
# 8 Occ: 1.7589 Atom: 6Au 8Au 10Au 16Au
# 7 Occ: 1.7589 Atom: 6Au 7Au 11Au 12Au
# 6 Occ: 1.7589 Atom: 7Au 8Au 9Au 19Au
# 5 Occ: 1.7589 Atom: 5Au 6Au 13Au 17Au
# 4 Occ: 1.8400 Atom: 4Au 12Au 13Au 14Au
# 3 Occ: 1.8400 Atom: 1Au 15Au 16Au 17Au
# 2 Occ: 1.8400 Atom: 3Au 18Au 19Au 20Au
# 1 Occ: 1.8400 Atom: 2Au 9Au 10Au 11Au
其中1至4號、5至10號是占據數簡并的軌道。通過選項8來預覽這些候選軌道,會知道前四個對應于Au20四個頂角的四中心軌道,后六個對應于四面體團簇棱邊的四中心軌道。現在我們要用選項0把這些軌道取出來作為AdNDP軌道。但是不要直接把10個都取走,這點十分關鍵,因為,AdNDP方法搜出的軌道彼此間并不是正交的,而是有交疊的,彼此間可能共享一部分密度。如果將10個一下都取出,就會令軌道的占據數偏高了(即overcounting)。通常,我們會按照占據數從高到低,將簡并軌道一批一批取出,每次取出后,它們的密度將會被扣除,剩下的候選軌道的形狀和占據數會被重新計算,這樣就可以較大程度避免overcounting問題。如果將占據數較高的軌道依次取出后,剩下的軌道中占據數最高的也已經小于了期望的值,比如1.7,即不能再被近似視為雙電子軌道,那么就不理它們了,而接著搜索更多中心的軌道。(注:我們取單中心軌道時總是可以一次都取出來。盡管可能有多個單中心軌道都處在同一個原子上,但同個原子上的候選軌道,或者廣義來說,相同原子組合上的數個候選軌道都是彼此間正交的,所以同時取走而不必擔心overcounting問題)
對于本例,我們先選0,輸入4將前四個候選軌道取出作為AdNDP軌道。由于它們的密度被扣除了,因此剩下的6個候選軌道的占據數有所降低,成為1.6913。我們再選0并輸入6將它們都取出。此時剩下的電子還有4.825個,雖然表面上可能還能搜出兩三個雙電子軌道,但經筆者嘗試,發現已經做不到了,分析也就到此結束了。如果選擇5,可以將搜索過程中已經取出的所有AdNDP軌道的基本信息顯示出來。
按照上面的步驟,我們得到了4個占據數為1.8400的和6個占據數為1.6913的AdNDP軌道。如果我們先把后六個軌道取出(即選0之后輸入5-10),然后再取出前四個,我們將會得到6個占據數為1.7589的和4個占據數為1.7581的AdNDP軌道。本例先取出占據數高的再取出占據數低的做法是一般性做法,讓高占據軌道有更高優先級顯得較為合理,但是這對于低占據數軌道多少有些不公平,尤其是占據數非常接近時,我認為這是AdNDP方法的不足之一。不過盡管兩種取法得到的占據數不同,但從軌道圖形上看沒什么差異。
實際上,在上面Li5+例子中,那兩個4c-2e彼此間也有稍許交疊,因為它們都涉及到中間的三個Li,也因此它們的占據數被稍微高估了一點。但是我們卻不能先取出一個再取出另一個,因為這會破壞這兩個軌道的簡并性,即它們的占據數不再相同,將分別為1.9973和1.8699,軌道形狀也不再完全對應。AdNDP方法的原則是寧可忍受一點overcounting問題,也要讓AdNDP圖樣保持與分子結構一致的對稱性。從圖形上看Li5+的兩個4c-2e間交疊程度很小,因此可以忽略這問題;但如果從圖上看幾個簡并的候選軌道交疊十分明顯的話,那么overcounting就是不可忍受的了,遇到這種情形時這些候選軌道哪個都不能取。
本節的分析結果的結論雖然與JPCA,113,866的完全一致,軌道圖形看不出區別,但是在軌道占據數上稍有出入,Boldyrev他們得出的是4個占據數為1.72和6個占據數為1.98的4c-2e軌道。導致差異的目前原因不明,而對于硼團簇以及平面芳烴的分析,Multiwfn都能得到與他們文中完全一致的結果。雖然我沒有他們的程序沒法分析導致差異的細節原因,但筆者目前有充分自信認為Multiwfn的結果是正確無誤的。
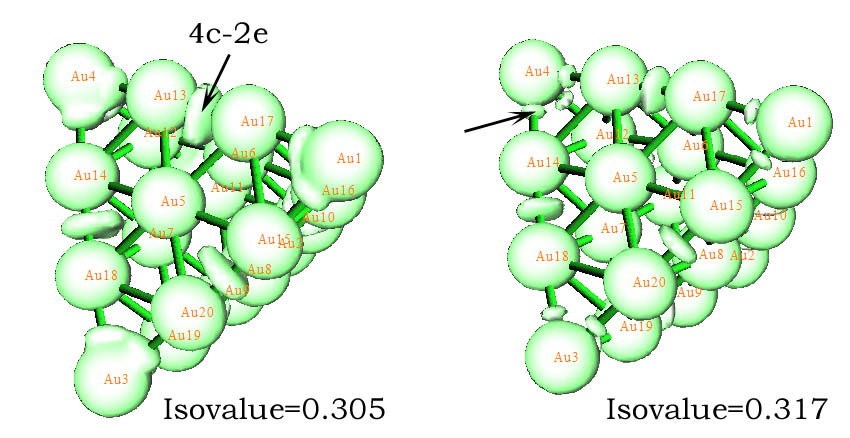
我們也可以試圖用ELF來分析Au20看看是否有相同的結論。但經過筆者嘗試,ELF并沒有得到期望的結果,看不出多中心鍵。而筆者使用類似于ELF的LOL(局域軌道定位函數)時,從等值面上確實可以看到四中心鍵的存在。下面左邊的圖中黑色箭頭指明了4c-2e軌道對應的電子高定域性區域。然而,無論怎么調節isovalue,也無法找到Au20頂角的4c-2e對應的獨立的高定域性區域,但每個頂角卻能找出三個小的定域性域,例如右圖黑色箭頭所示的,可以認為三個鄰近的這種域是和頂角的AdNDP 4c-2e軌道相呼應的。為了能讓這個體系當中的與4c-2e軌道對應的定域性域展露出來,我們必須將isovalue調到0.3多點,這是一個很低的值,也就是說,這個體系中的四中心軌道定域性并不很強(比Li5+的定域性弱多了,上節在ELF=0.99時都能清楚地看到對應的定域化域),某種程度也可以說,這樣的四中心軌道的電子占據數應該不是特別接近2才對,所以,從這個角度我認為Boldyrev他們得到的六條4c-2e軌道占據數高達1.98是有問題的,而Multiwfn得出的1.6913更合理。
在Multiwfn里只能一次觀看最多兩條軌道的等值面圖形,很多AdNDP文獻里都是將很多軌道做到一張圖上。作這種圖其實很容易,首先,在Multiwfn的AdNDP界面里選擇選項9,選擇一個合適的格點設置(這種大小的體系用Medium quality grid得到的圖像就足夠光滑),然后輸入要輸出的AdNDP軌道序號范圍,比如輸入101-110(101至110就是那10條AdNDP 4c-2e軌道,可以用選項5來查看序號),然后就能將它們的cube文件一次性導出到當前文件夾里,名字是AdNDPorbXXXX.cub,其中XXXX對應軌道序號。之后,就可以用支持cube格式且能夠同時顯示多個格點數據等值面的可視化軟件將這些軌道圖形同時顯示出來,筆者推薦使用VMD,如果不熟悉的話,可以參考《使用Multiwfn觀看分子軌道》(http://sobereva.com/269)的第6節的做法。但是,手動操作一個一個顯示軌道終究費事一些。Multiwfn程序包里的examples\AdNDP\plotAdNDP.vmd是寫的VMD腳本,可以直接將一批AdNDP軌道的cube文件的等值面圖一下都作出來。腳本內容如下,拷貝進VMD的命令行窗口直接運行即可:
#0 blue, 1 red, 2 gray, 3 orange, 4 yellow, 5 tan, 6 silver, 7 green, 8 white, 9 pink, 10 cyan, 11 purple
set posclr 1
set negclr 0
set istart 1
set iend 17
set isoval 0.06
set idinit 0
color Display Background white
display depthcue off
display rendermode GLSL
for {set i $istart} {$i<=$iend} {incr i 1} {
set idx [format %04d $i]
set name "D:\\CM\\my_program\\Multiwfn\\AdNDPorb$idx.cub"
puts Loading\ $name
mol new $name type {cube}
mol modstyle 0 $idinit Isosurface $isoval 0 0 0 1 1
mol modcolor 0 $idinit ColorID $posclr
mol modmaterial 0 $idinit Transparent
mol addrep $idinit
mol modstyle 1 $idinit Isosurface -$isoval 0 0 0 1 1
mol modcolor 1 $idinit ColorID $negclr
mol modmaterial 1 $idinit Transparent
incr idinit 1
}
腳本中開頭的posclr和negclr代表軌道正值和負值部分的顏色,常用的顏色代碼在#后面的注釋里已經標注了。istart和iend代表要載入的格點文件編號始末,比如分別設為101和104就會把AdNDPorb0101.cub、AdNDPorb0102.cub、AdNDPorb0103.cub、AdNDPorb0104.cub都依次載入。isoval是等值面數值大小。idinit是要載入的這些格點文件在VMD中的ID號,需要正確設定。設為0時,101號至104號軌道cube文件載入后就會在VMD中依次作為第0號、1號、2號、3號體系。如果你想再通過這個腳本再載入其它一批cube文件,那么idinit應該改為4,后續載入的格點文件將依次作為4號、5號...。set name命令的路徑需要用戶改成自己的實際路徑,注意Windows下的路徑里的斜杠必須寫成兩道斜杠。這個作圖腳本默認情況下繪制的等值面圖是透明度,透明效果可以在Graphics-Materials里選擇Transparent后做細致調節。如果想讓等值面變成不透明(如此例),需要將mol modmaterial前面加上井號來注釋掉。
例如,我們這里要把Multiwfn輸出的Au20的頂角的4個4c-2e軌道繪制為紅色(注:本節找到的4c-2e軌道沒有負值部分),即AdNDPorb0101.cub到AdNDPorb0104.cub,在把istart和iend分別設為101和104,把set name后的文件路徑設為實際路徑并啟動VMD后,直接在命令行窗口里運行上面的腳本即可。如果我們還想把另外六個在棱上的4c-2e用桔黃色繪制上去,即AdNDPorb0105.cub到AdNDPorb0110.cub,那么把istart和iend分別設為105和110,把posclr設為3,把idinit設為4,之后運行腳本即可。為了使體系中的原子也顯示在圖上,可以隨便在VMD Main窗口里選一個體系(坐標都是相同的),點Graphics-representations,在里面點Create Rep,然后在Drawing method里選一個合適的顯示方式,比如CPK,此時我們將得到下圖。
3.3 分析B11-團簇
B11-團簇就是AdNDP原文中分析的一個體系,這一節我們來重現他們的分析。這個體系的Gaussian輸入和輸出文件以及相應的fch文件在Multiwfn壓縮包里examples\AdNDP目錄下就能找到。為了與原文一致,對這個體系僅用了STO-3G基組生成波函數。實際上AdNDP對于基組依賴性很小,在STO-3G這樣很低級別的基組下也能得到定性合理的結果,這一點與NBO分析很像。使用太大基組不僅增加了Gaussian計算耗時,也增加了AdNDP搜索多中心鍵的耗時,因為基組越大則對每種原子組合所要構建的密度矩陣塊就越大,在對角化時會耗費更多時間。
將B11-.out載入并進入Multiwfn的AdNDP模塊后,還是先通過選項2搜索孤對電子,并且一無所獲。然后再選2來窮舉搜索雙中心軌道,這回找出9個候選:
# 9 Occ: 1.9727 Atom: 6B 10B
# 8 Occ: 1.9727 Atom: 5B 11B
# 7 Occ: 1.9742 Atom: 7B 9B
# 6 Occ: 1.9742 Atom: 7B 8B
# 5 Occ: 1.9869 Atom: 2B 6B
# 4 Occ: 1.9869 Atom: 3B 5B
# 3 Occ: 1.9871 Atom: 9B 11B
# 2 Occ: 1.9871 Atom: 8B 10B
# 1 Occ: 1.9942 Atom: 2B 3B
為了避免overcounting問題,我們不一次將它們都取出而是逐步將它們取出。由于第一個和第四個有交疊(都涉及3號原子),而前三個候選之間沒有交疊,所以選0然后輸入3先將前三個取走。然后再取走4個(雖然7-8和7-9都涉及7,但是為了避免破壞其簡并性,故同時被取走),最后把剩下的2個取走。此時就有了10條2c-2e AdNDP軌道,目前殘余電子是16.307.
接下來選2窮舉搜索三中心軌道,得到9個候選。先取走前2個(1-8-10和4-9-11),再取走一個(1-4-7),再取走兩個(1-2-6和3-4-5),此時剩下的四個軌道中占據數最高的僅為1.4085,沒法再被近似認為是3c-2e了,就不再理會它們。目前已得到的5個3c-2e AdNDP軌道圖形一起作在了同一張圖上,占據數都是1.85上下(圖中綠色文字是相應的三中心鍵級,見后文):
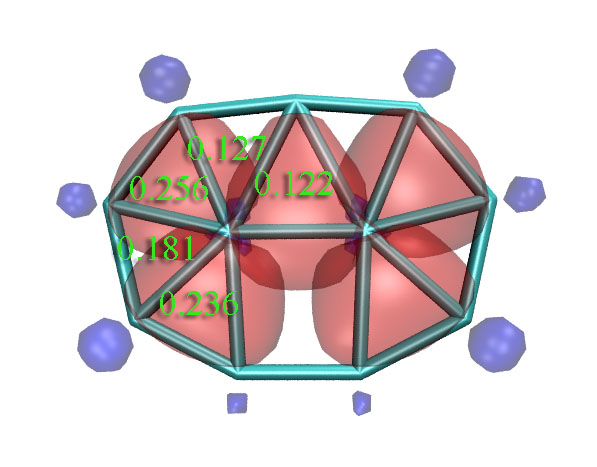
我們也可以做ELF平面圖進行對比(對此體系LOL圖和ELF圖的結論完全一致),在Multiwfn里做平面圖的方法可以參考手冊4.4節的幾個例子。從這圖上我們可以看到AdNDP和ELF的結論還是比較吻合的,兩側的四個3c-2e AdNDP軌道對應ELF的四個處在三個原子之間的高定域性區域。只是中間的3c-2e AdNDP軌道和ELF圖上的稍有不同,從ELF圖上看那更像是普通雙中心鍵,而AdNDP軌道則是向上方的一個原子偏一些而被算作三中心鍵。不過,畢竟AdNDP和ELF在理論基礎上還是有很大差異的,我們也不能強求它們總是完全一致。
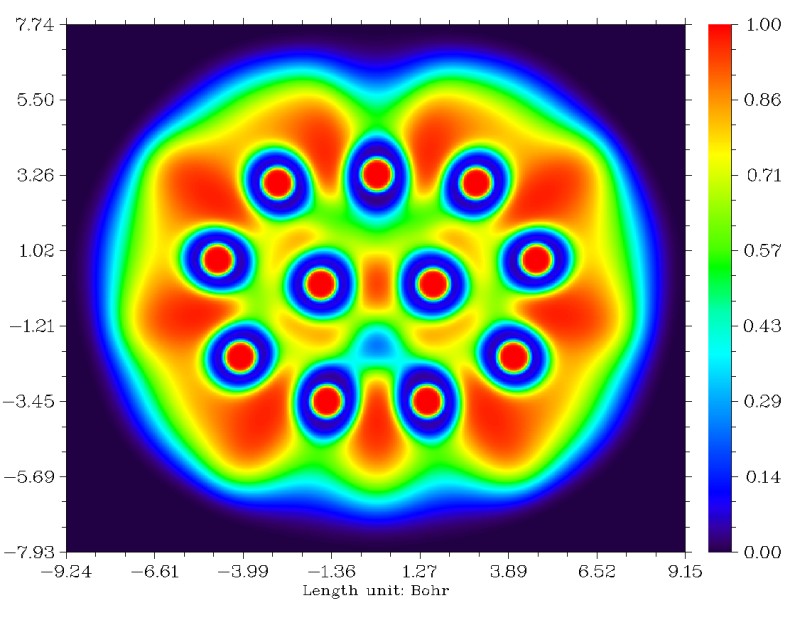
研究多中心鍵的主要方法中,除了ELF/LOL和AdNDP以外,還有多中心鍵級。多中心鍵級雖然不能給出具體的圖形,但是可以給出定量的鍵的強度。在Multiwfn中計算多中心鍵級需要以.fch、.mwfn、.molden等包含基函數信息的文件作為初始輸入文件,載入文件后進入主功能9,之后選2進入多中心鍵級分析功能,然后輸入環中原子的序號即可得出結果,輸入的序號應當按照原子的連接順序輸入。注意多中心鍵級有個特點,即有的時候,沿著環順時針輸入序號還是逆時針輸入序號有時會影響結果,比如1,2,3和3,2,1的結果有時并不完全相同,不過一般差異不大而可以忽略(更嚴格一點的做法是兩種情況取平均,懶得手動取平均的話也可以將Multiwfn的settings.ini里的iMCBOtype設為1,此時輸出的直接就是平均值了)。多中心鍵級計算結果在AdNDP那張圖上用綠色文字已經標出了。可見,兩側的四個3c-2e AdNDP軌道對應的三中心鍵級明顯比較大。但是,中間那個3c-2e AdNDP軌道對應的鍵級卻只有0.122,甚至還不如沒出現3c-2e AdNDP軌道的區域的三中心鍵級大。結合ELF和多中心鍵級的結果,我認為對這個體系,AdNDP方法給出的中間的三個原子之間有3c-2e軌道的結論未必合理。所以,大家也不要只拿AdNDP的分析結果說事,建議結合多中心鍵級以及ELF/LOL圖形做比較研究(實際上,ELF/LOL的結果和多中心鍵級結果也并非總一致)。
搜索完B11-的3c-2e后,目前還剩下7.029個電子,因此可能還能找到一些雙電子軌道。這個體系比Li5+要復雜很多,雖然原子數比Au20少,但是對稱性明顯比它要低,因此分析這個體系的更多中心的軌道并非易事,光靠窮舉搜索的方式往往并不奏效,不僅耗時而且未必能得到希望的結果。如果你不確定下一步該怎么做,建議先選11,將當前的密度矩陣和AdNDP軌道列表臨時保存到內存中,接下來如果選擇了不合適的軌道,那么還可以通過選擇12將密度矩陣和AdNDP軌道列表恢復回來。
假設我們繼續窮舉搜索4中心軌道,占據數最高的候選軌道的占據數也僅為1.7135,這可能并不是我們想要的,先不取它。接下來窮舉搜索5中心軌道,這時候發現前兩個候選軌道的占據數都為1.89,表面上看不錯,可認為是5c-2e。然而,此時取走它們卻未必是最佳選擇,因為完全有可能搜索出占據數更接近2.0的更多中心的軌道。這就是為什么我說使用AdNDP方法并不容易,用戶往往需要自己判斷下一步怎么走,不同的走法又影響到后續搜索,經常得得反復試驗很多次才能最終找到最理想的AdNDP圖樣。
實際用AdNDP分析復雜體系時,我們往往需要借住一些其它依據引導我們搜索軌道的過程。ELF圖的確可以作為搜索3、4乃至5中心軌道時的參考,然而從ELF圖通常并不能對涉及到很多中心、離域尺度很大的情況對AdNDP軌道的搜索過程做出指導,光從ELF圖上很難看出什么來。這個時候我們可以參考一下體系的正則分子軌道。通過檢查正則分子軌道,只發現3個軌道和pi電子有關,如下圖所示,而且這三個軌道是整體離域的。通過圖形檢驗可知,我們之前所找出的AdNDP軌道都不涉及到pi密度,pi密度還“閑著”,剩下的三個多中心雙電子AdNDP軌道很可能就是由這三個整體離域的表現pi密度的正則分子軌道以某種方式組合所給出。于是,我們直接去搜索11中心軌道。
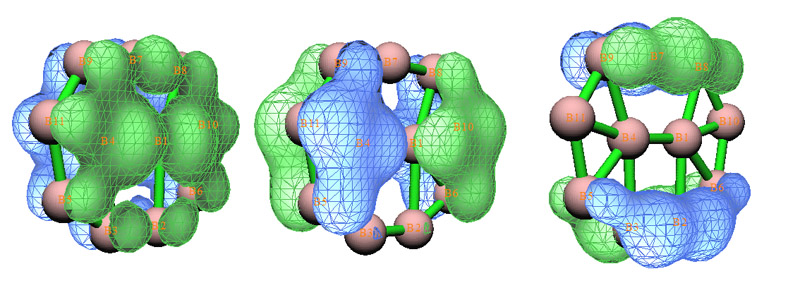
選擇3,然后輸入11,這表明接下來選2進行的窮舉搜索是搜索11中心軌道。由于體系總共就11個原子,所以Multiwfn實際上只進行了一次嘗試。這下找出來三個候選軌道,占據數都達到理論最高值2.0,很理想,通過圖形觀察,其實它們和原先的那三個pi型正則分子軌道是一樣的。把它們取出作為11c-2e AdNDP軌道后,剩下的電子只有1.029了,遠小于2,宣告AdNDP分析結束了。對照AdNDP原文給出的這個體系分析結果可以看到用Multiwfn得出的占據數和原文幾乎完全一致。
3.4 分析菲
這是最后一個例子,通過這個例子大家將了解如何使用用戶導向搜索(user-directed search)方式搜索AdNDP軌道。Gaussian的輸入文件如下,請自行生成輸出文件和fch文件。對于普通的有機小分子,3-21G級別的基組產生的波函數對AdNDP分析來說已經基本夠,使用更大基組也不會看到最后軌道形狀和占據數有很明顯的變化。不過為了避免審稿人嫌基組太low,建議實際研究中使用不低于6-31G*的基組。
%chk=C:\gtest\adndp\phenanthrene.chk
# b3lyp/3-21g pop=nboread
[空行]
b3lyp/3-21g opted
[空行]
0 1
C 0.00000000 3.56061700 -0.29722900
C 0.00000000 2.83932500 0.87979300
C 0.00000000 1.42361400 0.86771500
C 0.00000000 0.72986200 -0.38070000
C 0.00000000 1.49924400 -1.56931800
C 0.00000000 2.88151800 -1.53149000
C 0.00000000 0.67926500 2.09717700
C 0.00000000 -0.72986200 -0.38070000
C 0.00000000 -1.42361400 0.86771500
C 0.00000000 -0.67926500 2.09717700
C 0.00000000 -2.83932500 0.87979300
H 0.00000000 -3.34927800 1.83744900
C 0.00000000 -3.56061700 -0.29722900
C 0.00000000 -2.88151800 -1.53149000
C 0.00000000 -1.49924400 -1.56931800
H 0.00000000 1.23356400 3.02968400
H 0.00000000 4.64400700 -0.27588200
H 0.00000000 3.34927800 1.83744900
H 0.00000000 1.00203800 -2.53051300
H 0.00000000 3.44643700 -2.45642700
H 0.00000000 -1.23356400 3.02968400
H 0.00000000 -4.64400700 -0.27588200
H 0.00000000 -3.44643700 -2.45642700
H 0.00000000 -1.00203800 -2.53051300
[空行]
$nbo dmnao aonao $end
進入AdNDP分析界面后,即便還沒有搜索任何軌道,但也可以直接選7或者8來看一下分子結構,并記住原子編號:
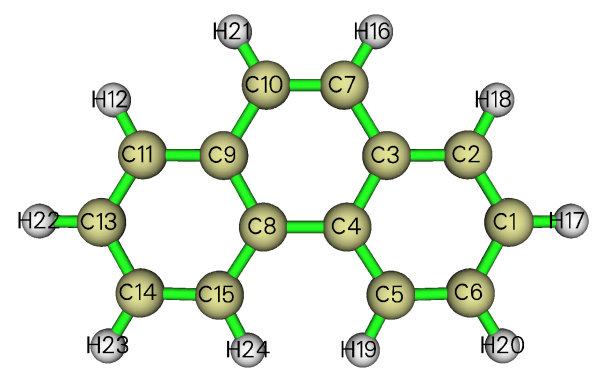
還是照例先窮舉搜索單中心軌道(一無所獲),然后窮舉搜索雙中心軌道。總共找出31個候選軌道,其中10個是C-H鍵對應的軌道。建議這里先把這10個候選軌道挑出來,也就是選0,輸入8-15,再選0,輸入9,10。接下來,依次挑出C-C間的候選軌道。軌道比較多,可能看著眼花,建議每次只挑出前幾個簡并的候選軌道,可以按照0 2 0 1 0 2 0 1 0 2 0 2 0 2 0 2 0 2的輸入來做,空格代表回車。挑出16個C-C候選軌道后,剩下的一個占據數最高的候選軌道是1.8033,通過選8來預覽它的軌道圖形可知它是C8-C10間的pi軌道,由于占據數還算比較高,也把它挑出。剩下的四個占據數只有1.7192的候選軌道就不管了。
這個體系中的原子并沒有構成類似B11-團簇那種局部的小三角形的排布,因此,直覺告訴我們此體系里應該沒有三中心鍵。那么接下來怎么搜索?我們可以看看剩余密度在每個原子上的布居數,選13,我們看到這樣的表
1C : 1.0250 2C : 1.0370 3C : 1.0280 4C : 1.0414
5C : 1.0339 6C : 1.0262 7C : 0.1322 8C : 1.0414
9C : 1.0280 10C : 0.1322 11C : 1.0370 12H : 0.0117
13C : 1.0250 14C : 1.0262 15C : 1.0339 16H : 0.0121
17H : 0.0113 18H : 0.0117 19H : 0.0126 20H : 0.0111
21H : 0.0121 22H : 0.0113 23H : 0.0111 24H : 0.0126
凡是殘余電子布居數接近0的原子,比如所有的氫和C7、C10,它們通常不會再涉及到其它多中心軌道(除非它們恰處于某些多中心軌道的節點上),我們可以暫且無視它們。而剩下的碳原子占據數都在1左右,正對應于還沒被用到的垂直于菲平面的p軌道單電子,它們可能會構成離域pi軌道。這些原子都分布在菲的左右兩個環上,因此極有可能左右兩個環都能構成像苯環一樣的六中心離域體系,因此我們應當把窮舉搜索范圍先設在其中一個環上。Multiwfn的窮舉搜索涉及的范圍只是搜索列表里的原子,搜索列表默認是整個體系。我們選-1進入搜索列定義界面,輸入clean清除默認內容,然后輸入a 1-6,就把搜索列表設為1至6號原子了,輸入x保存列表并退回。此時會看到剩余電子數目也變成了6.191,這個值顯示的只是當前搜索列表里原子上的剩余電子之和。
選3并輸入6,再選2,就會對搜索列表里原子的進行6中心軌道的搜索,這里找出來了三個候選軌道,占據數在1.82至2.0范圍,是比較合適的6c-2e中心軌道,將它們取出。通過預覽其圖形可知它們的形狀和苯分子的占據的pi正則分子軌道十分相似,見下圖,這也表明了菲分子當中兩側的環構成了局部的類似苯的六中心共軛體系。
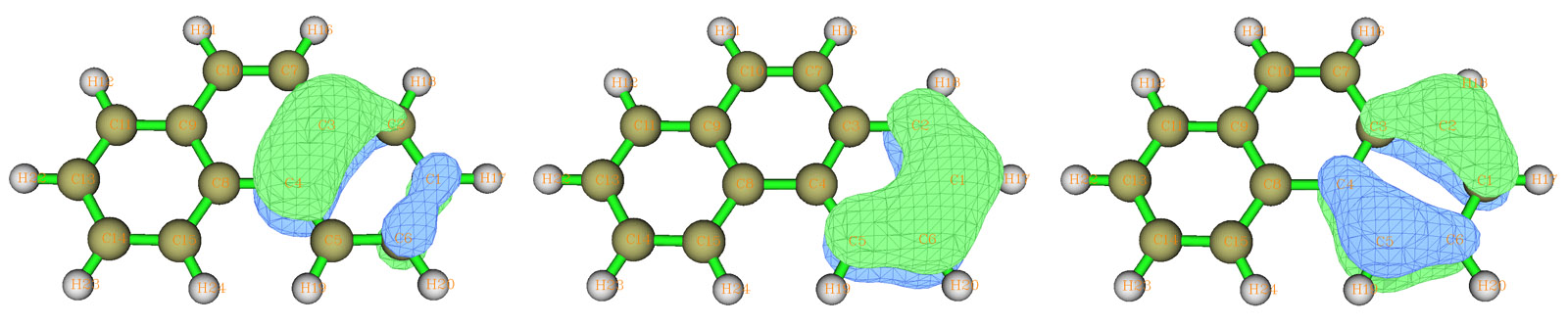
接著,再進入-1,輸入clean清空列表,輸入a 8,9,11,13,14,15,然后輸入x保存并退出,選2對另一個環進行六中心軌道搜索,也是搜索出三個候選軌道,將它們取出。我們再次進入定義搜索列表的界面,輸入addall將整個分子原子都加進去,保存并退回,此時看到整個分子只有1.152個剩余電子了,不可能找出其余雙電子軌道了,這標志著菲的AdNDP分析已經完成了。(注:對于某些分子,即便剩余電子還大于2不少,但可能也已經找不出占據數較高的軌道了,此時只能作罷)
兩側的環的多中心鍵級是0.0616,而中間的由3,7,10,9,8,4號原子構成的環的多中心鍵級僅為0.0261,這確實表明兩邊的環比起中間的環共軛性要強得多(再次提醒,計算多中心鍵級在輸入原子序號的時候必須按照連接關系輸入。對于此例的中間的六元環,如果不按順序,比如輸入成4,7,10,3,9,8,那么結果是沒意義的)。
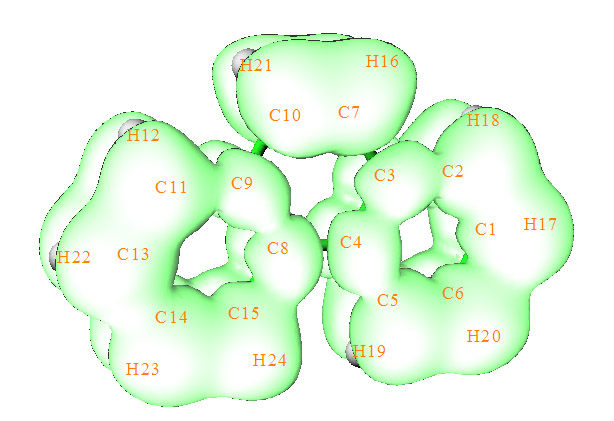
我們也可以通過ELF-pi來分析,ELF-pi的計算方法在《在Multiwfn中單獨考察pi電子結構特征》(http://www.shanxitv.org/432)里已經有詳述,這里不再累述。為了直觀,我們這里并不給出ELF-pi的(3,-1)臨界點的具體數值,而是直接給出只考慮pi電子的ELF=0.5的等值面圖。從下圖可見,在ELF=0.5的時候,兩側的六元環的ELF域各自是聯通的,然而兩側的環彼此間的等值面已經斷開了,而且也都不與C7-C10的ELF等值面相連。因此ELF-pi的分析也說明菲中包含了兩個顯著的六元環共軛結構。其它研究芳香性的方法,如NICS,也都能給出同樣的結論。
在前面的四個例子中有一個選項還沒被提到,就是選項1。這主要是給高級的用戶用的。用戶輸入一串原子組合,Multiwfn就會對這些原子構建相應的密度矩陣并對角化,所有本征向量無論占據數是多少,都會被納入候選軌道列表中。
3.5 分析AdNDP軌道成份
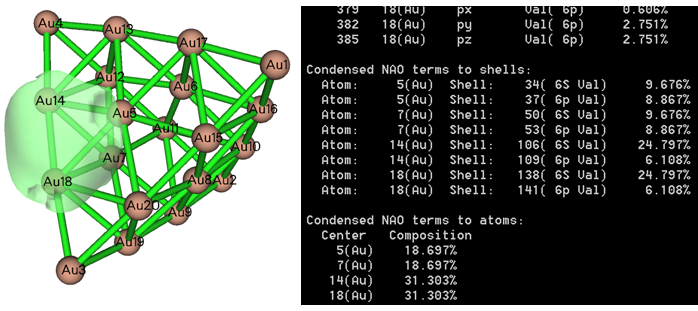
用Multiwfn還可以對AdNDP軌道成份進行分析。通常建議選擇AdNDP界面里的選項15,之后輸入AdNDP軌道序號,就可以通過NAO方法給出這個軌道的成份,下面是個輸出例子,可見原子軌道的貢獻、殼層的貢獻、原子的貢獻都非常清晰給出了,而且和軌道等值面圖對應很好。如果你不懂軌道成份分析方法,看《談談軌道成份的計算方法》(http://www.shanxitv.org/131)。
還可以用Multiwfn的主功能8里的其它方法分析AdNDP軌道成份,比如Hirshfeld、Becke、Mulliken等。做法是在AdNDP界面里選擇選項14,Multiwfn會讀取體系的.fch文件,然后在當前目錄下輸出AdNDP.mwfn文件(這種.mwfn文件也是一種通用的記錄波函數信息的格式,在Multiwfn中可以當成.fch來用)。假如當前體系有N個基函數,AdNDP分析過程中共挑出了M個AdNDP軌道,那么此文件里也包含N個軌道,但是只有前M個軌道是與挑出的M個AdNDP軌道相對應的,其余N-M個軌道都沒有意義,不用去管。將AdNDP.mwfn作為Multiwfn啟動后的輸入文件,然后利用主功能8的各種選項就可以分析AdNDP軌道成份了,操作和平時分析分子軌道成分完全一致。
3.6 計算AdNDP軌道能量
Multiwfn可以對已經挑出的AdNDP軌道計算軌道能量。在挑出一些軌道后,可以選"16 Evaluate and output energy of AdNDP orbitals",此時Multiwfn就會讓你輸入一個含有Fock/KS矩陣元數據的文本文件,Multiwfn會從中讀取矩陣并計算出各個AdNDP軌道能量。第i個AdNDP軌道能量計算公式是<i|F|i>,其中F是Fock/KS算符。輸入的文件的格式要求在手冊3.17.2節已經說明了。
對于Gaussian用戶,雖然可以用IOp(5/33=3)把Fock/KS矩陣輸出出來然后寫入到一個文本文件里提供給Multiwfn,但是每輪SCF迭代都會輸出一堆矩陣,輸出文件會很大,而且這樣操作步驟也多。Gaussian用戶得到AdNDP軌道能量最簡單的方式是產生.47文件(GENNBO輸入文件),也就是寫pop=nboread,然后輸入文件末尾空一行寫比如$NBO archive file=C:\YOSORO $END,則計算完成后就會產生C:\YOSORO.47文件。在進入AdNDP模塊的選項16之后,把這個.47文件路徑輸入進去,Multiwfn就會從中讀取Fock/KS矩陣,并輸出所有AdNDP軌道的能量。如果你看了以上文字還搞不懂的話,可以看Multiwfn手冊4.14節的例子,其中計算了AdNDP軌道能量。
4 總結
AdNDP是一個很有價值的分析多中心軌道的方法,理論依據也比較明確,但絕對不是一個黑盒子,而是需要經過實踐練習才能熟練、合理運用。另外AdNDP方法不是沒有缺點的,在挑軌道上有著一定含糊性,需要用戶干預,引入了主觀成分,如果用不好,可能結論還會與體系實際電子結構特征不符。可是AdNDP的相關文獻中基本只提AdNDP的優點卻很少提及它的缺點。
雖然AdNDP軌道搜索過程沒唯一準則可循,但可以總結出六點:1.剩余電子數越少越好 2.每個AdNDP軌道占據數越接近2.0越好(對于閉殼層而言) 3.挑出的軌道的中心數應盡量低(否則就又成了正則分子軌道了) 4.盡量避免占據數的overcounting 5.AdNDP軌道分布需滿足分子對稱性 6.挑出的軌道能較合理地展現出體系中實際的強共軛區域。其中5是必須遵循的,其它要求有時候會產生矛盾,只能自己看著辦了。
AdNDP、ELF/LOL、多中心鍵級都是目前研究多中心鍵的方法,它們各有優點和缺點,而且也都有個別失敗的例子。AdNDP需要做搜索,對復雜體系較費事,結果一定程度受人為因素的影響,但好處是可以給出軌道的圖形、能量、成份,而且可以自發地把sigma和pi多中心軌道分離描述。ELF/LOL圖分析方法雖然快速直觀,分析三、四中心鍵尤為合適,但是難以直接展現出體系內在的涉及原子數很多的多中心鍵(不過大范圍pi共軛的情況仍可以通過ELF-pi或LOL-pi體現,看《在Multiwfn中單獨考察pi電子結構特征》http://www.shanxitv.org/432);多中心鍵級分析不能給出圖形,但可以給出多中心鍵定量的強度數據,往往和能量有很好相關性(如J.Mol.Struct(Theo.),370,45),它也已經成為衡量芳香性的一個指標(如PCCP,2,3381及JPCA,109,6606)。筆者建議將AdNDP、ELF/LOL和多中心鍵級結合使用,取長補短。