使用Multiwfn+VMD快速地繪制高質量AIM拓撲分析圖(含視頻演示)
注:本文介紹的操作有對應的視頻演示,按照本文敘述的步驟操作還做不出來者務必觀看:https://www.bilibili.com/video/av33724816/
使用Multiwfn+VMD快速地繪制高質量AIM拓撲分析圖
文/Sobereva @北京科音
First release: 2018-Oct-13 Last update: 2020-Dec-2
1 前言
AIM(Atoms-In-Molecules)是Bader發展的極其知名、重要的考察電子結構的一套方法,相關信息見《AIM學習資料和重要文獻合集》(http://bbs.keinsci.com/thread-362-1-1.html)。Multiwfn(http://www.shanxitv.org/multiwfn)在AIM分析方面極度靈活、強大,已被大量研究文章廣泛使用,在Multiwfn中做AIM拓撲分析的操作在此文里介紹得非常明白:《使用Multiwfn做拓撲分析以及計算孤對電子角度》(http://www.shanxitv.org/108)。Multiwfn目前的觀看搜索出來的AIM臨界點以及鍵徑的界面中體系沒法通過鼠標隨意縮放和旋轉,體系大的時候可能也不方便尋找感興趣的臨界點的序號。之前筆者寫過一篇文章《Multiwfn結合VMD繪制AIM拓撲分析圖》(http://www.shanxitv.org/207)介紹了怎么把Multiwfn找出的臨界點和鍵徑放到強大而且免費的VMD程序中顯示得到更好的效果,但是那篇文章手動操作步驟較多,估計一些用戶嫌麻煩不愿意去實踐,或者由于閱讀理解能力差而無法成功實現。這里介紹一種基于腳本的將Multiwfn和VMD相結合的繪制AIM拓撲分析圖(包括臨界點、鍵徑、分子結構)的做法,此方法繪圖效果很好,過程極其簡單,完全零基礎的初學者應當也能瞬間學會。而且在VMD的圖形窗口里找出自己感興趣的臨界點編號也很容易,之后就可以直接根據Multiwfn輸出文件查詢到選定的臨界點上的各種屬性。但也并不是說有了本文,之前那篇博文就沒意義了,因為那里面詳細交代了VMD里的操作細節,弄懂的話就可以根據自己需要調整顯示效果。
本文內容對應于Multiwfn官網上最新版本,不要用太老版本。不了解Multiwfn的話參看入門貼《Multiwfn入門tips》(http://www.shanxitv.org/167)。本文使用VMD 1.9.3版,可在http://www.ks.uiuc.edu/Research/vmd/免費下載。這里假定用戶使用的是Windows系統,筆者用的是Win7-64bit。如果讀者沒有用過Multiwfn做過AIM拓撲分析的話,強烈建議閱讀《使用Multiwfn做拓撲分析以及計算孤對電子角度》(http://www.shanxitv.org/108)了解基礎知識。本文的做法在Linux下同樣可以輕易實現,只不過需要適當的變通。
2 本文涉及的文件和準備工作
首先,把Multiwfn的examples\scripts目錄下的AIM.bat和AIM.txt拷到Multiwfn可執行文件所在目錄。用文本編輯器編輯AIM.bat,把其中Multiwfn后面的文件名設成你要做AIM分析的文件的實際路徑,把里面的VMD路徑改為本機里VMD的實際路徑(如果路徑里含有空格則必須加雙引號,如move /Y *.pdb "D:\study\VMD 193")。然后把AIM.vmd這個VMD繪圖腳本文件拷到VMD目錄下,在VMD目錄下的vmd.rc文件末尾加入一行proc aim {} {source AIM.vmd},從而使得僅輸入aim就可以調用AIM.vmd來繪圖。準備工作就這些。
前述的AIM.bat是Windows下的批處理文件,會啟動Multiwfn并按照AIM.txt里的命令對指定的文件進行計算。AIM.txt是輸入流文件,里面每一行對應在Multiwfn的界面里要敲入的一個命令。如果你不知道Multiwfn怎么通過命令行調用的話,建議看手冊5.2節,看了之后就會明白AIM.bat是如何工作的了。
AIM.txt指揮Multiwfn干的事有以下這些:
(1)進入拓撲分析功能,通過依次使用選項2、3、4、5來搜索臨界點,絕大多數情況這樣搜索后都可以找齊臨界點。然后通過選項8產生拓撲路徑。
(2)把找到的臨界點導出到當前目錄下的CPs.pdb,把產生的拓撲路徑導出到當前目錄下的paths.pdb。
(3)對找到的所有臨界點計算各種屬性(為省時間,靜電勢不被計算,如果想算的話把AIM.txt里的-1改為1),然后導出到當前目錄下的CPprop.txt。
(4)將當前體系結構導出到當前目錄下的mol.pdb。
AIM.bat會把這些產生的.pdb都自動拷到VMD目錄下。
所有Multiwfn支持的含有波函數信息的文件都可以用作輸入文件,見《詳談Multiwfn支持的輸入文件類型、產生方法以及相互轉換》(http://www.shanxitv.org/379)。
3 例:Guanine-Cytosine二聚體
這里我們用Multiwfn文件包里examples目錄下的GC.wfn作為例子。把這個文件拷到Multiwfn可執行文件所在文件夾,編輯AIM.bat,把里面的輸入文件改為GC.wfn的路徑,然后雙擊AIM.bat。由于Multiwfn做AIM分析效率奇高,幾秒鐘就算完了,然后窗口也自動關閉了。如果當前目錄下出現了CPprop.txt而且內容不為空就意味著正常算完了,這個文件里記錄了每個AIM臨界點的各種信息。然后啟動VMD,在命令行窗口輸入aim,此時AIM.vmd就被激活,它自動載入VMD目錄下的CPs.pdb、paths.pdb和mol.pdb并顯示出來,馬上就看到了下圖。注意下圖不是直接從VMD的圖形窗口中截的圖,而是用VMD自帶的Tachyon渲染器渲染的,這樣有抗鋸齒效果,Tachyon使用很簡單,此文提了:《用Multiwfn+VMD做RDG分析時的一些要點和常見問題》(http://www.shanxitv.org/291)。
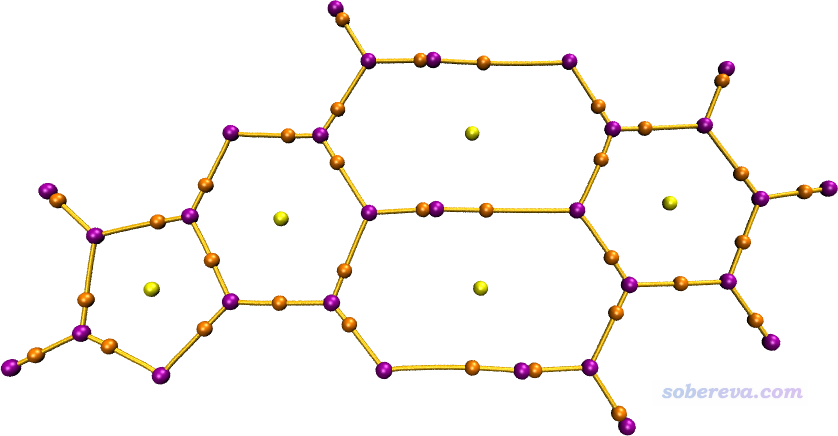
上圖中紫、桔黃、黃分別代表電子密度的(3,-3)、(3,-1)和(3,+1)臨界點。此體系沒有(3,+3)臨界點,如果有的話是用綠球表示。桔黃色的曲線對應的是鍵徑。如果你想修改臨界點和鍵徑的粗細、顏色,就編輯AIM.vmd即可,里面注釋寫得非常清楚。
實際上分子結構文件也被載入到了VMD,只不過沒有顯示出來。想顯示的話,雙擊下面的D,使之變成黑色
然后分子結構也顯示出來了,如下所示
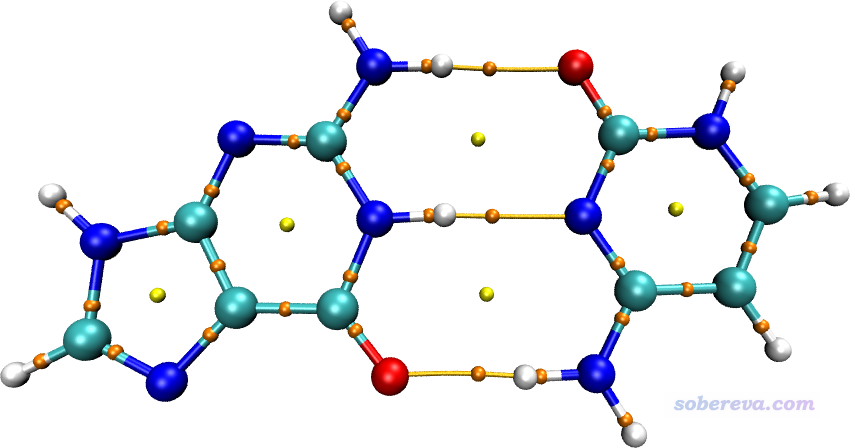
如果想獲知某個臨界點的性質,比如上圖中G-C之間最下面那個氫鍵對應的臨界點,就先在VMD Main窗口里在paths.pdb左邊的D標簽上雙擊,從而取消顯示鍵徑以免礙事。然后點擊VMD的OpenGL圖形窗口激活之,按鍵盤上的0鍵進入查詢模式(之后想返回默認的旋轉模式就按鍵盤上的r鍵),點擊那個臨界點的中心,此時VMD的命令行窗口里就顯示了以下信息
Info) molecule id: 0
Info) trajectory frame: 0
Info) name: N
Info) type: N
Info) index: 42
Info) residue: 0
...[略]
由于VMD的index是從0開始記的,因此這個臨界點在Multiwfn中的序號應該是42+1=43。打開CPprop.txt,在里面找43號CP就看到這個臨界點上的各種信息了,如下所示。初學者若看不懂這些輸出的含義的話就仔細看Multiwfn手冊2.6、2.7節關于實空間函數的介紹。
================ CP 43, Type (3,-1) ================
Position (Bohr): 3.50762367463684 -2.05977861892542 0.00000000000000
Density of all electrons: 0.2608153147E-01
Density of Alpha electrons: 0.1304076574E-01
Density of Beta electrons: 0.1304076574E-01
Spin density of electrons: 0.0000000000E+00
Lagrangian kinetic energy G(r): 0.2053077113E-01
G(r) in X,Y,Z: 0.1152158405E-01 0.7936470033E-02 0.1072717046E-02
Hamiltonian kinetic energy K(r): -0.1212608211E-03
Potential energy density V(r): -0.2040951031E-01
...略
AIM.vmd里還定義了批量顯示臨界點序號的命令labcp,也是在VMD命令行窗口里輸入的,用法為:
labcp [類型] [文字尺寸] [偏移量X] [偏移量Y]
類型可以為all、3n3、3n1、3p1、3p3、no,其中n和p意為臨界點符號中的-和+。文字尺寸可省,默認為1.8。偏移量X和Y也是可省的,默認分別為-0.1和0.0,可以用來控制標簽相對于臨界點的位置,前者越正則標簽越靠左,后者越正則標簽越靠上。大家還可以自己修改AIM.vmd來修改標簽的顏色和粗細。這里給出labcp命令的幾個例子:
labcp all:顯示所有臨界點編號
labcp no:刪除所有臨界點編號
labcp 3n1 1.3:僅顯示所有(3,-1)臨界點編號,文字尺寸用1.3
labcp 3p3 1.8 -0.05 0.1:僅顯示所有(3,+3)臨界點編號,文字尺寸用1.5,X,Y偏移量分別為-0.05和0.1
使用labcp命令顯示的序號和CPprop.txt文件里的序號是直接對應的,不需要手動加1。
AIM.vmd里還定義了標記特定序號的臨界點的命令labcpidx,用法為labcp [序號] [文字尺寸] [偏移量X] [偏移量Y]。例如labcpidx "3 6 to 8 12 15" 2.1就代表把3、6、7、8、12、15號原子的標簽以2.1的字號顯示出來。
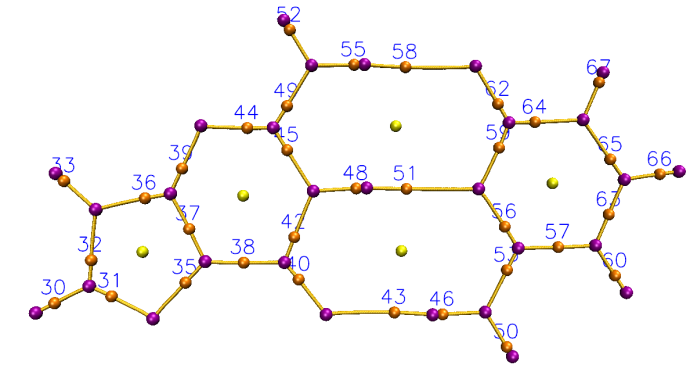
對Guanine-Cytosine二聚體顯示出所有(3,-1)標簽時的效果如下所示,筆者用的命令是labcp 3n1 1.5 -0.1 0.1
如果有的時候標簽位置不太好、或者被原子擋住而看不清楚序號,可以縮放/旋轉視角來解決,或者繪制標簽的時候調整位移值、用更大的標簽尺寸。如果想讓標簽完全不被繪制的物體擋住,那就只能自行用photoshop之類程序解決了,也沒什么復雜的。
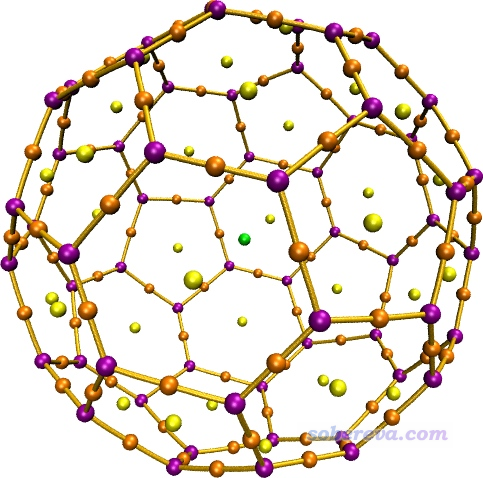
作為練習,大家可以自己算個C60,然后按照如上方法繪制拓撲分析圖,應該得到如下效果
4 總結&其它
本文介紹的方法使得繪制高質量AIM拓撲分析圖變得超級簡單,值得大力推廣。而且單從做AIM拓撲分析的角度,此文也把傳統流程大為簡化,操作效率大為提高,給初學者解釋起來能少費很多口舌。
在《一篇最全面、系統的研究新穎獨特的18碳環的理論文章》(http://www.shanxitv.org/524)介紹的筆者的論文中,我將本文的方法應用于了電子結構很特殊的18碳環的二聚體,并且為了效果更好,在VMD里做了精細的調節,得到了非常漂亮的圖像,建議感興趣的讀者看看。
對于大體系,如果想節約一些拓撲分析時間,可以把AIM.txt的第4、5行都給刪掉,此時Multiwfn不會試圖從每三個原子中心和每四個原子中心作為初猜去找臨界點了(而只通過原子核位置和每兩個原子中點作為初猜找臨界點),這在少數情況下可能會導致漏掉個別臨界點,但一般化學上感興趣的臨界點很少有可能會因此漏掉。
如果你想只讓某一類臨界點顯示而不想所有的都顯示,可以利用Multiwfn的拓撲分析界面里的選項-4里的子選項2 Delete some CPs刪除特定類型的臨界點之后再導出。更簡單的做法是進入VMD的Graphics - Representation界面,在Selected Molecule一欄里選擇CPs.pdb,然后就會看到下面的界面
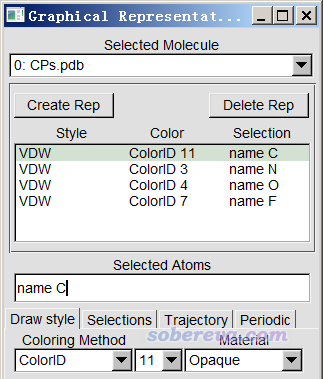
C、N、O、F分別對應(3,-3)、(3,-1)、(3,+1)、(3,+3),因此如果你不想顯示(3,+1)和(3,+3),就雙擊對應name O和name F項,使之變成紅色來取消顯示即可。另外,比如(3,-1)中你只想顯示1,4,6,7,8號臨界點,那么在Selected Atoms文本框里把name N改為name N and serial 1 4 6 to 8即可。如果你不想顯示這幾個(3,-1)臨界點,那就寫成name N and not {serial 1 4 6 to 8}。
類似地,也可以設置只顯示或不顯示哪些鍵徑。從paths.pdb的內容可知,不同的拓撲路徑對應于不同的殘基號,因此在VMD里可以通過殘基號來對顯示情況進行設置。比如你不想顯示3號和7號鍵徑,就進入Graphics - Representation界面,在Selected Molecule一欄里選擇paths.pdb,然后在Selected Atoms文本框里輸入not resid 3 7即可。查詢鍵徑序號的做法是:按鍵盤上的0進入查詢模式,然后點擊鍵徑上任意一個點,此時在文本窗口里resid:后面的數值就是這個鍵徑對應的殘基號。