Multiwfn結合VMD繪制AIM拓撲分析圖
注:強烈建議讀者先閱讀筆者后來寫的《使用Multiwfn+VMD快速地繪制高質量AIM拓撲分析圖》(http://www.shanxitv.org/445),其中將本文的操作過程通過腳本極大地簡化,只需要不到1/20的時間就可以繪制出本文相同效果的圖像。
Multiwfn結合VMD繪制AIM拓撲分析圖
文/Sobereva @北京科音
First release: 2013-Nov-5 Last update: 2018-Feb-27
實空間函數的拓撲分析是Multiwfn(http://www.shanxitv.org/multiwfn)的重要功能之一,電子密度及其拉普拉斯函數的拓撲分析是AIM分析中的核心,而ELF、LOL的拓撲分析在分析成鍵方式、芳香性等問題上也有重要應用。在Multiwfn中這些都可以通過主功能2來實現。關于實空間函數的拓撲分析的基本概念和在Multiwfn里的操作在此帖都有介紹:《使用Multiwfn做拓撲分析以及計算孤對電子角度》(http://www.shanxitv.org/108)。
雖然Multiwfn的拓撲分析功能本身就能繪制出臨界點和拓撲路徑,也足夠滿足通常的需要,但是如果想得到更漂亮的效果,或者更自由地控制顯示方式,則可以將分析結果從Multiwfn導出并通過VMD程序來繪制。此文就介紹下過程。這里以苯酚的電子密度拓撲分析(或稱AIM拓撲分析)為例來說明。
首先我們還是先照常在Multiwfn里找出體系的各種臨界點并生成鍵徑。啟動Multiwfn,然后依次輸入
examples\PhenolDimer.wfn
2 //主功能2
2 //以原子核位置作為搜索臨界點的初猜
3 //以每兩個原子的中點作為搜索臨界點的初猜
0 //查看結果
從命令行界面中可以看到找出了61個臨界點,并且Poincare-Hopf關系已經滿足了。同時從蹦出來的圖形窗口中也可以憑直覺看到該有的臨界點也都有了,說明臨界點都找全了。于是關閉窗口,然后選8,就把鍵徑都生成了,這即是(3,-3)和(3,-1)臨界點之間的拓撲路徑。雖然也可以再選9把(3,+1)和(3,+3)之間的拓撲路徑生成出來,但沒太大價值,這里就不做了。然后選0,可以看到臨界點和鍵徑都出現在圖中了,紫/橙/黃/綠小球分別代表核臨界點/鍵臨界點/環臨界點/籠臨界點。如果點擊窗口右側"Paths labels"復選框,還可以看到每條鍵徑的序號,如下所示
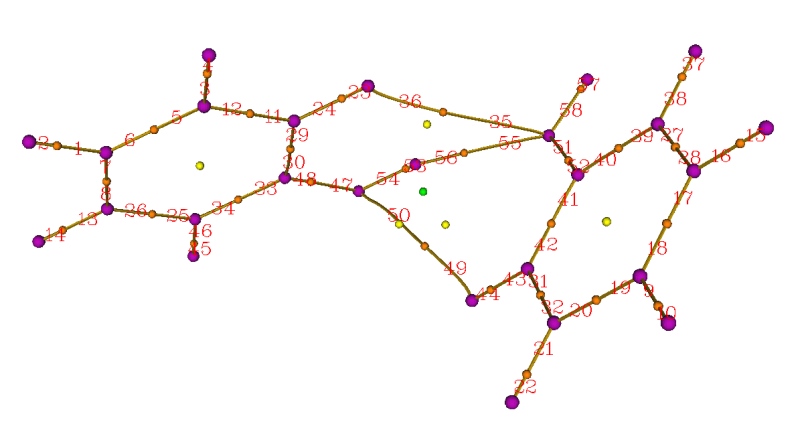
關閉圖形窗口,然后將臨界點和拓撲路徑都導出為.pdb文件。具體做法是選-4進入臨界點修改/導出界面,然后選6 Export CPs as pdb file in current folder,臨界點就被導出到當前目錄下的CPs.pdb里了,每個臨界點對應此文件中的一個原子,并且如屏幕提示所示,不同元素的原子對應不同類型臨界點,即碳/氮/氧/氟分別對應(3,-3)/(3,-1)/(3,+1)/(3,+3)。選0退回到拓撲分析主菜單,再進入選項-5,即修改、顯示、導出以及計算拓撲路徑上的性質的界面。然后選6 Export paths as paths.pdb file in current folder,這樣拓撲路徑就被導出到當前目錄下的path.pdb里了。實際上拓撲路徑就是由一連串緊挨著的點來表示的,整體來看就連成了曲線。在path.pdb里,這些構成鍵徑的點對應于一個個碳原子,每個原子所屬殘基編號就對應于拓撲路徑的編號。
下面我們就要在VMD中繪圖了。VMD是個十分靈活、效果出色的免費的分子可視化軟件,下載地址見http://www.ks.uiuc.edu/Research/vmd/。這里用VMD1.9版。
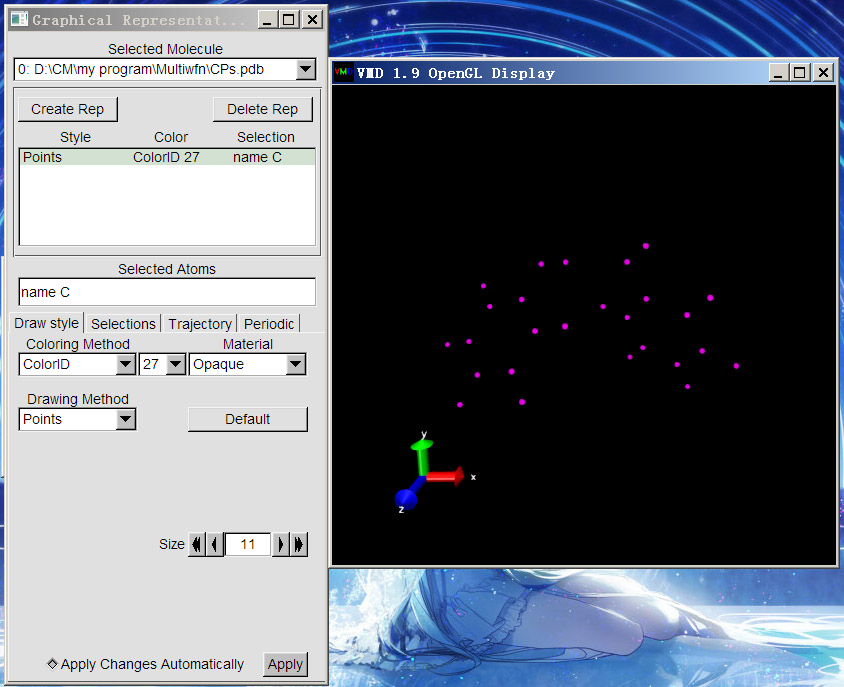
啟動VMD,將CPs.pdb拖到主窗口VMD Main中,然后就會看到一副雖然有點藝術感,但是連線亂七八糟的圖,這是因為VMD自動判斷了原子間連接關系。我們選Graphics-Representation來改變顯示方式。這里讓(3,-3)臨界點都用紫色圓點顯示,就改成下圖中這樣,即Selected Atoms輸入name C,Drawing Method用Points,Coloring Method用ColorID并自己指定顏色,大小通過Size來調
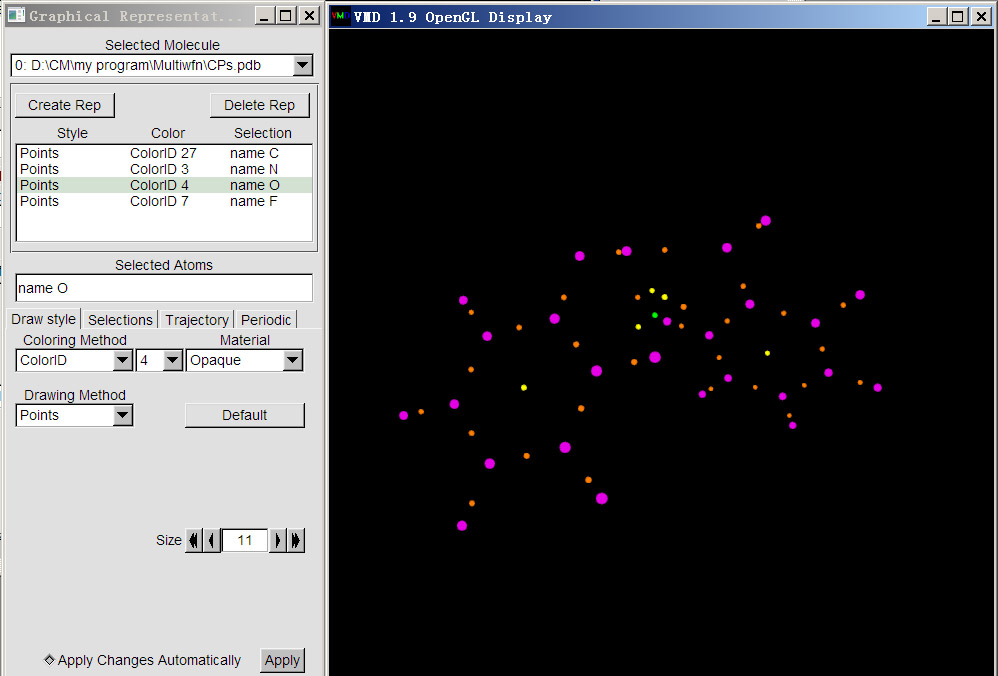
然后把(3,-1)也顯示。點擊Create Rep新建顯示方式,輸入name N,然后設定好繪制方法、顏色和尺寸。然后再類似地顯示出(3,+1)和(3,+3),它們用name O和name F選擇。最后繪制的效果如下。為了凸顯(3,-3)臨界點,它的size由默認的11改為了20。
上圖用的是黑色背景,臨界點位置看得很清楚,但是如果用白色背景(命令行窗口輸入color Display Background white),臨界點的顏色應該用得深一些。
接下來顯示鍵徑。我們把paths.pdb拖進VMD Main窗口,進入Graphics-Representation后確認Selected Molecule已經切換到paths.pdb了。將Drawing Method改為Points,Size改為1,顏色改為青色。這時圖像看起來效果已經很好了。
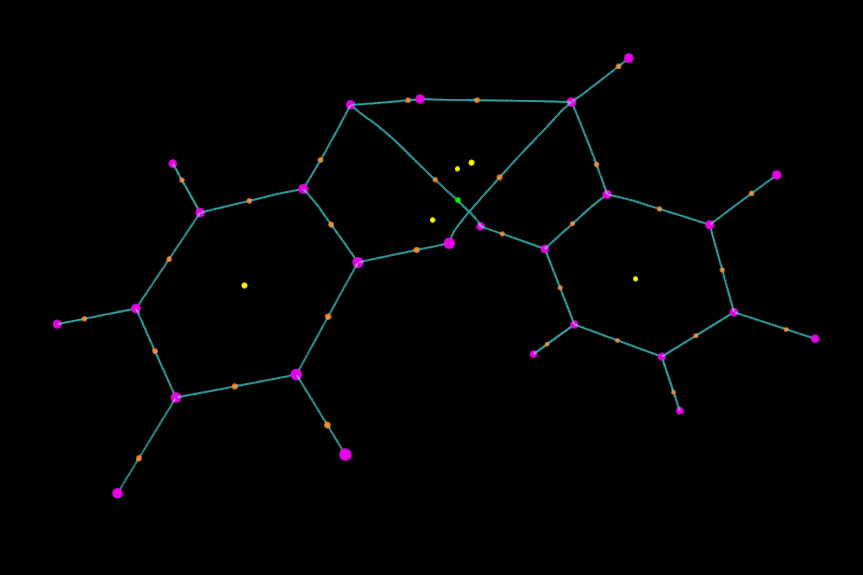
我們還可以對圖像進行更進一步修改。比如,我們想讓兩個苯酚之間的鍵徑和分子內的鍵徑區分開以便觀察。為此,我們先從本文的第一張圖找出分子間鍵徑的編號,可以看出編號是35 36 55 56 49 50。然后我們在把鍵徑對應的selected atoms框的內容由默認的all改為all not resid 35 36 55 56 49 50,這時由圖可見只剩下分子內的鍵徑顯示在圖中了。然后點擊Create Rep,將新的顯示方式的所選原子設為resid 35 36 55 56 49 50,然后把顏色設為紅色。我們再做些調整,比如這里把分子內的鍵徑加粗(即設大size),相應地把(3,-3)和(3,-1)臨界點的size也加大一些,并且不讓(3,+3)顯示出來,最終的圖像如下所示
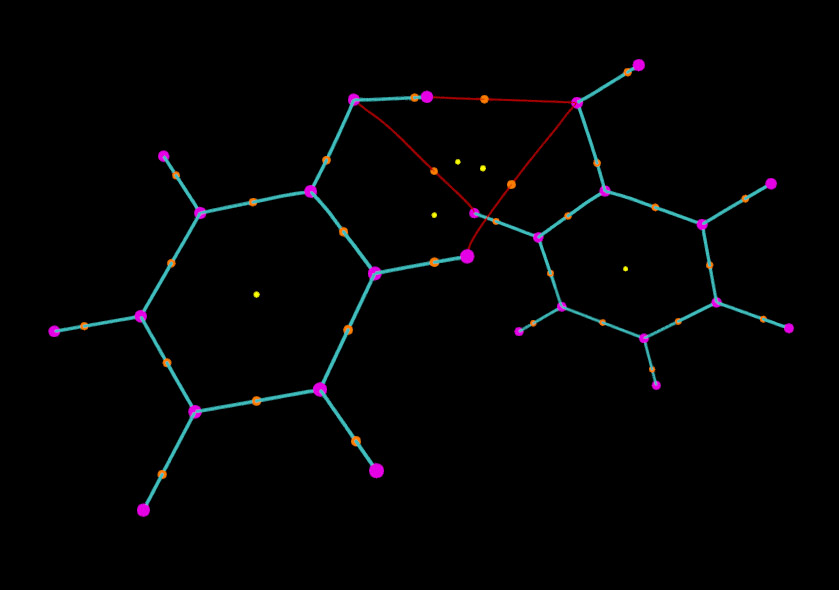
可見顯示效果非常不錯。依照這種方法,還可以繪制諸如ELF、LOL等實空間函數的拓撲路徑,操作過程一樣,這里就不累述了。
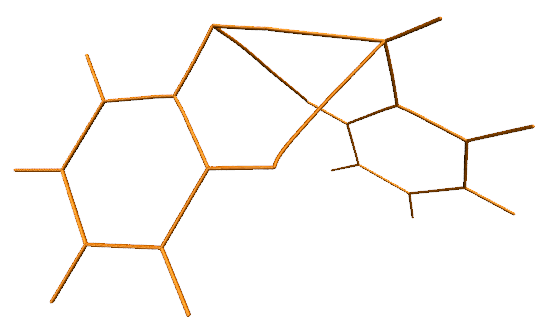
我們也可以改用VDW球方式顯示鍵徑的每個點,這樣鍵徑更有質感。具體來說,在顯示鍵徑的那個Rep里把Drawing Method改為VDW,然后把界面中Sphere Scale選項修改得很小。但是會發現,最小也只能設為0.1,但此時鍵徑顯得還是太粗。為了設得更小,我們需要利用命令行。為了獲取對應的命令行指令,我們選File - Log Tcl Commands to Console,然后隨便修改一下Sphere Scale,文本窗口會出現比如mol modstyle 0 0 VDW 0.200000 12.000000。其中的0.2對應當前的Sphere Scale,將之改為較小的值,然后把命令粘貼到文本窗口里,比如用mol modstyle 0 0 VDW 0.02 12.000000,則顯示鍵徑的那個Rep對應的效果就是這樣了:
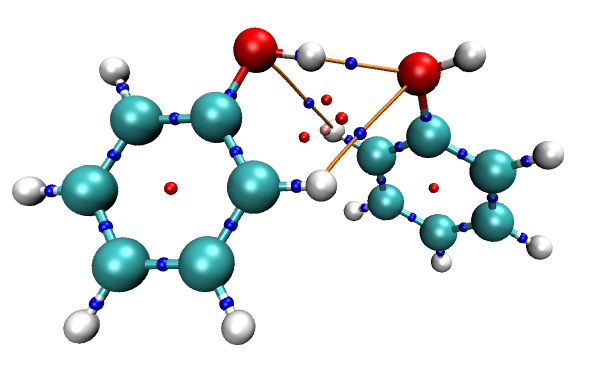
對于考察分子間相互作用的目的,其實顯示化學鍵的鍵徑沒什么大用,可以把體系結構以CPK方式顯示,這樣化學鍵的鍵徑就會被遮蓋住,而只顯示出分子間相互作用的鍵徑(體系的pdb結構文件可以通過Multiwfn主功能100的子功能2的相應選項來導出,導出后拖入到VMD里,設定顯示方式即可)。同時,我們也把臨界點也用VDW方式顯示出來,并用上述方法把其Sphere Scale設為0.07,最終得到的圖像如下。
Multiwfn雖然內建了很方便的可視化結果的功能,但為了靈活性考慮很多地方都留出了專門的選項用于導出數據,以便通過外部程序諸如VMD、sigmaplot等程序繪圖,以滿足一些有個性用戶的需求。使用Multiwfn時應當多嘗試,對照著手冊相應章節領會各個選項的含義。Multiwfn的靈活性與VMD的靈活性相結合使得繪制許多需要用戶高度自定義的圖像成為了可能,這是其它任何程序都無法代替的。其它的Multiwfn與VMD相結合進行繪圖的例子見《通過鍵級曲線和ELF/LOL/RDG等值面動畫研究化學反應過程》(http://www.shanxitv.org/200)和《使用Multiwfn結合VMD分析和繪制分子表面靜電勢分布》(http://www.shanxitv.org/196)。
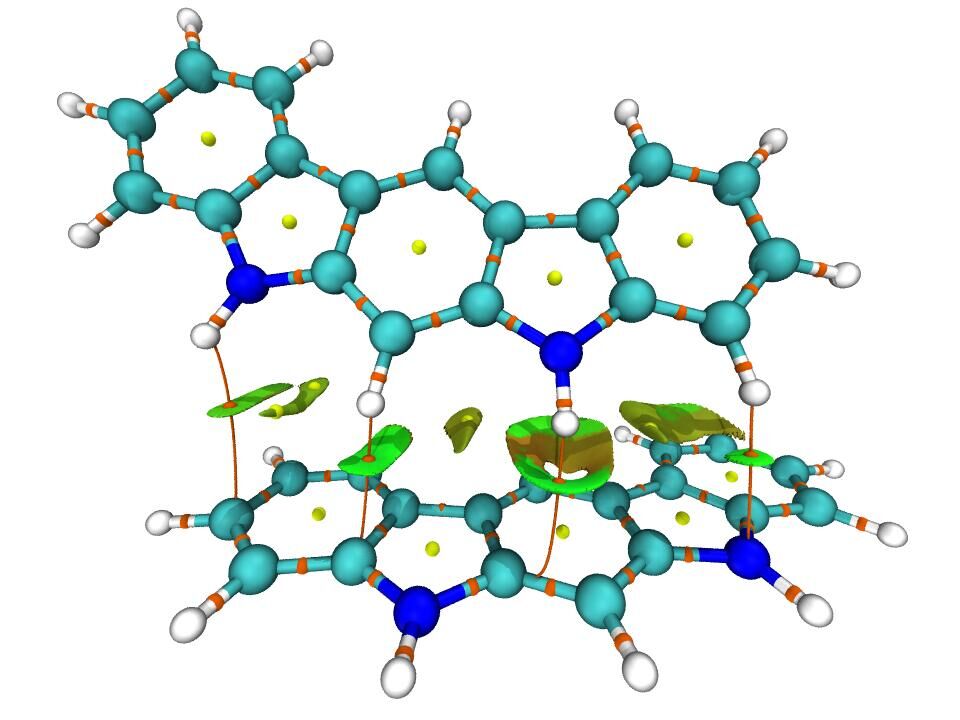
值得一提的是,在VMD中還可以把RDG填色等值面圖和臨界點、鍵徑圖顯示在一起,使得弱相互作用信息展現得更豐富。RDG填色等值面圖的介紹看Multiwfn手冊3.22.1節,繪制這種RDG+AIM圖的例子看Multiwfn手冊4.20.1節。下面是思想家公社QQ群里的群友“葉”繪制的這種圖: