使用Multiwfn結合VMD分析和繪制分子表面靜電勢分布
注:筆者后來又寫了《使用Multiwfn+VMD快速地繪制靜電勢著色的分子范德華表面圖和分子間穿透圖》(http://www.shanxitv.org/443),是繪制分子表面靜電勢圖最完美的解決方案。其中通過腳本,把此文的繪制標準靜電勢填色的范德華表面圖的步驟簡化到了極致,只需要用不到本文1/10的步驟和耗時就可以達到比本文更好的效果,而且完全避免了用戶對VMD操作不熟導致畫不出來圖。強烈建議讀者先閱讀443這篇文章后再讀本文,兩篇文章有互補性。
使用Multiwfn結合VMD分析和繪制分子表面靜電勢分布
文/Sobereva @北京科音
First release: 2013-Jul-28 Last update: 2022-Feb-24
1 前言
分子表面靜電勢圖經常在文獻中出現,不同表面區域靜電勢大小通過不同顏色展現,使分子表面上靜電勢的分布一目了然。分子表面一般都用Bader定義的范德華表面,即電子密度為0.001 e/Bohr^3的等值面。只要提供所需的輸入數據,這種圖在許多程序中都可以作。本文介紹的通過Multiwfn的定量分子表面分析功能結合VMD和Photoshop作分子表面靜電勢圖步驟相對步驟較多,但是優點十分顯著,就是可以在分子表面上顯示出靜電勢極值點位置,可以十分靈活地調節顯示效果,而且還可以通過相同的方法繪制出分子表面上靜電勢以外的實空間函數的分布,比如平均局部離子化能、局部電子親和能、Fukui函數等等。另外還可以順帶著獲得許多其它信息,比如原核與分子表面的距離,實空間函數在分子表面上分布的統計數據等等。只要了解每一步操作的意義,就會覺得其實根本不復雜,而且思想上獲得極大的解放。
本文將以一個簡單體系呋喃為例進行說明,波函數文件在B3LYP/6-31G**下由Gaussian產生。雖然本文主要是介紹作圖方法,但也會順便介紹一下定量分子表面分析的一些功能。閱讀本文前建議先看看《使用Multiwfn的定量分子表面分析功能預測反應位點、分析分子間相互作用》(http://www.shanxitv.org/159),和本文有互補性。更多關于靜電勢的相關文章看《靜電勢與平均局部離子化能相關資料合集》(http://bbs.keinsci.com/thread-219-1-1.html)。
Multiwfn可在http://www.shanxitv.org/multiwfn上下載,不了解此程序的話看《Multiwfn FAQ》(http://www.shanxitv.org/452)和《Multiwfn入門tips》(http://www.shanxitv.org/167)。VMD為1.9版,可在http://www.ks.uiuc.edu/Research/vmd/上免費下載。Photoshop為CS2版。如果你的研究中使用Multiwfn根據本文方法繪制和考察了靜電勢,請在發表的文章中提及并務必引用Multiwfn啟動時提示的程序原文。也建議同時按照《Multiwfn使用的高效的靜電勢算法的介紹文章已于PCCP期刊發表!》(http://www.shanxitv.org/614)末尾的說明引用介紹Multiwfn中靜電勢計算算法的文章。
2 在Multiwfn中計算、統計、導出數據
怎么產生可以給Multiwfn用的波函數文件在《詳談Multiwfn支持的輸入文件類型、產生方法以及相互轉換》(http://www.shanxitv.org/379)里明確說了。這里用呋喃為例子。啟動Multiwfn后依次輸入
furan.wfn
12 //定量分子表面分析
0 //開始計算
默認就是在電子密度為0.001 a.u.的表面上計算靜電勢,并且格點間距為0.25 Bohr。格點間距可以通過選項3來設定,格點間距越小,分子表面就會用越多的頂點來描述,定量統計值、極值點位置也會越精確,之后作出的分子表面填色圖的色彩過渡也越光滑,但是計算耗時將越長。如果你的目的僅僅是繪制分子表面靜電勢圖,可以忽略以下內容而直接跳到本節最后一段。
計算完畢后會看到分子表面上靜電勢極大、極小點的坐標和數值,并且輸出大量統計數據,比如分子表面上靜電勢的最大/最小值、平均值、方差、電荷平衡度、表面積等等,它們對于了解分子特征、建立QSPR/QSAR方程預測分子理化性質和生物活性等問題都十分有用,見《使用Multiwfn預測晶體密度、蒸發焓、沸點、溶解自由能等性質》(http://www.shanxitv.org/337)和Multiwfn手冊3.15.1節的介紹。
此時會看到后處理菜單。選選項0進入圖形界面,并且將Ratio of atomic size設為4.0就可以清楚看到分子表面上靜電勢極大點(紅點)和極小點(藍點)的位置。如果點擊Minimal/Maximum label,就會顯示出極值點的編號,可以和命令行窗口顯示的極值點信息相對照,得知極值點上的具體數值。點擊右上角Return可以關閉圖形窗口。
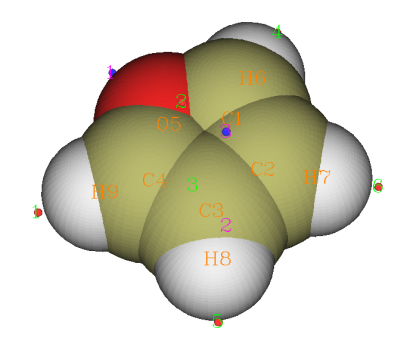
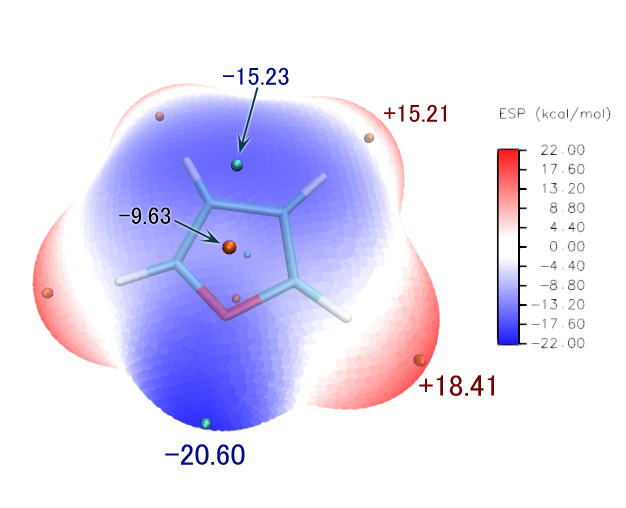
此例中各個極值點靜電勢數值如下(由于格點精度有限,所以等價的極值點的數值不完全一致,這里取了平均)
極小點1(最小點):-20.60 kcal/mol 體現氧的孤對電子對靜電勢的負貢獻
極小點2、3:-15.23kcal/mol 在兩個beta碳(鄰位碳)正上方,體現pi電子對靜電勢的負貢獻
極大點1、4(最大點):18.41kcal/mol 體現alpha位的氫原子所帶的正電
極大點5、6:15.21kcal/mol 體現beta位的氫原子所帶的正電
極大點2、3:-9.63kcal/mol 雖然是極大點,但是靜電勢數值為負,所以化學意義不大,可無視之
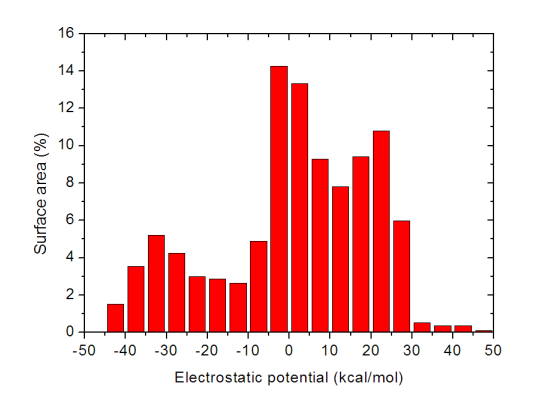
我們可以查看一下不同靜電勢區間內分子表面積,這對于了解分子表面靜電勢的定量分布很有益。做法是在后處理菜單中依次輸入
9
all //考慮所有原子對應的范德華表面
-25,22 //統計范圍。當前體系分子表面靜電勢范圍為-20.60~18.41kcal/mol,這里將范圍稍微擴大并取整數來定義范圍
15 //將-25~22kcal/mol均勻分為15個區間獲得表面積
3 //輸入的單位為kcal/mol
立刻屏幕上就輸出了不同靜電勢區間內的表面積,我們將Center這一列和Area這一列的數據分別從屏幕上拷下來(不會拷的讀者見手冊5.4節),并粘貼到諸如Origin等作圖工具里并作成條形圖,就可以得到諸如以下圖像
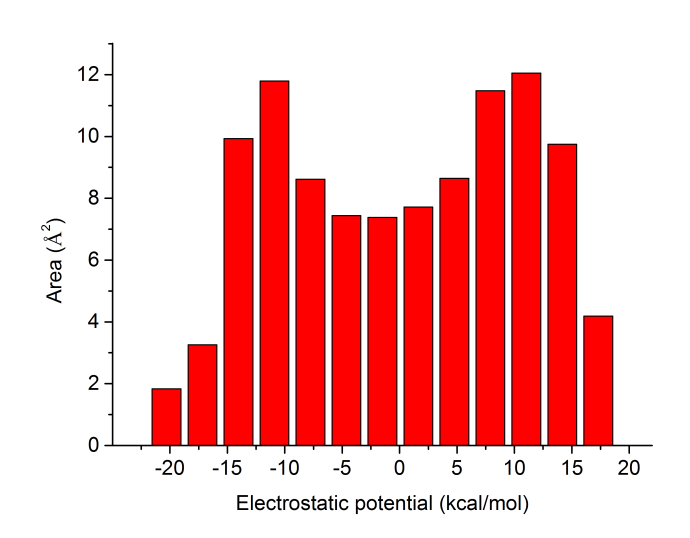
靜電勢的定量分布在這張圖上一目了然。此分子不同靜電勢區間內的表面積分布還是相對比較均勻的。這種靜電勢分布柱形圖統計是在后文提到的筆者發表的J. Phys. Org. Chem., 26, 473-483 (2013)和Struct. Chem., 25, 1521 (2014)兩篇文章里首次提出和使用的,如果大家在自己的文章里也用這種圖,除了引用Multiwfn原文外也請引用這兩篇文章。
如果選擇選項11,則程序會輸出每個原子對應的局部分子表面上的靜電勢統計數據,對于了解原子在此分子中的特征很有用,這個功能的詳細介紹見《談談怎么計算“原子的靜電勢”》(http://www.shanxitv.org/641)。例如Multiwfn顯示的局部表面靜電勢平均值
Atom# All/Positive/Negative average
1 -10.08997 NaN -10.08997
2 -12.59141 NaN -12.59141
3 -12.64250 NaN -12.64250
4 -10.04843 NaN -10.04843
5 -13.54925 NaN -13.54925
6 6.87357 9.80155 -3.13475
7 4.84667 8.19922 -4.56994
8 4.84069 8.21673 -4.50535
9 6.88875 9.78376 -3.16678
氧原子(5號)附近的分子表面靜電勢平均值最負(-13.55kcal/mol),這也容易理解,畢竟其孤對電子對靜電勢有很大的負貢獻。由于其附近分子表面上沒有正值區域,所以正值區域的靜電勢平均值顯示的是NaN。呋喃中碳原子裸露在分子表面上的區域主要就是體現pi電子特征的區域,由于pi電子云使這部分區域靜電勢為負,所以碳原子附近靜電勢平均值也都為明顯負值,并且無正值區域。同時從靜電勢平均值上也體現出beta碳(2、3號)比alpha碳(1、4號)的pi電子云更富集因而靜電勢更負。這種討論分子表面上對應不同原子區域的定量數據的方法和分子表面極值點分析往往會得到共通的結論,但此方法可以得到更多的信息,比如alpha碳上沒有出現極值點(因此對它沒有任何描述),而通過分析它對應的分子表面上的局部區域的靜電勢平均值等數據就可以定量考察它的特征。這種分析方法是筆者獨創的,也僅有Multiwfn支持。另外,通過選項12還可以查看分子表面上對應于指定分子片段區域的定量性質。
選過選項9之后會問你是否輸出locsurf.pdb文件,通過此文件可以利用VMD查看不同原子對應的分子表面區域。由于這不是本文的重點,所以輸入n不讓程序輸出。
利用選項10可以查看分子表面相對于指定坐標點或指定原子核的最遠和最近距離,這對于討論分子間相互作用導致分子表面的穿透距離很有用。此功能也可以用于計算分子半徑或直徑,見《談談分子半徑的計算和分子形狀的描述》(http://www.shanxitv.org/190)的討論。
現在選擇2將分子表面極值點導出到當前目錄下surfanalysis.pdb文件中,此文件中碳和氧原子分別對應分子表面靜電勢極大點和極小點,pdb文件的B因子那一列的數據是靜電勢數值(kcal/mol)。然后再選擇6將所有分子表面頂點導出到當前目錄下vtx.pdb文件中,B因子是每個頂點的靜電勢數值。另外,如果你還沒有當前分子的幾何結構文件,應再選擇5然后輸入furan.pdb把當前體系的坐標導出到當前目錄下furan.pdb中。
3 在VMD中作圖
由于Multiwfn所用的圖形庫的限制,Multiwfn自身無法直接產生不同顏色填充的分子表面圖,所以需要借助VMD來實現。下面的操作步驟對于當前體系比較適合,對于其它體系,應舉一反三,根據實際情況進行略微調整。
啟動VMD,然后把furan.pdb、surfanalysis.pdb和vtx.pdb按順序依次拖動到VMD主窗口里(VMD Main),它們的ID將分別是0、1、2。選擇Display-Depth Cueing將它關掉,否則圖像會有些朦朧。進入Graphics-Colors,選Display-Background-White將背景改為白色,并且在此界面的Color-Scale標簽頁里選擇BWR,使分子表面的色彩根據數值范圍由小到大以 藍-白-紅 的方式變化。選Display-Axes-Off不讓坐標軸顯示出來。
注:如果希望每次啟動VMD時都自動做如上操作,可以在VMD目錄下vmd.rc文件末尾添加以下4行
display depthcue off
color scale method BWR
color Display Background white
axes location Off
進入Graphics-Representations,然后執行以下3步來分別設定分子結構、分子表面極值點、分子表面頂點的顯示方式。(如果只是想簡單地看一下表面靜電勢分布,只要做第3步即可)
(1)在Selected Molecule一欄里選擇furan.pdb,Drawing Method選Licorice,Bond Radius減小到0.1。
(2)將Selected Molecule一欄切換到surfanalysis.pdb,在控制臺輸入mol modstyle 0 1 VDW 0.06 (0和1是顯示方式編號和體系的ID,此命令代表用大小為0.06的VDW球顯示。由于GUI中VDW球最小只能設到0.1,故這里用命令行來實現)。然后在Selected Atoms里輸入carbon并回車,然后將Coloring Method選為ColorID,并且在右邊新出現的框里選Orange2。此時分子表面極大點就通過橙色小圓球顯示出來了。點擊Create Rep按鈕創建新顯示方式,在Selected Atoms里輸入oxygen并回車,然后將ColorID右邊的框設為Cyan,此時分子表面極小點就通過青色圓球顯示出來了。
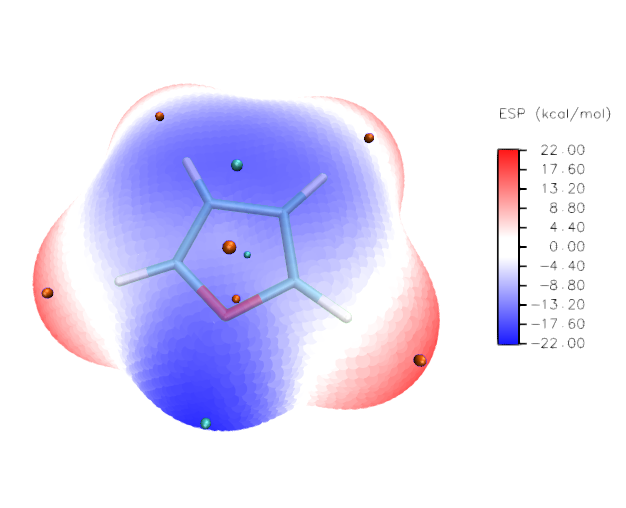
(3)在Selected Molecule一欄里選擇vtx.pdb,Drawing Method選Points,Size設為25(設多大合決于視角的遠近,在當前視角下應當讓size恰好足夠大,使分子表面上的頂點緊密相連,不留明顯空隙),Coloring Method選Beta(根據pdb文件里B因子那一列的數據,此例即靜電勢數值進行填色),在Trajectory標簽頁里將Color Scale Data Range填上-22和22并點擊Set,代表色彩刻度設為-22~20kcal/mol(其實此例用默認的色彩刻度范圍就可以,這里只是為了取個整)。現在分子表面填色圖就出現了。越藍的區域靜電勢越負,越紅的區域越正,白色區域的靜電勢數值在0附近。
之后給圖上加上色彩刻度軸。選Extensions-Visualization-Color Scale Bar,Color bar width設為0.08,Display title選on并且將Color bar title里寫上ESP (kcal/mol),Minimum和Maximum scale value分別填-22和22,Number of axis labels輸入10,Color labels選Black,Label format選Decimal。然后點Draw Color Scale Bar按鈕,色彩刻度就出現在畫面中了,并且VMD Main窗口中多出了一個名為Color Scale Bar的一項。然后調整它的大小和位置,即雙擊VMD Main窗口中Color Scale Bar那一項當中的F標簽使之變為紅色(即不讓色彩刻度軸在畫面中的位置凍結),而雙擊其它項目的F標簽使它們的F變為黑色(讓它們的位置凍結住)。然后點擊VMD的OpenGL圖形窗口激活之,按t鍵進入平移模式,然后拖動鼠標將色彩刻度軸放置到合適位置,并且用鼠標滾輪調整它的大小。調合適之后再按r鍵恢復旋轉視角模式,并且在VMD Main里將Color Scale Bar那一項的F重新雙擊成黑色,而其它三項的F重新雙擊為紅色。
當前的顯示效果如下
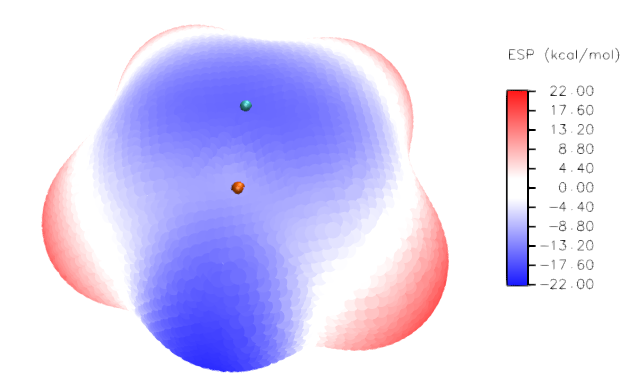
此圖還有許多地方值得進一步調整,也就是讓分子結構顯示出來(現在都被表面頂點掩蓋了)、給分子表面極值點標上具體數值、讓表面極值點更清楚地顯示,特別是藏在分子表面后方的極值點也顯示出來。為達到這些目的,需要利用photoshop。由于步驟比較細碎,筆者不把所有具體操作都敘述一遍,否則太羅嗦。只要會基本的photoshop操作的人都應該能理解應該怎么實現。
4 通過Photoshop改進作圖效果
將分子調整到一個合適的角度,然后在VMD main窗口里把所有條目的F標簽都雙擊成黑色來將它們固定住,以免隨后的操作過程中不慎旋轉了體系。
在VMD main窗口里面雙擊與furan.pdb和surfanalysis.pdb對應的條目的D標簽使其變紅,此時窗口內就只有分子表面和色彩刻度軸顯示了出來。然后按Alt+Printscreen鍵將窗口截圖,在photoshop里按Ctrl+N然后按OK,再用Ctrl+V把截的圖粘貼進去。此時圖像大小正好和VMD窗口大小完全一致。
在VMD main窗口里面只讓分子結構顯示出來,將背景改為藍色(只要不是白色就行,否則會和氫原子的白色連在一起),然后將窗口截圖并且粘貼進之前的ps窗口里成為新的圖層。選擇魔棒工具,Tolerance設0,Contiguous的對勾取消,然后點擊圖中藍色區域把背景區域選上,之后按delete鍵去除背景。之后將圖層的不透明度(Opacity)改為40%,這樣分子結構就會以半透明方式疊加到分子表面圖上了,而且疊加的位置完全精確。ps窗口里的圖像目前如下所示
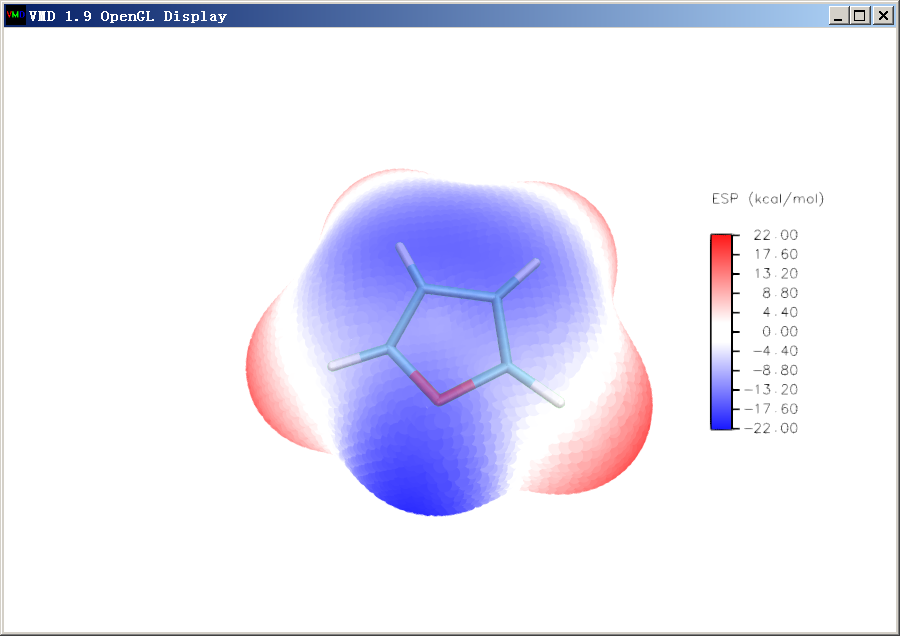
讓VMD窗口里只顯示出表面極值點,然后將窗口截圖也粘貼進ps里成為新圖層,同樣將藍色的背景選中并刪掉。然后用矩形選框工具恰當地把圖像主體和色彩刻度軸圈上,構圖滿意后點Ctrl+Q將多余的區域裁掉,目前圖像如下
可見目前這些表面極值點無論是正面還是背面的都是以完全不透明方式展現,看不出前后。因此我們把那些明顯處在背面的極值點都用矩形選框圈上,注意圈的時候一直按著shift鍵使選區依次累加。然后在圖上點右鍵選Layer via Cut,這樣處在圖中正面(或者邊緣)的極值點與處在圖中背面的極值點就分別用兩個圖層來儲存了。將后者的圖層的不透明度設為50%。
最后,在圖上用文本工具標上一部分極值點的靜電勢數值,數值在Multiwfn當中已經輸出過了。如果極值點比較密,可以同時繪制箭頭避免混亂。如果箭頭或者文字疊加在分子表面上,為了讓其邊緣清楚好看,建議在圖層混合選項中設定外發光。最終圖像如下所示
注:如果體系太大,不好把VMD里顯示的極值點和Multiwfn輸出的極值點數值對應上,就在VMD里面按鍵盤上的0,然后點擊一個極值點,此時這個極值點的信息就會輸出在VMD的文本窗口里。其中index序號是從0開始計的。對于極大點,index+1就是它在Multiwfn里的極大點序號;而對于極小點,index+1再減去極大點總數目就是它在Multiwfn里的極小點序號。對照一看一下就知道各個極值點的靜電勢值是多少了。
在筆者的J. Phys. Org. Chem., 26, 473-483 (2013)(https://onlinelibrary.wiley.com/doi/abs/10.1002/poc.3111)一文中,馬兜鈴酸的分子表面靜電勢圖也是通過類似方法繪制的,比此文的例子更復雜,在這里一起貼出
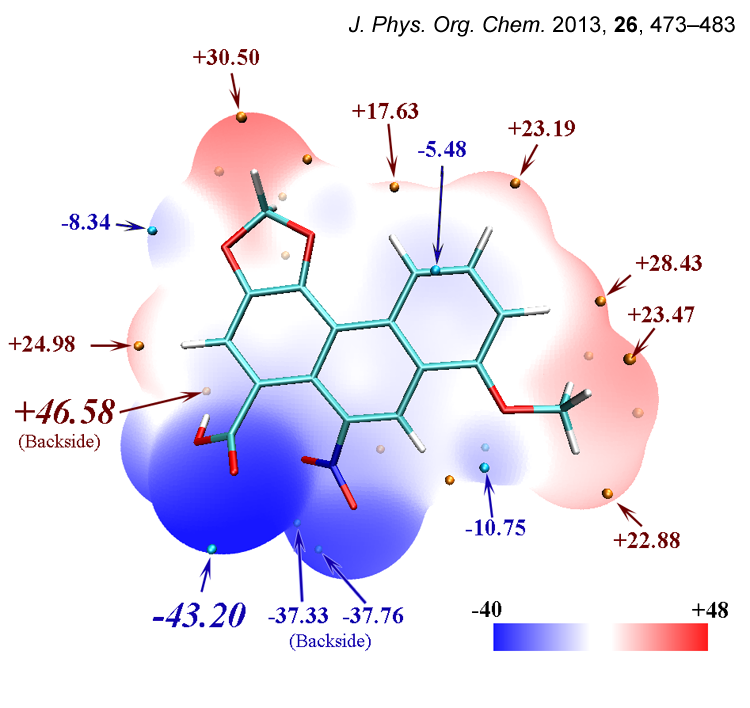
這是馬兜鈴酸的靜電勢定量分布圖,顯然沒有呋喃分布得均勻。正值區域、靜電勢接近0的區域占大部分分子表面,但是也有不小面積靜電勢非常負,這主要是羧基和硝基的氧的明顯負電荷導致的。
筆者在Struct. Chem., 25, 1521 (2014)(http://link.springer.com/article/10.1007/s11224-014-0430-6#)中給出了致癌物苯并[a]芘二醇環氧化物的分子表面靜電勢圖,也是通過類似方法繪制的
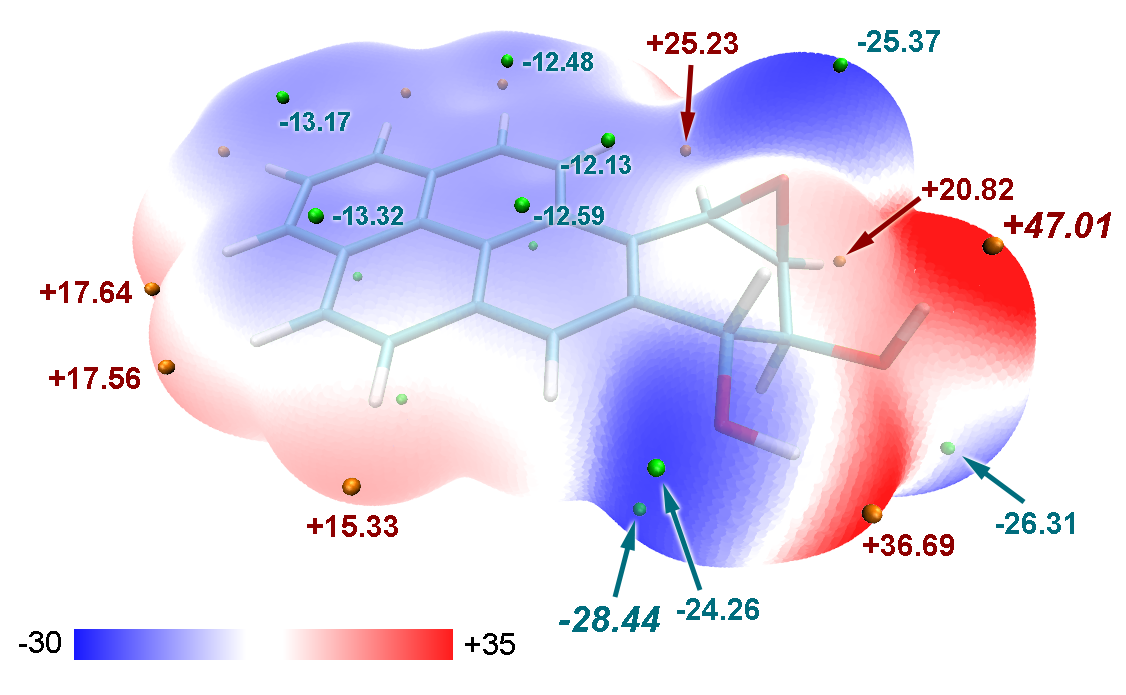
文中利用Multiwfn強大的局部定量分子表面分析功能,分別給出了此體系多環芳烴區域和其它部分分子表面上的靜電勢定量分布圖。可見在多環芳烴部分靜電勢分布廣度比起其它部分要窄,沒有靜電勢特別正也沒有靜電勢特別負的區域。繪制此圖很簡單,把第2節例子里輸入all的地方改為輸入多環芳烴區域的原子序號范圍,就能得到這部分的靜電勢分布統計情況,然后再用這個功能對其它部分也統計靜電勢分布,然后把兩部分靜電勢分布統計值都弄到Origin里一起繪制柱形圖即可。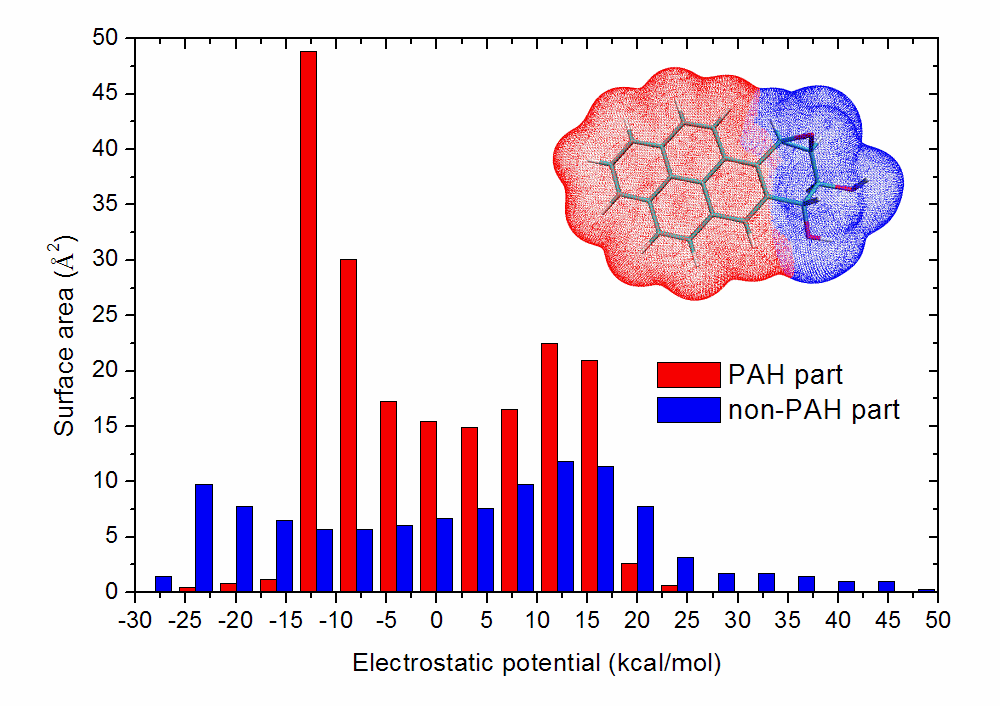
5 總結
本文介紹的繪制分子表面靜電勢圖的步驟比較瑣碎,需要很多手動操作,但這也是好處,使得我們可以很精細、隨意地調節顯示方式,而不受制于可視化程序所支持的選項。本文介紹的只是一般過程,建議大家多摸索以使顯示效果更好。利用本文相同的步驟,可以繪制各種各樣的實空間函數在分子表面的分布。例如繪制平均局部離子化能的圖,只要在Multiwfn的定量分子表面分析功能當中選選項2,然后再選2 Average local ionization energy,之后再選0啟動分子表面分析,隨后的步驟和前文都一樣。Fukui函數、雙描述符之類實空間函數并沒有出現在選項2給出的列表里,但是依然可以通過定量分子表面分析功能對它們在分子表面上的分布進行分析,并且隨后結合VMD繪制成填色圖,只不過操作過程稍微特殊一些,見手冊4.12.4節。
如果對分子表面分析有更多興趣,可以參看《使用Multiwfn的定量分子表面分析功能預測反應位點、分析分子間相互作用》(http://www.shanxitv.org/159)。
6 其它1:分析范德華表面穿透程度
將Multiwfn和VMD相結合,充分開動腦筋,還可以做很多有趣、有用的分析。例如,對水的二聚體中兩個水分別做定量分子表面分析,將得到的兩套表面頂點都放到VMD里同時顯示出來,就可以看到如下圖像
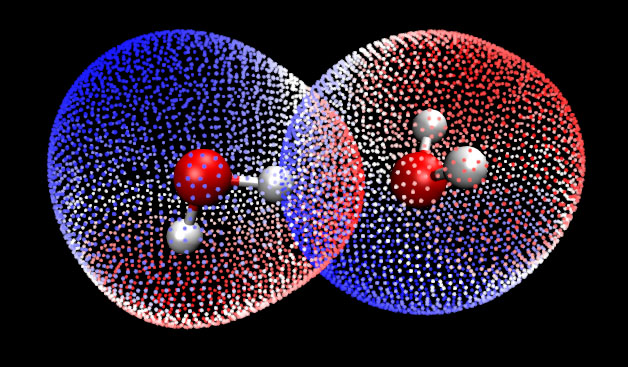
從圖中可以看到兩個水的范德華表面在形成二聚體后相互有明顯穿透,并且是靜電勢明顯為正(紅色)區域與靜電勢明顯為負(藍色)區域相互穿透,這表明水二聚體間的氫鍵作用本質主要是由于靜電吸引所導致的。
我們將視圖放大,并且不讓左邊的水顯示出來(因為它遮擋了表面頂點),然后找出兩個位置比較能反映穿透程度的表面頂點,按2鍵,然后分別點擊這兩個點,這兩個表面頂點間的距離就出現在圖上了,如下圖所示,其距離是1.12埃。這表明形成二聚體后,范德華表面總共被穿透了2*1.12埃,這叫做相互穿透距離(mutual penetration distance),其值越大通常說明弱相互作用越強。
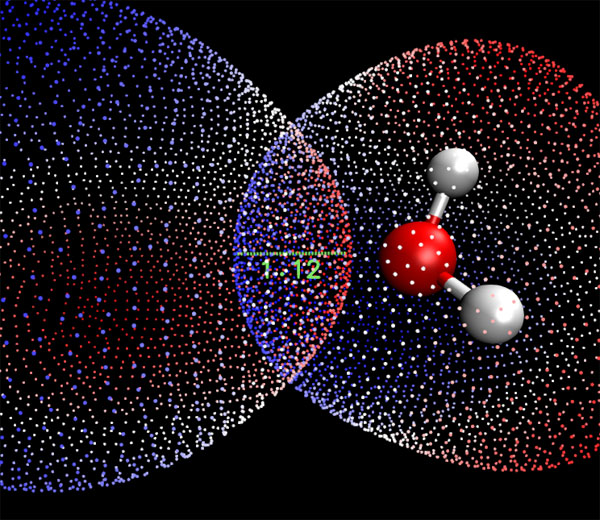
注:Mutual penetration距離其實有不同具體計算方法,最簡單粗糙的是直接拿兩個原子的范德華半徑和減去原子間距。而本文這種計算方法無疑是最準確的,因為精確反映了范德華表面的實際形狀。一個簡單點的但也比較準確的方法是在Multiwfn中做完定量分子表面分析后選10得到原子的非鍵原子半徑(non-bonded atomic radius),也就是其原子核距離范德華表面最近的距離。將相互作用的兩個原子的非鍵半徑相加減去它們的實際距離即是范德華表面相互穿透距離。
7 其它2:只繪制指定數值范圍的分子表面
得益于VMD極其靈活的范圍選擇功能,可以在VMD里只顯示指定數值范圍的分子表面,一些看似麻煩的分析也變得極為簡單。例如有的文章對鹵鍵定義了ω角度,如下圖所示,其中陰影覆蓋的弧面代表鹵原子分子表面靜電勢為正的區域。
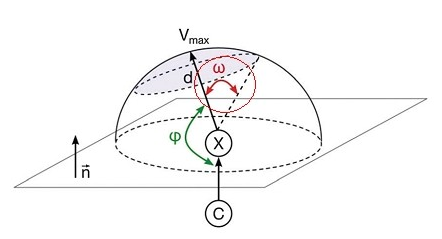
這里我們對Cl2來得到ω。按照前文所示的方法在VMD里繪制出分子表面和靜電勢極值點后,在對應于分子表面頂點的那個顯示方式里把Selected Atoms從默認的all改為beta > 0。如前所述beta值代表相應頂點的函數值,因此這樣設就會只顯示出分子表面上靜電勢為正的部分了。然后按數字鍵3進入角度測量模式,依次點擊靜電勢極大點、Cl原子,以及靜電勢為正的區域的邊緣的任意一個頂點,圖中就顯示角度了,如下所示(可以在Graphics-Labels里把原子標簽關掉并調整字體大小和粗細)。
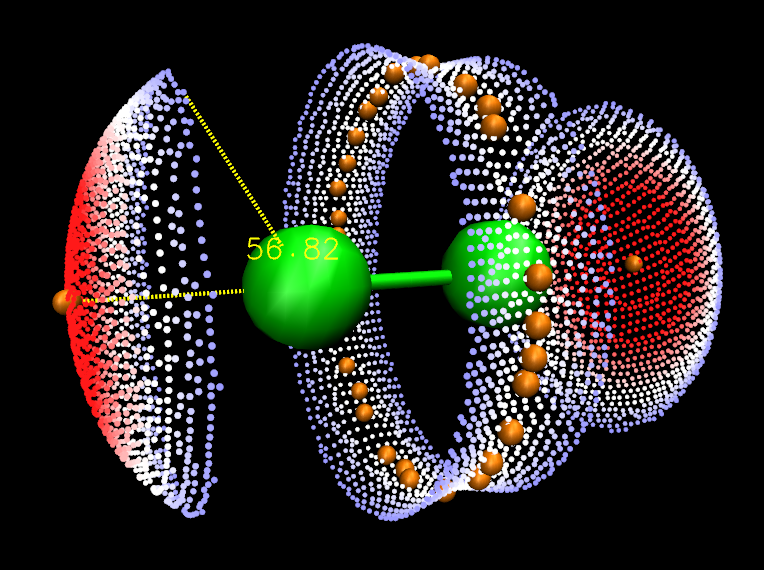
可見,對于與分子表面相關的分析來說,Multiwfn+VMD=むてき!
8 其它3:解決VMD繪制表面頂點時的一些顯示問題
在VMD繪制表面頂點時可能會看到圓點的邊緣透露出背景色,如下所示,圓點邊緣是背景色黑色,導致很不美觀
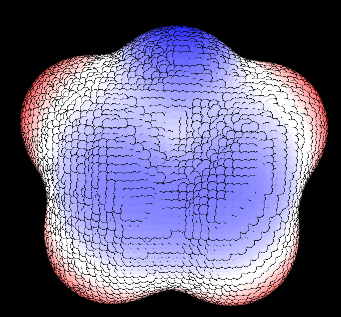
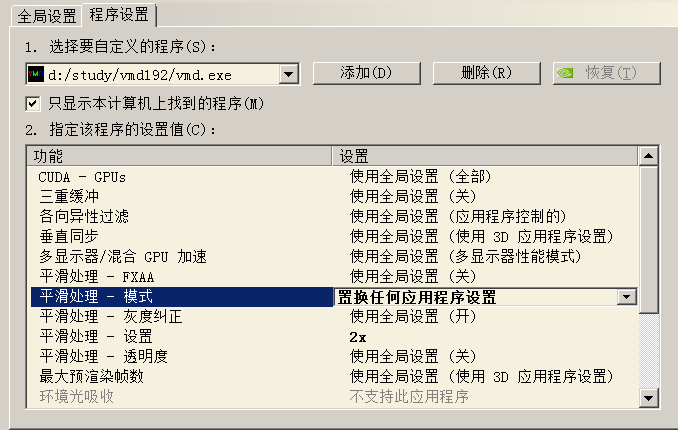
對于nvidia顯卡,解決方法是在驅動控制面板里對VMD強制開啟抗鋸齒,并且別用FXAA方式的抗鋸齒,如下所示。這么設過就和上文的圖像效果一樣了。
另外,當用point方式繪制表面頂點時,如果顯示的是立體的大圓球,這是開了GLSL導致的,應當改用Display-Rendermode-Normal。