談談分子半徑的計算和分子形狀的描述
經常有人談及和分子形狀有關的話題,本文就說說常用的三種模型怎么討論和計算,即球形模型、圓柱模型和矩形盒子。
1 球形模型
用球形模型,相當于討論怎么計算分子的半徑。首先要認識到,分子半徑不是一個可觀測量,在計算方法上也不可能有唯一的定義,因此“怎么計算分子半徑”這個問題本身就是不嚴格的。這段話和筆者在《談談分子體積的計算》(http://www.shanxitv.org/102)當中文首說的話幾乎如出一轍。對于半徑、體積這類沒有唯一定義的問題,應該先想清楚計算原理是什么,不要盲目計算。
談到分子半徑,這就自然已經假設了分子必須能較好作為球形來描述,如甲烷、球蛋白,至少也不能偏離球型太遠。如果偏離球形很遠,例如分子是大平面,像是卟啉或多環芳烴,或者是長鏈,如直鏈烷烴,那么就不可能靠球形模型來描述,也因此討論半徑對于這種問題是無意義的。
計算分子半徑的做法有很多,下文將列舉一些,但肯定還有不少方法本文中沒有提及。
1 尋找分子內相距最遠的兩個原子距離,然后加上相應兩個原子的范德華半徑,得到分子直徑,除以2即是分子半徑。
2 尋找與分子幾何中心最遠的原子距離,加上其范德華半徑,得到分子半徑。(更嚴格地說,應該是找出加上相應范德華半徑之后幾何中心離哪個原子最遠,不過本文就不考慮這點了)
上面這兩種方法計算起來比較容易,特別是方法1,直接在分子可視化工具里測量一下就行,不過原子很多的時候測量起來可能困難些。這兩種方法的計算在Multiwfn程序(http://www.shanxitv.org/multiwfn)里可以直接實現。這里以水分子為例。首先載入Multiwfn支持的含有分子結構信息的文件,如.pdb、.xyz、.mol等都可以,然后進入主功能100,然后選21,再輸入all。屏幕上馬上出現基于分子結構計算出的各種信息,其中可看到幾何中心坐標
Geometry center (X/Y/Z): 0.00000000 0.00000000 -0.27838535 Angstrom
下面會看到
Maximum distance is 1.517906 Angstrom, between atom 2(H ) and 3(H )
代表2H和3H距離最遠,相距1.52埃。H的bondi定義的范德華半徑為1.2埃(見JCP,68,441(1964)),因此按照方法1計算的分子半徑即為(1.52+2*1.2)/2=1.96埃。
從輸出信息中還看到
The atom farthest to geometry center is 2(H ) Dist: 0.784570 Angstrom
說明與幾何中心距離最遠的是2H且相距0.784埃,加上H的范德華半徑,0.784+1.2=1.984埃即是方法2算得的分子半徑。
如果用的是2018-Jun-6之后更新的Multiwfn,以方法1計算半徑還有更省事的辦法,即進入主功能100的子功能21之后,輸入size,屏幕上直接就會輸出Radius of the system: 1.959 Angstrom這樣的信息。
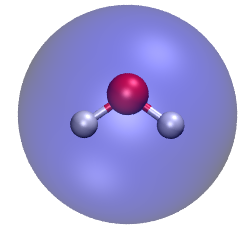
可以利用VMD程序來畫出球形,來直觀地比較分子結構和圓球的定義。將水分子結構文件載入VMD,在控制臺輸入以下命令,來繪制一個透明的半徑是1.984埃的圓球,其球心在幾何中心(上述過程中Multiwfn的輸出信息中已包含了幾何中心位置)。draw material Transparent
draw sphere { 0 0 -0.278 } radius 1.984 resolution 30
color Display Background white
然后在Graphics - Representation里把顯示方式改成CPK,最后效果如下,可見這個圓球比較好地包裹了整個水分子。
對于凝聚相體系,由于分子間互相擠壓或者存在諸如氫鍵這種相互作用,原子的范德華半徑會在一定程度上被穿透,所以,上述方法估算的分子半徑可能有所高估。因此,加上原子范德華半徑時可以考慮給范德華半徑乘個<1的刻度因子來體現這個效應。
上述兩種方法結果往往相差不大,由于計算簡便,適合計算很大體系的半徑,如蛋白質。
3 尋找分子表面最遠的兩個點的距離作為分子直徑,除以2即是分子半徑。分子表面的定義很多,見《談談分子體積的計算》的討論,不同的表面適用于不同情況,例如Bader建議以電子密度0.001/0.002 a.u.的等值面分別作為氣相/凝聚相時的范德華表面。這種方法類似方法1,但更嚴格、準確。
4 尋找分子幾何中心距離分子表面最遠處的距離作為分子半徑。這個方法類似方法2,但比它更嚴格、準確。
方法3、4可以通過Multiwfn的定量分子表面分析功能實現。做法是啟動Multiwfn后,載入相應體系的含有波函數信息的文件(如.wfn、.fch、.molden),然后進入主功能12,選0開始構建分子表面并進行表面性質的分析(默認是0.001 a.u.等值面作為分子表面,可用選項1修改)。然后在后處理菜單中選10,再輸入g,屏幕上就會輸出分子表面上距離幾何中心最近和最遠的距離;如果輸入f,程序會計算出分子表面上最遠兩個點的距離,以及這兩個點及其中點的坐標,之后可以像前例一樣通過中點位置以及最遠距離的一半作為半徑在VMD中畫出透明的球面。
(注:主功能12默認分析的是分子表面上的靜電勢,而靜電勢計算對大體系會比較慢。由于我們的目的只是得到表面頂點而非分析表面的性質,所以可以進入主功能12之后先選擇2設置被映射的函數,再選-1,然后選0開始分析。這里0代表考察的是用戶自定義函數,這個函數默認情況下是個常數,沒有任何計算耗時。)
對于水分子,在0.001 a.u.等值面作為范德華表面的情況下,利用Multiwfn程序得出的方法3定義的半徑為1.99埃,方法4定義的半徑為2.15埃。如果使用0.002 a.u.等值面當做范德華表面,則方法3、4定義的分子半徑將縮小一些,分別為1.85埃和2.02埃。
由于方法3、4都依賴于電子密度信息,所以用不同理論方法、基組計算波函數文件,所得半徑結果會有些許差異。
對于很大體系,利用方法3、4來計算半徑總耗時會頗長,所以此時還是建議用方法1、2,盡管原則上精度會差一些,但是對于體系這么大的情況,那點精度的損失是完全可以忽略的,更何況分子半徑的定義本身就是含糊的。
5 計算分子體積V,然后根據V=4/3*pi*r^3來解出分子半徑r。例如水分子,電子密度0.002 a.u.的等值面內的體積約為20.92埃^3,因此r=1.71埃。如果用0.001a.u.等值面當做范德華表面,則體積約為26.36埃^3,因此r=1.85埃。通過電子密度等值面定義的體積可以利用Multiwfn程序得到,見《談談分子體積的計算》。
之所以我們用這個方法算得的半徑比前述的方法略小,這并不難理解,因為此方法相當于把任意形狀的分子給攢成了理想球形,所以看起來當然變小了。如果分子偏離球形更多,那么此方法所得半徑將小得更多。
6 假定分子之間是無縫隙地堆積起來的,根據物質的密度推出分子體積和半徑。可用先用此公式解出分子體積:M/(V*10^-27*NA)=rho,即V=M/rho/6.02*10000,其中M是分子量,rho是密度(g/L)。例如水的rho=1000g/L,M=18.0,故V=29.9埃^3。將分子當成理想球形解得的分子半徑即為r=1.92埃。
7 對于AHn類分子(如H2O、NH3、CH4等),將重原子作為球中心,逐漸增加半徑,當半徑增大到一定值時球內電子密度恰包含了體系內98%的電子密度時,將這個半徑作為分子半徑。這個定義是JCP, 56, 4419(1972)提出的,如今很少有人用,算起來也不方便,也不是很普適。此文算出的水的半徑是1.67埃。
除上述這些,分子半徑/直徑還可以根據某些實驗來給出。比如可以根據能否通過指定孔徑的分子篩來確定分子直徑。這和動力學直徑關系密切,用Multiwfn可以容易地算出動力學直徑,見《使用Multiwfn計算分子的動力學直徑》(http://www.shanxitv.org/503)。順帶一提,Multiwfn還有計算分子孔洞直徑的功能,見《使用Multiwfn計算分子和晶體中孔洞的直徑》(http://www.shanxitv.org/643)。
對于聚合物,也包括蛋白質等體系,經常討論回轉半徑用來討論分子構象延伸的廣度。比如在一個蛋白去折疊過程中蛋白質鏈會伸展得越來越廣,回轉半徑也因此逐漸增大。但有人以為回轉半徑可以當成分子半徑,這是明顯錯誤的。例如,我們用一個酪氨酸激酶作為示例,總共有1806個原子,我們用前述方法2得到的半徑是30.638埃,而回轉半徑是16.842埃。以幾何中心作為球心,用方法2所得半徑和回轉半徑繪制的球分別以藍色和黃色表示,所得圖像如下所示
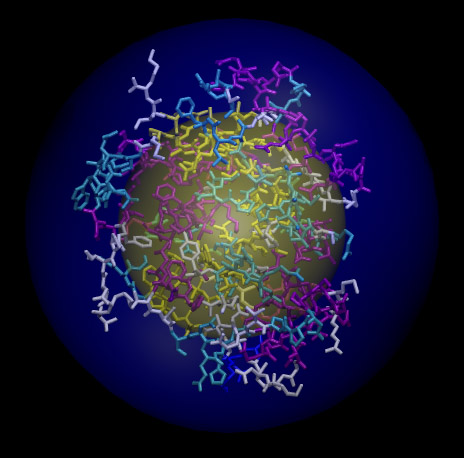
可見回轉半徑根本沒有包住整個體系。所以說,回轉半徑適合衡量結構的延展廣度,但不適合作為分子半徑的定義。回轉半徑也可以通過Multiwfn得到,依然是進入主功能100里的功能21,輸入all之后輸出的信息里就有整個體系的回轉半徑,即Radius of gyration:后面的數值。
2 圓片/圓柱模型
對于大平面分子,我們描述其分子形狀顯然就不適合再用圓球模型了。諸如卟啉這樣的分子,我們適合使用扁圓片模型來描述它的形狀。圓片的厚度可以簡單定義為碳原子范德華半徑的二倍,而圓片的半徑可以直接使用前述的方法1~4來得到。
對于長鏈體系,其形狀適合用圓柱模型來表示。我們可以沿用前述的方法1~4來得到圓柱的長,而圓柱的半徑需要根據實際結構來判斷。
一種更為嚴格、自動化的確定圓柱參數的方法是不斷調整圓柱的參數,使得圓柱能夠包圍所有原子且圓柱的尺寸最小。這個方法可以借住CYL程序來完成。此程序可以在這里免費下載到Linux和Mac OS X版:http://petitjeanmichel.free.fr/itoweb.petitjean.freeware.html
CYL程序下載后直接用諸如./bin.CYL.1.linux64.intel命令運行即可,然后輸入輸入文件的路徑。輸入文件格式類似這樣:
63 0 0 0 //63代表有63個點。后面的三個零不用管,一般都需要寫。
-12.235 -1.599 -0.445 //各個點的XYZ坐標,對于本文的情況也就是各個原子坐標
-10.905 -1.894 -0.414
-9.920 -0.847 -0.263
-10.369 0.509 -0.148
-11.788 0.778 -0.186
-12.685 -0.238 -0.329
-8.546 -1.127 -0.228
...略
對于點比較多的情況,計算要花不少時間。算完后屏幕上會看到諸如
Axis : 0.996020E+00 0.491685E-01 0.743408E-01
Center: 0.529569E+00 0.669861E+00 0.659797E+00
Radius : 0.305604E+01
Half-height: 0.143397E+02
Axis就是圓柱的軸的矢量,Center是圓柱中心,Radius是圓柱半徑,Half-height是圓柱長度的一半。利用這樣的信息,我們可以將圓柱的圖形畫出來,以便和分子結構相比較。我們使用VMD程序內建的繪圖命令來畫圖。
在啟動VMD后,先載入分子結構文件,然后在控制臺中將以下內容復制進去來定義molcyl繪圖命令
proc molcyl {cenx ceny cenz axisx axisy axisz rad halfh} {
draw material Transparent
set startx [expr $cenx-$axisx*$halfh]
set starty [expr $ceny-$axisy*$halfh]
set startz [expr $cenz-$axisz*$halfh]
set start "$startx $starty $startz"
set endx [expr $cenx+$axisx*$halfh]
set endy [expr $ceny+$axisy*$halfh]
set endz [expr $cenz+$axisz*$halfh]
set end "$endx $endy $endz"
draw cylinder $start $end radius $rad filled yes resolution 40
}
然后利用molcyl命令就可以根據CYL的結果畫出圓柱的圖了,此命令的參數順序是:柱中心XYZ坐標、軸矢量的XYZ分量、圓柱半徑、圓柱半高。對于上例,我們輸入molcyl 0.529569 0.669861 0.659797 0.996020 0.0491685 0.0743408 3.05604 14.3397之后就可以在屏幕上看到如下效果:

可見圓柱恰好套住了整個長鏈分子,所以CYL的結果是合理的。但要注意CYL給出的圓柱的參數沒有把范德華半徑考慮進去,而從圖中可看到,與圓柱邊緣相接的都是H,所以圓柱的半徑和長度應當分別加上氫的范德華半徑的一倍和二倍。
值得一提的是,長鏈體系往往具有很強柔性,不同構象下描述它的圓柱的參數可能有巨大差異,所以要注意說明是什么構象下計算的。
3 矩形盒子模型
一些體系適合矩形盒子模型,用長、寬、高來描述,比如下面的蒽,可以被矩形盒子很好擴住,比起圓球和圓柱模型都更合適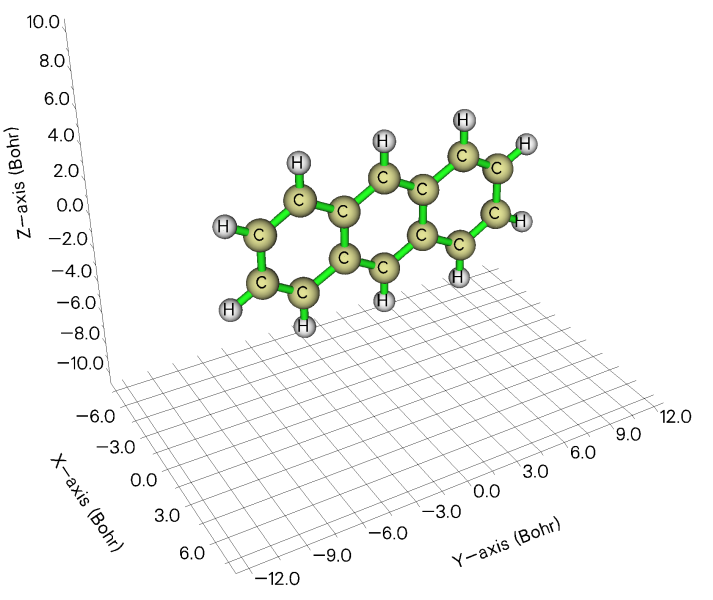
注:如果你的分子是復雜、歪斜的而非上圖那么正,或者你想純粹基于幾何坐標快速計算長寬高,務必看后來寫的《使用Multiwfn計算分子的長寬高》(http://www.shanxitv.org/426)。
我們需要獲得能夠擴住這個分子范德華表面(這里用電子密度0.001等值面來定義)的最小盒子尺寸。這可以通過Multiwfn來做。先用Gaussian計算出這個分子的wfn或fch文件,載入進Multiwfn后選
12 //定量分子表面分析
2 //修改被映射的函數
-1 //改為用戶自動以函數(計算沒有耗時)
0 //開始分析
然后從屏幕的輸出中能夠找到ρ=0.001定義的分子表面的頂點的X、Y、Z方向上的最小和最大值:
Min-X: -2.0419 Max-X: 2.0420 Angstrom
Min-Y: -5.9055 Max-Y: 5.9055 Angstrom
Min-Z: -3.7916 Max-Z: 3.7916 Angstrom
比如盒子的Z長度就是3.7916*2=7.58埃。
我們可以用VMD來繪制出這個盒子。首先把以下內容復制到VMD的控制臺里定義一個新命令drawbox
proc drawbox {xmin ymin zmin xmax ymax zmax} {
draw color blue
draw line " $xmin $ymin $zmin " " $xmax $ymin $zmin "
draw line " $xmin $ymin $zmin " " $xmin $ymax $zmin "
draw line " $xmax $ymin $zmin " " $xmax $ymax $zmin "
draw line " $xmin $ymax $zmin " " $xmax $ymax $zmin "
draw line " $xmin $ymin $zmax " " $xmax $ymin $zmax "
draw line " $xmin $ymin $zmax " " $xmin $ymax $zmax "
draw line " $xmax $ymin $zmax " " $xmax $ymax $zmax "
draw line " $xmin $ymax $zmax " " $xmax $ymax $zmax "
draw line " $xmin $ymin $zmin " " $xmin $ymin $zmax "
draw line " $xmin $ymax $zmin " " $xmin $ymax $zmax "
draw line " $xmax $ymin $zmin " " $xmax $ymin $zmax "
draw line " $xmax $ymax $zmin " " $xmax $ymax $zmax "
}
然后輸入drawbox -2.0419 -5.9055 -3.7916 2.0420 5.9055 3.7916,就可以根據前面得到的數據繪制出相應的盒子。可見盒子能很好地擴住分子。
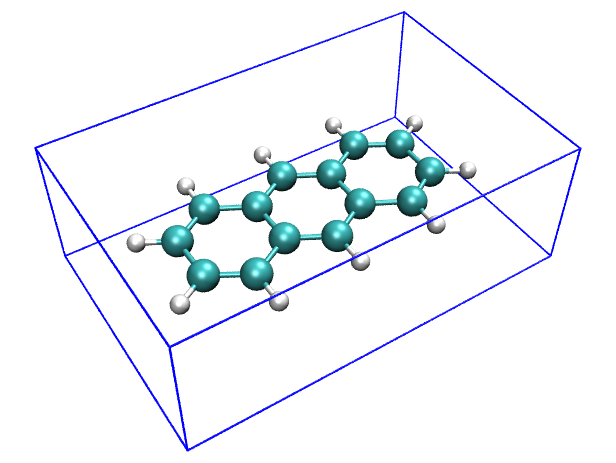
對于結構很復雜的體系,想要比較準確地且解析地描述它的形狀,無論是圓球、圓片/圓柱或矩形盒子模型都已經不再適用,這種情況可以用球諧函數展開來表現,這在本文就不談了。