使用Multiwfn計算分子的動力學直徑
使用Multiwfn計算分子的動力學直徑
文/Sobereva@北京科音
First release: 2019-Aug-19 Last update: 2022-Jun-17
1 相關知識&計算原理
分子直徑有很多不同定義。在分子分離研究領域中經常用動力學直徑(kinetic diameter)體現分子尺寸,這個量沒有一般性的方法,直到現在,主要來源還是Breck很老的1974年的Zeolite Molecular Sieves; Structure, Chemistry and Use一書,數值是通過氣體在不同溫度和壓力下的第二維里系數實驗和Lennard-Jones勢的形式推出來的,動力學直徑對應LJ作用為0的情況。
在J. Phys. Chem. A, 118, 1150 (2014)文中,作者提出了基于分子電子密度等值面的估計分子動力學直徑的方法。對于文中測試的一系列小分子,計算結果和被普遍采用的Breck書里的那些值有不錯的線性相關性,因此此文的做法可以作為一個普適型的算動力學直徑的做法。
分子表面可以通過電子密度等值面來定義,這點我在很多博文里提過,比如《使用Multiwfn和VMD計算分子表面積和片段表面積》(http://www.shanxitv.org/487)、《談談分子體積的計算》(http://www.shanxitv.org/102)、《使用Multiwfn的定量分子表面分析功能預測反應位點、分析分子間相互作用》(http://www.shanxitv.org/159)等。JPCA文中的思想是以這種方式定義分子表面,然后測量最短方向的等值面兩端的距離,即下面這些例子中的箭頭夾著的距離。
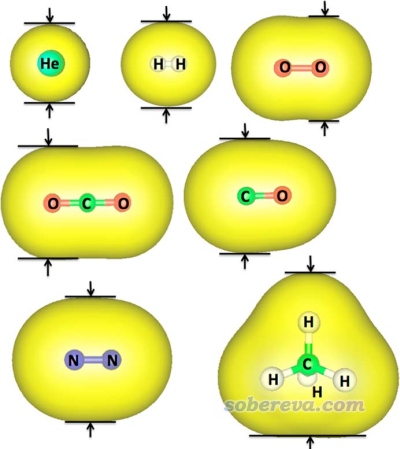
前述JPCA文中在PBE0/def2-TZVP級別下考察了不同電子密度下算的動力學直徑與Breck的值的線性關系,發現用0.0015 a.u.作為等值面定義效果最好。如下所示,這樣計算的幾個小分子的動力學直徑(縱坐標)與Breck書中的動力學直徑(橫坐標)的線性關系不錯。
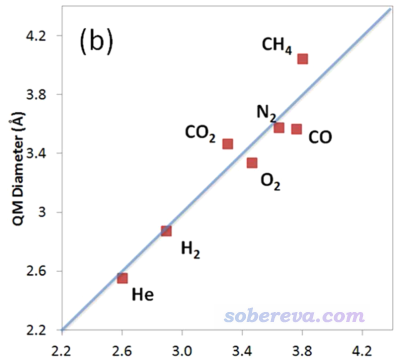
如JPCA文中表2所示,用0.0015 a.u.作為等值面數值計算后,原理上最好再乘上擬合出來的系數1.025。
此文的方法思想非常簡單,也沒有考慮分子在實際環境中電子的可極化效應導致對動力學直徑產生的影響。但不管怎么說,這個方法還是比較有實用性的,如果被考察的分子查不到現成的動力學直徑,不妨用這個方法粗略估計一下。
JPCA這篇文章的方法可以通過Multiwfn的定量分子表面分析功能非常容易地實現,下面就以CO2分子為例講一下如何計算。對Multiwfn完全不了解者參看《Multiwfn FAQ》(http://www.shanxitv.org/452)、《詳談Multiwfn支持的輸入文件類型、產生方法以及相互轉換》(http://www.shanxitv.org/379)。
2 計算實例:CO2
這里我們首先用Gaussian程序優化CO2并產生它的波函數。筆者用的是Gaussian16 A.03,計算級別用的和JPCA里相同,即PBE0/def2-TZVP。輸入文件如下
%chk=C:\CO2.chk
# PBE1PBE/def2TZVP opt
[空行]
Title Card Required
[空行]
0 1
C 0.00000000 0.00000000 0.00000000
O 0.00000000 0.00000000 1.25840000
O 0.00000000 0.00000000 -1.25840000
將CO2.chk用formchk轉換成CO2.fch,之后就可以用Multiwfn計算了,此文件可以從這里下載:http://www.shanxitv.org/attach/503/file.rar。用Multiwfn載入CO2.fch,然后輸入以下命令
12 //定量分子表面分析
1 //選擇表面的定義
1 //用電子密度等值面作為表面
0.0015 //用0.0015 a.u.作為等值面數值
6 //開始分析(但不考慮映射的函數,因為這不是我們目前感興趣的)
6 //將表面頂點坐標導出為當前目錄下的vtx.pdb
現在,通過vtx.pdb我們就可以考察動力學直徑了,有兩種方法可以實現。下文的“方法1”最普適,可以用于任意形狀體系,但需要借助VMD,且眼力不能太差,要有耐心;而“方法2”的做法省事得多,不過沒那么靈活,不適合形狀復雜的情況。
方法1:通過VMD測量表面頂點間距離
在http://www.ks.uiuc.edu/Research/vmd/下載VMD并安裝,啟動后將vtx.pdb拖入其中,在Graphics - Representation里將Drawing Method設為Points。然后在VMD Main窗口選擇Display - Orthographic使用正交視角以便于選取頂點。將圖形窗口拉大,按鍵盤上的2進入距離測量模式,恰當旋轉分子使得要選的頂點處在容易被選中的位置,然后按照JPCA文中的示意圖,點擊能反映動力學直徑的兩個位置的表面頂點。恰當選擇后,該體系兩個角度的圖像如下所示

從VMD的文本窗口中可以讀到精確數值:
Info) Added new Bonds label MOL1:C/MOL1:C = 3.385527
即動力學直徑是3.385埃,和Breck書中的3.31埃相符很好,和JPCA文中表1中的3.469埃也基本吻合(JPCA這篇文章沒說具體怎么測量的,但至少我們的測量方法是肯定恰當的)。
用此方法選取頂點時往往會碰到一個問題,就是從某個角度看選取的頂點似乎很合適,但換一個角度看又不那么合適。選取的技巧只可意會不可言傳,試多了自然就懂了。點上去之后如果發現不合適想刪除標簽,可以進入Graphics - Labels,在Atoms和Bonds里圈上條目后點Delete即可刪除。
方法2:通過Multiwfn直接統計表面頂點的最大、最小坐標
用這個方法前,必須先保證當前體系的最短方向沖著X、Y、Z坐標軸之一。比如對于CO2.fch,利用Multiwfn主功能0一看就可以看出最短軸是X或Y方向,如下所示,因此這個體系可以用下面的方法考察動力學直徑。
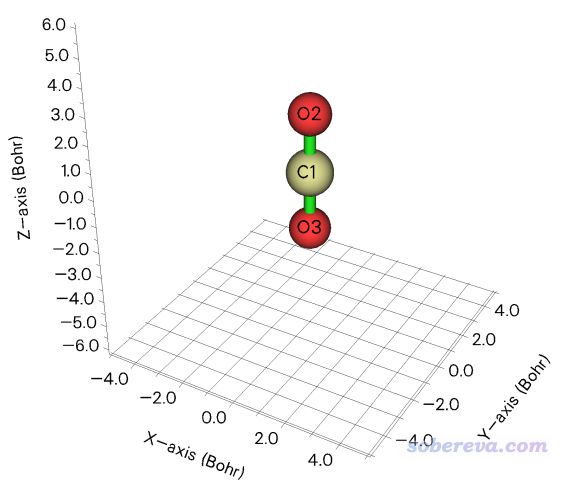
啟動Multiwfn并載入vtx.pdb,進入主功能100,然后進入子功能21,這個功能專門考察體系的幾何信息。然后輸入all,從輸出文件中可以找到以下信息
Minimum X is -1.69400000 Angstrom, at atom 134(C )
Minimum Y is -1.69400000 Angstrom, at atom 2112(C )
Minimum Z is -2.75500000 Angstrom, at atom 2177(C )
Maximum X is 1.69300000 Angstrom, at atom 4249(C )
Maximum Y is 1.69400000 Angstrom, at atom 2244(C )
Maximum Z is 2.75600000 Angstrom, at atom 2179(C )
由于Y軸方向原子位置(當前語境下對應表面頂點位置)最小值是-1.694,最大值是1.694,因此CO2的動力學直徑是1.694*2=3.388埃。此數值和“方法1”肉眼測量的3.385埃很接近,顯然用這種方法比方法1省事得多,還可以避免肉眼測量可能造成的不準確。但實際研究的分子往往比此例復雜得多,此方法未必總適用。有時雖然可以用,但Gaussian算之前可能需要將分子先恰當旋轉到合適朝向,并且計算時加上nosymm關鍵詞避免自動被旋轉,詳見《談談Gaussian中的對稱性與nosymm關鍵詞的使用》(http://www.shanxitv.org/297)。
值得一提的是,利用Multiwfn和VMD,不僅可以像上文這樣考察分子的外徑,還能通過測量表面頂點距離的方式考察環狀、籠狀分子的內徑。在《一篇最全面、系統的研究新穎獨特的18碳環的理論文章》(http://www.shanxitv.org/524)介紹的筆者的論文中就用Multiwfn+VMD測量了18碳環的內徑,并討論什么樣的分子有可能穿過去,感興趣的讀者建議看看。
Multiwfn還專門有計算分子孔洞直徑的功能,見《使用Multiwfn計算分子和晶體中孔洞的直徑》(http://www.shanxitv.org/643),只不過這是基于原子坐標和范德華半徑,而非基于電子密度等值面算的。