談談分子體積的計算
我曾在網上數次看到有人問怎么算分子體積,從提問以及很多人的回答上看有不少人對這問題存在錯誤的認識,本文就來談談這個問題,既會說明基本原理,也會說明怎么具體計算。本文用到的Multiwfn程序可以在http://www.shanxitv.org/multiwfn免費下載,相關知識見《Multiwfn FAQ》(http://www.shanxitv.org/452)。
1 分子體積的定義
首先要認識到,分子體積不是一個可觀測量,在計算方法上也不可能有唯一的定義,因此“怎么計算分子體積”這個問題本身就是不嚴格的。分子體積就是分子表面內部空間的體積,由于分子表面也沒有唯一的定義,所以不同的分子表面的定義就會給出不同的分子體積定義。首先看一個簡單分子的例子
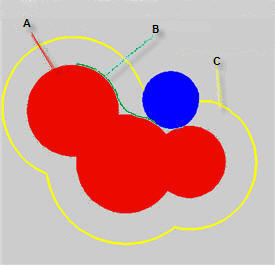
紅色區域是每個原子的范德華球(以原子核為中心,半徑為范德華半徑的球體)的疊加,這片區域就是分子的范德華體積,其表面也就被稱作范德華表面。圖中藍球代表作為探針的溶劑分子(顯然溶劑實際形狀并不是球形,所以這個藍球半徑是“等效”的溶劑半徑,在計算程序中通常是可調參數),讓這個藍球緊貼著分子范德華表面在各處滾一遍,就產生了圖中綠色的軌跡,對應的表面叫做Connolly表面,由于溶劑分子不能觸及到這個表面內的空間,所以也被叫做solvent-excluded表面,其內部區域的體積就叫做solvent-excluded體積。圖中黃色是藍球滾動時藍球中心經過的表面,這個表面叫溶劑可及表面,其表面積就是所謂的SASA。
平時討論化學問題時最常用的體積就是范德華體積。實際上范德華體積的定義并不唯一,上面說的原子范德華球疊加是比較簡單的定義方式。這種定義存在兩個缺點:
(1)原子范德華半徑沒有唯一定義,不同研究者給出的值往往存在很大分歧,有些研究者給出的半徑只包含很少量元素。關于原子半徑的知識,看《簡談原子半徑》(http://www.shanxitv.org/255)
(2)沒有考慮到電子效應。因成鍵導致的電子轉移、電子云變形效果都被忽略了。AIM理論的提出者Bader在J. Am. Chem. SOC., 109, 7968 (1987)提出過一種范德華體積的定義,消除了前述基于原子球疊加定義的弊端,并已被廣泛接受,也就是若分子處于氣相,則將電子密度為0.001 a.u.的等值面作為范德華表面,這種表面通常能夠囊括分子98%以上的電子,這種范德華體積通常比范德華球疊加得到的范德華體積要大一些。而對于處于凝聚態的分子,考慮到分子間擠壓、各種形式的相互作用,會使得范德華表面有一定穿透,Bader建議用電子密度為0.002 a.u.的等值面作為范德華表面(顯然其體積小于0.001等值面對應的體積)。
用Bader的定義計算范德華體積由于涉及到計算很多點的密度,所以比使用范德華球疊加方式的定義要慢很多,對于大分子,Bader的定義會因為太昂貴而無法使用。如果你是做量子化學計算的人,我總是建議用Bader的定義,因為只要常用的DFT計算能算得動,Bader定義的范德華體積在Multiwfn中也總是能算容易地算出來的。筆者在不少文章里都使用了Bader的這種范德華定義,大家可以在發文章時引用來支持這種定義的使用:
Carbon, 171, 514-523 (2021)
J. Mol. Graph. Model., 38, 314-323 (2012)
J. Phys. Org. Chem., 26, 473-483 (2013)
Struct. Chem., 25, 1521-1533 (2014)
如果你要算特大體系,比如蛋白質什么的,就只能算原子球疊加定義的范德華體積了。
下面第2、3節分別介紹兩種計算分子體積的方法,在Multiwfn程序里都可以輕松實現。蒙特卡羅法可以用于原子球疊加和Bader定義的范德華體積的計算,MT方法只能用于計算Bader定義的范德華體積。計算Bader定義的范德華體積時我一般建議用MT方法,直接看第3節即可。
2 蒙特卡羅方法計算體積
計算范德華體積常用的方法之一是蒙特卡羅方法。首先,設立一個矩形盒子,將整個分子擴住,并且各個方向上都預留一定空間以避免將范德華表面截斷,記這個盒子的體積為V_box。然后,在盒子里隨機分布m個點,依次檢驗這些點是否符合條件。對于范德華球疊加方式的定義,如果當前點與任何一個原子核的距離小于相應原子的范德華半徑,則認為此點符合條件;而對于Bader的定義,若當前點的電子密度大于閾值(0.001或0.002 a.u.),就認為符合條件。假設最后有n個點符合條件,那么分子的范德華體積就是n/m*V_box。
如果測試點數m較少,那么算出來的體積是不精確的,想要增加精度,就必須增加m。這就像人口普查,統計的人數越多結果越能反映實際國情。當然,分子越大,就需要越多的m,才能保證平均單位體積內隨機點的數目不變,即保證統計精度不變。由于每次用蒙特卡羅方法計算體積時隨機分布的點的位置都是不同的,因此符合條件的點數也會不同,故算出來的范德華體積的數值每次肯定會不同。m越大,由于統計誤差約小,每次計算的結果的波動就會越小,但記住計算耗時與m是成正比的。如果你不知道m設多大的話,可以做個測試,如果算幾次結果差異都不大的話,當前結果的數值誤差就是較小的,可以用了,反之需要再增加m數。
曾有人問什么程序計算的范德華體積更精確。這個問題太含糊,沒法回答。只能籠統地說,如果想較好地計算范德華體積,應當使用Bader的定義,用較高的隨機點密度去做蒙特卡羅計算(或者用下一節的MT方法),并且用比較合理的方式產生用于計算電子密度的波函數。
Multiwfn的主功能100的主功能3是用來基于蒙特卡羅方法算分子體積的。在Multiwfn程序的settings.ini文件里,如果MCvolmethod被設為了1,代表使用范德華球疊加方式定義的體積,此時可以用Multiwfn支持的任意包含原子坐標信息的文件作為輸入文件,如pdb/xyz/fch/gjf/mol/mol2/molden等等。如果MCvolmethod被設為了2(默認情況),代表使用電子密度等值面定義的體積,因此可以用來算Bader定義的范德華體積,此時需要用Multiwfn支持的含有體系電子波函數信息的文件作為輸入文件,比如wfn/wfx/fch/molden/mwfn等(下文統稱為波函數文件)。關于Multiwfn支持的文件格式詳細介紹,以及含有波函數信息的文件如何生成,詳見《詳談Multiwfn支持的輸入文件類型、產生方法以及相互轉換》(http://www.shanxitv.org/379)。
來看具體怎么用Multiwfn算Bader定義的范德華體積。假設MCvolmethod為默認的2,啟動Multiwfn,載入體系的波函數文件,然后選100,再選3。程序會提示你輸入i、x和k值。這里i代表將會有100 * 2^i個隨機點分布在盒子內,x對應等值面的電子密度數值,k代表盒子在分子周圍留出的距離為k乘以內置的原子范德華半徑。通常情況下,輸入9,0.001,1.7就可以了,對于較大的分子,建議將i值設大來得到更準確的結果。輸入后程序會立刻給出結果,比如:
The molecular volume: 171.982 bohr^3, ( 25.485 angstrom^3, 15.347 cm^3/mol)
如果要用原子范德華球疊加方式算范德華體積,先把MCvolmethod設為1,然后啟動Multiwfn,載入含有體系結構信息的文件,然后選100,再選3。程序會提示你輸入i,含義同上,也是越大越準確,輸入完畢后就會看到結果。
3 基于MT算法計算體積
Marching Tetrahedron (MT)算法簡單來說就是一種構建實空間函數等值面的方法,基于格點數據。很多可視化軟件讀取cube文件后顯示的等值面實際上就是用MT方法產生的。Multiwfn的主功能12是做定量分子表面分析的,所涉及的分子表面就是靠MT算法基于電子密度格點數據產生的。程序在輸出分子表面統計信息的同時也會一起把體積信息輸出出來。如果用的是Multiwfn的默認設置,即使用0.001 a.u.電子密度等值面來定義分子表面,最后輸出的體積顯然就是Bader定義的范德華體積。
MT方法算的分子體積的精度取決于電子密度格點數據的格點間隔,間隔越小精度越高,但需要算的點數也越多,因此計算耗時越高、越消耗內存。在Multiwfn默認的格點間距下計算精度就已經足夠高了。原理上,只要蒙特卡羅方法用的采樣點數無窮多,MT算法用的格點間隔無窮小,兩種方法的結果是精確相同的。在實際中,我更建議用MT方法來計算Bader的范德華體積,因為這沒有蒙特卡羅方法的那種隨機性,不用進行測試來判斷采樣點數是否夠多。MT方法的缺點是對大體系耗內存較高,因為需要儲存電子密度格點數據。
這里舉一個簡單的例子說明怎么用Multiwfn通過MT算法計算Bader的范德華體積。啟動Multiwfn后,依次輸入
examples\phenol.wfn //程序自帶的苯酚的波函數文件
12 //主功能12
6 //做定量分子表面分析,但不考慮被映射的函數
算完后就會看到輸出的范德華體積
Volume: 838.20232 Bohr^3 ( 124.20880 Angstrom^3)
同時還輸出其它一些信息,包括根據范德華體積和分子質量簡單換算出來的密度(這和真實密度相差肯定很大,不要太當回事),以及范德華表面積
Estimated density according to mass and volume (M/V): 1.2582 g/cm^3
Overall surface area: 477.44282 Bohr^2 ( 133.69762 Angstrom^2)
默認情況下Multiwfn對分子表面的定義用的是電子密度0.001 a.u.等值面,比如想改成0.002 a.u.等值面來得到凝聚相下分子范德華體積,在選6之前先選1設定表面定義,選擇Isosurface of electron density,然后輸入0.002即可。
如果你想得到數值精度更高的結果,可以進一步改小格點間距。做法是在選6之前先選3定義格點間距,然后輸入一個比屏幕上提示的當前值更小的值,比如0.2或0.18。注意格點間距越小,要算的點數就越多、越耗時,對內存需求也越高。用默認的格點間距得到的數值精度就已經足夠好了,一般不需要改。
Multiwfn的主功能12在《使用Multiwfn的定量分子表面分析功能預測反應位點、分析分子間相互作用》(http://www.shanxitv.org/159)里有專門的介紹,手冊3.15節有更多原理細節的說明。非常詳細的關于MT方法的介紹見筆者提出的改進版MT算法的文章J. Mol. Graph. Model., 38, 314 (2012),這也是Multiwfn的主功能12實際用的算法。大家用主功能12發文章時除了引用Multiwfn啟動時屏幕上提示的原文外也建議同時引用這篇文章。
4 其它問題
有人在某些數據庫上查到“分子體積”或“范德華體積”,這里要強調的是,不要盲目地相信這些查到的數據!很多這些數據庫里的數據僅僅是通過非常簡單的模型(比如不同元素的原子數和連接關系)估算出來的,顯然不如前文所述的方法算出來的體積更有合理性。使用、討論這些查到的數據之前,一定先搞清楚這數據是怎么得來的!
雖然范德華體積沒有唯一的定義,但在實驗上一種相對有意義的范德華體積的定義是從物質的密度和分子質量上反推出來的分子體積(下面簡稱“實驗范德華體積”。注意通過其它實驗手段也可以定義不同的實驗范德華體積)。有人會發覺并且抱有疑問,怎么這種實驗范德華體積和電子密度0.002等值面包圍的體積數值差距很大?其原因也是很明顯的。前文計算分子體積得到的是對單個分子的描述,是單分子的性質。而密度是物質的宏觀性質,由此反推出來的實驗范德華體積本質上也是宏觀性質。分子間的各種各樣復雜的相互作用直接影響物質的宏觀性質(范德華作用、靜電作用、分子間擠壓、構象變化等),也因此影響實驗范德華體積的因素十分復雜,原理上就不可能僅僅基于單個分子的性質就準確得到這種實驗范德華體積,否則就相當于只需要算個單分子就能準確得到物質的密度了。但是,通過前面的方法計算出的分子范德華體積,必然和實驗范德華體積有相關性,并且對于同一類物質,偏差會是較為系統的。因此,可以自行擬合一個方程(通常線性的就可以),來將二者關聯起來。
Gaussian程序里有個volume關鍵詞也可以算分子體積,這本質上用的是第2節介紹的蒙特卡羅方法基于電子密度等值面算的體積。我很不建議用這個關鍵詞!很不方便靈活,采樣點數和等值面數值得通過抽象、很難記憶的IOp才能改,而且在默認設置下算的結果極度不準確(差到根本不能用的程度),而把采樣點數改得足夠大使得結果誤差足夠小時耗時又特別高,遠不如用Multiwfn根據本文第3節的方法來算。volume關鍵詞輸出的分子體積一例:
Molar volume = 532.998 bohr**3/mol ( 47.564 cm**3/mol)
注意這里bohr**3/mol明顯是錯的,應為Bohr**3。另外括號里的數據只是通過簡單單位換算得到的密度,而并非是通過什么經驗公式得到的能和實際物質密度相比較的值。
PS:Volume關鍵詞還會輸出估計的Onsager半徑,即Recommended a0 for SCRF calculation后面的數。我發現給出的以埃為單位的這個半徑用的公式是:(V/pi)**(1/3) + 0.5,其中V是電子密度0.001 a.u.等值面內包圍的以埃^3為單位的分子體積(顯然,如上所述,Multiwfn也可以直接給出)。至于這個式子怎么來的我不清楚,手冊里也沒交代出處,應該有很強的經驗性,可能根本都不靠譜,絕對不要當回事,而且Onsager模型對于研究實際問題早就過時了,切勿拿這里給出的半徑說事。如果你要算分子半徑,看《談談分子半徑的計算和分子形狀的描述》(http://www.shanxitv.org/190)和《使用Multiwfn計算分子的動力學直徑》(http://www.shanxitv.org/503)。
值得一提的是,利用Multiwfn給出的分子范德華體積以及與靜電勢有關的一些輸出,可以預測特定類型分子晶體的密度,見《使用Multiwfn預測晶體密度、蒸發焓、沸點、溶解自由能等性質》(http://www.shanxitv.org/337)。
筆者還寫過一篇文章,與本文有一點關系,感興趣的讀者可以看看:《使用Multiwfn可視化分子孔洞并計算孔洞體積》(http://www.shanxitv.org/408)。
如果你想計算分子表面積,看這篇文章:《使用Multiwfn和VMD計算分子表面積和片段表面積》(http://www.shanxitv.org/487)。