使用Multiwfn的定量分子表面分析功能預測反應位點、分析分子間相互作用
使用Multiwfn的定量分子表面分析功能預測反應位點、分析分子間相互作用
文/Sobereva @北京科音
First release: 2012-Aug-6 Last Update: 2023-Jul-5
0 前言
定量分子表面分析對于預測反應位點、預測分子間結合模式、預測分子熱力學性質有重要意義。本文簡要介紹定量分子表面分析的概念和意義、它在Multiwfn程序中所用的數值算法,并通過實例說明怎么用Multiwfn的這個功能分析實際問題。實際上本文很多內容在Multiwfn手冊3.15節和4.12節中都已經涵蓋,數值算法在我發表的J. Mol. Graph. Model., 38, 314 (2012)一文中有十分完整、詳盡的說明,使用Multiwfn此功能時除了引用Multiwfn的原文J. Comput. Chem., 33, 580 (2012)外也請同時引用此文章。
Multiwfn可以在http://www.shanxitv.org/multiwfn免費下載。如果對此程序不了解,建議閱讀《Multiwfn入門tips》(http://www.shanxitv.org/167)和《Multiwfn FAQ》(http://www.shanxitv.org/452)并且關注《量子化學波函數分析與Multiwfn程序培訓班》(http://www.keinsci.com/workshop/WFN_content.html)。
《使用Multiwfn通過局部電子附著能(LEAE)考察親核反應位點、難易及弱相互作用》(http://www.shanxitv.org/676)里介紹的LEAE與本文介紹的平均局部離子化能有非常密切的聯系和高度互補性,請閱讀本文后記得閱讀。
1 定量分子表面分析的概念和意義
定量分子表面分析主要分析的是靜電勢(ESP)和平均局部離子化能(ALIE)在分子范德華表面的分布。分子范德華表面的定義非常多,最常用的是Bader的定義,也就是對于氣相分子,使用電子密度為0.001 e/bohr^3的等值面作為分子范德華表面,這種定義物理意義明確,而且可以反映局部電子特征產生的影響,比如孤對電子、pi電子。本文所說的分子表面都是指Bader的這種定義。
靜電勢是大家很熟悉的實空間函數,對于分子體系定義如下
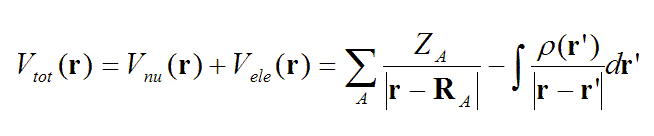
Z代表核電荷數,R是原子核坐標。一個分子在r處的靜電勢,等于將一個單位正電荷放在r處后它與此分子產生的靜電相互作用能,注意這里假定這個單位正電荷的出現對分子的電荷分布不產生任何影響。靜電勢由帶正電的原子核電荷產生的正貢獻和帶負電的電子產生的負貢獻構成。在r處如果靜電勢為正,說明此處的靜電勢是由原子核電荷所主導,如果為負,說明電子的貢獻是主導。在原子核附近,包括價層區域,由于離核較近,靜電勢都是正值,這部分通常不是我們感興趣的(盡管分析它們也有一些特殊用處,比如獲得共價半徑)。而在分子表面,電子的貢獻和核的貢獻達到可以抗衡的程度,電子密度分布的不均勻性會導致分子表面靜電勢有正有負。對中性體系,通常負值出現在高電負性原子的孤對電子、pi電子云以及碳-碳強張力鍵(如環丙烷)附近,因為這些區域電子比較富集。
對范德華表面靜電勢的研究已有幾十年的歷史,最常見的有兩類應用:
(1)在化學反應初期分子間通過靜電吸引而相互接近的過程與分子對其外部(范德華表面及更遠區域)產生的靜電效應密切相關,因此可以通過分析分子范德華表面上靜電勢的分布來預測在什么位點上最有可能發生何種化學反應,通常認為靜電勢越負(越正)的地方對應的原子越容易發生親電(親核)反應。實際上,通過原子電荷來預測反應位點在本質上是這種靜電勢分析方法的抽象、簡化。
(2)若已知某復合物是以靜電為主導的方式結合而成(如氫鍵、二氫鍵、鹵鍵等形式),利用靜電勢可以預測、解釋復合物中分子的相對朝向如何、結合的強度大概有多大,這對于研究分子晶體堆積方式、受體配體結合模式、分子吸附等問題很有用。其依據是分子之間容易以靜電勢互補方式接觸,即分子表面靜電勢正值區域傾向于接觸負值區域,而且正值越正、負值越負時這種傾向越強,這樣能最大程度降低整體能量。
大家已經十分習慣繪制分子表面的靜電勢填色圖來分析這些問題,用不同顏色代表靜電勢正負和大小(例如《談談Molekel做靜電勢填色等值面圖的方法及誤區》,見http://www.shanxitv.org/98)。這種圖形化分析雖然直觀,但是有很大局限性,也就是圖形化的研究不夠精確,討論問題時只能定性地分析,上升不到定量的高度。而且當比較一系列分子,或者當靜電勢分布復雜或體系稍大時,圖形觀察也很費力、麻煩。定量分子表面分析的主要目的之一就是根據靜電勢在分子表面上的極值點位置和確切數值對反應位點、結合模式等問題給出更準確可靠的分析結論。
另外,分子的凝聚相性質,比如蒸發/升華焓、密度、沸點、粘度、表面張力、LogP等等,都與分子間相互作用密切相關,對于極性(或具有局部極性)的分子,靜電相互作用通常比范德華相互作用大很多,因此通過分析在分子表面上的靜電勢,對于預測這些分子的這些性質十分有益。Politzer等人提出的General interaction properties function(GIPF)就建立了這么一套關系,見J.Mol.Struct.(THEOCHEM),307,55。GIPF的本質上含義是,依靠基于分子表面上靜電勢分布定義的分子描述符,通過擬合QSPR方程,可以很好地預測上述性質。這些分子描述符包括分子整體/靜電勢為正/靜電勢為負區域的表面積,表面上靜電勢最大值、最小值,表面上靜電勢的整體/正值/負值部分的平均值/平均偏差/方差,以及它們進一步相互運算得到的幾個描述符(具體公式見Multiwfn 2.5手冊3.15.1節)。計算并利用這些描述符也是定量分子表面分析的主要組成部分。
如果對靜電勢有興趣,建議看此帖的靜電勢綜述文章:http://bbs.keinsci.com/forum.php?mod=viewthread&tid=219
平均局部離子化能可能很多人還比較陌生,它在Can.J.Chem.,68,1440 (1990)一文中提出,這個函數寫為
ALIE(r)=∑[i]ρ_i(r)*|ε_i|/ρ(r)
其中ρ_i(r)是r處第i個分子軌道的電子密度,|ε_i|是第i個分子軌道能量的絕對值,可以視為i軌道的電子的離子化能,ρ(r)是r處總電子密度。r處ALIE值越小,代表在此處的電子平均能量越高,被束縛得越弱。ALIE有很多應用,比如衡量電負性、表征局部極化率、預測pKa,展現原子殼層結構(在《使用Multiwfn繪制原子軌道圖形、研究原子殼層結構及相對論效應的影響》一文中有實例,見http://www.shanxitv.org/152)。在ALIE的諸多應用中,最為重要的就是它預測反應位點的能力。在分子表面上,ALIE越小處,由于電子束縛得越弱,或者說電子活性越強,就越容易發生親電及自由基反應。通過定量分子表面分析算法,找出分子表面上ALIE最小的幾個點,看這幾個點離哪些原子比較近,就能判斷哪些原子最可能是反應活性位點。ALIE的極小點容易出現在pi電子、孤對電子附近,因為這些區域的電子被束縛得較弱。可以認為ALIE越小處,局部極化率越大,這與密度泛函理論中的局部軟度/硬度概念也是密切相關的。
關于平均局部離子化能的更多介紹,見《靜電勢與平均局部離子化能相關資料合集》(http://bbs.keinsci.com/thread-219-1-1.html)。
需要特別說明的是,通過靜電勢和ALIE在預測反應位點上不僅不矛盾,反倒是互補的。單靠靜電勢很多情況都不能正確預測反應位點;而單靠ALIE,在一些情況下也是失敗的(而且ALIE也不能用于預測親核位點),此時必須將靜電勢綜合考慮。這是因為,兩個分子相互反應的完整過程分為以下兩個步驟。注意這兩個步驟沒有明確界限,是平滑過渡的。
1 兩個分子從相距很遠逐漸接近:這個過程中分子的電子分布并不發生明顯改變,頂多是稍微被另一個分子的電場所極化。長程靜電相互作用在這個過程中是關鍵,通過分析分子表面的靜電勢,可以很好地估計反應物容易被拉到哪里。也因此,光靠分析ALIE來預測反應位點往往不行,因為即便靠ALIE預測出某些原子容易發生反應,但是若反應物不容易靠靜電作用被拉到這里,則在這里發生反應的幾率還是不大。
2 范德華表面快要接觸的兩個分子進一步靠近,通過成鍵、斷鍵這樣的電子結構重排過程完成化學反應:這一步在哪里容易進行,關鍵看哪個原子附近電子活性大,用ALIE最適合研究這個問題。這也表現出單純靠靜電勢預測反應位點的不可靠性,因為比如某原子附近靜電勢越負,只能說明親電試劑越容易被拉到這原子附近,卻不代表這原子附近的電子越活潑因而越容易參與反應。再者,當分子間接近到快要反應時,分子的實際電子分布與它在孤立時的電子分布已有很大差異,前面介紹靜電勢時提到的“對分子的電荷分布不產生任何影響”的前提條件已經不能滿足,所以用分子孤立狀態的靜電勢已經不能很好說明問題。
綜上所述,由于分子間反應涉及到了兩個步驟,而兩個步驟本質不同,分別適合用靜電勢和ALIE來分析,因此需要將它們在分子表面上的分布綜合考慮以判斷反應位點。通常可以先用ALIE判斷,如果遇到困難,比如好幾個原子都有對應的ALIE極小點(而且數值和最小點數值差不多),或者極小點介于兩個原子之間,那么可以再考察靜電勢的分布,看看反應物更容易被拉到哪個原子附近。另外,有時需要根據分子三維結構,將位阻效應對試劑接近過程的影響考慮進去。
判斷反應位點還有很多辦法,比如概念密度泛函理論中著名的福井函數(前線軌道理論判斷反應位點本質上是福井函數的近似),以及之后提出的雙描述符。這兩種方法實際上和ALIE一樣,著眼的也是反應過程的第二步。相關知識參看筆者寫的《親電取代反應中活性位點預測方法的比較》(http://www.whxb.pku.edu.cn/CN/abstract/abstract28694.shtml),福井函數和雙描述符可以用Multiwfn非常容易地計算,看《使用Multiwfn超級方便地計算出概念密度泛函理論中定義的各種量》(http://www.shanxitv.org/484)。更多知識看《概念密度泛函綜述和重要文獻合集》(http://bbs.keinsci.com/thread-384-1-1.html)。
2 定量分子表面分析在Multiwfn中的基本算法
定量分子表面分析算法的詳盡的技術細節不在本文討論,這并非一般用戶關心的,有興趣者可參看 J. Mol. Graph. Model., 38, 314-323 (http://dx.doi.org/10.1016/j.jmgm.2012.07.004)。但基本原理還是應當了解的,只有這樣面對一些實際問題時才能明白如何通過Multiwfn提供的一些選項自行解決。
既然分析的是分子表面,就必須先把分子表面構建出來。Marching Tetrahedra(MT)方法是計算機圖形學上最主流的基于格點數據(也叫體數據,詳細介紹見《談談體數據1:介紹體數據》http://www.shanxitv.org/127)構建等值面的算法,許多可以顯示等值面的分子可視化軟件都是用的這個方法。MT的流程可以簡要用下圖表示
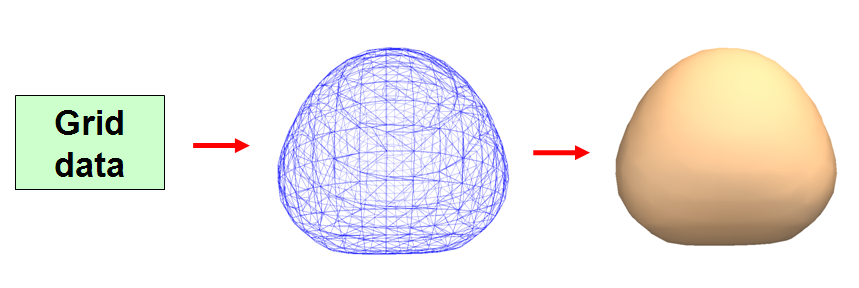
第一步,輸入格點數據。若要構建電子密度等值面,就必須先計算出至少能涵蓋等值面空間范圍的立方區域內的電子密度格點數據。
第二步,將格點數據轉化為等值面,等值面用一堆連續排布的三角形來描述。格點數據的間隔越小,則這個等值面越光滑,越接近精確等值面,頂點數也越多。
第三步,將每個三角形用一般的計算機圖形學的方法渲染出來,就是我們在可視化軟件中看到的等值面。
Multiwfn的定量分子表面分析模塊主要流程是:
(1) 根據分子幾何坐標,向其四周延展一定距離,以確定電子密度格點數據的空間范圍。再根據設定的格點間距,計算出電子密度格點數據。這一步耗時占總耗時很少,因為Multiwfn計算電子密度速度極快,而且支持并行。
延展的距離通過定量分子表面分析模塊中的選項4中的子選項1來設定,比如設為n,那么格點數據空間范圍在K方向的最大值就是在K方向原子坐標的最大值基礎上延展n乘以此原子范德華半徑的距離。默認值對于生成0.001 e/bohr^3等值面來說已經足夠,如果要生成的電子密度等值面數值比這更小,則等值面范圍就會更大,這時就需要將延展距離進一步設大以免生成的等值面被截斷。
(2) 利用MT方法得到分子表面的頂點坐標、頂點之間的連接關系,并記錄每個三角形與哪三個頂點相對應。這一步的耗時可以忽略不計。
MT過程涉及到插值問題,Multiwfn用的是二分法,迭代次數越多,所得頂點位置越精確,也因此定量分子表面分析結果越精確,但耗時也越多。默認是做3次迭代,已足夠精確,沒必要修改,想調整的話可以用選項4的子選項3。
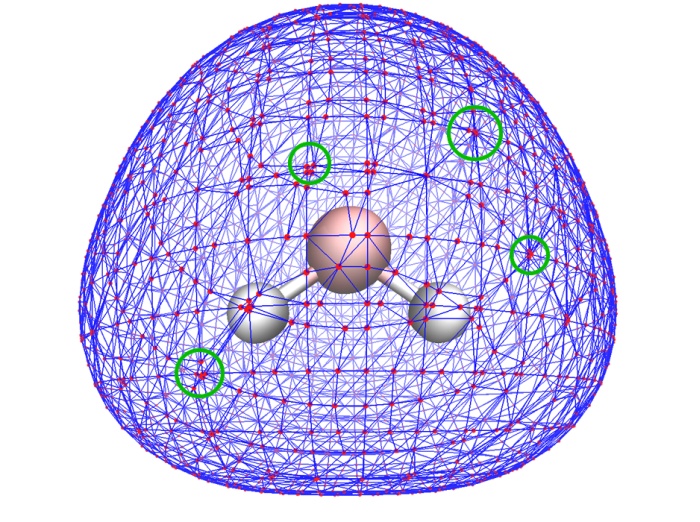
(3) 對分子表面的頂點中的多余頂點進行消除。之所以要做消除,是由于MT方法產生的表面頂點分布很不均勻,經常出現大量頂點聚集在很小區域內(如下圖綠圈標注的區域),實際上這些嚴重聚集的點只需要用一兩個點表示就足夠了,多余的頂點可以消除掉,以降低下一步的耗時。Multiwfn會檢測每一對兒相連頂點間的距離,如果距離小于閾值,就會消除其中一個,而將另一個的位置挪到它們的平均位置上。這個距離閾值可以用選項4中的子選項2來設,假設設的是x,那么閾值就是x乘以格點間距。默認值大致會消除掉60~70%的頂點,對于分析靜電勢,這也將節省約60~70%的總耗時。
(4) 計算每個表面頂點上的函數值。函數可以是靜電勢或ALIE,通過選項2來選擇,默認是計算靜電勢。當計算的是靜電勢時,由于計算靜電勢需要算復雜的電子積分,這一步的耗時幾乎等于整個定量分子表面分析的耗時。
(5) 尋找表面上的極小點和極大點。根據頂點間的連接性關系,如果一個頂點的函數值比它的所有的第一層和第二鄰居頂點的函數值都小(都大),那么這個頂點就被認為是極小點(極大點)。
(6) 計算前面提到的各種分子表面描述符。例如分子表面靜電勢平均值的計算方式為(1/S_tot)*∑[i]S_i*ESP_i,其中S_tot是分子總表面積,也就是分子表面上所有三角形面積的加和。S_i是第i個三角形的面積。ESP_i是第i個三角形的靜電勢,它是這個三角形的三個頂點上的靜電勢的平均值。
對于極個別體系,使用默認的格點設定得到的極值點分布可能出現一些假象,通常有四種情況:
(1) 極值點分布與分子結構對稱性不符。當然,嚴格相符是不可能的,因為表面頂點的分布并不滿足分子對稱性,但是合理情況下找出的極值點位置應該基本和分子結構對稱性偏差不大。如果比如一個Cs對稱性平面分子,在其中一側有個極值點,而在另一側對應的位置卻沒有,說明要么是這個找出來的極值點本不該存在,也有可能是另一側少找出一個極值點。
(2) 憑經驗斷定某處本該出現極值點,但是實際卻沒有找出來,或者本該出現幾個距離比較近的極值點,但是實際上只找到了一兩個。
(3) 某個極值點旁邊出現一個本不該出現的離它很近的極值點。
(4) 找出的極值點的位置和真實極值點位置距離偏離較大。
出現這些假象的主要原因是格點數據間隔偏大。將格點間距通過選項3調小,就可以有效避免這些假象,格點間距無限小的時候結果將無窮精確。默認的格點間距是權衡精度與計算量和資源占用量得到的值。想要更精確的結果可以適當調低它,但由于會造成分子表面頂點數迅速增多,故計算這些頂點的函數值的耗時也越長(這是對于分析靜電勢來說,計算ALIE的耗時可忽略不計)。調低到默認值的60%時已經非常精確了,不宜再調低。
當然,表面描述符的計算精度也是隨格點間距減小而增加。計算表面描述符對格點間距的要求并不像搜索極值點那么高,如果只是想得到表面描述符的話,將格點間距設為默認值的兩倍時結果依然是可靠的,而計算量卻能減少幾倍。
3 分子表面的靜電勢分析實例
這里我們先分析苯酚。任何Multiwfn所支持的含有波函數信息的文件都可以作為此任務的輸入文件,比如.wfn、.fch、.molden等,詳見《詳談Multiwfn支持的輸入文件類型、產生方法以及相互轉換》(http://www.shanxitv.org/379)。用Gaussian運行下面的輸入文件就可以得到B3LYP/6-31G**級別下苯酚的.wfn文件。通常B3LYP/6-31G**級別的波函數已經能定性正確重現分子表面的靜電勢分布了,進一步提高基組和理論級別對靜電勢影響一般不會特別顯著,本文例子若未注明均是在此級別下計算的。
# b3lyp/6-31g** opt out=wfn
[空行]
phenol
[空行]
0 1
C 0.02049800 -1.85749600 0.00000000
C 1.22053900 -1.14011200 0.00000000
C 1.21812700 0.25255100 0.00000000
C 0.00000000 0.94134000 0.00000000
C -1.20588400 0.23260100 0.00000000
C -1.18935500 -1.16319600 0.00000000
H 0.02983500 -2.94340300 0.00000000
H 2.16972400 -1.66964600 0.00000000
H 2.14221700 0.82214600 0.00000000
H -2.15252200 0.77084100 0.00000000
H -2.13105300 -1.70584400 0.00000000
O 0.05149400 2.30933400 0.00000000
H -0.85371200 2.65710700 0.00000000
[空行]
C:\phenol.wfn
[空行]
[空行]
啟動Multiwfn,依次輸入
C:\phenol.wfn //文件名
12 //定量分子表面分析功能
0 //開始分析。默認的是分析靜電勢
由于計算靜電勢較為耗時,所以需要耐心等一會兒。目前主流的四核機子花費1分鐘左右。之后,屏幕上會出現以下信息:
================= Summary of surface analysis =================
Volume: 840.76822 Bohr^3 ( 124.58902 Angstrom^3)
Overall surface area: 478.87396 Bohr^2 ( 134.09839 Angstrom^2)
Positive surface area: 236.05203 Bohr^2 ( 66.10131 Angstrom^2)
Negative surface area: 242.82193 Bohr^2 ( 67.99708 Angstrom^2)
Overall average value: 0.00006729 a.u. ( 0.04222108 kcal/mol)
Positive average value: 0.01773134 a.u. ( 11.12570370 kcal/mol)
Negative average value: -0.01710428 a.u. ( -10.73225294 kcal/mol)
Overall variance (sigma^2_tot): 0.00038566 a.u.^2 ( 151.83843791 (kcal/mol)^2)
Positive variance: 0.00028503 a.u.^2 ( 112.21695420 (kcal/mol)^2)
Negative variance: 0.00010064 a.u.^2 ( 39.62148371 (kcal/mol)^2)
Balance of charges (miu): 0.19285271
Product of sigma^2_tot and miu: 0.00007438 a.u.^2 ( 29.2824550 (kcal/mol)^2)
Internal charge separation (Pi): 0.01741439 a.u. ( 10.92683016 kcal/mol)
Molecular polarity index: 0.47384207 eV ( 10.92707 kcal/mol)
Nonpolar surface area (|ESP| <= 10 kcal/mol): 73.31 Angstrom^2 ( 54.67 %)
Polar surface area (|ESP| > 10 kcal/mol): 60.79 Angstrom^2 ( 45.33 %)
第一行是分子范德華體積,后面的都是前面提到過的分子表面描述符,在手冊3.15.1節有詳細說明。其中包括分子整體表面積,靜電勢正值/負值區域表面積,整體/正值/負值區域的靜電勢平均值和方差(后者用于衡量靜電勢的波動程度)、靜電勢平衡程度等等。這些量對于構建QSPR模型來預測分子凝聚相性質(如沸點、密度、LogP)很有意義,這種基于分子表面靜電勢性質的QSPR關系也被特稱為GIPF (general interaction properties function)。筆者專門在另一篇文章中有詳談:《使用Multiwfn預測晶體密度、蒸發焓、沸點、溶解自由能等性質》(http://www.shanxitv.org/337)。輸出的分子極性指數、極性和非極性表面積對于討論分子的極性非常有用,見《談談如何衡量分子的極性》(http://www.shanxitv.org/518)。
然后還會看到這些分子表面靜電勢極值點信息:
Number of surface minima: 3
# a.u. eV kcal/mol X/Y/Z coordinate(Angstrom)
1 -0.02768237 -0.753276 -17.369580 0.108653 -1.078708 -1.896855
2 -0.02767035 -0.752948 -17.362035 0.186791 -0.994546 1.876438
* 3 -0.04167254 -1.133968 -26.147853 1.461372 3.343026 -0.003233
Number of surface maxima: 5
# a.u. eV kcal/mol X/Y/Z coordinate(Angstrom)
1 0.02070473 0.563404 12.991390 -3.401136 -2.205347 0.018349
* 2 0.08186700 2.227714 51.368267 -1.953807 3.103544 0.023402
3 0.01735680 0.472303 10.890700 0.051211 -4.308061 0.028541
4 0.01776816 0.483496 11.148810 3.364233 -2.325592 0.023073
5 0.01379213 0.375303 8.654010 3.475490 1.099533 0.016113
這表明表面靜電勢極小點和極大點分別有3個和5個,靜電勢的數值用三種常用單位分別列出,它們的坐標也一并給出。打星號的那兩個極值點分別是最小點和最大點。如果想把這些信息從屏幕上拷到文本編輯器等地方,可參見手冊5.4節介紹的方法。
此時屏幕上可以看到很多后處理選項,比如將極值點位置輸出到文本文件或pdb文件、將表面頂點輸出到pdb文件、將函數值在一定范圍內的極大/極小點刪除(利用此功能,可以只將函數值較大的極大點以及函數值較小的極小點保留而去掉其它意義不大的極值點)、計算各個原子或片段對應的分子局部表面的統計特征。若選擇選項0,就會蹦出圖形窗口,顯示分子結構和極值點位置,如下所示
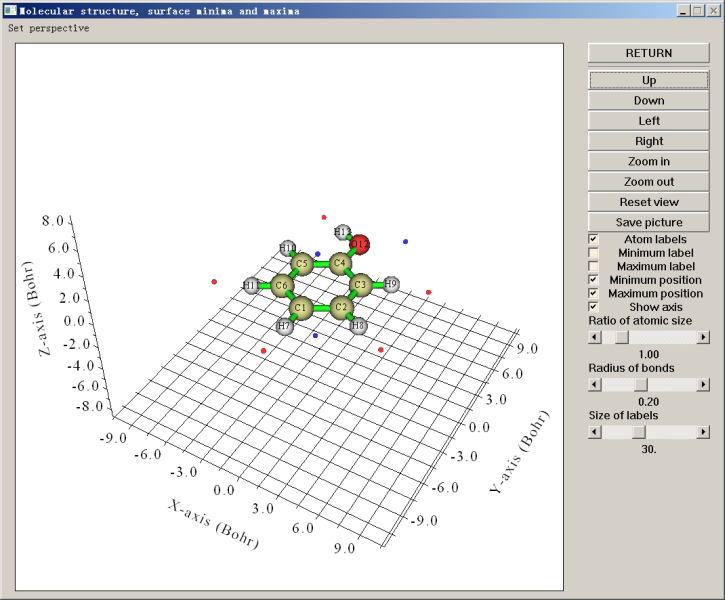
圖中藍點代表極小點,紅點代表極大點。利用右側的復選框可以控制是否讓它們顯示出來。從這個角度看,臨界點與原子的對應關系不是很清楚。我們將Ratio of atomic size拉到4.0,此時每個分子球的半徑就成了原子范德華半徑,放到一起就表現出了近似的分子范德華表面,由于這種近似的范德華表面比0.001 e/bohr^3定義的范德華表面一般略小,所以不會淹沒臨界點。與此同時,利用up down left right按鈕將視角調成垂直于分子平面,并且選上Minimum label和Maximum label將每個極值點的編號顯示出來,就能得到下面的圖,極值點與原子的對應關系已能看得很清楚了
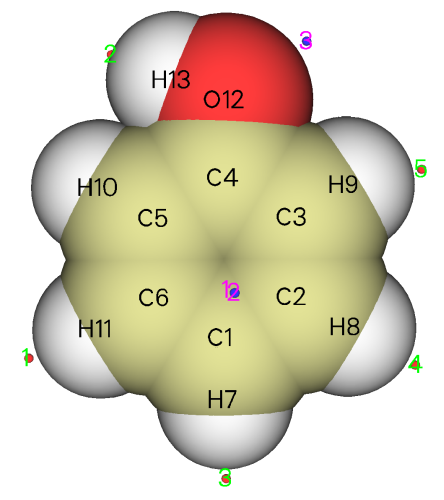
綠字標注的是極大點的序號,可見在氫附近的分子表面上都有靜電勢極大點,這是因為氫的電負性小于碳、氧,因此此體系中氫原子都顯正電性。從前面輸出的極大點數據看,羥基氫對應的極大點是最大點,其值51.368267 kcal/mol比其它各極大點都大好幾倍,這是由于氧比碳吸電子能力明顯更強,故羥基氫的正電性更顯著。H10不像其它氫那樣有對應的極大點,這也是因為羥基氫的正電性太強了,造成其附近一大片分子表面上的靜電勢都較大,淹沒了H10附近區域,因此H10較弱的正電性對靜電勢的影響體現得就不明顯了,無法造成極大點了。用粉字標注的3號極小點是由于羥基氧的孤對電子密集的電子云產生的,它也是最小點,靜電勢值為-26.147853 kcal/mol。1號和2號極小點體現的是豐富的pi電子云對靜電勢的顯著負貢獻,二者是以分子平面為對稱的,所以在當前視角下它們是重合的。從這兩個極小點位置和原子球涵蓋的區域可以看出,在羥基對位原子附近分子表面的靜電勢比在其它碳原子附近更負,因此更容易被親電試劑所進攻,這也在一定程度上解釋了羥基是鄰對位取代基。此例也同時體現出了靜電勢分析反應位點的局限性,靜電勢沒能預測出羥基也可以使鄰位碳變得易于被親電試劑進攻,而且,氧的孤對電子對應的是靜電勢最小點,因此貌似最容易發生親電反應,但事實上反應并不發生在這里。
通過靜電勢極值點,我們可以解釋苯酚二聚體為何是下面這種結構
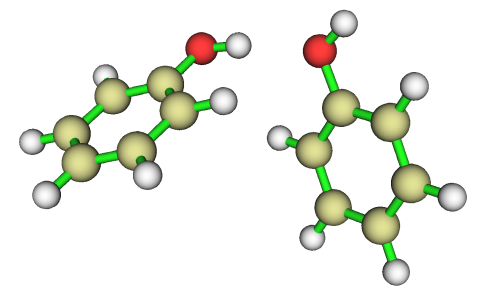
從極值點分析可知,羥基氫對應表面靜電勢最大點,而羥基氧對應最小點,因此這兩個原子最容易靠靜電相互作用結合到一起,而且是最大點正沖著最小點的這種朝向,這可以使二聚體能量最大程度降低。這也間接說明了,常規氫鍵的本質很大程度上可以解釋為靜電相互作用(但對于強氫鍵,共價成份不可忽視)。
這里再來看兩個例子。下圖左邊是環丙烷的結構和分子表面靜電勢極值點,右邊是Multiwfn的主功能5計算的它的電子密度拉普拉斯函數0.35等值面圖,藍色為負值(電子聚集區域)。
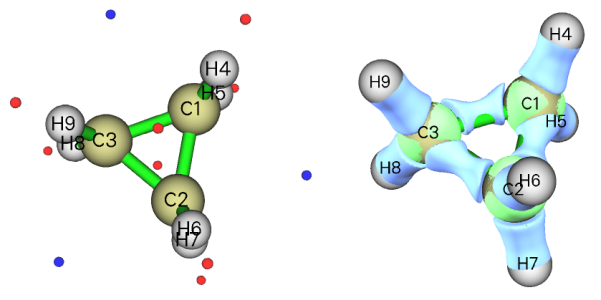
正如前文提到過的,在強張力的C-C鍵附近的分子表面會出現靜電勢極小點。之所以會出現這種情況,通過電子密度拉普拉斯函數等值面圖可以解釋。由圖可見,由于環存在很強張力,導致C-C之間的電子密度聚集區域一定程度上被往外“擠”出去了一點,而不是完全處在C-C連線上。正是這部分的電子密度對靜電勢的負貢獻使分子表面產生了靜電勢極小點。
在WIREs Comput Mol Sci,2,1一文中有下面的圖,左邊是F2---HF二聚體的真實結構,表面上看起來出現這樣的結構有點難以理解。作者將原因解釋為在這種結構下F的孤對電子與HF反鍵sigma軌道間的相互作用會造成能量降低,使結構穩定,如右圖所示
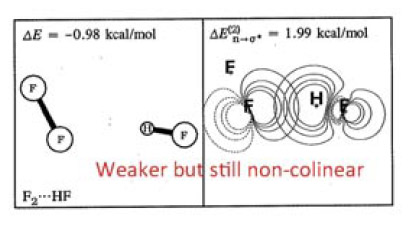
實際上,通過靜電勢分析,也同樣可以解釋出現這種結構的原因,而且通過靜電勢來解釋明顯更有意義、原理上嚴格清楚得多得多,被接受程度更高。下圖是F2和HF的各自分子表面上靜電勢極值點分布。
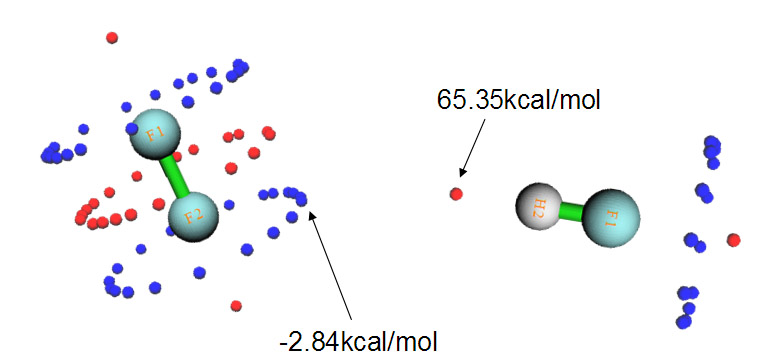
F2中很多點組成了三個環形,這是因為F2是軸對稱的,每個環上的點的數值都是相同(簡并)的。藍色那圈圓環對應于F的孤對電子造成的靜電勢極小點,而分子軸線兩端的兩個極大點對應于所謂的“sigma穴”,它是指某些第四至第七主族原子在某些環境下形成sigma鍵后,在sigma鍵軸末端會出現一小塊靜電勢比旁邊更正的區域。在HF中,F附近的情況和F2中類似,與氫對應的靜電勢最大點數值高達65.35 kcal/mol(因此容易形成很強的氫鍵)。如果將F2和HF的圖按照實際二聚體中它們的相對朝向擺放,也就是如上圖那樣,就會發現,HF的氫對應的靜電勢最大點正好對著F2的靜電勢極小點,當它們以這種朝向接近后,靜電勢的正負區域的互補必會使能量很大程度降低。因此,通過靜電勢極值點分布,可以很好地解釋為什么F2---HF二聚體是這種結構。
這種靜電勢極值點分析方法對于研究通過sigma穴以及近來提出的pi穴相互作用的體系極其有用,前者就是鹵鍵出現的根本原因。Politzer等人利用這種方法分析了大量這類體系,有興趣的讀者可參考以下文獻,
J.Mol.Model.,13,305、J.Mol.Model.,15,723、J.Mol.Model.,13,291、J.Mol.Model.,18,541、IJQC,107,3046、PCCP,12,7748
在其中對一系列分子的研究中會看到靜電勢最大值和最小值與分子間相互作用能有很強相關性。例如IJQC,107,3046一文中,討論了NH3通過氮的孤對電子與SeF2及SF2上的Se和S的sigma穴形成的復合物。SF2和SeF2的表面靜電勢最大值(對應于S和Se的sigma穴)分別為34.4和41.8 kcal/mol,顯然后者更易于與Lewis堿結合,這解釋了為什么H3N---SF2的相互作用能-7.7kcal/mol比H3N---SeF2的相互作用能-12.2kcal/mol要小。
需要注意的是分子間并非僅存在靜電作用,而且分子間靜電相互作用涉及到所有原子,另外當分子接近過程中幾何結構、電子分布相對于自由狀態會有一定調整,所以僅靠靜電勢大點、極小點的位置和數值的分析方法不可能對于各種復合物體系都完全適用。當你發現這種靜電勢極值點分析結果合乎預期,能說明問題,那么就可以從這個角度去討論;如果和期望的不符,那么說明還有更多因素需要考慮,當前研究方法過于粗糙。
4 分子表面的平均局部離子化能分析實例
由于ALIE依賴于分子軌道能量,所以必須用HF或DFT波函數。對于典型的雜化泛函,如B3LYP,計算的軌道電離能(相應分子軌道能量的絕對值)有這樣的趨勢:DFT計算的i軌道電離能 < 實驗得到的i軌道電離能 < HF計算的i軌道電離能。雖然DFT計算的軌道電離能與實驗值有一定偏差,但是對每種雜化泛函,這個偏差都是呈系統性的。根據ALIE定義可知,如果對每個軌道電離能加上一個常數,等于ALIE值加上這個常數。由于研究分子表面上ALIE時主要研究的是它的相對值而非絕對值,因此DFT的軌道電離能的系統偏差對分析結果帶來的影響可以較好抵消,故使用DFT波函數研究ALIE是可靠的。像計算靜電勢一樣,使用B3LYP/6-31G**等級的波函數就可以得到不錯的結果。HF軌道電離能比實驗值高估的程度的系統性則相對來說不很好,從這個角度講不如DFT適合計算ALIE。
這里還是以分析苯酚為例進行說明。啟動Multiwfn后依次輸入
C:\phenol.wfn
12
2 //設定映射到分子表面上的函數
2 //將函數選為平均局部離子化能
0 //開始分析
由于計算ALIE非常快,整個分析經過幾秒鐘就運算完畢了。屏幕上列出了所有分子表面上ALIE的極值點位置和數值,由于ALIE在分子表面上分布比靜電勢復雜得多,所以找出的極值點也明顯多得多。同時屏幕上也列出了分子體積和分子表面積,因此,這個Multiwfn的功能其實可以單純地作為計算分子體積和分子表面積的工具來用。Multiwfn的主功能100里的子功能3也是用于計算范德華體積的,但用的是蒙特卡羅方法,結果和當前方法得到的十分一致,可參考《談談分子體積的計算》(http://www.shanxitv.org/102)中的討論。
算完后,還是通過選項0來觀看極值點分布,如下所示
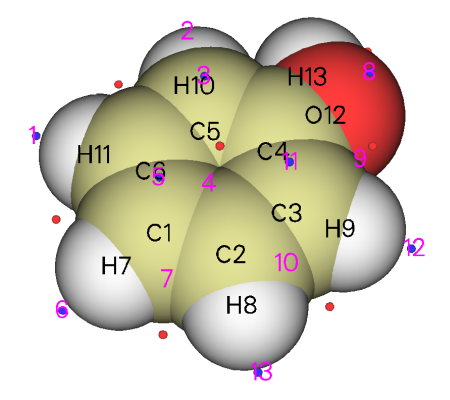
可看到,鄰、對位碳原子都明顯有對應的極小點,ALIE值都差不多8.7~8.8eV左右,都可算作是最小點,這就說明,鄰對位碳原子附近的電子被束縛得最弱,最有可能是反應活性位點,這很好地解釋了羥基是鄰對位取代基。在氧旁邊也有兩個ALIE極小點,可認為是氧的容易極化的孤對電子產生的,它們的值為10.49eV,明顯大于最小點的值,這也很好地說明親電反應并不會發生在羥基氧上。
我們再看幾個例子,下圖左邊和右邊分別是間二氯苯和硝基苯的ALIE極值點分布。為了清楚起見,沒有讓極大點顯示出來。
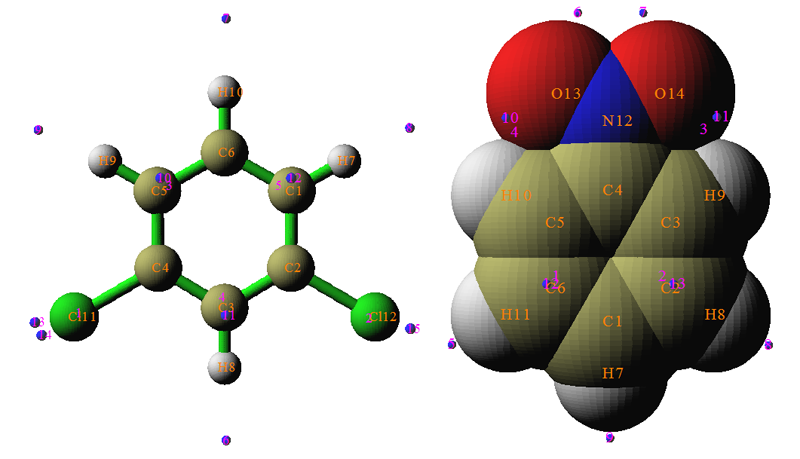
氯是鄰對位定位基,對于間二氯苯,兩個氯的鄰對位定位效應正好能夠相疊加,所以理應C1、C3、C5是活性位點。ALIE的極小點分布也確實驗證了這一點,在這三個原子上都有極小點出現,為9.7 eV左右。在氯旁邊也出現了數值很小的極小點,數值為9.85 eV,看似比9.7 eV大不了多少,所以ALIE分析認為親電反應也容易發生在氯上,不過實際上顯然這并不會發生,這算是ALIE的一點小缺陷,并無大礙。
對于硝基苯,在間位碳上出現了ALIE最小點,其值為9.94 eV,比起其余極小點的值都小不少,這很好地表現出硝基是間位定位基,反應容易發生在間位碳上。
由以上例子可見,用ALIE判斷反應位點可靠性很高,比使用靜電勢分析反應位點往往更可靠、結果更清晰。但之前也提到了,ALIE和靜電勢是互補的,單靠ALIE判斷往往是不行的,這里也舉一個例子,預測吡咯的反應位點。
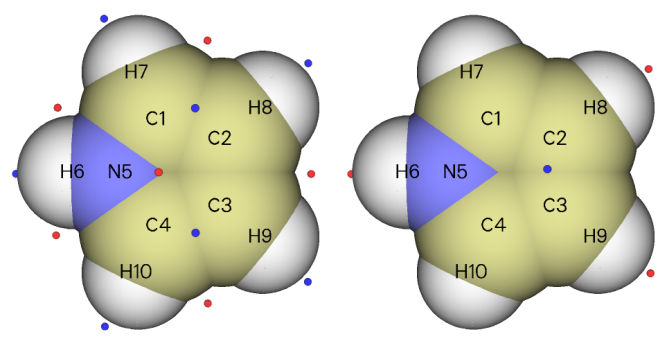
上圖左邊是ALIE極小點分布。可見,極小點落在了鄰位和間位碳之間,因此我們沒法判斷到底是鄰位碳容易反應還是間位碳容易反應,或者說,當分子離得比較近后它們發生反應的可能性一樣大。那么,預測實驗上的反應位點就還得看親電試劑更容易接近哪個碳。上圖右邊是靜電勢極小點圖,此圖表明在兩個間位碳之間靜電勢最小,容易吸引親電試劑。將ALIE和靜電勢的分析結合起來就能得出吡咯的間位碳最有可能是反應位點的結論。實際上,氣相實驗結果也確實證明了這個預測是正確的。
使用ALIE分析呋喃時會發現ALIE極小點也是落在鄰位和間位碳之間,只是稍微偏鄰位碳一點,因此也很難單純靠ALIE分析下定結論親電反應最可能發生在鄰位還是間位碳上。而通過分子表面靜電勢極值點分析,會發現在氧的孤對電子附近的分子表面上有一個數值為-20.6 kcal/mol的靜電勢最小點,在兩個間位碳之間有一個-15.2 kcal/mol的極小點,顯然親電試劑更容易被拉向氧的那邊,因此比起間位碳更容易接觸到鄰位碳,故可以預測反應位點是在鄰位碳上,事實也確實如此。
5 使用VMD程序可視化靜電勢和平均局部離子化能
基于Multiwfn輸出的文件,利用VMD,可以非常容易、快速地繪制效果極好的靜電勢、ALIE著色的分子表面圖,而且表面極值點位置也可以顯示在上面、可以方便地查詢它們的數值,這部分內容請參見
使用Multiwfn+VMD快速地繪制靜電勢著色的分子范德華表面圖和分子間穿透圖(含視頻演示)
http://www.shanxitv.org/443(http://bbs.keinsci.com/thread-11080-1-1.html)
使用Multiwfn和VMD繪制平均局部離子化能(ALIE)著色的分子表面圖(含視頻演示)
http://www.shanxitv.org/514(http://bbs.keinsci.com/thread-14632-1-1.html)
巨大體系的范德華表面靜電勢圖的快速繪制方法
http://www.shanxitv.org/481(http://bbs.keinsci.com/thread-13140-1-1.html)
使用Multiwfn結合VMD分析和繪制分子表面靜電勢分布
http://www.shanxitv.org/196