使用Multiwfn超級方便地計算出概念密度泛函理論中定義的各種量
注:軌道權重福井函數/雙描述符特別適合前線軌道簡并或近簡并體系,也是使用本文提到的功能來計算,但本文沒涉及,在另一篇博文里專門作了介紹《通過軌道權重福井函數和軌道權重雙描述符預測親核和親電反應位點》(http://www.shanxitv.org/533),非常建議看過本文后閱讀。
注:本文對應2021-Jul-15及以后更新的Multiwfn及手冊,不要用老版本!
使用Multiwfn超級方便地計算出概念密度泛函理論中定義的各種量
文/Sobereva@北京科音
First release: 2019-May-21 Last update: 2022-May-30
1 前言
由Parr最初開始發展的概念密度泛函理論(Conceptual density functional theory,CDFT)也稱密度泛函反應性理論(DFRT),是量子化學和波函數分析領域中的一個重要組成,專門用來預測和解釋化學物質的反應活性、反應位點等問題,對于研究化學反應有重要意義。相關介紹以及筆者寫過的相關博文見《概念密度泛函綜述和重要文獻合集》(http://bbs.keinsci.com/thread-384-1-1.html)。
CDFT框架里有大量概念,包含很多實空間函數、局部指數和全局指數,它們經常出現在各種文獻里用來討論反應問題。只要有N、N+1、N-1電子態的波函數和能量,就可以計算它們中的絕大部分。N指的是體系的電子數,比如某個分子最穩定狀態是中性的,則N就是指中性時的電子數,N+1就是指帶一個負電荷的陰離子狀態的電子數,N-1就是指帶一個正電荷的陽離子態的電子數。其實計算CDFT里的那些量并不復雜,手動算起來沒什么難度,然而如果把所有量全都一一算出來,還是比較費事,而且容易弄錯數據。另外,網上也經常有人問我怎么去算那些量,由于初學者的量子化學基礎知識往往很薄弱,經常解釋起來很費勁,甚至怎么說也說不明白。于是,筆者決定在Multiwfn里直接加入一個功能,通過極簡單的步驟,就可以一次性把所有CDFT框架內常見的量全都算出來,過程完全傻瓜化。筆者相信這個功能有極高的實用性,經常要計算CDFT里涉及的量的讀者務必要重視此功能。
這個功能是Multiwfn的主功能22。Multiwfn最新版可以在官網http://www.shanxitv.org/multiwfn免費下載。相關入門知識看《Multiwfn入門tips》(http://www.shanxitv.org/167)、《Multiwfn FAQ》(http://www.shanxitv.org/452)。
使用本文介紹的Multiwfn的功能發表文章時,除了要按照Multiwfn程序啟動時的提示引用Multiwfn的原文外,也請同時引用下面的書籍章節,其中專門介紹了Multiwfn程序中的概念密度泛函理論分析的功能和實現:
Tian Lu, Qinxue Chen, Realization of Conceptual Density Functional Theory and Information-Theoretic Approach in Multiwfn Program. In Conceptual Density Functional Theory, WILEY-VCH GmbH: Weinheim (2022); pp 631-647. DOI: 10.1002/9783527829941.ch31
2 能計算的量
這個功能能直接計算的量主要包含下面這些,分為三大類,具體定義在這里就不寫了,因為在Multiwfn手冊3.25節都已經明確給出了。
? 全局指數(即對整個體系而言的數值)
垂直電離能(Vertical ionization potential,VIP)
垂直電子親和能(Vertical electron affinity,VEA)
Mulliken電負性(Mulliken electronegativity)
化學勢(Chemical potential)
電子的硬度(Hardness)
電子的軟度(Softness)
親電指數(Electrophilicity index)
親電指數ωcubic(在ω之后提出,形式更嚴格)
親核指數(Nucleophilicity index)
? 實空間函數(即變量是三維空間坐標的函數)
福井函數(Fukui function)
雙描述符(Dual descriptor)
局部軟度(Local softness)
局部親電指數(Local electrophilicity index)
局部親核指數(Local nucleophilicity index)
? 原子指數(即每個原子各有一個數值)
簡縮福井函數(Condensed Fukui function)
簡縮雙描述符(Condensed dual descriptor)
簡縮局部軟度(Condensed local softness)
相對親電指數(Relative electrophilicity index)
相對親核指數(Relative nucleophilicity index)
簡縮局部親電指數(Condensed local electrophilicity index)
簡縮局部親核指數(Condensed local nucleophilicity index)
簡縮局部ωcubic親電指數
上述原子指數都可以寫為基于原子電荷計算的形式。根據一些文章的測試,Hirshfeld電荷非常適合這種目的(不建議用NPA等其它電荷),所以Multiwfn自動計算它們都是用的Hirshfeld電荷,相關討論見《TCA上的一篇對比不同原子電荷預測反應位點、親電/親核性的文章》(http://bbs.keinsci.com/thread-15512-1-1.html)。
3 實例:對苯酚計算CDFT定義的各種量
這個而功能的用法和細節在手冊3.25節介紹了,本文就不再累述,下面直接看一個具體使用例子,對苯酚計算各種量。看完例子后建議再看一下手冊這一節。
3.1 準備.wfn文件
用本文介紹的功能計算各種量之前需要先在當前目錄下給出N.wfn、N+1.wfn和N-1.wfn,分別記錄當前體系三個電子態的波函數信息。可以自行提供它們,做法見《詳談Multiwfn支持的輸入文件類型、產生方法以及相互轉換》(http://www.shanxitv.org/379);但如果你是Gaussian用戶,按照下面的流程操作省事得多。
計算前述所有量要用的幾何結構都是相同的,都是N電子態的極小點結構。對于苯酚這個例子而言,我們需要對苯酚中性狀態進行幾何優化,這一步是用戶自己手動做的,用什么程序、什么級別無所謂,靠譜就行。普通有機體系用常用的B3LYP-D3(BJ)/6-31G*級別一般就夠了,如果想更便宜一些,用半經驗、GFN-xTB之類也可以。在B3LYP/6-31G*下優化中性苯酚得到的結構已經直接給了,是Multiwfn目錄下的examples目錄中的phenol.xyz。
啟動Multiwfn,依次輸入
examples\phenol.xyz //一開始載入的文件只要含有中性苯酚優化后的結構即可,其它格式如pdb/fch/gjf/mol/mol2等也都可以
22 //計算CDFT里定義的各種量
1 //產生波函數文件
[直接按回車] //這一步讓你輸入gjf文件里對應的計算單點任務的關鍵詞。按回車代表用默認的B3LYP/6-31G*,此級別雖然很便宜,但對于考察CDFT定義的量來說一般基本已可以滿足需要
[直接按回車] //這一步讓你輸入計算N、N+1和N-1態用的電荷和自旋多重度。按回車代表用對三個態分別用默認的0 1、-1 2和1 2
現在當前目錄下已經有了N.gjf、N+1.gjf和N-1.gjf,是用于產生.wfn文件的單點任務的Gaussian輸入文件。如果你不自己改gjf里的.wfn文件路徑的話,把這些gjf都用Linux下的Gaussian執行后,N.wfn、N+1.wfn和N-1.wfn會產生在當前目錄下,而如果用Windows下的Gaussian執行則會產生在臨時文件目錄下(比如D:\study\G16W\Scratch下)。如果你當前機子里已經裝了Gaussian,而且之前Multiwfn的settings.ini里的gaupath已經被你設為了實際的Gaussian可執行文件路徑,那么建議此時直接在Multiwfn窗口里輸入y,這樣Multiwfn就會自動調用Gaussian計算這個三個gjf文件,并在當前目錄下產生相應的三個wfn文件,然后gjf和out文件會自動被刪掉。顯然,如果你當前的機子里沒Gaussian,應當把gjf放到有Gaussian的機子上算,算完之后再把得到的.wfn文件拷回來。
注:讓Multiwfn順利調用Windows版Gaussian需要做額外的設置,見Multiwfn手冊Appendix 1。
計算這些gjf文件過程中,特別是計算N+1.gjf(通常對應陰離子)時,可能出現SCF不收斂問題,屆時按照此文加上幫助SCF收斂的關鍵詞即可:《解決SCF不收斂問題的方法》(http://www.shanxitv.org/61)。
三個態的波函數文件也不是必須叫做N.wfn、N+1.wfn和N-1.wfn并處在當前目錄下。用選項2或3開始計算時,若Multiwfn發現當前目錄下缺少這些文件的一個或多個,程序就會讓用戶直接輸入相應電子態的.wfn或.wfx或.fch或.mwfn文件的路徑。因此,比如你想用自行算出來的N.fch、N+1.fch、N-1.fch做后面的計算也完全可以。
另外,如果你沒有Gaussian而只有ORCA,也可以選擇-2 Choose the quantum chemistry program used in option 1之后再選2。之后再選選項1產生輸入文件時,Multiwfn產生的就是三個態的ORCA的輸入文件N.inp、N+1.inp和N-1.inp,手動算完了之后就會在當前目錄下產生N.wfn、N+1.wfn和N-1.wfn(其它產生的文件可刪掉)。如果你把settings.ini里的orcapath設為了實際ORCA可執行文件路徑,那么Multiwfn在產生輸入文件后會問你是否自動調用ORCA去計算它們并在計算結束后刪掉其它文件。
3.2 計算各種指數
現在當前目錄下已經有了N.wfn、N+1.wfn和N-1.wfn,可以開始算了。在當前模塊里選擇2 Calculate various quantitative indices,然后程序就會依次載入wfn文件,讀取其中的能量信息和波函數信息,自動計算Hirshfeld電荷,最后給出各種指數。對于6-31G*基組下的苯酚,在Intel 4核機子下不出10秒鐘就都算完了。算完的數據被輸出到了當前目錄下的CDFT.txt中,此文件內容如下:
Note: the E(HOMO) of TCE used for evaluating nucleophilicity index is the value evaluated at B3LYP/6-31G* level
Hirshfeld charges, condensed Fukui functions and condensed dual descriptors
Units used below are "e" (elementary charge)
Atom q(N) q(N+1) q(N-1) f- f+ f0 CDD
1(C ) -0.0587 -0.1185 0.0852 0.1439 0.0598 0.1018 -0.0841
2(C ) -0.0390 -0.1674 0.0268 0.0658 0.1284 0.0971 0.0626
3(C ) -0.0597 -0.1873 0.0319 0.0916 0.1276 0.1096 0.0360
4(C ) 0.0737 0.0216 0.1739 0.1001 0.0522 0.0762 -0.0480
5(C ) -0.0731 -0.1956 0.0090 0.0821 0.1225 0.1023 0.0404
6(C ) -0.0415 -0.1730 0.0336 0.0751 0.1315 0.1033 0.0563
7(H ) 0.0417 -0.0048 0.0993 0.0576 0.0464 0.0520 -0.0112
8(H ) 0.0450 -0.0193 0.0904 0.0455 0.0642 0.0548 0.0187
9(H ) 0.0473 -0.0160 0.0953 0.0480 0.0633 0.0557 0.0153
10(H ) 0.0387 -0.0230 0.0854 0.0467 0.0617 0.0542 0.0150
11(H ) 0.0444 -0.0207 0.0914 0.0470 0.0651 0.0561 0.0181
12(O ) -0.1977 -0.2443 -0.0517 0.1460 0.0465 0.0963 -0.0995
13(H ) 0.1789 0.1482 0.2294 0.0505 0.0307 0.0406 -0.0197
Condensed local electrophilicity/nucleophilicity index (e*eV)
Atom Electrophilicity Nucleophilicity
1(C ) 0.02576 0.45535
2(C ) 0.05533 0.20827
3(C ) 0.05496 0.28982
4(C ) 0.02249 0.31688
5(C ) 0.05280 0.25977
6(C ) 0.05665 0.23773
7(H ) 0.02001 0.18234
8(H ) 0.02767 0.14394
9(H ) 0.02729 0.15195
10(H ) 0.02659 0.14779
11(H ) 0.02807 0.14875
12(O ) 0.02005 0.46203
13(H ) 0.01325 0.15965
Condensed local softnesses (Hartree*e) and relative electrophilicity/nucleophilicity (dimensionless)
Atom s- s+ s0 s+/s- s-/s+
1(C ) 0.3761 0.1562 0.2661 0.4154 2.4076
2(C ) 0.1720 0.3356 0.2538 1.9510 0.5126
3(C ) 0.2393 0.3333 0.2863 1.3926 0.7181
4(C ) 0.2617 0.1364 0.1990 0.5211 1.9190
5(C ) 0.2145 0.3202 0.2674 1.4925 0.6700
6(C ) 0.1963 0.3436 0.2700 1.7500 0.5714
7(H ) 0.1506 0.1213 0.1360 0.8057 1.2412
8(H ) 0.1189 0.1678 0.1433 1.4116 0.7084
9(H ) 0.1255 0.1655 0.1455 1.3187 0.7583
10(H ) 0.1221 0.1612 0.1416 1.3211 0.7570
11(H ) 0.1228 0.1702 0.1465 1.3858 0.7216
12(O ) 0.3816 0.1216 0.2516 0.3186 3.1384
13(H ) 0.1319 0.0804 0.1061 0.6094 1.6409
E(N): -307.464860 Hartree
E(N+1): -307.383614 Hartree
E(N-1): -307.163438 Hartree
E_HOMO(N): -0.218913 Hartree, -5.9569 eV
E_HOMO(N+1): 0.161297 Hartree, 4.3891 eV
E_HOMO(N-1): -0.464864 Hartree, -12.6496 eV
Vertical IP: 0.301421 Hartree, 8.2021 eV
Vertical EA: -0.081246 Hartree, -2.2108 eV
Mulliken electronegativity: 0.110088 Hartree, 2.9956 eV
Chemical potential: -0.110088 Hartree, -2.9956 eV
Hardness (=fundamental gap): 0.382667 Hartree, 10.4129 eV
Softness: 2.613235 Hartree^-1, 0.0960 eV^-1
Electrophilicity index: 0.015835 Hartree, 0.4309 eV
Nucleophilicity index: 0.116285 Hartree, 3.1643 eV
上面的數據基本沒有什么需要特別解釋的,信息都非常易懂,也可以去對照手冊3.25節的式子。唯一值得特別一提的是親核指數(以及局部親核指數),這個量是依賴于TCE(四氰基乙烯)的HOMO能量定義的,TCE的HOMO能量按理說是應當使用和當前計算級別完全一樣的級別來計算的,這樣得到的全局和局部親核指數才是嚴格的,但Multiwfn在給出的時候是自動用筆者事先算好的B3LYP/6-31G*下的TCE的HOMO值-0.335198 Hartree算的。如果你當前用的不是B3LYP/6-31G*級別且追求更嚴格的結果,應當自行用當前級別去算TCE的HOMO值然后手動根據定義得到全局和局部親核指數。
在Multiwfn手冊里也有手動計算簡縮福井函數和簡縮雙描述符的例子,見4.7.3節,用的也是Hirshfeld電荷和B3LYP/6-31G*級別,因此結果和上面自動算出來的完全一樣。相比之下,用本文介紹的功能實在方便太多了,所有重要的量一口氣全都輸出了!
關于ωcubic的計算
前面例子中輸出信息里的親電指數是Parr早年提出的形式,也是目前用的最多的。后來在J. Phys. Chem. A, 124, 2090 (2020)又有人提出了新的而且更嚴格的形式,比Parr定義的考慮了更高階項,稱為ωcubic。文中發現對于R-X...NH3型鹵鍵二聚體(R為不同基團),鹵原子上的簡縮局部形式的ωcubic值與結合能有很不錯的線性關系,R^2達到0.94,比原本的Parr定義的ω的相關性明顯更好,因此ωcubic也很有意義,可以用于預測弱相互作用強度、解釋弱相互作用內在特征。但由于ωcubic還額外依賴于N-2態的能量,所以默認情況下Multiwfn不計算。如果想計算的話,對于本例應如下操作。載入文件后,依次輸入
22 //計算CDFT里定義的各種量
-1 //要求計算ωcubic
1 //產生波函數文件
[直接按回車] //用默認的B3LYP/6-31G*
[直接按回車] //對N、N+1、N-1、N-2態用默認的電荷和自旋多重度(對于中性閉殼層分子這是適合的)
此時當前目錄下就有了N.gjf、N+1.gjf、N-1.gjf、N-2.gjf,你可以讓Multiwfn直接調用Gaussian計算也可以自行計算。算完后就有了這四個態的.wfn文件。之后進入選項2,在輸出的CDFT.txt中可見不僅我們之前看到的那些量都給出來了,還給出了體系整體的ωcubic、各個原子的ωcubic。與此同時計算ωcubic中用到的第二垂直電離能也會輸出出來。
3.3 計算福井函數和雙描述符
接下來計算福井函數和雙描述符。在當前模塊里選擇3 Calculate grid data of Fukui function and dual descriptor,然后選擇一個合適的格點設定,對于當前這樣小體系選擇Medium quality grid就夠了(大體系應當用High quality grid或其它選項,詳見http://www.shanxitv.org/452中的Q39的討論),然后程序會依次載入當前目錄下的N.wfn、N+1.wfn和N-1.wfn并計算電子密度格點數據,之后會看到一個菜單。通過選擇相應選項,可以把各種類型的福井函數以及雙描述符直接顯示成等值面圖。比如此例我們依次選擇選項1、2、3、4來把f+、f-、f0福井函數和雙描述符都依次繪制出來,下圖是把圖像手動合并到一起的圖,等值面數值都是用的0.007:
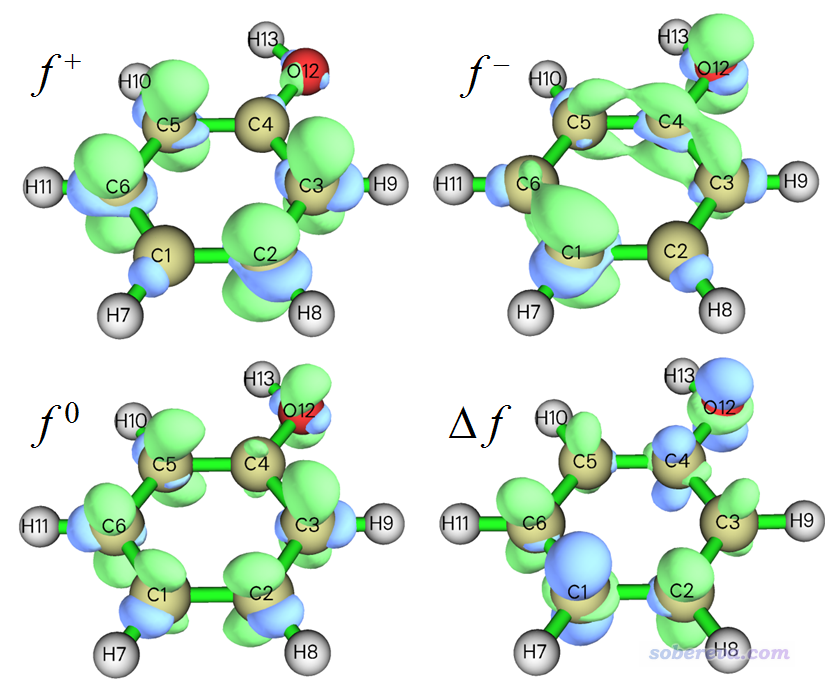
通過主功能5手動計算福井函數和雙描述符的方法在Multiwfn手冊4.5.4節已經詳細介紹了,得到的圖像和上圖完全一樣。使用本文介紹的步驟,明顯遠比手動操作方便得多得多得多!
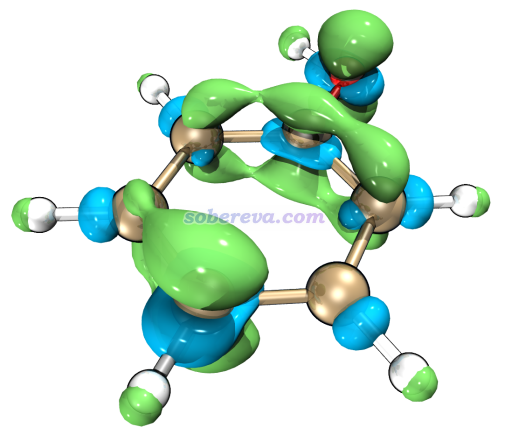
值得一提的是,如果你想要更好的顯示質量,可以借助VMD。比如我們選擇6 Export grid data of f- as f-.cub in current folder,然后把當前目錄下產生的f-.cub按照《在VMD里將cube文件瞬間繪制成效果極佳的等值面圖的方法》(http://www.shanxitv.org/483)里介紹的方法載入VMD并通過Tachyon渲染器渲染,只需要很簡單幾步就可以得到下圖,可見效果極好!
另外,當前功能也可以對局部軟度、局部親電/親核指數觀看等值面和導出cube文件,因為它們的定義都是對特定類型的福井函數乘上一個數值,而Multiwfn當前功能里可以設乘上的系數。比如局部軟度s-定義為全局軟度乘以福井函數f-,我們根據前面算出的數據已知苯酚的局部軟度是2.613235 Hartree^-1,因此若我們想導出局部軟度cube文件,就選-1 Set the value multiplied to all grid data,輸入2.613235,之后選6 Export grid data of f- as f-.cub in current folder導出的cube文件就對應于s-的格點數據了,若選2 Visualize isosurface of f-則看到的是s-的等值面。
4 總結&其它
本文介紹了Multiwfn的超級便利的一口氣算出來概念密度泛函理論里涉及到的各種量的功能,這使得初學者也可以非常順利、快速且不犯錯誤地計算這些量。相信此功能對于將概念密度泛函理論廣泛地應用于實際化學問題的研究有積極的促進作用,也鼓勵大家在日常研究中充分使用此功能,以從繁復的手動操作中解脫出來。
經常有用戶問本文介紹的方法是否只能用于閉殼層中性體系、用于其它體系時怎么對N、N+1和N-1狀態在Multiwfn里輸入凈電荷和自旋多重度。在這里我就再次著重強調一下:顯然本文的方法可以用于非中性、非閉殼層體系。例如,若當前體系是個中性自由基(并假定是普通有機類體系,沒什么復雜的情況),N狀態的凈電荷和自旋多重度是0 2,N+1時是-1 1,N-1時是1 1。原因很容易理解:N+1和N-1的時候分別比N狀態多一個和少一個電子,凈電荷自然分別是N狀態的減1和加1。N狀態原本有一個未配對電子,多一個電子時必然令其配對,少一個電子時它必然丟失,體系自然就成了閉殼層狀態,故N+1和N-1時的自旋多重度為1。注意這里假設N+1和N-1狀態時不涉及到分子軌道簡并因而變成高自旋的可能,一般有機體系都如此,而碰上過渡金屬配合物體系時,拿不準的話應當對N+1和N-1態分別計算最低幾種可能的自旋態的能量看哪種最低以判斷它們應當是什么自旋多重度。
類似地,對于普通有機類體系,其它幾種情況的凈電荷和自旋多重度為:
? 若當前體系是個+1電荷的陽離子自由基,N狀態是1 2,N+1狀態是0 1,N-1狀態是2 1。
? 若當前體系是個-1電荷的陰離子自由基,N狀態是-1 2,N+1狀態是-2 1,N-1狀態是0 1。
? 若當前體系是個+1電荷的閉殼層陽離子,N狀態是1 1,N+1狀態是0 2,N-1狀態是2 2。
? 若當前體系是個-1電荷的閉殼層陰離子,N狀態是-1 1,N+1狀態是-2 2,N-1狀態是0 2。
還值得一提的是,Multiwfn亦能夠計算親核和親電超離域度(superdelocalizabilities),類似于能量權重的基于軌道計算的福井函數。這個量最早由Schüürmann在Environ. Toxicof. Chem., 9, 417 (1990)和Quant. Struct.-Act. Relat., 9, 326 (1990)中提出,被廣泛用于構建QSAR方程。但原文的計算形式只適合基于半經驗方法進行計算,而Multiwfn使用的計算形式是筆者對之進行廣義化后的,也能夠用于HF和DFT波函數。具體細節見Multiwfn手冊3.25.5節,用戶只需要提供分子原本狀態的fch、molden、mwfn之類含有基函數信息的文件,進入主功能22后選擇選項8即可得到各個原子的超離域度。