談談Molekel做靜電勢填色等值面圖的方法及誤區
注1:閱讀此文不要斷章取義。要讀就全讀完,要么別讀。
注2:如果你的目的純粹是想繪制分子表面靜電勢填色圖,此文是絕對的最佳的做法:《使用Multiwfn+VMD快速地繪制靜電勢著色的分子范德華表面圖和分子間穿透圖(含視頻演示)》(http://www.shanxitv.org/443),而當前這篇文章就完全沒必要看了。
談談Molekel做靜電勢填色等值面圖的方法及誤區
文/Sobereva @北京科音 2011-Aug-2
1 前言
研究分子靜電勢(MEP)的時候通常研究的是它在分子范德華表面(一般取電子密度為0.001的等值面)上的數值分布,最直觀的方法是繪制填色等值面圖進行分析。這種圖可以通過很多軟件繪制,比如GaussView、ChemCraft、VMD等等,它們都需要載入兩套格點數據(一般為高斯cub格式),分別描述整個空間的靜電勢和電子密度分布。Molekel 4.x版也能做到,而且有獨特的優勢,就是能讓色彩刻度離散化,并且每種色彩過渡區域都能顯示黑線,類似于填色等值面圖上再加上等值線,這使得分析更容易定量化。然而Molekel 4.x程序本身的界面設計得極差,所以有必要專門介紹下做法。另外,Molekel程序本身可以計算分子靜電勢,而且速度很快,看似十分方便,但實際上這是通過Mulliken原子電荷計算的,這樣得到的靜電勢有嚴重的誤導性,我認為很有必要進行澄清。
由于Molekel 4.x版本已經很老了,目前的Molekel 5.x版本的界面與之相比有了很大變化,所以本文先順便說說用Molekel 5.x做填色等值面圖的方法。遺憾的是,5.x版本的代碼完全重寫了,已不支持色彩刻度離散化,所以不能替代4.x。
2 用Molekel 5.4繪制由Mulliken原子電荷產生的靜電勢填色等值面圖
Molekel目前最新版本是5.4,在這里下載http://molekel.cscs.ch。
先在Gaussian 03里計算一個分子(比如算單點能),計算的時候必須加上gfoldprint pop=full關鍵詞,這樣才會輸出足夠的信息令Molekel能夠計算電子密度。然后在Molekel里選擇打開文件,文件類型選擇g03 output,然后選剛才得到的G03輸出文件。分子載入后,選Surfaces-Electron Density,把Density Matirx(這里實際上就是指電子密度)和Map Molecular Electrostatic Potential都打上勾,Isosurface Value輸入0.001。Bounding Box里面x,y,z代表的是格點范圍的中心,dx,dy,dz是往相應方向延展的距離,將之調大以避免等值面被截斷,調整的時候注意3D窗口中的虛線框,它描述的是格點數據的范圍。都設好后,點Generate就生成了填色等值面圖。如果想把背景調為白色,選Display-Background,然后設成白色。如果想把分子周圍的方框去掉,選Display-Hide Bounding Boxes。如果想顯示色彩刻度條,選Display-Show MEP Scalar Bar,可以用鼠標拖動色彩刻度條移動之。若想讓等值面變得透明,可以再次選Surfaces-Electron Density,點Transparency,把Density Matrix的數值調大點。
最終得到的圖像如下(雙氧水)
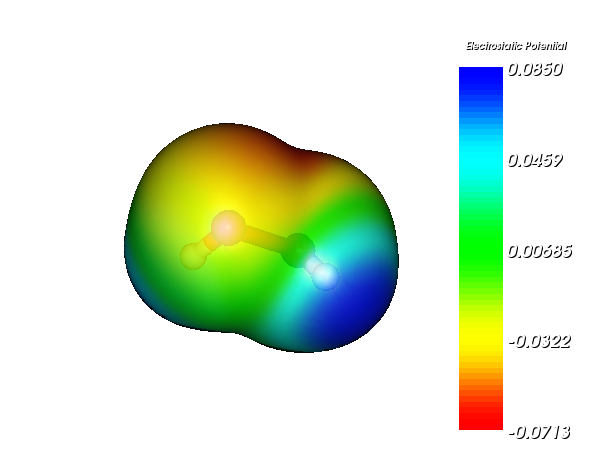
3 用Molekel 4.3繪制由Mulliken原子電荷產生的色彩刻度離散的靜電勢填色等值面圖
4.x版在Molekel網站上已不提供下載,4.3的windows版可以在這里下載/usr/uploads/file/20150610/20150610202037_66558.rar,在Windows XP下可以正常使用,雙擊molekel4.3.win32.exe即可啟動。4.3版的手冊找不到,4.1版的見此http://www.chm.tu-dresden.de/edv/molekel/manual.4.1.html。
在4.x版里面縮放分子可以按住ctrl+鼠標中鍵拖動,或者按住Ctrl+Shift+鼠標左鍵拖動。注意關閉任何子窗口的時候不要按右上角的X,而要點cancel,否則關了之后就再也打不開那個子窗口了。
和Molekel 5.x類似,Gaussian03的輸入文件中必須包含gfoldprint pop=full關鍵詞。由于Molekel 4.x面向的是Gaussian98,在G03中輸出文件信息有了些變動,所以必須手動修改。首先把輸出文件開頭的比如
******************************************
Gaussian 03: IA32W-G03RevE.01 11-Sep-2007
01-Aug-2011
******************************************
里的"Gaussian 03"改成"Gaussian 98",否則載入分子后會發現立體的分子變成了平的。然后把布居分析中的Mulliken atomic charges當中的"Mulliken"改為"Total",否則Molekel找不到原子電荷條目,會認為原子電荷都是0,Molekel也就無法計算靜電勢。如果是多步任務,比如幾何優化,要改的是最后一次出現的Mulliken atomic charges。
這里以乙酸為例介紹作圖方法,筆者的Gaussian03是windows E.01版本。把已經改好的Gaussian03輸出文件放到Molekel所在目錄下(后綴名必須是.out或.log,注意大小寫),在Molekel主界面點右鍵,Load里選gaussian log,選擇那個輸出文件,點Accept。此時分子出現在主窗口中。
然后生成電子密度格點數據,步驟是在主窗口點右鍵選Compute-El. density,直接點OK,然后程序問你生成的電子密度格點數據存在哪里,隨便輸入一個名字,點Accept(后綴是macu,這是Molekel私有的格點數據格式,是二進制文件)。此時主窗口中出現了白色的電子密度等值面,如下圖所示

然而這個默認等值面的數值并非0.001。在主窗口點右鍵選Surface,把cutoff設為0.001,再點Create surface,原來的等值面就被更新為數值為0.001的等值面了。也許你會發現等值面在邊緣有窟窿,這表明默認的格點數據的空間范圍太小,把等值面截斷了。解決方法是主窗口點右鍵-Compute-Adjust box,把x,y,z的上下限都調大些,然后再按上述步驟重新生成電子密度格點數據。
主窗口點右鍵-Compute-MEP,選map to surface,點OK,立刻主窗口中的等值面上出現了靜電勢的色彩投影,同時還出現一個小窗口讓你設定色彩刻度上下限,一般直接點OK用默認即可。
若想將背景設為白色,在主窗口點右鍵,選Color,Diffuse color下面的r,g,b都設為1,此時左上角的球會變成白色。然后點background字樣下面的top和bottom按鈕。如果還想把色彩刻度條的文字改成黑色,把Diffuse color下面的r,g,b都設為0,然后點label按鈕即可。結果如下圖所示
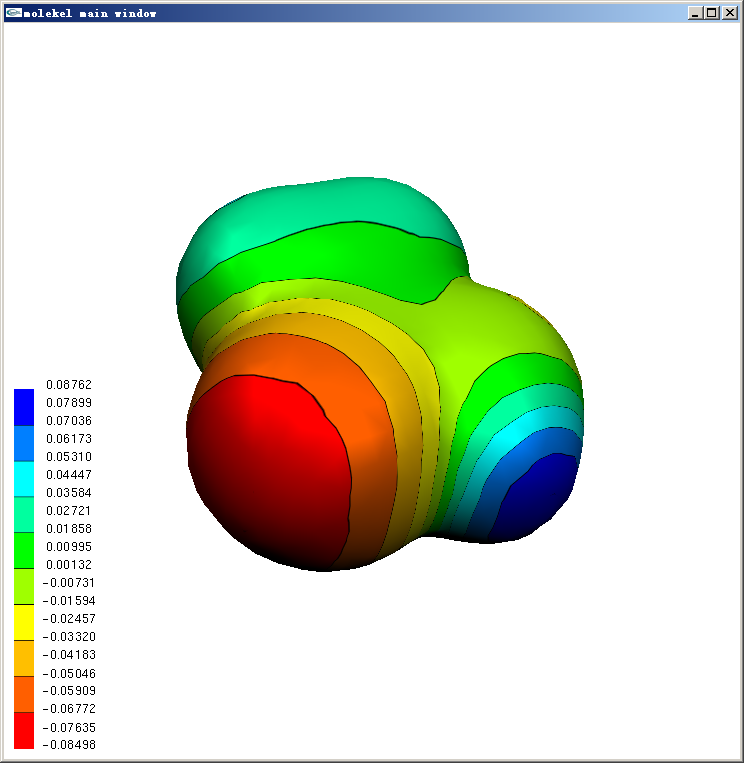
等值面上用黑線分隔的離散的色彩刻度的表現方式在Molekel里叫做Texture。如果想把色彩連續化,即類似在Molekel 5.4中那樣,把主窗口右邊的main interface窗口中的texture的對鉤去掉即可。Texture也允許設定成不同類型,在主窗口點右鍵,選Texture,點擊tex list up/down按鈕就可以切換預設的四種texture,選成想要的texture后,在add texture to下面點擊surface,再點擊主窗口中的等值面,就可以將之套用在相應等值面上了。texture properties框里面可以調texture細節,右下角的start/end texture可以設定只讓某個刻度區間內的texture顯示出來,也是選完之后在add texture to下面點擊surface再點相應等值面以生效。經過簡單調節,上面的圖可以變成下面這樣
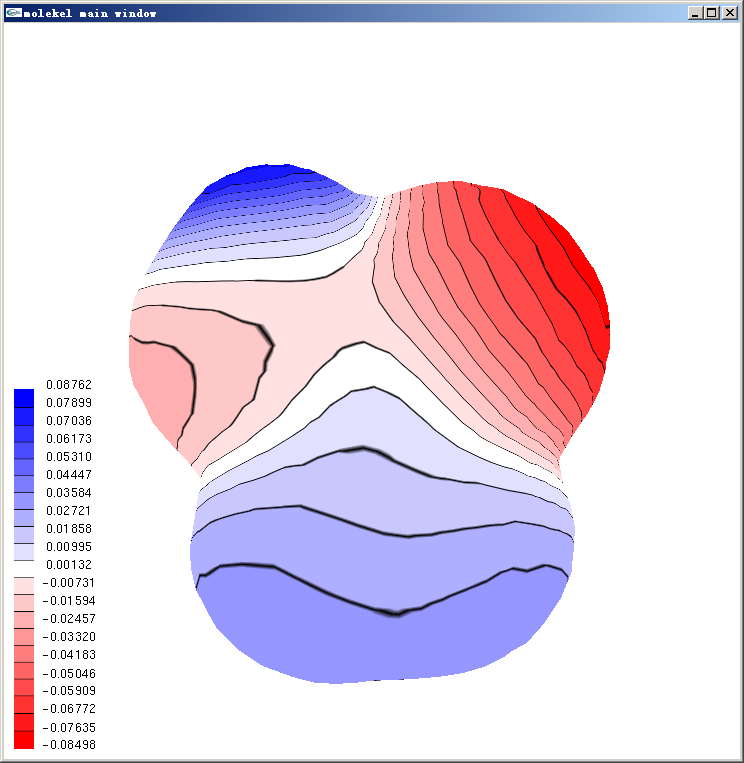
4 在Molekel 4.3里顯示精確的靜電勢圖
在第三節里,靜電勢數據是通過高斯輸出文件里的Mulliken原子電荷生成的,在下一節將會談到這樣生成的靜電勢是很糟的。精確地繪制上一節的那種圖,就需要從外部載入一個電子密度格點文件和一個精確計算的靜電勢的格點文件,注意兩個格點文件的格點設定必須完全一致。這兩個格點文件可以用高斯自帶的cubegen來生成,也可以用筆者開發的Multiwfn來生成(http://www.shanxitv.org/multiwfn),見Multiwfn手冊里的實例,這里就不累述了。注意Multiwfn計算靜電勢用的是核吸引勢積分的方法,是完全精確的。cubegen用的算法不明,有可能用的是近似方法,如多級展開、泊松方程方法,在單核的機子上生成靜電勢的速度比Multiwfn快一些,但是不像Multiwfn那樣支持并行。
把算好的電子密度和靜電勢格點文件放在Molekel所在目錄,后綴改為cube。在Molekel的主窗口里點右鍵,選Load-gaussian cube,然后選電子密度或靜電勢格點文件,點Accept。實際上此時并沒有把格點數據讀進去,只是把cube文件里記錄的分子結構信息讀入了。然后點右鍵選Surface,把類型選為gaussian cube,再點Load,選擇電子密度格點文件,點Accept。把cutoff設為0.001,點create surface。此時電子密度0.001的等值面生成好了。再點Load,選擇靜電勢格點文件,點Accept,然后再點grid value,點OK直接用默認的色彩刻度。此時靜電勢數據就被投影到剛才生成的等值面上了,圖像和第3節的效果類似。
5 原子電荷生成的靜電勢與精確靜電勢的關系
原子電荷這種模型本身有很大的局限性,它產生的靜電勢相當于一個個點電荷產生的球型勢的疊加。然而在分子體系中,當原子核附近電子分布的各向異性十分明顯時,比如出現pi鍵、孤對電子的地方,這種模型就顯得太粗糙,很多電子密度分布產生的效應就被抹殺掉了,原子電荷產生的靜電勢很可能連定性正確都稱不上。F2是個很好的例子,下面是使用Multiwfn繪制的截面的靜電勢圖,粗黑線是范德華面,因此對應分子內部的灰色區域不必關心。
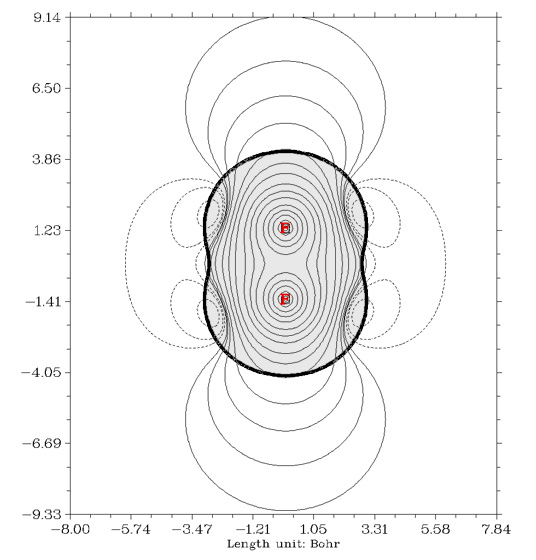
從圖上看,在分子軸線的兩端有靜電勢正值區域(實線處,被稱為sigma穴),由靜電勢互補規則可知,F2可以通過軸向與Lewis堿形成復合物,這被稱作鹵鍵(相關概念見諸如J Mol Model,13,291、J Mol Model,13,305)。圖中虛線區域顯示F2在側面還有很大的靜電勢負值區域,因此能夠F2可以利用側面與Lewis酸復合。然而,無論哪種計算原子電荷的方法,由于F2的對稱性,得到的F的原子電荷一定是0。這也就是說,用原子電荷計算的靜電勢在整個空間處處為0,上圖中豐富的靜電勢信息全沒了!所以,千萬別用Molekel生成的靜電勢去分析電子效應過強的問題,否則結果可能是荒謬的。
在很多研究中,對于靜電勢的精度要求并不那么高,或者并不主要關注電子在原子核附近各向異性分布產生的效應,此時使用合適的計算方法得到的原子電荷是能夠滿足需要的,也因此大多數分子力場使用原子電荷模型描述靜電相互作用,并且能得到滿意結果。不同的方法計算的原子電荷對精確靜電勢(這里指與計算原子電荷相同理論級別產生的量子化學計算的靜電勢)的重現性相差極其懸殊,顯然,最好的是擬合靜電勢方法,擬合過程就是指最小化原子電荷產生的靜電勢與精確靜電勢的偏差。不過擬合靜電勢方法也有很多弊端,筆者會在其它文章中詳細討論。而常見的Mulliken電荷的靜電勢重現并不好,偏差通常是擬合靜電勢電荷的2~3倍。而且Mulliken電荷還有很多問題,諸如交叉項平分缺乏物理意義、結果不隨基組質量提高而收斂、在含有彌散函數的基組下容易出現荒謬結果等等。因此,很不建議使用Mulliken電荷來產生靜電勢,也因此Molekel自己產生的靜電勢數據得到的填色等值面圖頂多用來做為定性參考,在文章中對靜電勢進行簡要分析時,起碼應該把高斯輸出文件中Mulliken電荷的數值替換成擬合靜電勢電荷的,然后再讓Molekel計算靜電勢并生成填色等值面圖。如果文章專門探討某些分子的靜電勢,尤其涉及到定量問題時,一定要按第四節的方法繪制精確的靜電勢圖,而不能用原子電荷產生的靜電勢,否則文章會產生嚴重的誤導。