巨大體系的范德華表面靜電勢圖的快速繪制方法
重要提示:下文是很早以前寫的,如今已經沒意義了!如今使用Multiwfn官網上的最新版本,結合下文提到的xtb產生的molden文件作為輸入文件,直接用《使用Multiwfn+VMD快速地繪制靜電勢著色的分子范德華表面圖和分子間穿透圖》(http://www.shanxitv.org/443)介紹的ESPiso腳本繪制才是最好的做法。因為后來得益于《Multiwfn使用的高效的靜電勢算法的介紹文章已于PCCP期刊發表!》(http://www.shanxitv.org/614)中介紹的Multiwfn計算靜電勢速度的巨大提升,以及《巨幅降低Multiwfn結合VMD繪制分子表面靜電勢圖耗時的一個關鍵技巧》(http://www.shanxitv.org/602)所描述的巨幅降低耗時的技巧,用443號文中的做法比下文的做法不僅更省事、省時,還不需要調用收費的Gaussian里的cubegen。
提示:如果你要繪制1000原子以上的特大體系的表面靜電勢圖,應當使用《基于原子電荷極快速繪制超大體系的分子表面靜電勢圖》(http://www.shanxitv.org/639)里的做法,用Multiwfn基于原子坐標和原子電荷來計算,耗時超級低!
巨大體系的范德華表面靜電勢圖的快速繪制方法
文/Sobereva@北京科音
First release: 2019-May-7 Last update: 2020-Apr-26
有人問我,怎么繪制他的含有多達336個原子的分子的分子表面靜電勢圖。對這么大體系,繪制這種圖看似極其困難,很難吃得消,但其實只要方法得當,哪怕只有臺普通四核機子,也完全沒有壓力,而且繪制的圖像效果極佳,本文就來介紹一下流程。繪制用的步驟和《使用Multiwfn+VMD快速地繪制靜電勢著色的分子范德華表面圖和分子間穿透圖》(http://www.shanxitv.org/443)里介紹的基本一樣,但牽扯到一些其它過程和技巧,沒讀過此文的讀者一定要先閱讀一下。本文用到的Multiwfn可以在其官網http://www.shanxitv.org/multiwfn上免費下載,讀者用的Multiwfn版本應是2019年4月或以后更新的。
本文筆者的條件是:CPU為Intel i7-2630QM(4核),操作系統是Win7-64bit,Multiwfn是2019-May-5版,xtb是2019-Apr-16版,Gaussian是G16 A.03版,VMD是1.9.3版,這幾個程序里只有Gaussian是收費的。筆者跑xtb時是在VMware裝的CentOS 7.4虛擬機下跑的,因為此程序目前只有Linux版,不會裝Linux的話看《在VMware 15中安裝CentOS 7.6的完整過程視頻演示》(http://www.shanxitv.org/454)。
要繪制分子表面靜電勢圖,不管通過什么途徑,肯定得先產生波函數,至少得算一次單點任務。但如果你只有一臺四核機子,遇見300多個原子的體系,用Gaussian算DFT就甭想了,哪怕在ORCA里用純泛函開RI,照樣很難算得動。雖然用半經驗方法如PM6算幾百個原子很快,但是一般的程序都不支持基于半經驗方法得到的波函數去算靜電勢。碰到這種情況,最佳的出路就是用Grimme的xtb程序。此程序的安裝和基本用法在《將Gaussian與Grimme的xtb程序聯用搜索過渡態、產生IRC、做振動分析》(http://www.shanxitv.org/421)已經介紹了,不會用xtb的人一定要看一下。
先把那個336原子的大分子的結構文件載入Multiwfn(支持mol、mol2、pdb、xyz、gjf等眾多格式),進入主功能100的子功能2,選擇相應選項導出為xyz文件,比如叫336.xyz。然后把此文件隨便放到一個空目錄下,在此目錄下運行此命令通過xtb做單點計算:xtb 336.xyz --molden(如果這個結構是未經優化過的,可再加上--opt讓xtb優化此結構)。在筆者的4核機子上只花了20秒鐘就算完了,在當前目錄下出現了molden.input,這是Molden輸入文件,可以被Multiwfn讀取并做波函數分析。
把Multiwfn目錄下的settings.ini里的nthreads設為機子里實際的CPU物理核心數,使得CPU運算能力能在接下來的計算中充分發揮。
下面產生此體系的電子密度格點數據。啟動Multiwfn,載入molden.input,然后依次輸入以下命令:
5 //計算格點數據
1 //計算電子密度
3 //高質量格點
計算過程花費僅不到1分鐘,然后選2導出格點數據,此時當前目錄下出現了density.cub,將之改名為density1.cub備用。
然后我們要計算靜電勢格點數據,用Multiwfn調用Gaussian目錄下的cubegen速度會很快,但是這要求必須以fch作為輸入文件,所以我們要把當前的波函數導出為fch文件。接著在Multiwfn里輸入
0 //返回主菜單
100 //其它功能part 1
2 //導出文件
7 //導出fch文件
336.fch //輸出文件名
馬上當前目錄下就有了336.fch。現在退出Multiwfn。
我們修改Multiwfn目錄下的settings.ini,將其中的cubegenpath路徑設為你機子里實際的Gaussian目錄下的cubegen可執行文件的路徑,比如D:\study\G16W\cubegen.exe。關于這點,在http://www.shanxitv.org/435里有更多說明。
當前這個體系很大,當Multiwfn調用cubegen去處理這個文件時,會由于G16的cubegen默認可用內存上限不夠而報錯。為解決這個問題,對于Windows用戶,進入操作系統的控制面板,選擇“系統”-“高級系統設置”-“高級”-“環境變量”,然后在用戶變量里點擊“新建”,變量名填GAUSS_MEMDEF,變量值填1400MB,然后點“確定”。之后cubegen處理fch文件時最大可用內存就為1400MB了,對當前體系完全夠了(如果你的Gaussian是64bit版本,GAUSS_MEMDEF也可以設更大,比如可以填4GB。而對于32bit版Gaussian,GAUSS_MEMDEF通常最多只能設1400MB,再大往往就無法運行了)。如果你用的是Linux版Gaussian,應當運行比如export GAUSS_MEMDEF=4GB命令來讓cubegen可以最大調用4GB內存(建議把這行命令也加入到~/.bashrc文件中,否則每次開啟終端后都得再運行一次此命令)。
啟動Multiwfn,依次輸入
336.fch
5 //計算格點數據
12 //計算靜電勢
3 //高質量格點
現在會看到Multiwfn正在調用cubegen計算靜電勢。這一步是整個過程最耗時的一步,花了50分鐘(而在筆者的2*E5-2696v3 36核服務器上三分多鐘就能算完)。然后選2導出格點數據,此時當前目錄下出現了totesp.cub,將之改名為ESP1.cub。
現在把density1.cub和ESP1.cub都拷到VMD目錄下,把Multiwfn目錄下的examples\drawESP目錄下的ESPiso.vmd也拷到VMD目錄下。啟動VMD,在文本窗口輸入source ESPiso.vmd并回車,馬上就可以看到下圖:
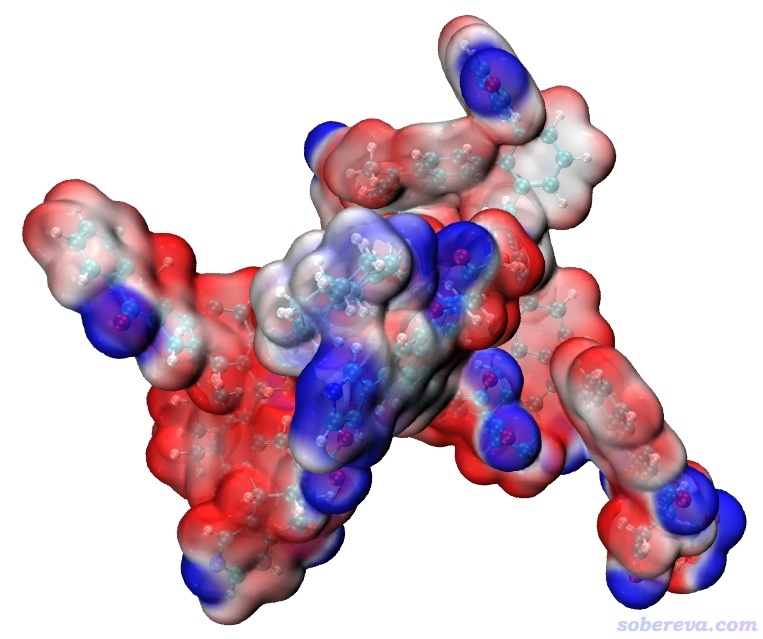
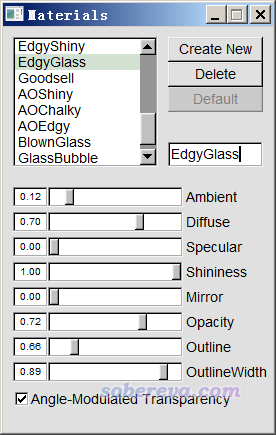
這圖雖然效果已經很好了,但看著還有點亂,因此我們進入Graphics - Materials,選里面的EdgyGlass材質(這是用于顯示當前分子表面用的材質),然后適當調節各個參數令效果盡可能好,最后改成下面這樣:
然后進入Graphics - Representation,點擊對應顯示分子結構的rep,把CPK顯示方式切換為Licorice,并讓鍵的粗細從默認的0.3改為0.2。然后點擊對應Isosurface顯示方式的那個rep,選擇Trajectory標簽頁,把Color Scale Data Range范圍設成比默認-0.03~0.03更寬的-0.04~0.04使得顏色變化更柔和些。最后圖像效果如下,兩個視角都給了,可見效果相當令人滿意!(可能有人覺得這個圖還沒一開始看到的酷炫,但此時的圖更容易分辨分子表面上不同區域靜電勢的差異)
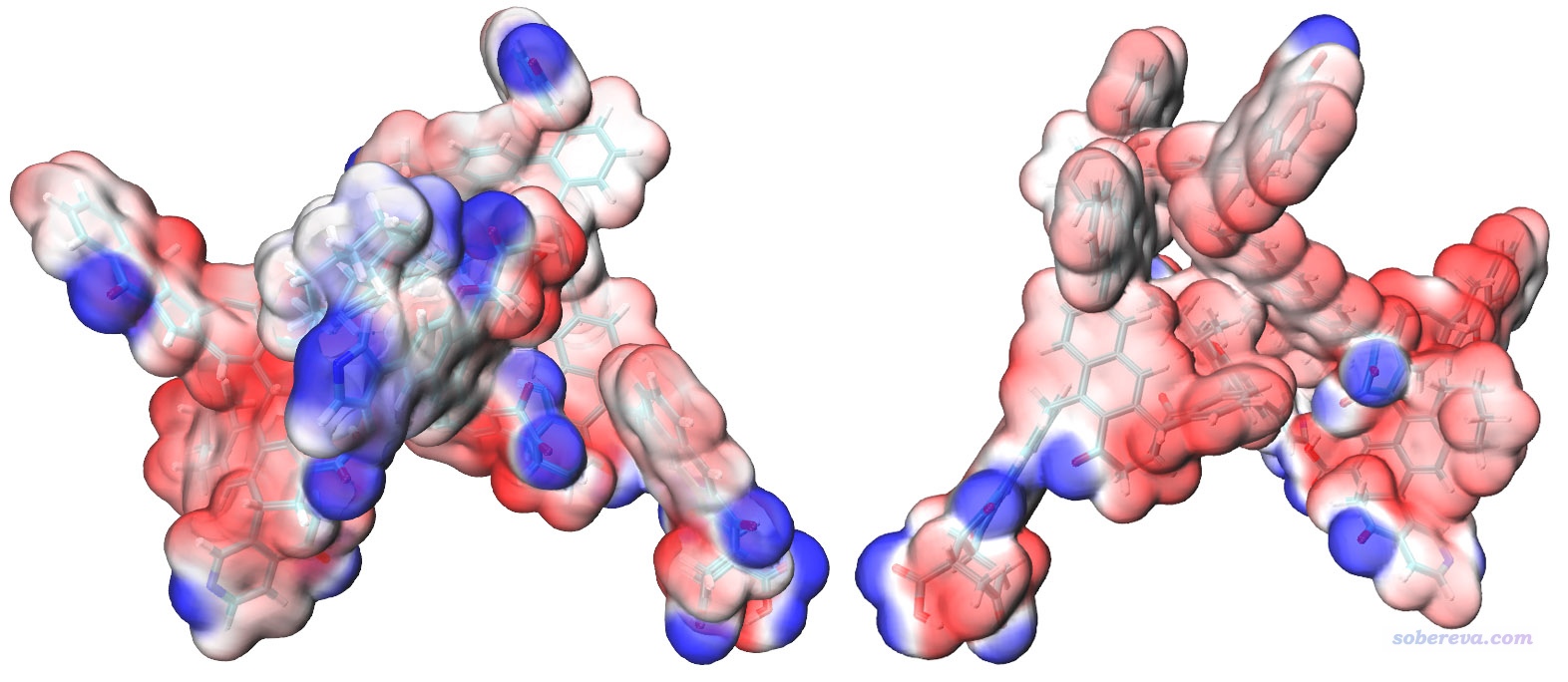
圖中紅色和藍色分別對應于靜電勢為正和為負的區域。如果你想把色彩刻度軸加上去也很容易,做法在http://www.shanxitv.org/443文中已經介紹了。
本文的做法繪制甚至四五百個原子的體系的靜電勢圖也是完全沒問題的,哪怕只有一臺四核機子,也完全可以算得動,只不過是時間長一些而已。本文給出的靜電勢分布其實算不上很準確,畢竟xtb程序做的GFN-xTB計算只相當于半經驗層面的DFT方法,用的是極小基,不過對于幾百個原子的靜電勢圖的繪制,誰也不會那么關注定量大小,只要定性正確就足夠了,當前繪制的圖完全可以滿足這個需要。本文的例子也充分體現出,對于好幾個百原子的精度要求不很高的波函數分析,xtb+Multiwfn可謂是黃金組合。
下面是xtb產生的波函數與精度理想的B3LYP/def2-TZVP波函數繪制的分子表面靜電勢圖的對比,可見差異不是很大,基于xtb的結果已經完全可以滿足需要了。
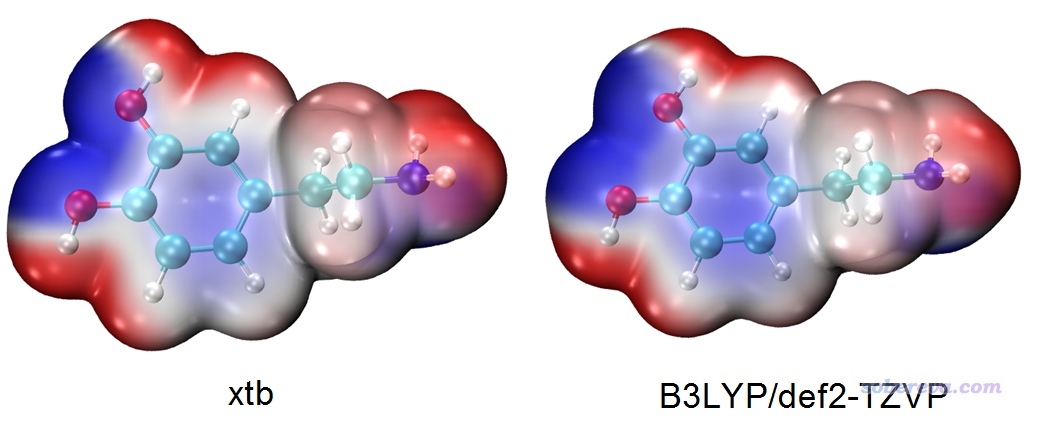
不過,如果你想基于xtb的波函數按照《使用Multiwfn的定量分子表面分析功能預測反應位點、分析分子間相互作用》(http://www.shanxitv.org/159)去定量考察分子表面靜電勢特征,那么xtb的波函數質量還是差點意思。比如對于上面這個多巴胺的分子表面靜電勢最大、最小點數值,基于xtb算的結果與基于B3LYP/def2-TZVP波函數算的結果甚至能差出20 kcal/mol的程度。而對于計算擬合靜電勢電荷,比如CHELPG,基于xtb波函數算的結果與基于B3LYP/def2-TZVP算的結果差異最大的可達0.23,這也不是個小數目。所以,xtb波函數對應的靜電勢可以滿足肉眼觀看、定性分析需要,但定量層面上還明顯達不到準確的標準。