基于原子電荷極快速繪制超大體系的分子表面靜電勢圖
基于原子電荷極快速繪制超大體系的分子表面靜電勢圖
文/Sobereva@北京科音 2022-Apr-13
0 前言
筆者之前寫過很多和分子表面靜電勢分析相關的文章,匯總見http://bbs.keinsci.com/thread-219-1-1.html。其中,《使用Multiwfn+VMD快速地繪制靜電勢著色的分子范德華表面圖和分子間穿透圖》(http://www.shanxitv.org/443)介紹的分子表面靜電勢圖繪制方法已經廣為流行。這種做法需要依賴于體系的電子波函數信息來計算電子密度和靜電勢。由于Multiwfn本身計算的高效率和《巨幅降低Multiwfn結合VMD繪制分子表面靜電勢圖耗時的一個關鍵技巧》(http://www.shanxitv.org/602)里介紹的特殊加速手段,在一般計算條件下對于好幾百個原子的體系繪制表面靜電勢圖都不難。但是,對于超大體系,諸如動輒上千原子的生物分子體系,且不說表面靜電勢圖的繪制,就連量子化學計算得到波函數的過程都幾乎算不動(除非是用比如半經驗層面的GFN-xTB理論)。對這類體系的表面靜電勢圖的繪制,一個可行的方法是通過原子坐標計算準分子(promolecular)密度來構造近似的電子密度的0.001 a.u.等值面,并將基于原子電荷計算的靜電勢投影上去。當然,這樣的表面靜電勢圖的質量肯定不如基于波函數計算得到的,因為準分子密度等同于各個原子孤立狀態下電子密度的簡單疊加,沒有考慮電子轉移、成鍵、極化等效應造成的電子密度改變,而且沒有哪種原子電荷能精確重現分子表面靜電勢分布。不過,對于特大體系,通常對于表面靜電勢圖的精度要求也不高,能半定量正確也就夠了。本文就結合具體例子講解怎么通過Multiwfn結合VMD程序以上述方式對特大體系超快地繪制出表面靜電勢圖。這種做法是完全普適的,同樣的做法顯然也可以直接挪用到其它體系上,請讀者在理解例子里每一步含義的基礎上舉一反三。
1 關于計算分子表面靜電勢用的原子電荷
需要注意的是,計算分子表面靜電勢用的原子電荷應當是能夠對靜電勢較好重現的原子電荷,比如擬合靜電勢電荷(如MK、CHELPG、RESP等),用ADCH也可以。參考《原子電荷計算方法的對比》(DOI: 10.3866/PKU.WHXB2012281)文中第7節的討論。然而,這些基于波函數信息計算的原子電荷對于巨大體系由于太昂貴而無法得到。
那么,用于繪制表面靜電勢圖目的的原子電荷怎么來得到比較合適?這要看具體體系。對于蛋白質和核酸,在分子動力學模擬程序比如GROMACS里用pdb2gmx處理后,或在AMBER程序里用leap處理后,會自動賦予特定的分子力場定義的原子電荷,這些原子電荷對靜電勢的描述都不錯(要不然這些力場也不可能合理地描述分子動力學模擬過程中的靜電相互作用)。而對于其它雜七雜八的大型體系,諸如有機聚合物,原子電荷可以用Multiwfn計算EEM電荷,例子看手冊4.7.5節,它只需要依賴幾何結構,能瞬間對上千原子體系算出來所有原子的電荷,而且結合Multiwfn默認的參數得到的電荷對靜電勢的描述也不錯,只不過此方法支持的元素種類有限,詳見Multiwfn手冊3.9.15節的說明。
如果你對靜電勢的準確度要求很寬松,那也可以用半經驗(如PM6)或者GFN-xTB方法進行計算,特別是后者支持周期表絕大多數元素,對于個人電腦算個兩、三千原子的單點任務無壓力,可以用順便得到的Mulliken電荷來計算靜電勢。但半經驗和GFN-xTB方法本身對電子結構的描述就很糙,再加上Mulliken電荷對靜電勢重現性很一般,這么得到的表面靜電勢圖的可靠性就不敢恭維了,但如果實際作出來的圖你覺得沒大問題那也可以湊合用。
2 例子體系
本文要對下面這個crambin蛋白質繪制分子表面靜電勢圖。這個蛋白質是使用GROMACS在水環境中做過限制性動力學的結構,質子化態對應pH=7的狀態,總共642個原子。
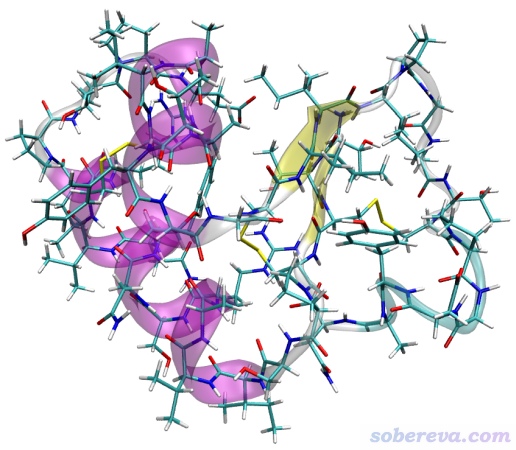
下面例子涉及到的所有文件(除了Multiwfn自帶的)都可以在這里下載:http://www.shanxitv.org/attach/639/file.rar。
讀者請使用2022-Apr-12及以后更新的Multiwfn,否則文件包里沒本文提到的腳本。Multiwfn可以在官網http://www.shanxitv.org/multiwfn免費下載。本文用的VMD是1.9.3 32bit Windows版,可以在http://www.ks.uiuc.edu/Research/vmd/免費下載。對于特別大的體系,按下文步驟產生的cub文件可能非常大,如果運行作圖命令后在VMD自動載入cub文件時VMD崩潰(通常由于可用內存不夠所致),應嘗試用64bit版VMD。本文所有操作假定在Windows下實現。
3 產生chg文件
chg文件是Multiwfn私有的記錄原子坐標和原子電荷的文件。為了用Multiwfn+VMD繪制基于原子電荷和準分子密度的表面靜電勢圖,應當先得到此文件。chg文件格式的定義非常簡單,例如水分子,此文件內容應為:
O 0.000000 0.000000 0.119308 0.301956
H 0.000000 0.758953 0.477232 0.150977
H 0.000000 0.758953 0.477232 0.150977
這5列分別是元素名,X、Y、Z坐標(埃),原子電荷。chg文件怎么準備看實際情況,只要你知道原子坐標和原子電荷,很容易就能手動通過文本編輯器或者依靠Linux下的awk等命令整理成這種格式。
上面的蛋白+水共9366個原子的GROMACS做完限制性動力學后即將做正式動力學的tpr文件是本文文件包里的md.tpr,用的力場是CHARMM36。對于此例,我們要把蛋白質部分的元素、坐標、原子電荷搞成chg格式,具體做法如下:
首先運行gmx editconf -f md.tpr -o all.pdb,得到的all.pdb里最后一列是元素,原子坐標也在文件中體現了。只保留ATOMS開頭的蛋白質原子對應的行而刪除其它的,最后剩下642行。然后Ultraedit等文本編輯器的列模式,把每一行整理成[元素名] [X坐標] [Y坐標] [Z坐標]的形式,命名為protein.chg。
然后按照《將GROMACS的原子電荷信息讀入VMD的方法》(http://www.shanxitv.org/365)里的方法,從tpr文件里提取各個原子的電荷到txt文件中,再用文本編輯器的列模式把原子電荷粘貼到protein.chg的最后一列,chg文件就準備好了,在本文的文件包里已經提供了。
4 計算電子密度和靜電勢格點數據
啟動Multiwfn,輸入以下內容
protein.chg //輸入實際路徑
5 //計算格點數據
1 //準分子密度
4 //設置格點間距
0.3 //0.3 Bohr
2 //導出準分子密度格點數據為當前目錄下的density.cub
0 //返回主菜單
5 //計算格點數據
8 //基于原子電荷算的靜電勢
4 //設置格點間距
0.3 //0.3 Bohr
2 //導出靜電勢格點數據為當前目錄下的nucleiesp.cub
然后把density.cub改名為density1.cub,把nucleiesp.cub改名為ESP1.cub,都挪到VMD目錄下。
上面輸入的格點間距越小圖像越光滑,但計算和導出格點數據的耗時越高、導出的cub文件越大、載入VMD后占內存越大。由于計算準分子密度和基于原子電荷算靜電勢很便宜,以上計算過程在我的8核筆記本上才花了不到15秒鐘!
如果嫌上述過程步驟多,可以把Multiwfn目錄下的examples\drawESP\atmchg目錄下的ESPiso_atmchg.bat和ESPiso_atmchg.txt拷到Multiwfn可執行文件所在目錄下,把bat文件里的輸入文件名和VMD目錄設成實際情況,然后雙擊ESPiso_atmchg.bat,即可自動完成上述的操作。
5 在VMD里繪制分子表面靜電勢圖
請讀者務必先閱讀《使用Multiwfn+VMD快速地繪制靜電勢著色的分子范德華表面圖和分子間穿透圖》(http://www.shanxitv.org/443)了解怎么依靠我提供的VMD作圖腳本十分便捷地在VMD里顯示出靜電勢著色的0.001 a.u.電子密度等值面圖,并恰當配置好vmd.rc文件。由于當前VMD目錄下density1.cub和ESP1.cub分別對應準分子密度格點數據和原子電荷計算的靜電勢格點數據,啟動VMD后在文本窗口里直接輸入iso命令,即得到我們想要的圖了,如下所示。
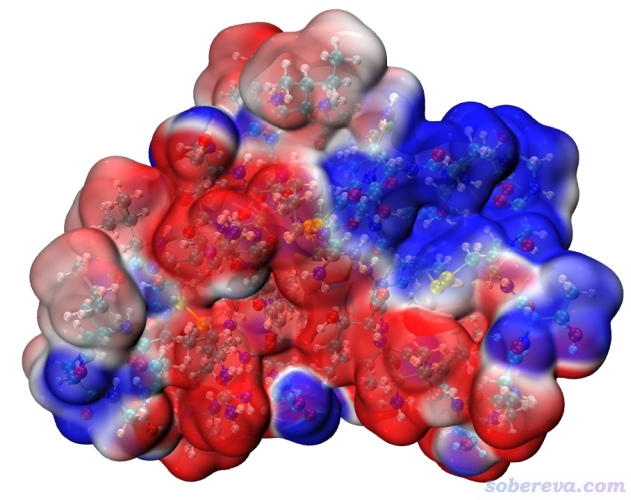
其中白色是靜電勢基本為0的區域。越紅靜電勢越正,越藍靜電勢越負。作圖腳本ESPiso.vmd里默認的色彩刻度范圍是-0.03 ~ 0.03 a.u.。
當前這個圖幾乎非紅即藍,顏色差異太大,導致難以分辨紅色區域以及藍色區域內不同位置的相對靜電勢大小。這主要是因為當前體系里有很多帶電殘基,對表面靜電勢貢獻很大,導致它們附近的表面靜電勢絕對值也往往很大。為了不同區域差異能從顏色上更好地分辨,在VMD的Graphics - Representation界面里選擇對應Isosurface的那項,在Trajectory標簽頁里的兩個文本框里分別輸入-0.06和0.06然后按回車。現在圖像如下,可見顏色的層次更清楚了
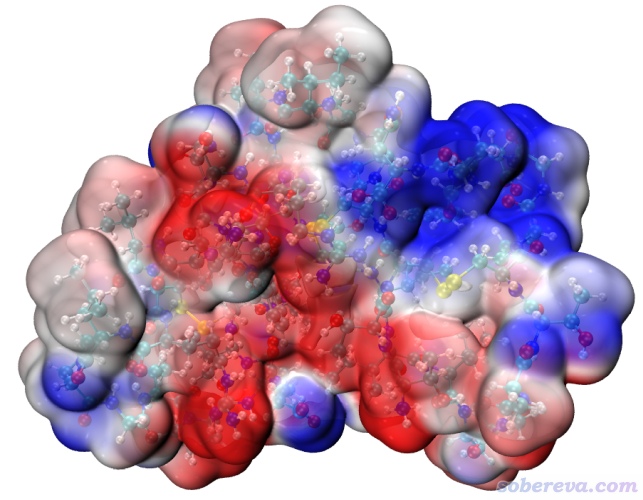
之后,可以自己把Graphics - Colors - Color Scale界面里的色彩刻度條挪到靜電勢圖上并標上上、下限的數值。
6 顯示表面靜電勢極值點位置
在《使用Multiwfn+VMD快速地繪制靜電勢著色的分子范德華表面圖和分子間穿透圖》(http://www.shanxitv.org/443)里演示過怎么把分子表面靜電勢極大點和極小點通過Multiwfn得到并在VMD中顯示出來。對于本文的情況也可以實現。把examples\drawESP\atmchg目錄下的ESPext_atmchg.bat和ESPext_atmchg.txt拷到Multiwfn可執行文件所在目錄下,把這兩個文件里的input.chg改成protein.chg,把.bat文件里的VMD目錄改成當前實際路徑。然后雙擊ESPext_atmchg.bat運行之,將會調用Multiwfn產生記錄表面極值點信息的surfanalysis.pdb文件并自動挪到VMD目錄下。注意這個腳本計算出來的極值點只包含靜電勢為正區域的極大點和靜電勢為負區域的極小點,而靜電勢為負的極大點和靜電勢為正的極小點沒輸出(因為本來大體系表面極值點就很多,所以意義不大的極值點就不輸出了,免得太亂)。
在VMD的文本窗口里輸入ext,之前窗口里顯示的靜電勢著色的分子表面圖上就出現了表面極值點了。在Graphics - Representation中的surfanalysis.pdb體系設置里,適當把VDW顯示方式的Sphere Scale改大到0.2,此時看到的圖如下,其中青色和黃色分別為表面靜電勢極小點和極大點。

可見,表面靜電勢極小點大多在去質子化的羧基氧附近,因為它帶顯著負電荷;而表面靜電勢極大點大多在結合質子的氨基以及羥基的氫附近,因為它們帶顯著正電荷。這很合乎常識,體現出當前圖像的合理性。
讀者之后還可以按照《使用Multiwfn+VMD快速地繪制靜電勢著色的分子范德華表面圖和分子間穿透圖》里說的方法去查詢表面極值點的靜電勢數值并自行標注在圖上。
順帶一提,ESPext_atmchg.bat腳本按照ESPext_atmchg.txt里的指令調用Multiwfn干的事情就是讓Multiwfn載入protein.chg,進入定量分子表面分析功能,把定義表面的函數切換為準分子密度,把映射的函數切換為基于指定的chg文件里的原子電荷計算的靜電勢,然后照常做定量分子表面分析。其實讀者還可以手動這么操作后,在后處理菜單做整體或片段的表面靜電勢分布統計之類的,參考《使用Multiwfn結合VMD分析和繪制分子表面靜電勢分布》(http://www.shanxitv.org/196),以及獲得分子表面靜電勢相關的各種描述符,參考《使用Multiwfn的定量分子表面分析功能預測反應位點、分析分子間相互作用》(http://www.shanxitv.org/159)。
7 總結
本文介紹了怎么僅僅基于原子坐標和原子電荷在極短的時間內獲得分子表面靜電勢圖。雖然此做法不及基于波函數來算準確,但圖像定性正確,且耗時極低。本文的做法若用于幾千個原子的情況,在普通個人計算機上的耗時也就是幾分鐘的事。
值得一提的是,還有一種獲得表面靜電勢的方法是基于原子電荷和半徑信息基于離散格點求解Poisson-Boltzmann (PB)方程,此時可以指定蛋白內和溶劑區域的介電常數,最終求解出的靜電勢格點數據可以明確考慮溶劑效應。這種做法的原理比本文的方法明顯更復雜,而且普適性明顯不及本文的做法。本文的例子沒有明確考慮溶劑效應,溶劑效應實際上已經等效地體現在了CHARMM36力場對氨基酸定義的原子電荷里(可以認為已經等效考慮了溶劑水的極化作用,也因此此力場適合模擬水中的蛋白質)。
本文的方法也完全可以用于周期性體系,做法和本文介紹的沒有絲毫的不同,也是提供記錄元素、坐標和原子電荷的chg文件就行了。