使用CP2K結合Multiwfn繪制密度差圖、平面平均密度差曲線和電荷位移曲線
使用CP2K結合Multiwfn繪制密度差圖、平面平均密度差曲線和電荷位移曲線
文/Sobereva@北京科音
First release: 2022-Mar-10 Last update: 2022-Mar-18
1 前言
電子密度差(electron density difference, EDD)是分析成鍵、電子轉移非常常用的手段。波函數分析程序Multiwfn(http://www.shanxitv.org/multiwfn)在繪制和分析密度差方面非常靈活、方便、強大,筆者之前有不少文章與此相關,見《使用Multiwfn作電子密度差圖》(http://www.shanxitv.org/113)、《使用Multiwfn考察分子軌道、NBO等軌道對密度差、福井函數的貢獻》(http://www.shanxitv.org/502)、《使用Multiwfn計算激發態之間的密度差》(http://www.shanxitv.org/429)、《使用Multiwfn做電子密度、ELF、靜電勢、密度差等函數的盆分析》(http://www.shanxitv.org/179)等。之前筆者寫的密度差有關的文章主要是對于孤立體系寫的,本文的目的之一是介紹怎么將非常流行、免費強大的CP2K第一性原理程序與Multiwfn相結合,對周期性體系繪制密度差圖。
本文另一個目的是介紹怎么通過Multiwfn繪制平面平均密度差(plane-averaged EDD)曲線,它是密度差的衍生。密度差是個三維空間函數,經常通過繪制平面圖、等值面圖方式圖形化考察。雖然這兩種圖很直觀,但平面圖會受到截面選取的影響,而等值面圖會受到等值面數值(isovalue)選取的影響,因此都有一定任意性,也不容易定量討論。對于電子轉移有比較明確方向的情況,可以繪制平面平均密度差曲線,由此可以清楚地考察垂直于選取的方向上每個截面上的密度差積分值,在定量討論、對比上比較便利。例如,固體表面上吸附一個小分子,吸附導致垂直于表面方向有明顯的電子轉移,就可以在垂直于表面的方向上繪制平面平均密度差曲線來準確、細致地考察不同截面處的電子凈增、減情況。還有一種圖叫做電荷位移曲線(charge displacement curve),它相當于平面平均密度差曲線的積分曲線,對于定量討論電荷轉移量情況很有幫助,本文也會介紹怎么通過Multiwfn繪制這種圖。
本文使用的例子是NaCl板吸附一個CO分子,NaCl和CO將被定義為兩個片段,通過密度差考察吸附導致的電子轉移情況。本文文件包里的opt目錄下的NaCl_CO-1.restart是CP2K在PBE-D3(BJ)/DZVP-MOLOPT-SR-GTH級別下對這個體系做優化產生的restart文件,我們將基于這個結構繪制密度差。為簡化問題,在優化的時候整個NaCl板的坐標被凍結為了晶體坐標的狀態,因此不涉及表面重構。
下文涉及到的相關文件可以在http://www.shanxitv.org/attach/638/file.rar下載。cub文件由于體積太大,文件包里就不提供了。
本文的計算使用CP2K 9.1,安裝方法見《CP2K第一性原理程序在CentOS中的簡易安裝方法》(http://www.shanxitv.org/586)。VMD使用的是1.9.3版,可在http://www.ks.uiuc.edu/Research/vmd/免費下載。讀者務必使用Multiwfn最新版,如果不知道是不是最新的就現在立刻去主頁http://www.shanxitv.org/multiwfn下載一個。如果對Multiwfn不了解,看《Multiwfn FAQ》(http://www.shanxitv.org/452)和《Multiwfn入門tips》(http://www.shanxitv.org/167)。不了解cub文件的話可以看看《Gaussian型cube文件簡介及讀、寫方法和簡單應用》(http://www.shanxitv.org/125)。
2 計算密度差格點數據
這一節我們要得到整個體系、NaCl板和CO的各自的電子密度的cub文件,然后由Multiwfn求差。
我們先創建一個計算整個體系的電子密度cub文件的CP2K輸入文件。啟動Multiwfn,然后輸入
NaCl_CO-1.restart //在本文文件包里
cp2k //創建CP2K輸入文件。在《使用Multiwfn非常便利地創建CP2K程序的輸入文件》(http://www.shanxitv.org/587)一文里有詳述
NaCl_CO.inp //產生的輸入文件的路徑
現在進入了創建CP2K輸入文件的界面。為了之后繪制的平面平均密度差曲線比較好觀看,最好把當前體系挪到盒子中央附近。先看一眼當前的結構是什么樣,輸入
-11 //進入幾何操作界面。此界面的詳細介紹見《Multiwfn中非常實用的幾何操作和坐標變換功能介紹》(http://www.shanxitv.org/610)
0 //觀看當前結構
現在出現了圖形窗口。點擊菜單上的Other settings - Toggle showing cell frame把盒子邊框顯示出來,再選Other settings - Toggle showing all boundary atoms把邊界原子顯示出來。現在看到的圖如下
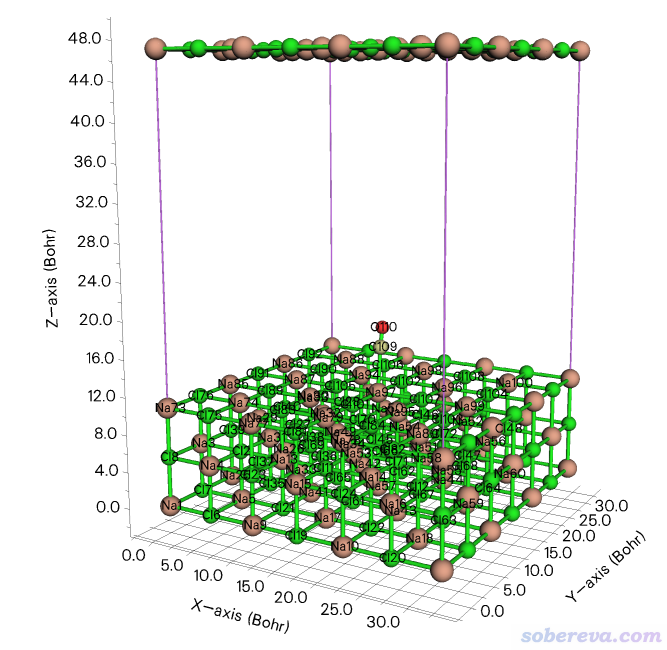
可見當前整個體系在盒子底部,NaCl有三層,由于最下層的原子正好在Z=0的位置,因此這些邊界原子在盒子頂端也顯示了出來。點擊右上角的RETURN按鈕返回。為了把體系挪到盒子中央去,輸入
24 //平移體系使得選中的部分在晶胞中居中
1-110 //當前體系里所有原子。也可以直接按回車選中整個體系
0 //再次觀看體系
可見此時體系確實在盒子中央了
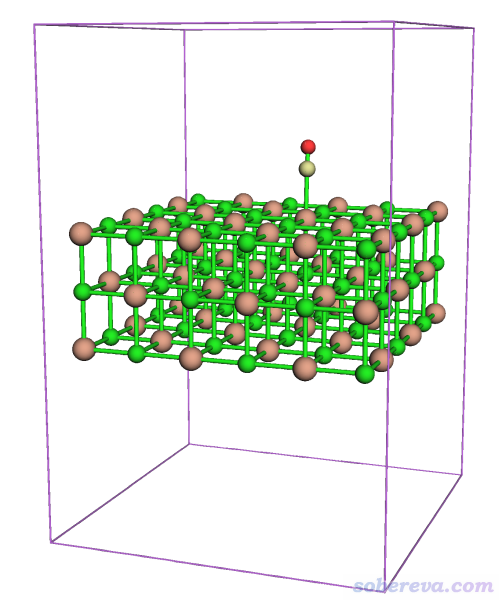
關閉圖形窗口,接著輸入
-10 //返回CP2K輸入文件創建界面
-7 //設置周期性
XY //XY方向二維周期性
-3 //要求產生cube文件
1 //電子密度
0 //產生CP2K輸入文件
現在當前目錄下就有了NaCl_CO.inp。讀者可根據要求修改里面的CUTOFF等設置,本例不那么講究,就用默認的。運行之,算完后當前目錄下就有了NaCl_CO-ELECTRON_DENSITY-1_0.cube,將之改名為NaCl_CO.cub。
將NaCl_CO.inp復制為NaCl.inp,用文本編輯器打開,把其中PROJECT NaCl_CO改為PROJECT NaCl,把&COORD里面C和O原子刪掉。用CP2K運行NaCl.inp,將得到的cub文件改名為NaCl.cub。
將NaCl_CO.inp復制為CO.inp,用文本編輯器打開,把PROJECT NaCl_CO改為PROJECT CO,把&COORD里面Na和Cl原子刪掉。用CP2K運行CO.inp,將得到的cub文件改名為CO.cub。
現在用Multiwfn對三個cub文件求差。啟動Multiwfn,然后輸入
NaCl_CO.cub //輸入此文件實際路徑
13 //處理格點數據
11 //格點數據運算
4 //減去另一個格點數據
NaCl.cub //輸入此文件實際路徑
11 //格點數據運算
4 //減去另一個格點數據
CO.cub //輸入此文件實際路徑
現在內存里的格點數據就已經是密度差格點數據了。想觀看一下等值面的話,可以進入選項-2 Visualize isosurface of present grid data。由于物理吸附對應的密度差的數量級往往很小,所以要用比較小的等值面數值才能清楚看到。在圖形窗口右側把等值面數值設為0.0005后,看到的圖像如下(點save picture按鈕保存的圖像比直接在圖形窗口里看到的更好)
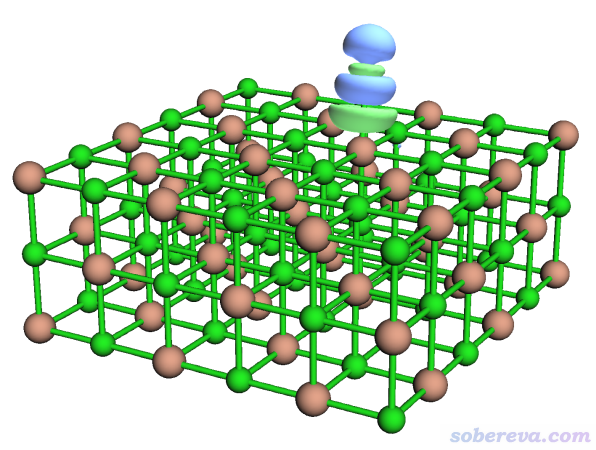
綠色和藍色等值面分別代表格點數據數值為正和為負的部分。當前密度差是按照ρ(NaCl...CO)-ρ(NaCl)-ρ(CO)算的,顯然綠色體現吸附后電子密度增加的部分,藍色體現電子密度減少的部分。點擊右上角RETURN關閉圖形窗口。
若想得到密度差的cub文件,就選0 Export present grid data to Gaussian-type cube file (.cub),輸入EDD.cub,然后內存里的格點數據就被導出為了當前目錄下的EDD.cub。
使用《在VMD里將cube文件瞬間繪制成效果極佳的等值面圖的方法》(http://www.shanxitv.org/483)里提供的我寫的腳本,在VMD程序里只需要輸入一行命令cub EDD 0.0004,就可以把EDD.cub的0.0004等值面顯示出來(如果沒顯示出來,一個字一個字看http://www.shanxitv.org/483這篇文章)。在文本窗口里輸入pbc box把盒子顯示出來,再改成正交視角,經過Tachyon渲染看到的就是下面的圖。圖中藍色小球是Na,棕色是Cl,等值面的藍色和綠色和前面Multiwfn里的定義相同。
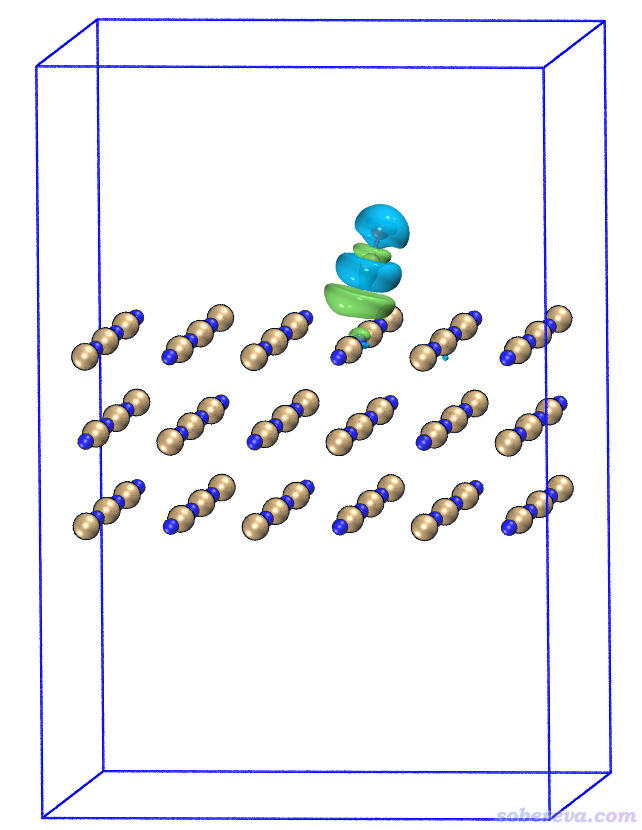
從此圖可以明顯看出,CO的碳結合到Na上后,C和O上的電子密度整體大幅減少,很大程度往Na的方向上轉移了。這也容易理解,當前體系里Na帶顯著的正電荷,CO結合上去,特別是C還帶著豐富的孤對電子,必然其電子會被往Na那邊顯著地極化。
再來看看把等值面數值設得更小的情況。在VMD文本窗口里輸入cubiso 0.0001把等值面數值調為0.0001,稍微調整視圖后看到的圖如下
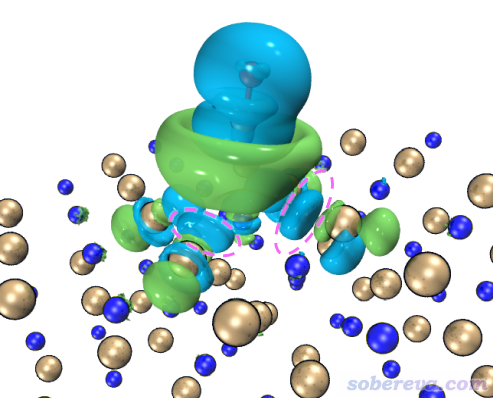
可見此時的密度差圖展現了比之前更多的信息。粉色虛線圈住的地方電子密度有一定降低,由于僅在等值面數值很小的時候才能顯現出來,所以這部分降低量很少(想通過積分得到確切值的話需要用Multiwfn的盆分析功能,前面http://www.shanxitv.org/179文中介紹過)。這部分電子密度降低是因為CO向Na這邊轉移了電子,由于交換互斥作用,使得原本這部分的電子被排斥開,向更遠處轉移。
3 繪制密度差局部積分曲線和平面平均密度差曲線
這一節繪制密度差局部積分曲線和平面平均密度差曲線,首先了解一下定義。某個三維空間函數的局部積分曲線(local integral curve)可以對任意方向繪制,如果對比如Z方向繪制,則表達式為
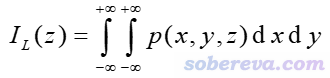
其中p是被考察的函數。如果p取為密度差的話,那么這個曲線就是前述的密度差局部積分曲線了。
還有個概念叫積分曲線(integral curve),就是對局部積分曲線再進一步積分。例如對Z方向如下所示
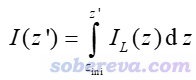
其中積分的起點z_ini由自己決定。如果被考察的函數是密度差,則這個積分曲線對應的就是前述的電荷位移曲線。
平面平均曲線(plane-averaged curve)定義如下,其中A_XY是格點數據對應的盒子在XY方向的面積。這種曲線體現的是被考察的函數在特定截面上的平均值。
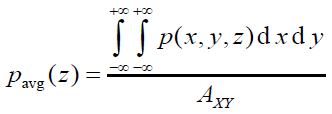
在Multiwfn里可以基于格點數據對任意函數繪制上述三種曲線,需要先把相應的格點數據存到內存里。比如可以通過主功能5格點數據運算功能算出來后自動存到內存里,也可以啟動時從cub等格點數據文件里直接載入,等等。
下面繪制密度差局部積分曲線。如果你之前已經關了Multiwfn,就再啟動Multiwfn,載入密度差格點數據文件EDD.cub,然后進入主功能13,再進入子功能18,這是用來繪制(局部)積分曲線和平面平均曲線的功能。如果你按照上一節操作之后還沒關Multiwfn,則內存里的格點數據正是密度差格點數據,因此直接進入子功能18就行了。
在子功能18里,Multiwfn先問你對哪個方向繪制,我們想垂直于NaCl界面繪制,因此輸入Z,因為NaCl界面平行于XY方向。之后Multiwfn問你繪制范圍的下限和上限位置是什么,此例想對整個Z范圍進行繪制,因此按照屏幕上的提示直接輸入a。之后會看到一個菜單,通過相應選項可以直接繪制積分曲線、局部積分曲線、平面平均曲線,可以保存它們的圖像,還可以把曲線數據導出為文本文件便于用Origin、gnuplot、qtgrace等第三方程序繪圖。
現在選擇屏幕上的選項2 Plot graph of local integral curve,由于當前內存里的格點數據是密度差,所以下圖就是密度差局部積分曲線
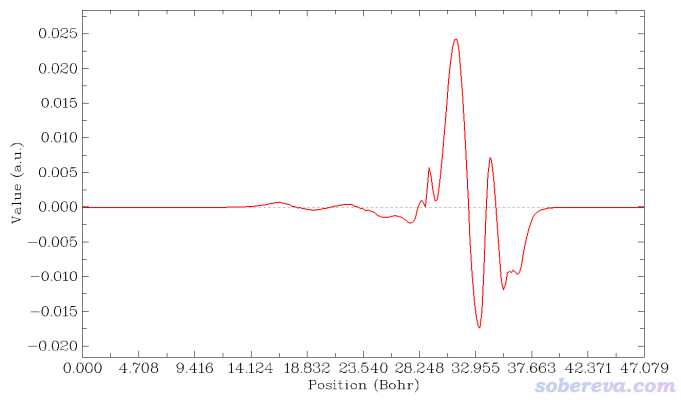
讀者如果想讓等值面圖和密度差局部積分曲線便于準確對照,可以用屏幕上相應選項讓Multiwfn導出局部積分曲線數據為當前目錄下的locintcurve.txt(每一列的含義看導出時屏幕上的提示),然后放到Origin等程序里作圖。之后在VMD里用正交視角,把盒子邊框顯示出來,把保存的等值面圖和局部積分曲線圖放到一起,把前者的白色背景設為透明色或者摳掉,再調節位置和大小讓兩張圖邊框位置正好對上,如下所示。不會用Photoshop的話用版本不很老的PowerPoint也可以完成
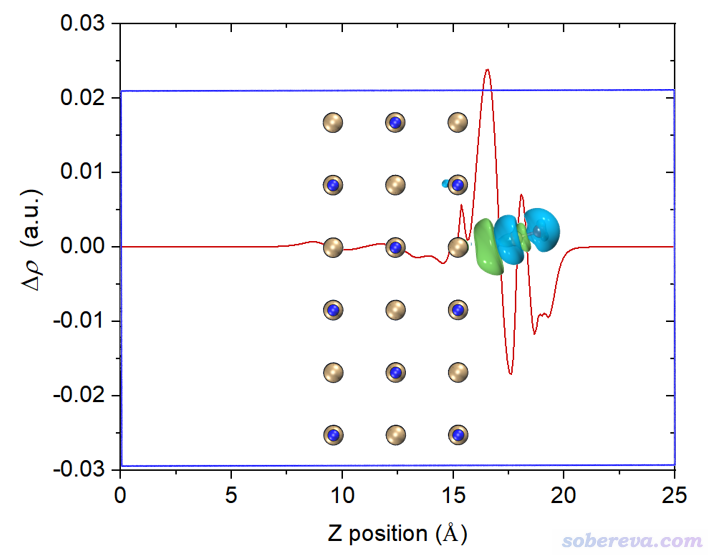
再改一下透明度,裁切一下邊緣去掉盒子邊框,最終得到下面的圖,效果很好。由這個圖可以比較準確地捕捉各個截面上電子的凈增減,可見在C與表層的NaCl相接的地方電子密度由于相互作用而增加明顯(但絕對的數量級并不算大)。
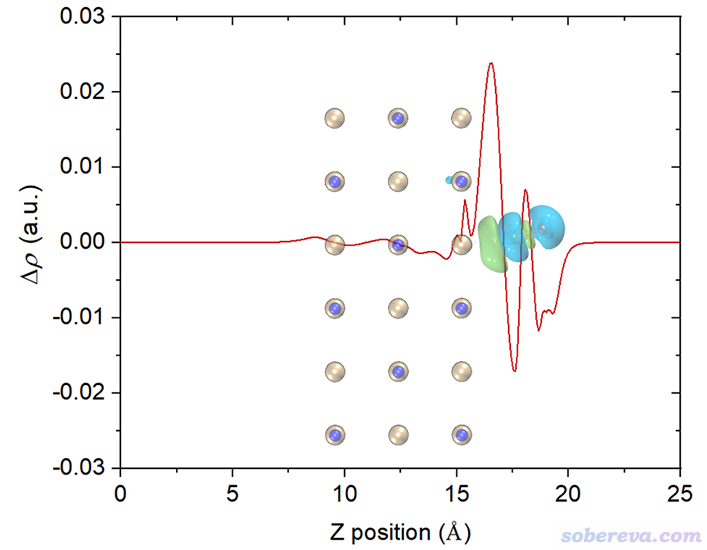
如果想要繪制平面平均密度差曲線,前面選2 Plot graph of local integral curve的地方改成選3 Plot graph of plane-averaged curve即可,其它不變。
4 繪制電荷位移曲線
在Multiwfn當前界面里選1 Plot graph of integral curve,由于當前內存里的格點數據是密度差,因此得到的積分曲線的就是電荷位移曲線了:
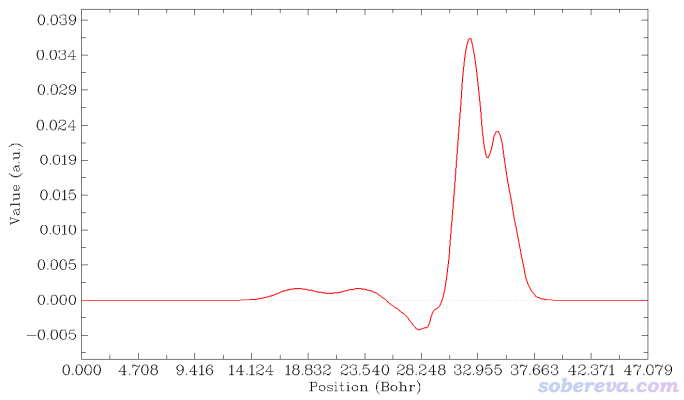
上圖橫軸左端對應于NaCl...CO體系Z=0的位置,橫軸右端對應于NaCl...CO體系Z=25埃(盒子Z尺寸)的位置。這個圖是對上一節的密度差局部積分曲線從左向右積分產生的,從肉眼可以清楚看出來對應關系。曲線最右邊數值為0,體現了密度差全空間積分為0,也即CO與NaCl結合沒有導致總電子數有凈變化。
為了便于觀看,將電荷位移曲線和結構圖+等值面圖疊加起來。我也把曲線最高的峰的橫、縱坐標位置標注上了。電荷位移曲線縱坐標是無量綱的,因為對應的是電子數。
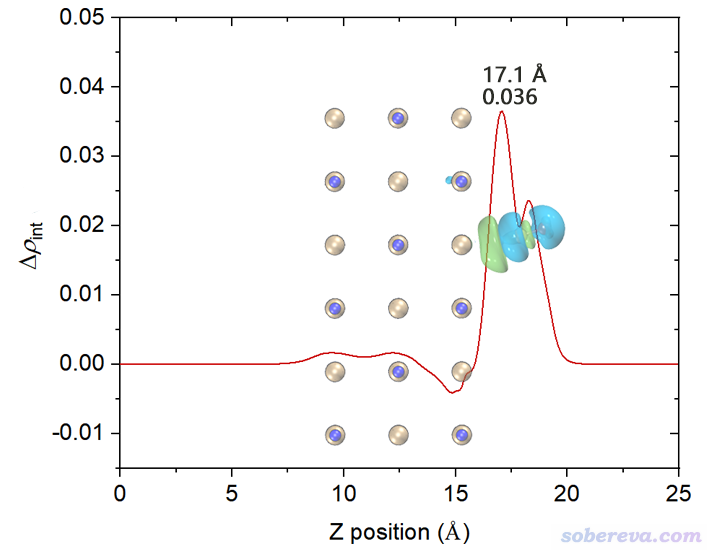
這張圖體現出,在Z<17.1埃的區域里,電子數凈增加了0.036。如果姑且把這個位置作為CO和NaCl的分界,那么說成是CO向NaCl凈轉移了0.036個電子是可以的。對于其它吸附體系,未必在表面和被吸附物之間恰好有一個峰,讀者可以自己決定哪個Z值可作為兩部分的分界面(這有一定任意性),從而從電荷位移曲線上讀出電子凈轉移量。考察電荷位移曲線顯然不是唯一的判斷電荷轉移量的方法,更普適、更常用的是計算片段電荷,即某個片段里所有原子電荷的加和(結果明顯受原子電荷計算方法的選取所影響。原子電荷計算方法有很多,比如基于CP2K的波函數Multiwfn可以計算CM5、Hirshfeld、Mulliken等電荷)。
5 對其它函數計算(局部)積分曲線
上面介紹的Multiwfn計算(局部)積分曲線和平面平均曲線對任何三維空間函數都適用,這里順帶再舉個例子。比如想考察NaCl...CO這個體系不同Z位置的電子密度分布,就啟動Multiwfn,依次輸入
NaCl_CO.cub
13 //處理格點數據
18 //繪制(局部)積分曲線
Z //順著Z方向繪制
a //整個Z范圍
-1 //將橫坐標單位切換為埃
2 //繪制局部積分曲線
由于載入的NaCl_CO.cub里記錄的是電子密度,所以此時看到的下面的局部積分曲線體現了對應不同Z值的XY截面上的電子量。中間三個很高的峰正對應于NaCl板的每一層,18.8埃附近的小峰來自于CO的電子。
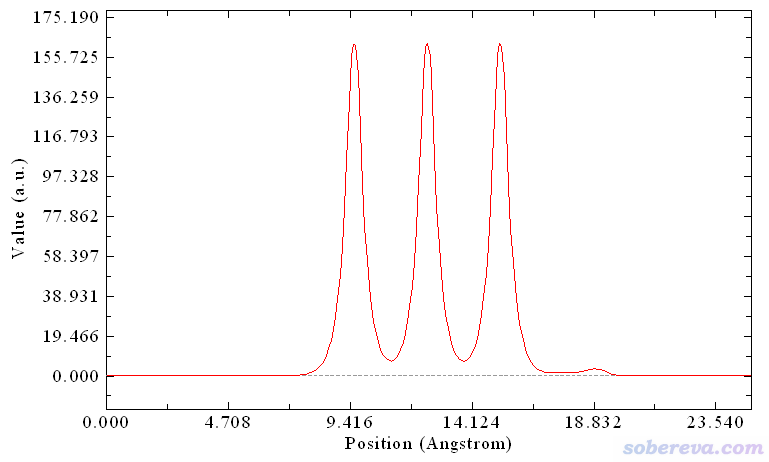
可見,不管什么函數,Multiwfn都能畫(局部)積分曲線或平面平均曲線,只要提供記錄相應函數的格點數據文件就可以。諸如自旋密度也同樣可以繪制這些曲線,比如你有個反鐵磁性體系,位于不同層上的原子自旋磁矩情況各有不同,就可以對自旋密度繪制局部積分或平面平均曲線,清楚地考察電子自旋分布情況。對于靜電勢也可以繪制平面平均曲線,特別是對于界面體系,在垂直于界面方向的這種圖是計算功函數的關鍵。用戶只要給Multiwfn提供靜電勢的格點數據文件即可,比如流行的CP2K就可以直接產生,即在Multiwfn產生CP2K輸入文件的界面里選擇導出cube文件的內容的時候選4 Hartree potential (negative of ESP)。
在http://bbs.keinsci.com/thread-29906-1-1.html一貼里有VASP用戶通過本文介紹的功能基于VASP計算的CHGCAR和LOCPOT文件,用Multiwfn分別考察了平面平均電子密度曲線和平面平均靜電勢曲線,和文獻里的結果相當一致,讀者可以看看。
6 總結&其它
本文介紹了(局部)積分曲線、平面平均密度差曲線、電荷位移曲線這些概念,并詳解介紹了如何將CP2K第一性原理程序與強大的Multiwfn波函數分析程序相結合繪制密度差圖以及這些曲線。從本文的例子可見,密度差等值面圖能很直觀地展現三維空間中各處電子密度的凈增減,而對于恰當的情況(電子轉移和極化以單一方向為主),若將之與平面平均密度差曲線、電荷位移曲線相結合,則能考察得更清楚、更便于定量討論,還便于準確地橫向對比。例如一個固體表面吸附不同分子,想考察電子轉移特征的差異,就可以把不同情況的平面平均密度差曲線同時繪制在一張圖上,差異一目了然。
本文的做法絕不僅限于CP2K用戶使用。記錄電子密度的cub文件用任何其它程序產生也都可以,cub是化學領域最常見的記錄格點數據的格式,因此被支持得很廣泛。Multiwfn支持的格點數據文件格式有很多,還包括dx、grd、vti、VASP的CHGCAR/CHG/ELFCAR/LOCPOT。對于VASP用戶,想得到密度差,可以先用Multiwfn載入CHGCAR,進主功能13用子功能0轉成cub文件,對整體和各個片段都這么做得到各自的電子密度cub文件后,再按上文方式進行操作。
Multiwfn也可以基于波函數文件直接用主功能5計算密度差格點數據。如果你是Gaussian、ORCA等主流量子化學程序用戶,一般都建議先計算出各個狀態的波函數文件(支持的格式和產生方法見《詳談Multiwfn支持的輸入文件類型、產生方法以及相互轉換》http://www.shanxitv.org/379),之后用主功能5的自定義運算功能可輕松得到密度差格點數據(參考《使用Multiwfn作電子密度差圖》http://www.shanxitv.org/113里使用主功能5的過程),然后直接就能去主功能13里的子功能18里繪制密度差局部積分曲線、平面平均密度差和電荷位移曲線。對于CP2K也可以如此,先對涉及的各個狀態產生記錄波函數信息的molden文件,按照《使用Multiwfn結合CP2K通過NCI和IGM方法圖形化考察固體和表面的弱相互作用》(http://www.shanxitv.org/588)里的說明自行把晶胞信息作為[Cell]字段寫進去,然后用主功能5做自定義運算就可以獲得密度差格點數據。
在《全面探究18碳環獨特的分子間相互作用與pi-pi堆積特征》(http://www.shanxitv.org/572)和《一篇文章深入揭示外電場對18碳環的超強調控作用》(http://www.shanxitv.org/570)介紹的我關于18碳環體系的研究中都使用了本文提到的功能繪制了密度差的局部積分曲線,由其中的例子可見這對于考察分子間相互作用和外電場對電子分布的影響特別有好處,非常直觀。
本文的做法是非常普適的,絕不僅限于展現片段間相互作用的密度差。對于電子激發導致激發態相對于基態電子密度分布改變的密度差、外加電場導致電子分布變化的密度差、體系電離或附著電子導致的密度變化的密度差等等,都可以按本文的方法繪制相應的(局部)積分曲線來研究。
使用本文的做法通過Multiwfn產生相關數據若用于發表,記得按照程序啟動時的提示恰當引用Multiwfn。