使用PSI4做對稱匹配微擾理論(SAPT)能量分解計算
使用PSI4做對稱匹配微擾理論(SAPT)能量分解計算
文/Sobereva@北京科音
First release: 2019-Dec-24 Last update: 2020-Oct-15
1 前言
能量分解方法之前我在《Multiwfn支持的弱相互作用的分析方法概覽》(http://www.shanxitv.org/252)中簡單介紹過。能量分解就是將片段間相互作用能分解成不同物理成分,從而能從能量角度更深入地了解相互作用的本質。其中最流行、被接受程度最高的一種是對稱匹配微擾理論(Symmetry-Adapted Perturbation Theory, SAPT),其物理意義較嚴格,也已經有很多程序支持。經常有人問SAPT怎么做,本文就通過例子簡單說一下如何通過免費的PSI4程序來實現。
本文不打算涉及太多SAPT的原理,這部分筆者會在北京科音高級量子化學培訓班里面專門去深入介紹。如果想自己多了解一些原理的話,可以看以下綜述:
Computational Molecular Spectroscopy (2000)第3章
Chem. Rev., 94, 1887 (1994)
WIREs Comput. Mol. Sci., 2, 254 (2012) DOI: 10.1002/wcms.86
WIREs Comput. Mol. Sci., 2, 304 (2019) DOI: 10.1002/wcms.84
WIREs Comput. Mol. Sci., 10, e1452 (2019) DOI: 10.1002/wcms.1452
筆者之前的一些論文使用了SAPT考察了不同問題,很建議一看,可以作為SAPT的應用例子進行參考,了解怎么對SAPT的結果進行分析討論,也非常歡迎大家引用:
? J. Comput. Chem., 40, 2868 (2019) DOI: 10.1002/jcc.26068。主要內容介紹:《透徹認識氫鍵本質、簡單可靠地估計氫鍵強度:一篇2019年JCC上的重要研究文章介紹》(http://www.shanxitv.org/513)
? Carbon, 171, 514-523 (2021) DOI: 10.1016/j.carbon.2020.09.048。文章內容詳細介紹+大量評注:《全面探究18碳環獨特的分子間相互作用與pi-pi堆積特征》(http://www.shanxitv.org/572)
? J. Mol. Model., 19, 5387 (2013) DOI: 10.1007/s00894-013-2034-2。主要內容介紹:《靜電效應主導了氫氣、氮氣二聚體的構型》(http://www.shanxitv.org/209)
? Comput. Theor. Chem., 1195, 113090 (2021) DOI: 10.1016/j.comptc.2020.113090。此文里筆者用SAPT結合def2-TZVPP基組計算了一大批AtXn(X=Cl, Br, I; n=1, 3, 5)???H2O/H2S類型的體系,是SAPT研究含重元素體系的很好的實例。
本文只涉及對單個結構下做SAPT計算,如果你要考察SAPT能量項隨分子間距離的變化,參看《考察SAPT能量分解的能量項隨分子二聚體間距變化的簡單方法》(http://www.shanxitv.org/469)。
2 一些SAPT相關的關鍵性的知識
SAPT在上世紀七十年代最早提出。它既是將片段間作用分解為不同物理成份的方法,本身也是一種計算片段間相互作用能的方法,而且沒有BSSE問題。SAPT只能用于研究弱相互作用,不能用來考察化學鍵這種強相互作用的物理成份,因為SAPT的基本思想是片段間微擾,而強相互作用已經違背了微擾理論的前提。
SAPT可以將相互作用能分解為四部分:
? 靜電(electrostatics):描述片段間經典庫侖作用,數值可正可負
? 交換(exchange):描述片段間近距離出現的交換互斥作用,數值為正(即不利于結合)
? 色散(dispersion):數值為負,起到吸引作用
? 誘導(induction):體現片段間電荷相互極化和相互轉移的作用,數值為負
如果你對分子間相互作用了解不多的話,建議看此文中的相關介紹:《談談“計算時是否需要加DFT-D3色散校正?”》(http://www.shanxitv.org/413)。
SAPT理論涉及片段內的微擾和片段間的微擾,隨著微擾階數增加結果也越來越好。高階SAPT可以達到接近金標準CCSD(T)的弱相互作用計算精度。原理上來說(按照考慮的微擾階數逐級增高),精度關系是SAPT0<SAPT2<SAPT2+<SAPT2+(3)<SAPT2+3。SAPT0對中等、中等偏大體系還能算得動,而SAPT2+(3)這樣的就只算得動小體系了。
為了讓SAPT算的相互作用能精度更好,PSI4程序中的SAPT0還包含了δHF項,它體現了高階項誘導作用。對于SAPT2+、SAPT2+(3)、SAPT2+3這樣的高階SAPT,還可以加上δMP2項來考慮誘導和色散間的高階耦合作用,諸如SAPT2+(3)結合δMP2被稱為SAPT2+(3)δMP2。但δ項的物理意義不是很清楚,也無法進一步劃分。δ項的數值通常較小,一般把它歸入誘導項。
在實際中,并非耗時越高的越高階SAPT結果就越好。在J. Chem. Phys., 140, 094106 (2014)中,通過不同基組結合不同級別SAPT方法的組合測試,發現主要有三個組合最值得推薦,并且用金屬貴重程度進行命名:sSAPT0/jun-cc-pVDZ(銅)、SAPT2+/aug-cc-pVDZ(銀)、SAPT2+(3)δMP2/aug-cc-pVTZ(金),耗時依次升高,精度也依次升高。除了這些以外的組合就完全沒必要考慮了。其中sSAPT0代表scaled SAPT0,是對SAPT0的交換項的經驗校正版本,耗時和SAPT0相同,但結合jun-cc-pVDZ時由于誤差抵消較好的原因,有明顯更好的精度(不要試圖將sSAPT0和其它基組結合,比如用更大的aug-cc-pVTZ。因為這樣會導致誤差抵消得沒那么好,結果反倒會整體變差)。在筆者的J. Comput. Chem., 40, 2868 (2019)中的氫鍵測試中,SAPT2+(3)δMP2/aug-cc-pVTZ計算的弱相互作用能的精度與公認的高精度CCSD(T)/jun-cc-pVTZ結合一半counterpoise校正的結果相比,相對誤差只有3 %,對多數體系絕對誤差小于0.1 kcal/mol,可見已經非常理想了。
能做SAPT計算的程序不少。PSI4是其中做得最好的,開源免費,速度快,輸入文件不復雜,還可以做到很高階的SAPT2+3。Molpro也能做SAPT,但只支持最低階的SAPT0,而且價錢很貴,如今還按年收費。SAPT最初提出者Szalewicz等人的SAPT代碼雖然功能也挺強,但是安裝和使用麻煩。阿Q只能做最低階的SAPT0,還要錢,沒有使用價值。
SAPT還有基于DFT描述單體內作用的變體,包括Molpro能做的DFT-SAPT,以及Szalewicz的SAPT程序和CamCASP程序能做的SAPT(DFT),它們的思想和結果相仿佛。它們比起原本基于微擾方式描述單體內作用的SAPT在原理上能以更低的耗時達到更高的精度,但麻煩的一點是需要額外提供分子的電離能來校正DFT交換勢的漸進行為。前述的J. Mol. Model., 19, 5387 (2013)文中就用DFT-SAPT考察了氫氣和氮氣二聚體,結果很不錯。目前PSI4的SAPT(DFT)搞得還不成熟,預計以后會完善。
PSI4的SAPT0速度很快(遠遠快于高階SAPT),但sSAPT0/jun-cc-pVDZ對很大體系照樣算不動。這種情況就別指望用基于量子化學的方式做能量分解了,此時最適合的是用Multiwfn做基于力場的能量分解,看《使用Multiwfn做基于分子力場的能量分解分析》(http://www.shanxitv.org/442)。
PSI4的各種SAPT方法中只有SAPT0支持開殼層分子間以及開殼層與閉殼層分子間的計算,但研究開殼層弱相互作用的場合不多,本文不舉例子。
有文章專門把SAPT里的極化作用和電荷轉移作用能拆分開,以討論更透徹。在PSI4里也支持,但此時沒法用δMP2改進結果,而且需要額外的計算耗時,因此一般不特意考慮這個。
PSI4里SAPT2+及以上的高階SAPT還可以用CCD方法算色散作用的多體項,需要更多的耗時,據說對于色散作用很強的時候會有改進,本文不做涉及。諸如PSI4里sapt2+(3)(ccd)dmp2關鍵詞就是指SAPT2+(3)δMP2結合這種處理。
PSI4還支持其開發者搞的原子SAPT(A-SAPT)、官能團SAPT(F-SAPT)以及I-SAPT,它們底層都是基于SAPT0的。這些變體不屬于本文內容范疇。
其它量子化學程序也支持一些五花八門的能量分解方法,但要么原理上不如SAPT嚴格(比如LMO-EDA里色散部分只是靠DFT-D色散校正能估算的),要么有這樣或那樣的缺點(諸如局限性大、昂貴,分解出的物理成分意義不清楚、缺乏主流程序支持等等),對于研究弱相互作用來說都不如SAPT理想。
值得一提的是在J. Comput. Chem., 40, 2868 (2019)中筆者發現對氫鍵作用體系,基于總的氫鍵作用能直接就能估計出SAPT各個物理成份的大小,并給出了擬合出來的公式。靠這個都能免得專門去做SAPT分析了,對氫鍵研究很有實際用處。詳見《透徹認識氫鍵本質、簡單可靠地估計氫鍵強度:一篇2019年JCC上的重要研究文章介紹》(http://www.shanxitv.org/513)。
3 PSI4的安裝
PSI4可以在官網http://psicode.org下載。可以直接下載installer包。如果你機子里已經有conda了,也可以按網站上的說明用conda裝。也可以下載源代碼包自行編譯,但沒必要這么折騰。
以下是筆者使用PSI4 1.3.2,用installer包的安裝過程:
下載Psi4conda-1.3.2-py36-Linux-x86_64.sh后,用chmod +x加上可執行權限,然后運行./Psi4conda-1.3.2-py36-Linux-x86_64.sh啟動之。然后輸入安裝路徑,比如/sob/psi4_132,之后開始安裝。等安裝好后輸入yes即可。安裝器會在~/.bashrc文件末尾寫入一大堆亂七八糟的東西,將之都刪掉,然后加上以下兩句
export PATH=$PATH:/sob/psi4_132/bin
export PSI_SCRATCH=/sob2/psi4scr
這里/sob2/psi4scr是筆者專門建立的存放PSI4運行過程臨時文件用的目錄。
之后重新進入終端,PSI4就可以通過輸入psi4命令用了。
4 SAPT計算實例
為了令初學者也能很容易地上手SAPT計算,筆者在Multiwfn中加入了相應的選項產生PSI4的SAPT任務的輸入文件,只需輸入兩個片段里原子序號、選擇計算級別,就可以馬上得到現成的SAPT任務的輸入文件。此功能從2019-Dec-23及之后更新的Multiwfn中才有,最新版本可以在官網http://www.shanxitv.org/multiwfn免費下載。對于這個功能,給Multiwfn用的輸入文件可以是Multiwfn支持的任何含有結構信息的文件,諸如.xyz、.mol2、.pdb、.fch等等,詳見《詳談Multiwfn支持的輸入文件類型、產生方法以及相互轉換》(http://www.shanxitv.org/379)。另外,在Multiwfn的主功能0里還提供了方便的獲得整個片段里原子序號功能,使得產生SAPT輸入文件時輸入原子序號非常容易。
本文用到的文件都可以在此下載:http://www.shanxitv.org/attach/526/file.rar。
4.1 例1:水-氨氣二聚體
這里首先我們用一個很簡單的體系,水-氨氣二聚體為例介紹演示SAPT的計算過程。這個體系是筆者的J. Comput. Chem., 40, 2868 (2019)文中計算的第24號體系,當時用B3LYP-D3(BJ)/ma-TZVP級別優化出來的xyz文件是本文文件包里的H2O_NH3.xyz。
啟動Multiwfn,然后輸入
H2O_NH3.xyz
100 //其它功能 (Part 1)
2 //導出文件或產生量子化學程序輸入文件
15 //產生PSI4的輸入文件
sapt.inp //產生的文件的名字
1 //SAPT任務
3 //當前體系很小,因此我們選最昂貴的,即“金”級別的SAPT2+(3)δMP2/aug-cc-pVTZ
1-3 //第一個片段(水分子)里原子的序號
4-7 //第二個片段(氨氣)里原子的序號
當前目錄下此時已經有了sapt.inp文件了,可以關閉Multiwfn窗口了。文件內容如下
memory 6000 MB
molecule dimer {
0 1
O -0.03836200 1.54613400 0.00000000
H 0.06356500 0.57566500 0.00000000
H 0.85354600 1.90026300 0.00000000
--
0 1
N -0.03836200 -1.37627100 0.00000000
H -1.03728700 -1.54652400 0.00000000
H 0.34780000 -1.83229000 0.81779500
H 0.34780000 -1.83229000 -0.81779500
}
set {
scf_type DF
freeze_core True
basis aug-cc-pVTZ
}
energy('sapt2+(3)dmp2')
這個文件里memory應根據自己機子的實際情況進行修改,有更大內存的話可以設得更大。Multiwfn產生的SAPT輸入文件里每個片段都假定電荷為0、自旋多重度為1,如果與實際情況不符需自行更改。PSI4默認不凍核,這里用freeze_core True啟動凍核近似可以令計算更快。scf_type DF代表利用密度擬合加速SCF過程(其實這行可以不寫,因為是默認的)。在官網的某些SAPT例子里還通過df_basis_sapt關鍵詞指定SAPT過程用RI輔助基組,其實是多余的,因為這本來就是默認的。
然后運行psi4 sapt.inp sapt.out -n 36執行這個輸入文件,得到sapt.out。-n后面接的是并行核數。在筆者的Intel 2*2696v3雙路36核機子下此任務用了24秒就算完了。
從末尾可以看到SAPT結果匯總:
SAPT Results
--------------------------------------------------------------------------------------------------------
Electrostatics -18.65467700 [mEh] -11.70598655 [kcal/mol] -48.97784771 [kJ/mol]
Elst10,r -19.13938171 [mEh] -12.01014335 [kcal/mol] -50.25043976 [kJ/mol]
Elst12,r 0.07639831 [mEh] 0.04794066 [kcal/mol] 0.20058373 [kJ/mol]
Elst13,r 0.40830641 [mEh] 0.25621614 [kcal/mol] 1.07200833 [kJ/mol]
Exchange 20.14110989 [mEh] 12.63873727 [kcal/mol] 52.88047673 [kJ/mol]
Exch10 18.33496859 [mEh] 11.50536649 [kcal/mol] 48.13845340 [kJ/mol]
Exch10(S^2) 18.10056896 [mEh] 11.35827850 [kcal/mol] 47.52303725 [kJ/mol]
Exch11(S^2) 0.43063271 [mEh] 0.27022611 [kcal/mol] 1.13062603 [kJ/mol]
Exch12(S^2) 1.37550859 [mEh] 0.86314467 [kcal/mol] 3.61139731 [kJ/mol]
Induction -6.63886139 [mEh] -4.16594842 [kcal/mol] -17.43032818 [kJ/mol]
Ind20,r -8.79354765 [mEh] -5.51803446 [kcal/mol] -23.08745617 [kJ/mol]
Ind22 -0.83815403 [mEh] -0.52594959 [kcal/mol] -2.20057310 [kJ/mol]
Exch-Ind20,r 5.13721139 [mEh] 3.22364881 [kcal/mol] 13.48774663 [kJ/mol]
Exch-Ind22 0.48965157 [mEh] 0.30726100 [kcal/mol] 1.28558002 [kJ/mol]
delta HF,r (2) -2.69965724 [mEh] -1.69406050 [kcal/mol] -7.08794911 [kJ/mol]
delta MP2,r (2) 0.06563457 [mEh] 0.04118632 [kcal/mol] 0.17232354 [kJ/mol]
Dispersion -5.16221417 [mEh] -3.23933830 [kcal/mol] -13.55339144 [kJ/mol]
Disp20 -5.91459173 [mEh] -3.71146234 [kcal/mol] -15.52875845 [kJ/mol]
Disp30 0.25910602 [mEh] 0.16259148 [kcal/mol] 0.68028275 [kJ/mol]
Disp21 0.10497528 [mEh] 0.06587299 [kcal/mol] 0.27561257 [kJ/mol]
Disp22 (SDQ) 0.06225733 [mEh] 0.03906707 [kcal/mol] 0.16345661 [kJ/mol]
Disp22 (T) -0.79231330 [mEh] -0.49718410 [kcal/mol] -2.08021828 [kJ/mol]
Est. Disp22 (T) -0.94442417 [mEh] -0.59263511 [kcal/mol] -2.47958531 [kJ/mol]
Exch-Disp20 1.27046309 [mEh] 0.79722763 [kcal/mol] 3.33560039 [kJ/mol]
Total HF -7.16040663 [mEh] -4.49322299 [kcal/mol] -18.79964501 [kJ/mol]
Total SAPT0 -11.80453526 [mEh] -7.40745771 [kcal/mol] -30.99280306 [kJ/mol]
Total SAPT2 -10.27049811 [mEh] -6.44483487 [kcal/mol] -26.96518908 [kJ/mol]
Total SAPT2+ -11.04768966 [mEh] -6.93252993 [kcal/mol] -29.00570521 [kJ/mol]
Total SAPT2+(3) -10.38027724 [mEh] -6.51372231 [kcal/mol] -27.25341413 [kJ/mol]
Total SAPT2+dMP2 -10.98205509 [mEh] -6.89134361 [kcal/mol] -28.83338166 [kJ/mol]
Total SAPT2+(3)dMP2 -10.31464267 [mEh] -6.47253599 [kcal/mol] -27.08109059 [kJ/mol]
Special recipe for scaled SAPT0 (see Manual):
Electrostatics sSAPT0 -19.13938171 [mEh] -12.01014335 [kcal/mol] -50.25043976 [kJ/mol]
Exchange sSAPT0 18.33496859 [mEh] 11.50536649 [kcal/mol] 48.13845340 [kJ/mol]
Induction sSAPT0 -6.15381951 [mEh] -3.86158004 [kcal/mol] -16.15685089 [kJ/mol]
Dispersion sSAPT0 -4.59412980 [mEh] -2.88285997 [kcal/mol] -12.06188612 [kJ/mol]
Total sSAPT0 -11.55236243 [mEh] -7.24921687 [kcal/mol] -30.33072337 [kJ/mol]
程序把SAPT2+(3)δMP2定義的所有項都輸出了,并且為了考察方便,把不同的項進行加和成為Electrostatics、Exchange、Induction、Dispersion四部分便于考察。其實有些項的歸屬是有任意性的,諸如Exch-Ind22這種交換和誘導的耦合項歸屬到交換也可以,歸屬到誘導也可以,PSI4的開發者將之歸到了誘導項里。
從上面的結果我們可知當前級別總相互作用能是-27.08 kJ/mol,即-6.47 kcal/mol,和J. Comput. Chem., 40, 2868 (2019)的表1中相應的數據(BE-2)完全一致,而文中用高精度的CCSD(T)/jun-cc-pVTZ結合一半counterpoise校正能的結果為-6.41 kcal/mol,可見我們當前用的SAPT級別相當精確。總相互作用中靜電作用貢獻了-48.98 kJ/mol,交換作用貢獻了52.88 kJ/mol,誘導作用貢獻了-17.43 kJ/mol,色散作用貢獻了-13.55 kJ/mol。可見此體系結合的主要貢獻者是靜電作用,而色散和誘導作用相對次要卻也不可完全忽略。
由于當前我們算的是高階SAPT,因此更低階的SAPT相互作用能也都順帶給出了。另外還給出了sSAPT0能量,由于這個結果肯定比我們當前的SAPT2+(3)δMP2糙得多,因此沒必要管它。
輸出文件末尾顯示Buy a developer a beer!,說明運行正常結束了。如果開發者讓你買咖啡,說明任務失敗了。
4.2 例2:尿素晶體中的相鄰尿素間的相互作用
此例我們考察尿素晶體中的相鄰尿素間的相互作用。之前筆者錄過一個視頻《基于分子晶體cif文件摳出分子團簇結構》(https://www.bilibili.com/video/av35864488/),按照視頻里的做法,基于尿素的cif文件我們可以得到下圖的團簇,其結構文件是本文文件包里的Urea_cluster.pdb。
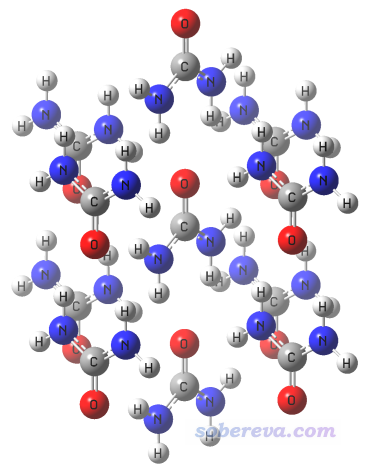
此例我們想獲得中間那個尿素與它上方那個尿素之間的相互作用能和各種能量成份。注意由于X光衍射一般沒法得到確切的氫的位置,做SAPT計算之前理應先固定此體系的重原子而優化所有氫原子,做法可參看《在Gaussian中做限制性優化的方法》(http://www.shanxitv.org/404)。但本例僅是示例目的,所有忽略掉了這一步。
由于此例的團簇中尿素很多,分子內原子序號又不連著,所以我們首先應當用Multiwfn把要考察的分子中的原子序號得到。啟動Multiwfn,輸入Urea_cluster.pdb的路徑,然后進入主功能0。在界面上方菜單欄中選Tools - Select fragment,然后輸入上圖的中間的尿素上隨便一個原子序號,比如2,之后會看到整個分子都被高亮顯示了,并且在文本框里顯示了其中原子的序號,如下所示(如果是Linux版,序號在文本窗口里顯示)。將顯示的原子序號2,13,16,34,36,55,58,77拷出來備用。
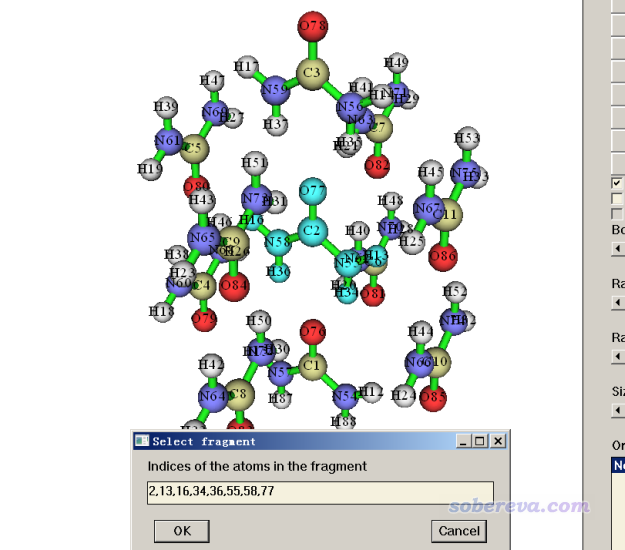
類似地,再次用上述功能,我們得到團簇最上方的尿素的序號,為3,14,17,35,37,56,59,78。
點擊Return按鈕返回Multiwfn主菜單,然后輸入
pi //進入產生PSI4輸入文件的快捷命令,意為PSI4 input
sapt.inp
1 //SAPT任務
1 //作為示例,這里我們用很便宜的sSAPT0/jun-cc-pVDZ級別
2,13,16,34,36,55,58,77 //中間尿素的原子序號
3,14,17,35,37,56,59,78 //上方尿素的原子序號
運行此命令開始計算:psi4 sapt.inp -n 36。用了15秒就算完了。像這樣不指定輸出文件名的話,程序會將輸入文件名自動加上.dat后綴,因此我們得到了sapt.inp.dat。其中SAPT部分的結果如下
Special recipe for scaled SAPT0 (see Manual):
Electrostatics sSAPT0 -24.34792103 [mEh] -15.27855111 [kcal/mol] -63.92545785 [kJ/mol]
Exchange sSAPT0 15.70145759 [mEh] 9.85281339 [kcal/mol] 41.22417122 [kJ/mol]
Induction sSAPT0 -5.93603581 [mEh] -3.72491871 [kcal/mol] -15.58505986 [kJ/mol]
Dispersion sSAPT0 -4.77805738 [mEh] -2.99827627 [kcal/mol] -12.54478793 [kJ/mol]
Total sSAPT0 -19.36055663 [mEh] -12.14893270 [kcal/mol] -50.83113444 [kJ/mol]
即相互作用能為-50.83 kJ/mol。這個結果是比較靠譜的,差不多是兩個中等強度氫鍵的能量。從前面的圖來看,中間和上頭的尿素間就是形成兩個氫鍵。
注意,上面我們得到的并不能認為是絕對嚴格的在晶體環境中的中間的尿素和上方的尿素的相互作用能,因為我們忽略了周圍尿素對它們電子結構的極化、電荷轉移等效應。實際上也沒有辦法絕對嚴格地去計算凝聚相環境中的兩個分子間的相互作用能,因為體系中存在不可分割的多體耦合作用。這種效應導致比如對于三聚體而言,三聚作用能E(ABC)-E(A)-E(B)-E(C)并不等于每一對二聚體作用能的加和,即[E(AB)-E(A)-E(B)] + [E(AC)-E(A)-E(C)] + [E(BC) - E(B) - E(C)]。尿素分子是顯著極性的,但是是電中性的,所以多體耦合作用對相鄰尿素之間的作用能的影響雖然不能完全忽略,但也不至于有定性程度的影響。但如果某一個二聚體旁邊有一個離子,那么耦合作用就強到不可忽略了,因為它與這兩個分子間會有顯著的電荷轉移,并且產生嚴重的電子密度的極化,進而明顯影響兩個單體間的相互作用能。
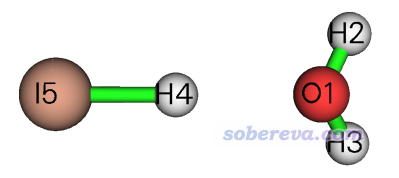
4.3 例3:碘化氫與水的相互作用
此例我們用SAPT2+(3)δMP2研究下圖的碘化氫與水的二聚體。之前筆者用Gaussian通過B3LYP-D3(BJ)結合def-TZVP(氧和氫)和Lanl08(d)(碘)對此體系已做了幾何優化,Gaussian輸入輸出文件已提供在了本文的文件包里。
用GaussView打開H2O_HI_opt.out,直接保存成optimized.gjf文件。此文件就包含了優化后的結構信息,并且可以直接被Multiwfn讀入。
啟動Multiwfn,輸入以下命令
optimized.gjf
pi
sapt.inp
1 //SAPT任務
3 //SAPT2+(3)δMP2/aug-cc-pVTZ級別
1-3 //水中的原子序號
4,5 //碘化氫中的原子序號
當前的sapt.inp不能像之前那樣直接跑,因為aug-cc-pVTZ對第四周期之后的元素都沒有定義!對于這種情況,最省事的做法就是把基組改成def2系列。如《談談贗勢基組的選用》(http://www.shanxitv.org/373)所述,def2對第四周期之后是贗勢基組。對于當前體系用def2系列的話,PSI4自動就會對碘用對應的贗勢。我們把當前輸入文件里的basis aug-cc-pVTZ改成basis def2-QZVP。之所以用def2-QZVP,是因為比它更低的def2-TZVP不帶彌散函數,質量比aug-cc-pVTZ差,因此我們得提高zeta數來彌補其不足。QZ檔次基組計算弱相互作用精度就已經相當高了,除非是超高精度計算或者是陰離子體系,否則完全可以不帶彌散函數(雖說aug-cc-pVTZ-PP對碘有定義,和aug-cc-pVTZ最搭,但在PSI4里沒有自帶,自定義用著麻煩,所以這里不用)。如果def2-QZVP算不動,改為def2-TZVPP也可以,耗時能低一個數量級甚至更多。
然后我們用PSI4對此體系進行計算,SAPT2+(3)δMP2/def2-QZVP計算的總作用能為-14.08 kJ/mol。靜電、交換、誘導、色散分別貢獻-31.67、45.99、-15.39、-13.02 kJ/mol。之前筆者用MP2/jun-cc-pVTZ(對碘用jun-cc-pVTZ-PP)結合counterpoise校正,結果是-13.6 kJ/mol,當前SAPT2+(3)δMP2/def2-QZVP的結果與其很接近(注:MP2算弱相互作用精度很平庸,但唯獨算氫鍵很好。當前數據從側面體現了SAPT2+(3)δMP2/def2-QZVP結果的合理性)。
5 計算CT對結合貢獻的方法
如果想把電荷轉移(CT)對結合能的貢獻單獨得到,把SAPT方法名后面加上-ct即可,在Multiwfn產生SAPT輸入文件的界面里也可以看到有兩個預置的:
10 SAPT0/aug-cc-pVDZ with explicit charge-transfer energy
11 SAPT2+(3)/aug-cc-pVTZ with explicit charge-transfer energy
如果對4.1節的例子選擇上面第11號選項的話,就得到了本文文件包里的sapt_CT.inp。運行后輸出的CT部分信息如下:
SAPT Charge Transfer Analysis
------------------------------------------------------------------------------------------------
SAPT Induction (Dimer Basis) -4.0048 [mEh] -2.5131 [kcal/mol] -10.5147 [kJ/mol]
SAPT Induction (Monomer Basis) -2.5741 [mEh] -1.6153 [kcal/mol] -6.7584 [kJ/mol]
SAPT Charge Transfer -1.4307 [mEh] -0.8978 [kcal/mol] -3.7563 [kJ/mol]
可見CT部分貢獻只有-3.76 kJ/mol,這對應于上面6.7584和10.5147的差值,也即在Monomer basis和Dimer basis下不考慮δHF貢獻的Induction項的差值。
當前任務實際上是在Dimer basis和Monomer basis下分別做了一次SAPT2+(3)計算,前者等于單獨做SAPT2+(3)給出的結果,其中induction作用能(含δHF在內)為-17.6 kJ/mol。CT貢獻的-3.76 kJ/mol只占其很小部分,可見對于弱相互作用二聚體來說,電荷轉移作用對結合的貢獻通常是非常小的。
6 其它
PSI4里一般自動用SAD方式產生初猜波函數,時候難收斂,此時可以在set { ... }里寫上guess GWH換初猜方式,可能就收斂了。不過,有時候GWH會收斂到非基態波函數導致算出的SAPT作用能明顯不對。為判斷是否有這種可能,可以和Gaussian等程序對比一下HF的計算結果進行判斷。另外,在難收斂時也可以嘗試加上soscf true和soscf_max_iter 30。
筆者發現在個別時候,用SAPT2+(3)δMP2結合凍核近似時,誘導項里的delta MP2,r (2)項的數量級異常之大,導致SAPT2+(3)δMP2的結合能明顯不合理,偏離SAPT2+(3)的很多。這種情況應當去掉freeze_core True避免用凍核(當然,代價是對于含有較多周期較靠后的原子的體系耗時會猛增N倍)。我認為這是PSI4的凍核考慮的bug所致。什么情況下會有這種問題筆者還沒摸索出規律,至少之前用SAPT2+(3)δMP2/def2-QZVP計算AtBr5與H2S形成的鹵鍵復合物的時候遇到過這個問題。
有人問做SAPT的時候怎么考慮溶劑效應,實際上SAPT不能直接結合隱式溶劑模型用。溶劑效應對結合能的貢獻應當估計為:使用常規的超分子方法(即二聚體減兩個單體能量之和)在氣相下和溶劑模型下分別計算結合能,二者求差,由此作為溶劑效應對結合能的影響。注意這是一個獨立的項,不要納入到SAPT的任何一個物理成分中。
如果Multiwfn產生SAPT輸入文件的功能對你的研究起到了幫助,希望在文章中提及,如The SAPT input files of PSI4 were generated with help of Multiwfn program,并引用Multiwfn程序啟動時顯示的原文。