談談范德華勢以及在Multiwfn中的計算、分析和繪制
談談范德華勢以及在Multiwfn中的計算、分析和繪制
文/Sobereva@北京科音
First release: 2020-May-2 Last update: 2022-Jul-5
0 前言
在研究分子間弱相互作用領域,靜電勢是經常被考察的量。通過靜電勢我們可以方便、直觀地了解分子的什么地方傾向于與帶有什么電荷的物質(或局部)相互作用,可以對結合強度、結合方式進行解釋和預測。關于靜電勢筆者寫過大量文章,Multiwfn程序在靜電勢分析方面極為靈活和強大,相關博文和資料見《靜電勢與平均局部離子化能綜述合集》(http://bbs.keinsci.com/thread-219-1-1.html)。
對于分子間弱相互作用來說,范德華作用與靜電作用有同等重要的地位,如果對弱相互作用本質不了解的話,建議看《談談“計算時是否需要加DFT-D3色散校正?”》(http://www.shanxitv.org/413)和《透徹認識氫鍵本質、簡單可靠地估計氫鍵強度:一篇2019年JCC上的重要研究文章介紹》(http://www.shanxitv.org/513)中的相關介紹。既然通過靜電勢可以用來估計某分子與其它物質之間的靜電作用特征,那么對范德華作用,是否也能定義一個“范德華勢”來簡單直觀地分析考察某個分子與其它物質之間的范德華作用呢?為此,筆者基于分子力場的思想明確定義了范德華勢的形式,在Multiwfn程序中加入了其計算和分析功能,還專門發表了一篇文章對此進行了詳細介紹,并通過實例展示了范德華勢的特征及其實際價值,請讀者們仔細閱讀,并在使用Multiwfn此功能時務必引用:
Tian Lu, Qinxue Chen, van der Waals Potential: An Important Complement to Molecular Electrostatic Potential in Studying Intermolecular Interactions. J. Mol. Model., 26, 315 (2020) DOI: 10.1007/s00894-020-04577-0
對于極性分子間的相互作用,由于靜電作用占主導,對結合強度、結合模式有決定性作用,因此范德華勢的意義相對弱一些。而對于弱極性分子間相互作用,范德華勢的重要性與靜電勢是相仿佛的,應當將二者同時納入考慮。對于非極性分子間的相互作用,由于范德華作用占主導,范德華勢的分析有著關鍵性的意義。為了充分凸顯范德華勢分析的價值,后文涉及的體系都是非極性分子體系。
下面將首先簡要介紹范德華勢,然后通過實例講解如何通過Multiwfn實現范德華勢的分析。讀者將體會到使用Multiwfn做這種分析非常快速方便,而且相當有實際意義。筆者希望通過本文以及上述論文里更具體的介紹,能令讀者們認識到分析范德華勢的重要性,并且將這種分析充分應用于日后實際問題的研究中。
1 范德華勢的定義和實現
三維空間某個位置r處的靜電勢衡量的是位于r處的一個單位點電荷與當前體系的相互作用能。對于范德華作用,如果以分子力場中常用的Lennard-Jones 12-6勢描述,范德華勢可以寫為下面這樣
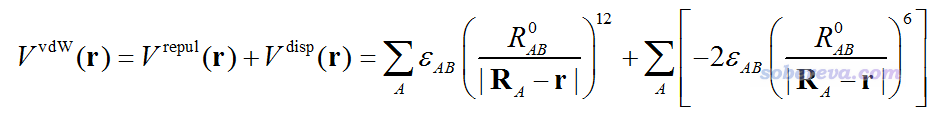
其中V_repul和V_disp分別是范德華勢當中的交換互斥勢和色散吸引勢,分別產生正貢獻和負貢獻。A循環所有原子,ε和R0分別是原子間LJ勢勢阱深度和平衡距離,R_A代表A原子的核坐標。在分子力場中,不同原子的范德華參數可能不同,因此ε和R0都是依賴于原子的。如上定義的范德華勢中,B原子相當于探針原子。探針原子類型選取得不同,范德華勢的分布也會不同。
在Multiwfn中,范德華勢用的參數是基于UFF力場的。之所以用UFF力場,是因為UFF支持的元素近乎涵蓋整個周期表(從H到Lr),而且對每個元素只有一種范德華參數,而不像GAFF那樣對同種元素的不同原子類型往往也有不同的參數,因此避免了指認原子類型的麻煩。也即是說,基于UFF力場分析范德華勢的時候,對探針原子只需指定其元素就夠了。根據筆者的對比,用UFF和用GAFF的參數得到的范德華勢的特征基本上是一致的,再加上通常范德華勢主要就是用來做個定性研究,對精度沒有特別高要求,用普適性的UFF力場的參數就夠了。
2 Multiwfn的范德華勢的分析功能
Multiwfn從2020年4月更新的版本開始全面支持了范德華勢的分析,可以在官網http://www.shanxitv.org/multiwfn免費下載。不熟悉Multiwfn者建議看《Multiwfn FAQ》(http://www.shanxitv.org/452)和《Multiwfn入門tips》(http://www.shanxitv.org/167)。用的輸入文件只要能給Multiwfn提供幾何結構信息即可,比如xyz、mol、mol2、pdb、gjf、fch、molden等等都可以,詳見《詳談Multiwfn支持的輸入文件類型、產生方法以及相互轉換》(http://www.shanxitv.org/379)。探針原子通過settings.ini文件里的ivdwprobe來設,設的是其元素的序號,比如氖就設10。
Multiwfn可以以不同形式做范德華勢的分析:
(1)計算格點數據:使用主功能20的子功能6實現。進入之后選擇格點設置,之后范德華勢以及構成它的交換互斥勢和色散吸引勢都會分別計算出來。計算范德華勢極其便宜,即便是好幾百個原子的體系也是瞬間就能算完。格點數據可以導出成cube文件,也可以選擇直接觀看等值面。
(2)盆分析:載入范德華勢的cube文件后,可以用Multiwfn強大、普適的盆分析功能尋找范德華勢的極大點和極小點和具體的數值,其中極小點尤為有意義,后文將會詳述。盆分析功能在此文里有詳細介紹:《使用Multiwfn做電子密度、ELF、靜電勢、密度差等函數的盆分析》(http://www.shanxitv.org/179)。
(3)拓撲分析:用于找范德華勢極小點位置并得到其函數數值,雖然盆分析也能實現此目的,但拓撲分析的定位精度明顯高于盆分析,而且幾乎不耗內存,因而強烈建議用它代替盆分析。此方法專門在《使用Multiwfn對靜電勢和范德華勢做拓撲分析精確得到極小點位置和數值》(http://www.shanxitv.org/645)進行了介紹,因此本文就不演示了。
(4)繪制成曲線圖和平面圖:范德華勢、交換互斥勢、色散勢分別對應于Multiwfn的自定義函數92、93、94,因此在settings.ini里把iuserfunc設為相應值使得用戶自定義函數(user-defined function)對應相應函數后,就可以通過Multiwfn利用主功能3繪制兩點連線之間的曲線圖、利用主功能4繪制各種風格的平面圖。這對于全面、準確考察這些勢在特定區域的分布特征很有意義。另外,還可以通過主功能1來考察任意坐標處的這些勢的具體值。注:從2022-Jun-4更新的Multiwfn版本開始,范德華勢可以直接在選擇函數的菜單里選25來使用,就不用先把iuserfunc設為92然后再在選擇函數的菜單里選User-defined function了。
Multiwfn還可以考察上述這些勢在分子表面上的定量分布、繪制這些勢在分子表面上的著色圖。但經過實測筆者感覺這兩種形式的分析對于這幾種勢效果不算特別好(明顯不如用于靜電勢時的效果),因此本文對此不再多提。
注意凡是和范德華勢有關的分析,Multiwfn給出的單位一律都是kcal/mol。下面的例子都是使用2020-May-2更新的Multiwfn實現的,不要用更老的版本。
3 實例1:螺烯
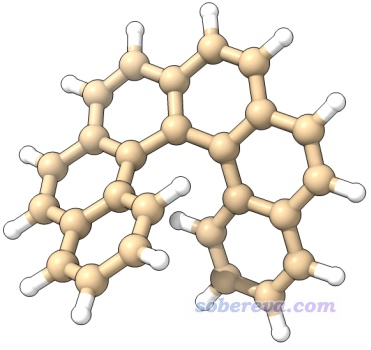
這個例子我們考察一下螺烯的范德華勢,具體來說是6-螺烯,由6個六元碳環并在一起構成的像海螺一樣的立體結構,如下所示。對這個體系考察范德華勢有助于我們了解小分子通過范德華作用傾向于與這個體系在什么區域結合
我們來繪制一下以He元素作為探針原子的范德華勢等值面圖。先把Multiwfn目錄下的settings.ini文件里的ivdwprobe設為2(代表周期表第2號元素,He),然后啟動Multiwfn,輸入
examples\helicene.xyz
20 //弱相互作用的可視化分析
6 //可視化范德華勢
3 //高質量格點(因為這個體系不算小,而且范德華勢計算很快,所以用高質量格點。用中等質量格點的話等值面會比較粗糙)
在進入范德華勢計算功能的時候屏幕上顯示了當前用的范德華參數值,參數在UFF原文里也可以查到
Element of probe atom: He
UFF atomic well depth: 0.056 kcal/mol
UFF atomic radius: 1.181 Angstrom
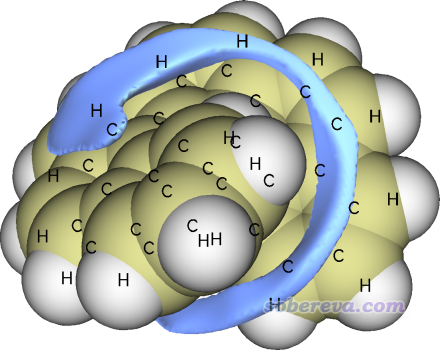
格點數據一眨眼就算完了,之后會看到一些選項,可以直接可視化交換互斥勢、色散勢、范德華勢,也可以將它們導出成cube文件。通常來說單獨考察交換互斥勢和色散勢意義不大,只有它們的加和,即范德華勢,才比較有實際化學意義。我們選擇選項3顯示范德華勢的等值面,把Isovalue改為0.6(當前情況下單位是kcal/mol),恰當旋轉圖像,再用Other settings - Set atom label type要求只顯示元素名,之后就可以看到下圖(a),綠色和藍色分別為正值和負值部分的等值面。等值面的形狀是顯著受Isovalue值的影響的,當前用的0.6能較為清楚地展現范德華勢的主體分布特征。
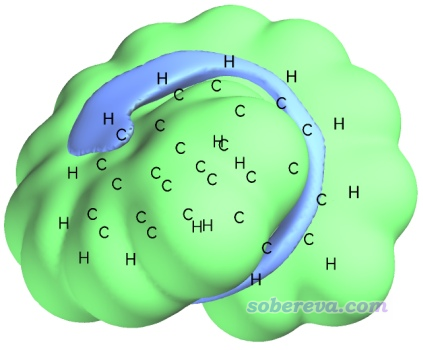
如上所示,在距離原子較近的區域范德華勢總是由交換互斥勢占主導(如同距離原子較近的區域靜電勢總是由核電荷的貢獻占主導),因此這部分通常沒什么可分析的,分析范德華勢主要關心的是負值區域,即色散吸引勢對應的負貢獻大于交換互斥勢產生的正貢獻的部分,探針原子也只能結合到這樣的區域。由上可見,負值等值面只在螺烯的溝壑區域出現,這體現出探針原子He,或者更廣義地說各種小分子(尤其是低極性的、不涉及pi氫鍵之類復雜情況的),傾向于結合到螺烯的溝壑區域。為什么這個區域范德華勢為較為明顯的負值很容易理解,因為在這樣的地方同時有多個周圍的碳原子對其有明顯的色散吸引作用。
為了讓負值區域看得更清楚,我們可以只顯示負值區域。具體做法是在Multiwfn圖形窗口里點擊Show both sign,然后把等值面數值設為-0.6,這樣負值區域就通過綠色顯示出來了。如果想讓其以藍色顯示,可以在菜單里選Isosurface style - Exchange positive and negative colors來讓正、負等值面顏色交換。我們還可以顯示出分子的范德華表面,這樣往往更便于考察,也就是把Ratio of atomic size設為4.0,此時原子球半徑正對應于范德華半徑。現在的圖像如下所示,明顯比上面的圖更容易分辨負值等值面的位置了
如上獲得的范德華勢是否真的能和一些現實情況相對應?大家感興趣的話可以用量子化學程序做一個螺烯+He復合物體系的幾何優化,看看都能得到哪些極小點結構、結合能都是多少。由于極小點可能有很多,建議用《genmer:生成團簇初始構型結合molclus做團簇結構搜索的超便捷工具》(http://bbs.keinsci.com/thread-2369-1-1.html)文中介紹的genmer+molclus的方式做二聚體構型搜索,以確保不會因為優化用的初始結構考慮不全面而有所遺漏。
為了考察范德華勢的分布與實際中He在螺烯附近的動力學行為是否有相關性,筆者對螺烯+He復合物體系用xtb程序在便宜的GFN0-xTB級別下做了2500 ps的動力學模擬。由于二者間的范德華作用很弱,為了模擬過程中He不會因為熱運動跑飛了,因此動力學是在很低溫(10 K)下做的。做這種模擬很容易,參考《使用Molclus結合xtb做的動力學模擬對瑞德西韋(Remdesivir)做構象搜索》(http://bbs.keinsci.com/thread-16255-1-1.html)里關于xtb做動力學的簡要介紹,筆者在北京科音高級量子化學培訓班里會深入全面講從頭算動力學模擬,其中也包括這個例子。動力學模擬得到的結果如下
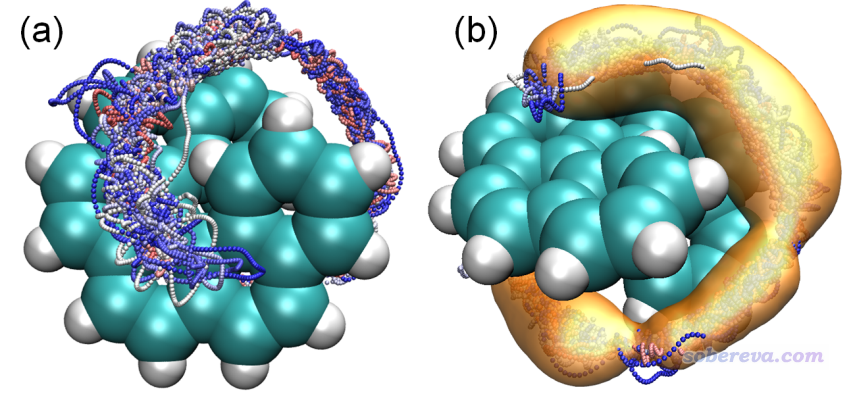
上圖(a)是在VMD程序里將所有幀一起疊加顯示出來的效果(螺烯部分已經做了align以消除其平動和轉動),并且根據模擬時間按照藍-白-紅進行著色。圖(b)是通過VMD的volmap插件計算的空間分布函數(sdf)的等值面。從上圖可以清楚地看到,在模擬過程中He原子在螺烯的縫隙反復來回穿梭,這是為什么不同顏色的小圓點交錯分布在一起。從sdf等值面圖可以更清楚地看到模擬過程中He主要出現的空間范圍,將之與范德華勢等值面圖相對照會發現He的主要出現位置和范德華勢為明顯負值的區域高度吻合,二者的等值面形狀基本一致,因此這個例子充分說明通過考察范德華勢可以便利、直觀地估計其它分子由于范德華作用傾向于被吸附到當前體系的什么區域,顯然這種圖對于研究范德華作用主導的物理吸附非常有價值。
PS:有范德華勢論文的讀者問我怎么繪制上面那種粒子位置疊加圖,我在這里做了詳細說明:http://www.shanxitv.org/wfnbbs/viewtopic.php?id=331。
4 實例2:18碳環
4.1 范德華勢等值面圖的繪制和極值點分析
對18碳環這個幾何和電子結構十分特殊的體系,筆者開展了大量研究,匯總見http://www.shanxitv.org/carbon_ring.html,對于其分子間相互作用特征筆者專門發表了一篇論文做了深入探討,內容介紹見《全面探究18碳環獨特的分子間相互作用與pi-pi堆積特征》(http://www.shanxitv.org/572),非常建議閱讀,其中就充分利用了范德華勢討論了18碳環與小分子作用的本質。在前述的范德華勢的論文里筆者還基于不同探針原子分別繪制了18碳環的范德華勢等值面圖,如下所示,其中只將最重要的負值等值面顯示了出來。圖中黃色小球對應于范德華勢全空間中的極小點位置。圖上將等值面數值和極小點數值都標注了出來,單位為kcal/mol。
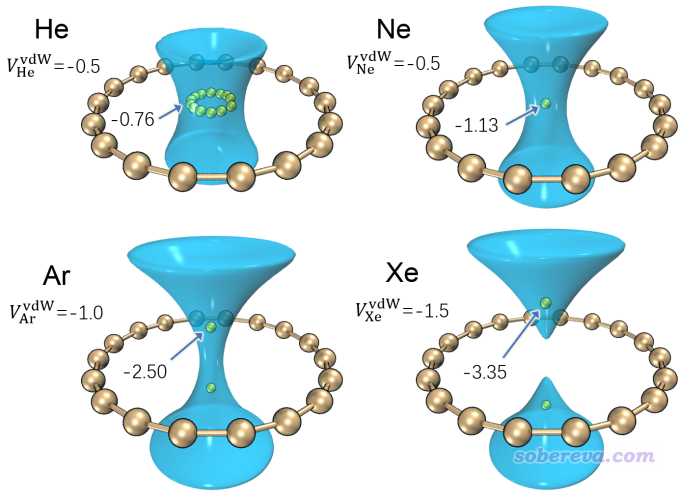
從上圖(a)、(b)中可以看到,以He或者Ne為探針的時候,范德華勢為明顯負值的區域(小于-0.5 kcal/mol)是在環中央及其上方和下方的一定區域。以He為探針的情況,極小點在體系中央連成一個環,數值都是-0.76 kcal/mol,而以Ne為探針的情況,極小點在體系正中央,而且比He的情況更負一些。至于為什么會出現這樣的情況,在范德華勢的論文里筆者做了詳細討論,推薦大家看看。簡單來說,從當前圖上可以判斷出18碳環對Ne的吸附能強于He,并且都是將它們吸附于環的中央。再來看以Ar為探針原子的情況,由于Ar的勢阱深度參數更大,為了便于從等值面上觀察范德華勢的主體分布特征,上圖將其等值面數值設為了比He/Ne更負的-1.0 kcal/mol。由圖可見此時范德華勢極小點不在體系中央了,而是對稱分布在環的上方和下方,這是由于Ar與碳的范德華作用曲線的平衡位置比環的半徑更大所致。再看以Xe為探針原子的情況,其極小點偏離環中央更遠了,等值面的負值區域甚至在環中央處斷開了,這暗示在環中央處要么范德華勢為正,要么就是比等值面數值(-1.5 kcal/mol)更小的負值。造成這種現象的原因在于Xe比Ar的范德華半徑明顯更大。對于甲烷這樣的分子,其整體半徑明顯大于Ne原子,因此可以預期當18碳環吸附它的時候,甲烷的出現位置應當像Ar/Xe的范德華勢的極小點一樣,在碳環的上/下方出現。筆者用量子化學方法做過18碳環與甲烷復合物的幾何優化,結果確實是如此。PS:分子的半徑可以用Multiwfn很容易地計算,可參考《談談分子半徑的計算和分子形狀的描述》(http://www.shanxitv.org/190)和《使用Multiwfn計算分子的動力學直徑》(http://www.shanxitv.org/503)。
下面來看如何繪制上面的圖,這需要借助VMD,可在http://www.ks.uiuc.edu/Research/vmd/免費下載,筆者用的是1.9.3版。這里就繪制以Ar為探針原子的情況。先將settings.ini里的ivdwprobe設為18,然后啟動Multiwfn,輸入
examples\C18.xyz //這是在wB97XD/def2-TZVP下優化得到的結構
20 //弱相互作用的可視化分析
6 //可視化范德華勢
3 //高質量格點
6 //將范德華勢格點數據導出為當前目錄下的vdW.cub
之后重啟Multiwfn,依次輸入
vdW.cub
17 //盆分析
1 //生成盆
2 //基于內存里的格點數據做盆分析
盆分析會把格點數據的正值區域的所有極大點和負值區域的所有極小點位置都找出來(統稱吸引子,attractor),并且確定三維空間每個點都歸屬于哪個吸引子,因此每個吸引子都有一個對應的盆(basin)。如果想看看吸引子和對應的盆的分布的話,可以進入選項0,圖形窗口顯示的圖像如下所示
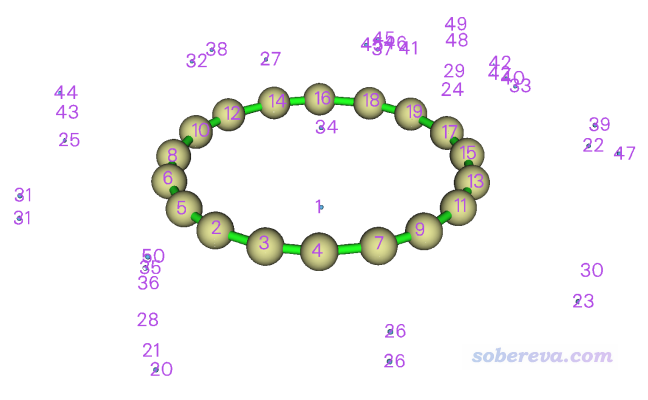
圖中藍色小球對應極小點位置,其中1、34號吸引子明顯就是我們要關注的全局最負的兩個點的位置。在每個碳原子中央也有吸引子,這對應的是范德華勢的極大點,顯然在每個原子的原子核位置必然會有一個。在環附近還有一大堆亂七八糟的吸引子,這些不用去管,是由于數值噪音所致的沒有化學意義的吸引子。
把圖形窗口關閉。接下來我們把吸引子導出成VMD可以認的格式,以便于在VMD里繪制。輸入以下命令
-4 //將吸引子導出
2 //導出為當前目錄下的attractors.pqr
pqr是生物體系分子模擬領域常用的格式,是文本文件。如Multiwfn在屏幕上的提示所示,這個文件里的殘基號對應自動歸簇后的吸引子的序號,簡單來說就是你在上圖中看到的序號。這個文件的倒數第三列(pqr格式原本用來記錄原子電荷的列)是吸引子處的格點數據的數值。這個文件里對應1、34號吸引子的是這兩列
HETATM 1 C ATT A 1 -0.013 0.048 -1.133 -0.25018300E+001 1.00 C
HETATM 35 C ATT A 34 -0.013 0.048 1.113 -0.25016000E+001 1.00 C
可見它倆的范德華勢的數值都是-2.50 kcal/mol,和我在前面的圖里用箭頭標注的一致。
現在我們用VMD將等值面和吸引子都繪制出來。怎么用VMD非常簡單地把cub文件繪制成效果極好的等值面圖我在《在VMD里將cube文件瞬間繪制成效果極佳的等值面圖的方法》(http://www.shanxitv.org/483)中已經詳細說了,細節不再累述。按文中的做法把VMD配置好,把vdW.cub挪到VMD目錄下,啟動VMD后輸入cub vdW 1,就會把vdW.cub對應的函數值為+1和-1的等值面分別用綠色和藍色顯示出來。之后在Graphics - Representation里看到有三項,分別對應顯示分子結構、顯示正值等值面、顯示負值等值面。雙擊第二項令其成為紅色,則正值等值面部分就不顯示了。然后我們把之前導出的attractors.pqr拖入VMD main窗口載入,在Representation界面里將Selected Atoms設為resid 1 34,這樣就只有1和34號吸引子被顯示了。再把Sphere Scale設為0.1減小球的尺寸,把Coloring Method設為Color ID,在旁邊選Yellow。之后,在Graphics - Materials里把Glossy材質(這是目前顯示等值面正在用的材質)的Opacity減小到0.4,使得等值面透明度更高避免遮擋吸引子。現在在VMD圖形窗口中看到的圖就和本節一開始的圖里面的(c)一致了。
2022-Jul-5重要補充:后來Multiwfn支持了對范德華勢做拓撲分析,找范德華勢極小點比用盆分析強得多得多,不僅精度高得多,而且不像盆分析那樣計算出的格點數據會占很大內存,還不像盆分析那樣由于數值噪音導致產生多余的極小點,因此強烈建議用拓撲分析代替上文的盆分析。具體做法見《使用Multiwfn對靜電勢和范德華勢做拓撲分析精確得到極小點位置和數值》(http://www.shanxitv.org/645)。
4.2 范德華勢曲線圖的繪制
為了透徹地研究垂直于環方向且穿過環中心的范德華勢的具體變化情況,可以用Multiwfn將范德華勢在這條直線上的變化繪制成曲線圖。下面這張圖是我原文里的,原點位置是環中心處,可見此圖把基于不同探針原子在不同位置上的范德華勢差異展現得非常清楚,圖中還標出了極大點位置。
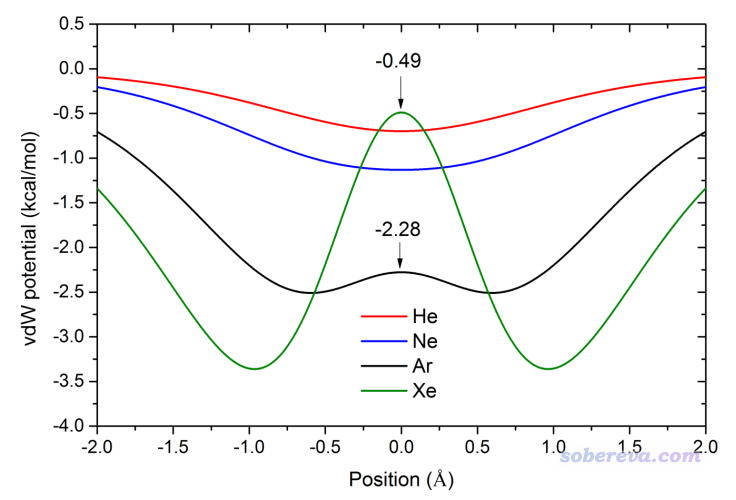
下面我們用Multiwfn繪制以Ar為探針原子的這種圖。將settings.ini里的ivdwprobe設為Ar的原子序數18,并且把iuserfunc設為92,使得用戶自定義函數對應范德華勢。然后啟動Multiwfn,輸入以下命令
examples\C18.xyz
3 //繪制曲線圖
100 //繪制用戶自定義函數(目前對應范德華勢)
2 //自定義兩個端點的坐標
0,0,-6,0,0,6 //第一個點為(0,0,-6) Bohr,第二個點為(0,0,6) Bohr。由于體系處在XY平面上,中心在(0,0,0)位置,因此這樣的定義的線段垂直于環且穿過環中心
馬上看到下圖,和前面的圖里Ar的情況一致
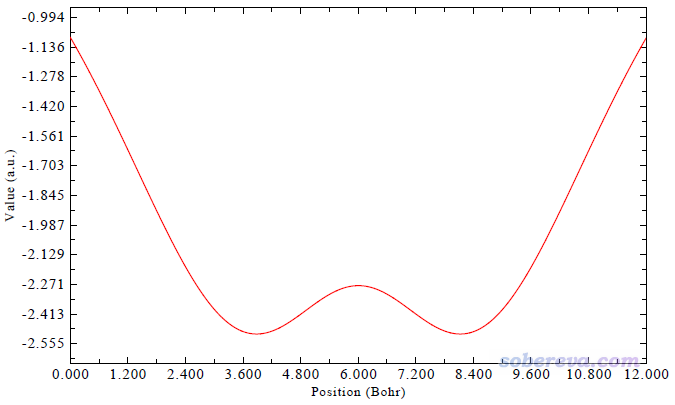
把圖像關閉后,選擇2就可以把曲線數據導出成為文本文件。我論文里的圖是將不同探針原子的情況都這樣產生曲線數據的文本文件,再把里面的數據都導入到Origin里一起繪制出來的。
4.3 計算原子對范德華勢的貢獻
在我的范德華勢的原文里還基于UFF力場考察了He原子與18碳環的各個原子的相互作用,由此可以展現各個原子對特定位置范德華勢的貢獻,如下所示,黃球對應于被考察的位置
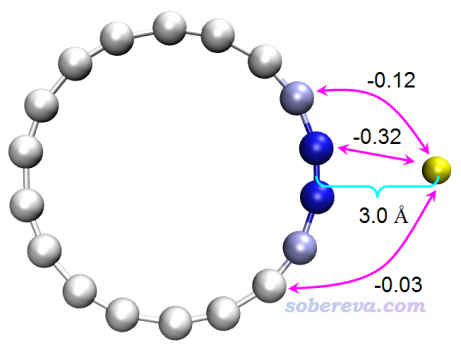
用Multiwfn做這個分析很容易,只要自己建立個18碳環和He原子的復合物結構文件(He的坐標設為被考察的那個點的坐標),按照《使用Multiwfn做基于分子力場的能量分解分析》(http://www.shanxitv.org/442)里說的做法做個EDA-FF能量分解即可。具體來說,考察以A原子為探針原子時各原子對r處范德華勢的貢獻,等價于在r處增加一個A原子,然后用EDA-FF方法考察當前體系各原子與A的相互作用。
比如要考察以He為探針原子時18碳環的各原子對(6.5408,1.1533,0.0)埃處范德華勢的貢獻,就把examples\C18.xyz的第一行的18改成19,然后在末尾加上一行:He 6.5408 1.1533 0.0,另存為C18_He.xyz。不了解xyz格式者參看《談談記錄化學體系結構的xyz文件》(http://www.shanxitv.org/477)。啟動Multiwfn,載入C18_He.xyz,然后輸入以下命令
21 //能量分解
1 //基于力場的能量分解(EDA-FF)
-1 //設置范德華作用計算方式
1 //UFF力場
2 //定義片段
2 //有兩個片段
1-18 //第一個片段(18碳環)的原子序號
19 //第二個片段(He原子)的原子序號
-3 //要求在分析時輸出片段間每一對原子的相互作用
1 //開始分析
剎那間,屏幕上輸出了以下信息
Interaction energy components between all fragments:
Electrostatic Repulsion Dispersion Total
Frag 1 -- Frag 2: 0.00 0.56 -1.51 -0.95
這即是18碳環與He原子的相互作用能信息,注意單位是kJ/mol。由于我們沒有設置原子電荷,因此原子電荷默認為0,故而兩個片段間靜電作用能為0。當前情況下,這里顯示的0.56、-1.51、-0.95分別對應于在(6.5408,1.1533,0.0)的位置上,18碳環以He為探針原子時的交換互斥勢、色散勢和范德華勢的值。
如屏幕上所示,兩個片段間每一對原子對相互作用能的貢獻都輸出到了當前目錄下的interatm.txt,內容如下
Atom_i Atom_j Dist(Ang) Electrostatic Repulsive Dispersion Total
...略
14 19: 3.934 0.00 0.01 -0.13 -0.12
15 19: 3.061 0.00 0.27 -0.59 -0.32
16 19: 3.061 0.00 0.27 -0.59 -0.32
17 19: 3.934 0.00 0.01 -0.13 -0.12
18 19: 5.089 0.00 0.00 -0.03 -0.03
這里15、16號原子是18碳環中與He挨得最近的兩個原子,19號原子就是He原子,可見C15-He和C16-He對范德華作用能的貢獻都為-0.32 kJ/mol,換句話說,C15和C16對(6.5408,1.1533,0.0)的位置上以He為探針的范德華勢的貢獻都為-0.32 kJ/mol,這也正是前面圖中我標記出的值。
在前面的圖里我還根據18碳環上的原子對范德華作用的貢獻進行了著色,越藍說明貢獻值越負(對結合起到越重要的作用)。怎么實現這點在《使用Multiwfn做基于分子力場的能量分解分析》里已經專門說了,就是導出pqr文件,在VMD里恰當設置一下著色即可,這里不再累述。
5 實例3:碳納米管片段
此例考察碳納米管片段,是一小截(6,6)手性的邊緣用氫飽和的碳納米管。下面這張圖是我范德華勢原文中的±0.7 kcal/mol等值面+極小點圖,探針原子為Ne。這張圖怎么繪制我就不再細說了,把前面的例子弄會了,再舉一反三就能得到。此體系有三類范德華勢極小點,圖中分別用黃色、粉色和橙色小球顯示以便區分。
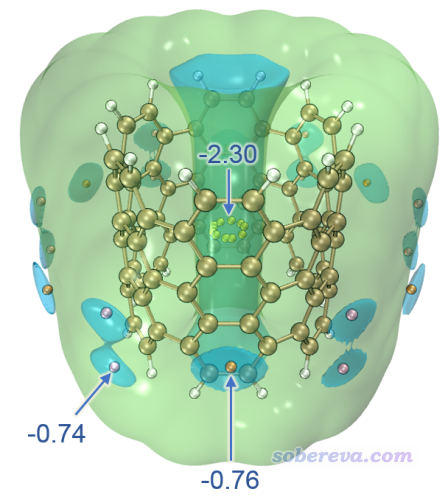
上圖體現出范德華勢在納米管的內側數值很負,最負達到-2.3 kcal/mol,即曰在UFF力場下Ne與這種納米管結合能最多為-2.3 kcal/mol。而且最負的區域是管中央由黃色小球示意的那一小圈,因此如果把Ne放在比如管口的位置,肯定在優化過程中Ne會自發運動到黃球所示的位置。為什么管里面范德華勢那么負很容易理解,畢竟周圍圍繞了一堆碳,肯定總的色散吸引作用很強。此體系在管外側也有范德華勢明顯為負的區域,藍色等值面展示的部分是相對來說最負的部分,由粉色和橙色標注的極小點處的數值可見最負的地方也不過只有-0.76 kcal/mol,所以管外側靠范德華作用對分子的吸附能力遠遠低于管內側。這張圖再次充分展現出分析范德華勢的價值,一張圖就非常直觀地把小分子(假定是無極性或弱極性)被物理吸附的最優位置體現出來了,而且還給出了具體數值便于定量對比不同區域的情況。
為了能更充分地把范德華勢的分布細節展現清楚,最好繪制平行于管的范德華勢的平面圖,下面說一下繪制過程。先在這里下載這個納米管片段的結構:http://www.shanxitv.org/attach/551/CNT66.xyz,此體系中心在原點,管子順著Z軸。將iuserfunc設為92,ivdwprobe設為10,然后啟動Multiwfn,載入此文件,之后輸入
4 //繪制平面圖
100 //用戶自定義函數
1 //填色圖
[按回車用默認的格點數]
0 //修改延展距離
10 //10 Bohr
3 //YZ平面
0 //Z=0
之后把蹦出來的圖關閉,做一下修改:
1 //修改色彩刻度范圍
-1.5,1.5 //下限和上限
4 //顯示原子標簽
2 //用綠色
17 //修改顯示標簽時原子與作圖平面距離的閾值
20 //20 Bohr。此值很大,可以確保所有原子標簽都顯示出來
n
8 //顯示化學鍵
14 //用棕色
2 //顯示等值線
3 //修改等值線設置
14 //修改負值的等值線風格
20,20 //虛線的長度和間隔
5 //線粗值(比默認的更粗)
1 //保存等值線設置并返回
19 //修改色彩變化方式
8 //藍-白-紅
選-1重新繪制圖像,如下所示
這張圖漂亮的圖展現出在距離原子較近的位置范德華勢都為很大的正值,在管內部范德華勢則相當負,在管的外圍雖然也為負,但程度低多了。
由于上面的操作較多,我建議把當前繪圖設置保存到文件里,以便以后需要重新繪制或者對作圖效果稍作調整的時候不用在后處理菜單重新敲一遍調整作圖設置的命令。具體來說,關閉圖像后輸入
-4 //保存或讀取所有作圖設置到外部文件
1 //保存到特定txt文件中
CNT66_plane.txt //輸出的文件名
在未來,重新用主功能4對此體系作圖后,在后處理菜單選擇-4,再輸入2,輸入CNT66_plane.txt的路徑,就可以恢復之前的作圖設定了。
6 實例4:卟啉
范德華勢的分析和操作在前面已經充分講解了。最后這個例子是我范德華勢原文里的一個例子,這里只是簡單說一下。下圖是卟啉分子的以Ne為探針原子的范德華勢的等值面圖和截面圖,可見這個體系范德華勢最負的地方是在環中心的上方和下方。
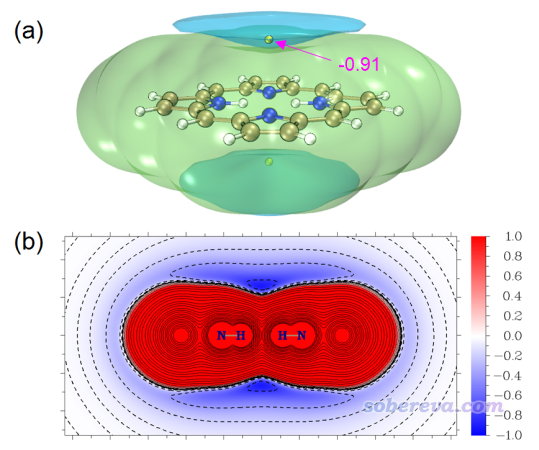
上面的范德華勢圖是否能與被吸附物的空間分布聯系起來?筆者用xtb在GFN1-xTB理論下跑了1 ns動力學,控溫在40 K(因為更高溫度難以束縛住Ne),并且在模擬時加了球形限制勢防止Ne偶爾因為熱運動飛走。模擬過程的Ne的軌跡疊加圖如下所示,給了兩個視角,其中(b)還添加了空間分布函數的一個等值面以勾勒出主體分布區域。
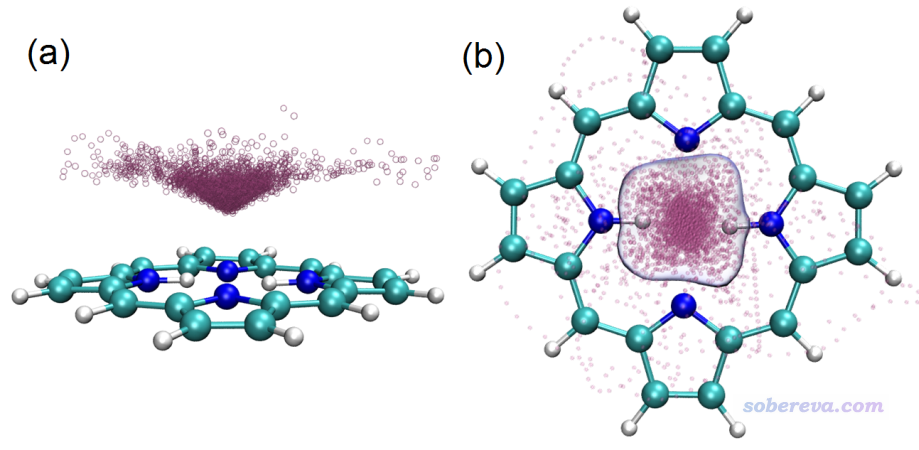
從上面兩張圖可以看到,范德華勢為明顯負值的區域呈現出錐形,軌跡疊加圖當中粒子的分布也同樣展現出錐形特征,而且越接近范德華勢最負的位置粒子出現密度越大。此例又一次體現出范德華勢可以用來預測和解釋由范德華作用主導的單原子(或小分子)的動力學行為、分布特征。
7 實例5:周期性體系的范德華勢分析--MOF
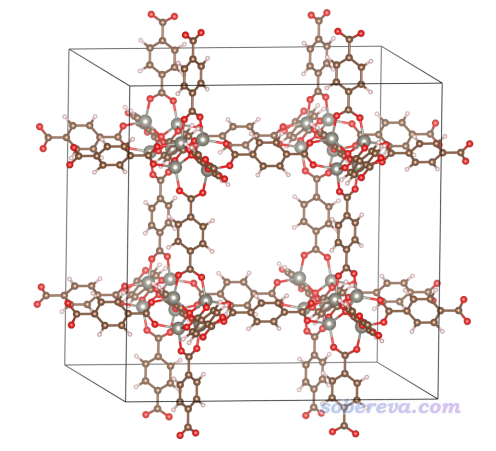
前面給出的例子都是分子體系的,從2021年2月更新的Multiwfn開始,還支持了周期性體系的范德華勢分析,輸入文件必須包含晶胞信息才行。Multiwfn支持的哪些格式包含晶胞信息,參見此文第2節:《使用Multiwfn非常便利地創建CP2K程序的輸入文件》(http://www.shanxitv.org/587)。這一節以一個MOF體系(金屬有機框架化合物)作為例子進行演示,下面用到的此體系的cif晶體結構文件可在http://www.shanxitv.org/attach/551/MOF5.cif下載,此體系的結構如下
啟動Multiwfn,然后輸入
MOF5.cif //cif文件可以給Multiwfn提供結構和晶胞信息
20 //圖形化考察弱相互作用
6 //范德華勢
9 //借助晶胞信息設置格點
[按回車] //使用0,0,0作為盒子原點
[按回車] /將晶胞邊長作為盒子邊長
0.3 //格點間距(Bohr)
算完后可以直接選3可視化范德華勢等值面圖。注意對于此例這種格子是正交的情況,可以在Multiwfn里正常觀看等值面,但如果此體系的晶胞是非正交的,按照上面方式操作的話,格子也將是非正交的,這時候等值面圖沒法在Multiwfn里正常顯示,必須導出cub文件后放到比如VMD或VESTA等支持非正交格子的程序里顯示成等值面(或者在格點設定界面里用其它模式定義格點)。
為了得到比較好的圖像效果,這里我們選6把范德華勢的格點數據導出成vdW.cub,挪到VMD目錄下,然后利用《在VMD里將cube文件瞬間繪制成效果極佳的等值面圖的方法》(http://www.shanxitv.org/483)里提供的腳本在VMD里顯示出等值面。之后再做如下調節:
? 在Graphics - Representation里把對應正值等值面的Rep刪掉,并把對應負值等值面的Rep里的Isovalue設為-1.2
? 在Graphics - Representation里把顯示體系結構的那個Rep的Drawing method改為Licorice,把Bond Radius改為0.2
? 在Graphics - Representation里新增一個Rep,顯示方式設為DynamicBonds,Bond Radius改為0.2,Distance Cutoff改為2.0。這個Rep使得MOF中所有鍵都能被顯示出來(原本有些金屬和配位原子的鍵沒有被識別出來)
? 在VMD文本窗口里輸入pbc box把盒子邊框繪制出來
? 在VMD Main窗口里選Display - Orthographic設為正交視角
? 為了讓圖像層次感清楚,使用距離霧化效果:確保VMD Main窗口里的Display - Depth Cueing處于已選中的狀態。然后進入Display - Display Settings,把Cue mode設為Linear,把Cue Start和Cue End分別設為1.5和3.5
此時看到的-1.2 kcal/mol的等值面圖如下,-1.5 kcal/mol的等值面的圖也一并給出了
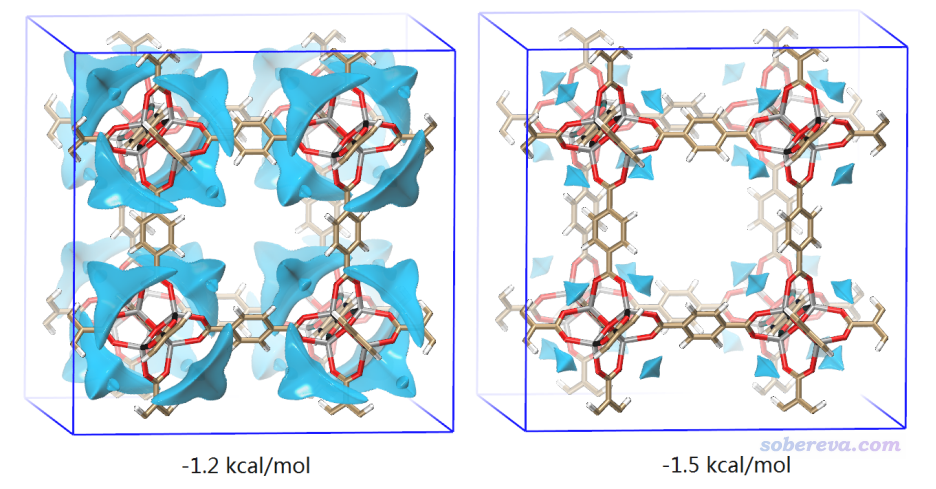
從上圖可以非常清晰地看出,只考慮范德華作用的話,這種MOF最傾向于把小分子吸附到骨架的邊角部分,因為如上圖藍色等值面所示,這種地方范德華勢最負。大家也可以像之前的例子那樣做盆分析獲得范德華勢極小點的具體數值。此例體現出范德華勢也很適合用于多孔體系吸附問題的研究。
Multiwfn做周期性體系的分析以及VMD對周期性體系的顯示還有更多的細節,在此文有所體現:《使用Multiwfn結合CP2K通過NCI和IGM方法圖形化考察固體和表面的弱相互作用》(http://www.shanxitv.org/588)。
8 總結
本文介紹了筆者對范德華勢的定義以及在Multiwfn程序中的實現,并通過多個體系,全面介紹了范德華勢如何分析、計算和繪制。范德華勢在研究分子間相互作用、考察物理吸附、估計復合物穩定性等問題上有重要價值,這無疑是弱相互作用分析的工具箱里又一重要成員。借助Multiwfn對范德華勢做計算、分析和繪制,可以輕松得到清晰、直觀、漂亮又非常能說明問題的圖像,筆者十分鼓勵大家在實際研究中在恰當的場合多利用此方法進行分析。在范德華勢的原文中筆者還有對范德華勢的更多的介紹和分析討論,很建議讀者們一看。使用Multiwfn做范德華勢分析的時候除了引用Multiwfn原文外也請注意引用范德華勢的原文J. Mol. Model., 26, 315 (2020)。
值得在最后強調的是,范德華勢和靜電勢在弱相互作用的研究上可以認為是互補的關系。范德華作用主導時應主要關注范德華勢,靜電作用主導時應主要關注靜電勢,兩類作用都重要時不能忽略任意一方。本文的例子用的分子都是無極性分子,探針原子用的也都是稀有氣體原子,因此靜電相互作用完全可以忽略不計,而實際情況往往復雜得多,需要根據理論知識、化學直覺以及對范德華勢原理的認識來判斷什么時候能用范德華勢說明問題、能說明什么,不要亂分析或者過度解釋。還值得一提的是,相對來說,范德華勢的分布特征不像靜電勢那么復雜,與范德華勢關系最直接的是體系的幾何結構,而靜電勢則對體系的電子結構很敏感(相對來說,電子結構對范德華勢的影響弱得多得多),在分析時要注重各自的內在特點。
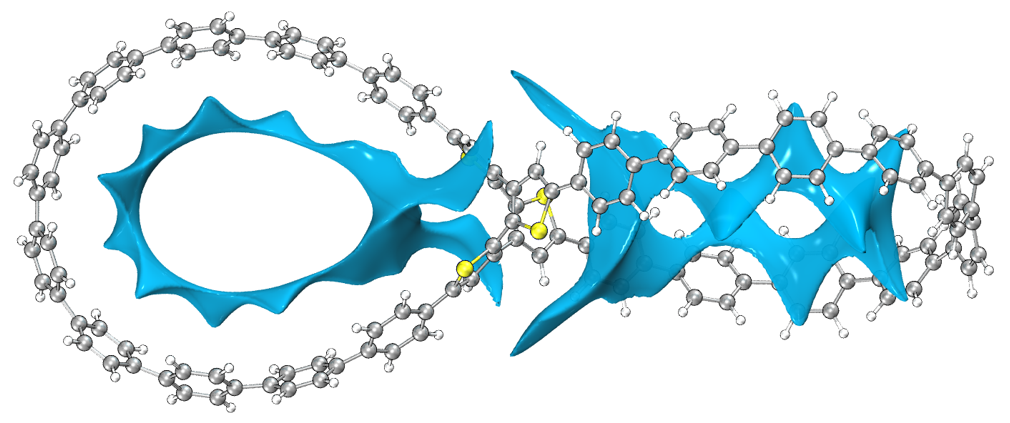
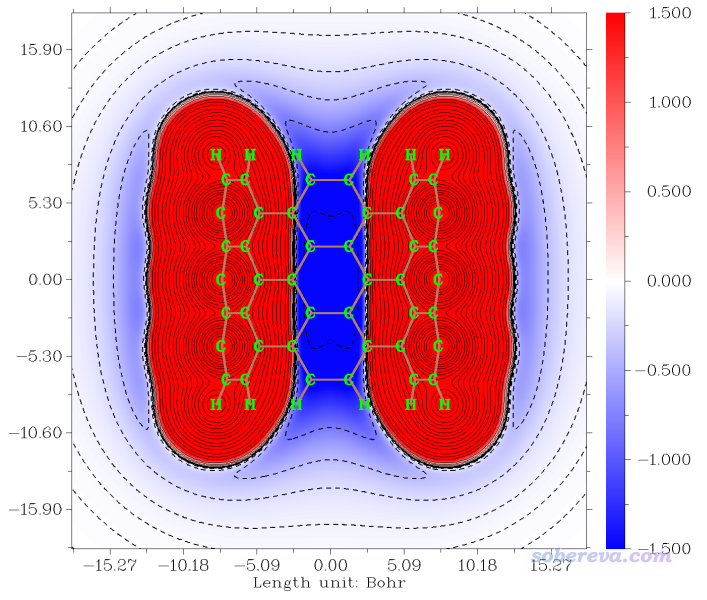
2023-Jun-30補充:《8字形雙環分子對18碳環的獨特吸附行為的量子化學、波函數分析與分子動力學研究》(http://www.shanxitv.org/674)一文中介紹了筆者研究OPP分子吸附18碳環的機制,其中使用了范德華勢進行分析,是個極好的通過范德華勢研究實際問題的例子,非常建議閱讀和引用此文。下圖是OPP分子的以碳原子為探針的范德華勢的-1.2 kcal/mol等值面,OPP大環區域的范德華勢等值面的出現區域與18碳環被吸附后出現的位置理想重合,很好地揭示了為什么OPP能對18碳環產生穩定的吸附作用。