On the new features and changes of ORCA 5.0
文/Sobereva@北京科音
First release: 2021-Jul-2 Last update: 2021-Jul-10
1 前言
2021年7月1日,量子化學程序ORCA 5.0發布了,造成了不小影響。這個程序現在流行度已經相當高,從《2021年計算化學公社論壇“你最常用的計算化學程序和DFT泛函”投票結果統計》(http://www.shanxitv.org/599)的投票結果上可見一斑。這里就簡單說下這個更新對于大多數用戶值得一提的新功能和改變,以及我的一些相關看法,便于讀者了解新版本都帶來了什么。更全面的更新說明見5.0版手冊1 ORCA 5.0 Foreword和2 ORCA 5 Changes部分。
5.0版手冊比4.2.1擴增了近300頁,達到了1300多頁的規模。我所知的手冊最厚的量化程序是Q-Chem(5.3.2版是1391頁)。按照ORCA的勢頭,據說將在2022年發布的ORCA 6的手冊厚度預計將超過Q-Chem。
2 比較重要的改進
5.0的一個很大進步是加入了全新開發的積分代碼SHARK。號稱對于低角動量和高角動量基函數比以前版本用的Libint電子積分庫更快。ORCA自動決定用SHARK還是Libint來達到最好的效率。據說此改變令解析Hessian、TDDFT、磁相關屬性速度提升較大。SHARK既適合片段收縮基組,對廣義收縮基組也專門做了優化,因此能算得動ANO那種完全廣義收縮的基組了。筆者實際測試了一下,ANO這種完全廣義收縮的基組確實在ORCA 5.0里能算得動了,而在4.2.1里哪怕用ANO-pVDZ算個很小的分子都極其吃力。我用帶有高角動量函數的基組cc-pVQZ也簡單測試了一下算一個小體系HF的耗時,5.0比4.2.1耗時低了百分之十幾。
DFT積分格點和COSX積分格點利用機器學習技術進行全新的設計。目前版本定義了三種格點的關鍵詞,defgrid1、defgrid2和defgrid3,控制所有涉及到積分格點步驟用的格點(COSX、CPSCF、TDDFT等)。默認是defgrid2,據稱沒有特殊情況這個檔次的格點質量就足夠了,而對積分格點要求特高的情況(近乎Gaussian里int=superfine的情況),比如VPT2做非諧振計算的時候,才需要defgrid3。另外,使用對積分格點要求較高的明尼蘇達系列泛函的時候程序會提示建議用defgrid3。defgrid1的大小近似相當于之前版本默認的挺糙的格點。由于默認的defgrid2比之前的積分格點明顯更好,因此DFT計算的數值精度比以往版本有明顯提升,而且這還令DFT的SCF收斂、幾何優化收斂等也往往更為容易。據說即便把之前版本的格點數設得和當前版本的差不多,新的積分格點的精度也更好,這是因為格點分布調教得更科學了,期間還利用了GMTKN55數據集進行了訓練和測試。defgrid系列格點還有一個特點就是有自適應能力,對于帶彌散的基組、帶緊s基函數的基組、帶核極化函數的基組,新版本在對格點剪裁的時候有相應的特殊考慮來保證數值精度。特別要注意的是,之前版本分別控制DFT和COSX格點的grid和gridx關鍵詞不僅不能用了,而且連出現都不行,這導致原先大量輸入文件都不能用了,這點很變態(哪怕自動無視也好)。
新版本里COSX計算解析積分的代碼效率得到了優化。結合新的COSX格點,令數值穩健性也顯著提升、SCF和幾何優化達到收斂的步數有可能更少。由于RIJCOSX已經非常成熟、理想了,因此從5.0開始RIJCOSX成為了雜化和雙雜化泛函計算時默認開啟的加速方法(以前版本只是純泛函默認用RIJ)。如果要關閉RIJCOSX的話,寫noCOSX。由于RIJCOSX已變得更完美,RIJK可以算是沒有任何用武之地了,而且用RIJK時功能限制很大。
提升了CPSCF的效率,因此做freq等需要求解CPSCF方程的任務耗時有所降低。
隱式溶劑模型得到了提升。一個很大的改變是5.0用的CPCM的默認設置是范德華型的溶質孔洞結合Gaussian分布的表面顯著電荷,而老版本默認用的設置是gepol_ses型溶質孔洞結合離散的表面點電荷,新版本默認的溶劑模型的數值穩定性更好,解決了原來版本溶劑模型下可能出現的勢能面不連續問題,這也使得從頭算動力學過程結合溶劑模型的時候更令人放心了。另外,新版本的溶劑模型能用的場合更多了,新版本可以在CPCM溶劑模型下算解析Hessian是個巨大的進步,不過尚不支持SMD下的解析Hessian。并且新版本的CPCM可以結合(DLPNO-)CCSD(T)使用。
由于上述數值方面的各種改進,估計計算Hessian容易出虛頻的毛病會得到明顯改善。
CPCM(線性響應的形式)正式支持了TDDFT。我發現之前版本的ORCA在溶劑模型下得到的TDDFT激發能不合理,和Gaussian的線性響應溶劑模型相差很大,貌似是因為之前版本沒有考慮隱式溶劑對電子激發的響應。我找了個D-pi-A體系做了個簡單測試(氨基-苯-硝基),用PBE0/def2-SV(P)結合SMD描述的水環境做TDDFT計算,考察最低激發能和振子強度。G16算出來是3.77 eV,f=0.44;ORCA 5.0算出來是3.76 eV,f=0.45;ORCA 4.2.1算出來是3.90 eV,f=0.36。明顯ORCA 5.0比4.2.1合理多了,和G16基本一致了。注:ORCA里的SMD的極性部分用的是CPCM,和Gaussian的SMD的極性部分用的IEFPCM有異,結果注定不可能非常相近,但差異不會太顯著。
ORCA作為MPI并行程序,內存消耗量問題一直為人所詬病。5.0在SCF和CPSCF過程中部分程度使用了共享內存方式節約內存消耗量。希望以后能想辦法把DLPNO的巨大內存花費降下來。
支持了VV10色散校正的解析梯度,wB97M-V、wB97X-V之類的帶VV10的泛函終于都可以用于優化了,以前這是Q-Chem的專利(阿Q一直都活在Gaussian的陰影下,現在免費的ORCA又來勢洶洶,把阿Q的賣點一個一個拔掉,在二者的夾縫中日子真不好過)。不過wB97M-V雖然支持了解析梯度優化,但我感覺也沒什么實際意義,畢竟還不支持解析Hessian。而優化完了不做個振動分析檢驗虛頻的話很難發文章。而且wB97M-V雖然能量計算精度很好,但并不代表幾何優化方面會有什么突出的。實際上B3LYP-D3(BJ)優化就已經挺理想了,這點我在《談談量子化學研究中什么時候用B3LYP泛函優化幾何結構是適當的》(http://www.shanxitv.org/557)中已經說過;而且就算遇到B3LYP-D3(BJ)不適合的時候,還有wB97X-D3或wB97M-D4可用,就算真的優化結果比wB97M-V差一些,也就是毫厘之間。
終于支持了meta-GGA、meta-雜化泛函的解析Hessian了,可以用諸如M06-2X在優化完之后做振動分析了。
加入了一些新泛函:B97M-D4、wB97X-D4、wB97M-D4。wB97M-D4的精度略遜于wB97M-V一丁點,但好處是有解析Hessian。不過我實測了一下,發現這泛函結合小基組下對苯分子這么簡單的體系做振動分析時CPSCF都特別難收斂,所以在opt freq方面我不覺得這泛函有什么實用性。雙雜化加入了wB88PP86、wPBEPP86、RSX-QIDH、RSX-0DH、PBE-QIDH、PBE0-DH,以及它們的為激發態計算優化參數的SCS、SOS形式的變體。不過這些雙雜化泛函在我來看并沒什么用處,算基態不靈,算激發態的話,對于普通價層激發肯定不如DSD-PBEP86,而算顯著的CT態、里德堡態比起之前就有的wB2GP-PLYP也不會有什么優勢。
還有一個新加入的雙雜化泛函是2009年提出的wB97X-2。這個泛函帶DFT-D3(BJ)的版本wB97X-2-D3(BJ)在《簡談量子化學計算中DFT泛函的選擇》(http://www.shanxitv.org/272)里介紹過,對于除了分子間弱相互作用以外,這個泛函通常是最佳選擇之一(雖然遜于后繼者wB97M(2),但目前wB97M(2)僅有Q-Chem一個程序支持)。不過我發現ORCA 5.0沒有自帶這個泛函的DFT-D3(BJ)參數,所以沒法直接用wB97X-2-D3(BJ),而必須自己定義參數才行。為了大家用起來方便,我在《詳談Multiwfn產生ORCA量子化學程序的輸入文件的功能》(http://www.shanxitv.org/490)介紹的Multiwfn的相應功能中已經直接加入了產生wB97X-2-D3(BJ)輸入文件的選項。
支持了剛提出不久的r2SCAN-3c。其構建思想類似于已經流行開來的B97-3c,而其中的純泛函部分改用了2020年底才提出的SCAN泛函的第二個修改版,即r2SCAN。雖然r2SCAN-3c耗時比B97-3c高百分之幾十,但精度有了全面提升,因此多付出的代價是劃得來的。建議對于單點能計算任務(幾何優化和振動分析這樣對計算級別要求不高的任務除外),r2SCAN-3c能算得動的情況一律用它代替B97-3c。由于其重要性,我在《詳談Multiwfn產生ORCA量子化學程序的輸入文件的功能》介紹的功能中也已經加入了產生其輸入文件的選項。
當默認的SCF solver收斂失敗或者表現不佳時,會默認切換為新加入的augmented Hessian trust radius SCF converger (TRAH)。TRAH比默認的SCF solver要慢,但據說收斂能力很強,號稱除非結構不合理、基組有問題、有線性依賴這些嚴重硬傷外幾乎總能收斂,并聲稱對付收斂普遍偏難的含過渡金屬的體系很好。至于實際效果如何筆者沒試過,但至少是又多了一個可能解決SCF不收斂的選擇。想關閉的話可以用noTrah。
NEB功能得到了改進,并且支持結合TDDFT對激發態勢能面找過渡態和反應路徑。
支持了共線形式的spin-flip TDDFT以及其解析梯度。這個挺有意義,一些圓錐交叉點的搜索都可以用這個做了。以前spin-flip TDDFT用得比較多的是GAMESS-US和Q-Chem,而前者用著麻煩,后者又收費。
改良了NEVPT2、CASPT2的計算形式,號稱對大活性空間情況提升了計算速度。
動力學模塊支持了一維和二維的metadynamics獲得自由能面。集合變量可以用距離、角度、二面角、配位數,還可以加諧振或高斯限制勢。支持了Nose-Hoover chain壓浴,支持了velocity-rescale熱浴(這個很重要,之前只能用Berendsen熱浴,很容易被瞎較真的審稿人吐槽)。新支持了消除質心運動的設置以避免模擬過程中漂移,手冊里搜CenterCOM(在之前版本中為了消除漂移,我都是靠質心約束來實現的)。
支持了兩層和三層ONIOM,并支持電子嵌入,可以結合到優化、掃描、NEB、IRC、振動分析等各種任務。并且改進了原先的QM/MM。不過我感覺,做ONIOM沒有個gview這樣的專門開發的界面還是不夠方便直觀。
DLPNO-MP2和DLPNO-雙雜化可以算NMR和極化率了。這對于需要算較大體系高精度NMR者是個福音,有測試表明DLPNO-DSD-PBEP86算NMR很好,不僅勝于普通泛函,比MP2也更好。相比之下,Gaussian的MP2或雙雜化算稍大一點體系的NMR都非常吃力。另外值得一提的是,當體系不大時,這種DLPNO形式和RI形式計算耗時相仿佛,而當體系很大時,DLPNO形式耗時顯著低于RI形式。
支持了Ionic-Crystal-QMMM和Mol-Crystal-QMMM以簇模型分別計算離子晶體和分子晶體中的局部區域,背景電荷可以自洽地自動確定(對于離子晶體的情況,中心部分和背景電荷之間還有一層capped ECP)。利用這種模型可以算晶體中的電子光譜、NMR,乃至算離子晶體的帶隙,還可以算表面吸附問題(這比起第一性原理程序里能用的理論方法豐富得多,還有準確得多得多的方法可用)。對Ionic-Crystal-QMMM方式的計算,ORCA中新加入了專門的orca_crystalprep工具用來構造模型。
在workflow控制方面做了加強,可以在輸入文件里控制多個任務的執行、數據提取(計算過程中涉及的大量信息都可以通過變量直接訪問)、輸出、數學運算,還可以用循環、條件判斷,相當于在輸入文件里可以直接用腳本語言進行非常靈活的計算流程的控制,也因此可以方便地實現熱力學組合方法,而不必依賴于外部的Shell、Python等腳本。甚至還可以靠腳本實現自動循環各個振動頻率,發現有虛頻則自動按虛頻模式調節結構,之后重新做優化,反復如此直到沒有虛頻為止。
支持了通過簡單的關鍵詞實現很多組合方法。比如寫compound[W2-2]就可以做W2.2高精度熱力學組合方法的計算。支持的組合方法關鍵詞看ORCA 5.0手冊9.47.3 List of known Simple input commands部分。
3 其它一些值得一提的改進或變化
新版本默認對雙雜化啟用RI,但做純粹的MP2則不默認用。可以noRI關閉RI。
DLPNO支持了multi-level,對不同片段內部和片段間的電子相關用不同的PNO閾值考慮,從而在不明顯損失精度的情況下節約耗時。
SARC ZORA/DKH基組支持了Rb-Xe,這是J. Comput. Chem., 41, 1842 (2020)中提出的。
已經可以直接使用月份基組了,現在ORCA的基組庫已經做得挺完善了。
加入了J. Chem. Phys., 152, 214110 (2020)中提出的CASPT2-K方法,入侵態問題比CASPT2更小,和IPEA shift一樣對CASPT2結果大有改進,而不需要IPEA shift那種經驗性的參數,因此從本質上更好。此方法算激發能不如CASPT2-IPEA和NEVPT2,不過算Cr2的解離曲線則比CASPT2加不加IPEA都好。此方法只能算是對CASPT2自身的一種改良,相對于NEVPT2并沒什么實際優勢。
Iterative configuration expansion (ICE)這個功能之前就有,屬于selected CI的一種,利用微擾方法篩選出對Full CI貢獻較大的組態用于計算,從而在精度犧牲很小的前提下大幅減少傳統做Full CI要考慮的組態數,因此比Full CI能算更大一些的體系。5.0對這個功能進行了強化(近期Neese在JCC和JCTC上發了相關文章)。做超高精度計算的人可以嘗試。
算激發態支持了SCS-CIS(D)和SOS-CIS(D),不過適用場合很有限。當各種泛函下TDDFT結果都不理想,而DLPNO-STEOM-CCSD又算不動的話可以試一把。
支持了DLPNO-NEVPT2-F12。支持了完全內收縮多參考耦合簇(FIC-MRCC)。
DLPNO系列方法支持了做PNO外推。DLPNO加速方法受TCutPNO參數影響,此外推是在一個較大(較便宜)和一個較小(較昂貴)的TCutPNO下分別計算能量,再根據簡單的公式經驗外推到TCutPNO=0的極限得到更精確的能量。寫compound[EXTRAPOLATE-PNO]關鍵詞的話會在DLPNO-CCSD(T1)/cc-pVTZ下做自動這種外推。
支持了VPT2非諧振模型得到振動平均的NMR化學位移和EPR超精細耦合常數,由此還可以體現不同同位素的差異。
加入了ANISO程序用來計算自旋哈密頓參數和屬性。
ab initio ligand field analysis (AILFT)做了改進。
支持了化學位移的定域軌道的分解分析,并給NBO的NCS(自然化學屏蔽)分析留了接口。
4 一些吐槽和局限性
windows版只有靜態庫版本,解壓完了竟然占24.8GB,而4.2.1版則只有9.2GB而已。
PWPB95、PW6B95、DSD-PBEB95沒解析梯度,這個之前版本的局限性到現在還沒解決。
可惜沒有直接支持比DSD-PBEP86-D3(BJ)精度更好的后繼者revDSD-PBEP86-D3(BJ)。它在現有雙雜化泛函中精度是名列前茅的。
雙雜化泛函依然不支持解析Hessian。
meta-泛函做TDDFT時沒有解析梯度。
算拉曼活性依然還得用數值Hessian,是個硬傷,算中等體系都超級耗時,和Gaussian完全沒法比。
TDDFT的解析Hessian什么時候ORCA才能支持是個未知數,不過我相信ORCA早晚會支持的。這對于一個普適型、大眾型,而且又對激發態計算特別重視的程序來說太重要了。
新版本振動分析的輸出格式發生了變化,迫使我今日更新了《OfakeG:使GaussView能夠可視化ORCA輸出文件的工具》(http://www.shanxitv.org/498)里介紹的OfakeG程序。除了這個外,新版本還不允許出現grid和gridx關鍵詞,也迫使我今日更新了Multiwfn的相關功能。據說2022年可能要出ORCA 6,但愿出下個大版本的時候別再折騰輸出格式,并且考慮對以往輸入文件的兼容性(之前ORCA 4出來的時候相對于3.x在基組寫法方面就變化甚巨)。老折騰的話對于寫輔助程序、寫相關教程、維護input library的人是很大負擔。
]]>Several methods to straighten molecular structures using Gaussian or ORCA program
文/Sobereva@北京科音
First release: 2021-May-4 Last update: 2021-Jun-15
1 前言
有人問怎么把一個分子從已有的較為卷曲的構象轉化成比較平直的構象。這有一些實際意義,比如通過genmixmem程序構造磷脂雙層膜、用packmol程序構造表面活性劑的囊泡,都需要用戶提供分子比較平直的構象的結構文件。一個非常簡單、直觀、效果又好的做法是使用SAMSON可視化程序里的twist工具,筆者在《生成混合組分的磷脂雙層膜結構文件的工具genmixmem》(http://www.shanxitv.org/245)里還提供了我錄的一段操作視頻。但可惜如今SAMSON程序免費版當中已經沒有twist工具了,需要買收費版才行(雖然收費版也可以申請免費試用)。還有一個辦法是在GaussView等程序里一個一個改二面角,但無疑這非常費時費力,特別是分子可旋轉的鍵很多的時候。
在此文,筆者介紹如何利用Gaussian和ORCA量子化學程序以不同方式實現把分子從卷曲的結構拉直,里面用到的做法對于讀者研究其它一些問題可能也會有啟發。本文使用的例子是《將Confab或Frog2與Molclus聯用對有機體系做構象搜索》(http://bbs.keinsci.com/thread-20063-1-1.html)中的例子,即Actos分子。通過Molclus程序(http://www.keinsci.com/research/molclus.html)搜索出的此體系的能量最低構象如下,我們后文將要把此結構拉直:
本文涉及的所有文件可以在http://www.shanxitv.org/attach/594/file.rar下載,上面的結構是文件包里的Actos.xyz。Gaussian使用的是G16 B.01版,ORCA是4.2.1版。
2 通過ORCA拉直分子:給兩個原子間加外力
筆者寫過不少與ORCA的安裝和使用有關的文章,參看http://www.shanxitv.org/category/ORCA/,相關常識性問題這里就不再說了。在ORCA中優化時可以給兩個原子在連線方向上加外力驅使它們遠離,很適合用于拉直分子。對于上面圖中展示的Actos分子,顯然我們應當把最末端的兩個重原子,即8和25號之間拉遠。為此,寫一個ORCA輸入文件:
! AM1 opt nopop
%geom
POTENTIALS
{ C 7 24 3.0 }
end
end
* xyz 0 1
[Actos.xyz里的坐標]
*
這里POTENTIALS下面花括號里的C代表constant force。注意原子序號是從0開始算的。3.0是把倆原子間拉遠的力的大小,單位是nN。數值大小不是很重要,一般個位數大小就可以。數值太小的話拉不開,而太大的話會最終把一些化學鍵過度拉長、鍵角被過度拉大(不過鍵伸縮、鍵角彎曲相對于二面角扭轉來說是很剛的,只要外力不是特別大這就不是明顯問題)。這里用的AM1半經驗方法是ORCA里能直接用的幾乎最便宜的方法(雖然ORCA也支持力場計算,但還得搞參數,這里不考慮),注意此方法不支持并行計算。如果嫌AM1太low,也可以用稍微好一點的HF-3c(Hartree-Fock結合額外校正項),對不太大的體系也非常便宜。
用ORCA運行上面的輸入文件,三分鐘就算完了,最終得到的結構如下(即file文件包里ORCA目錄下的Actos.xyz),可見完美地實現了我們的目的,分子被拉得很直
file文件包里的Actos_trj.xyz是優化軌跡,感興趣者可以將之拖到VMD程序(http://www.ks.uiuc.edu/Research/vmd/)里看看優化過程。順帶一提,考察加外力對化學體系的影響專門有人做過大量研究,參見Chem. Rev., 112, 5412 (2012)、Angew. Chem. Int. Ed., 58, 5232 (2019)。
3 通過Gaussian拉直分子
在Gaussian程序中不支持直接給原子加外力,但有其它的方法也可以實現把分子拉直,將在下面幾節介紹。
3.1 方法1:柔性掃描
Gaussian中做柔性掃描在《詳談使用Gaussian做勢能面掃描》(http://www.shanxitv.org/474)里筆者詳細介紹過。顯然,做兩端的兩個原子距離逐漸拉長的柔性掃描最終就可以得到被拉得平直的結構。寫一個這種任務的Gaussian輸入文件,如下所示
%nprocs=1
#T UFF opt(loose,nomicro,modredundant)
Generated by Multiwfn
0 1
[Actos.xyz里的坐標]
8 25 S 50 0.5
這里有幾個需要注意的地方:
(1)為了耗時盡可能低,這里使用了Gaussian支持的最便宜的方法,即分子力場。這里用的是較粗糙但支持元素最廣、用著最省事(不需要指定原子類型)的UFF力場。由于本例只需要得到一個較為平直的構象,對質量什么沒要求,所以UFF就夠了。
(2)柔性掃描每一步相當于一次限制性優化,為了節約時間,用了loose收斂限降低每一步掃描的耗時。
(3)opt里必須寫nomicro。因為分子力場的優化在Gaussian里有專用的代碼,不受modredundant設置的控制。而加了nomicro后,就會用通用的幾何優化代碼,雖然遠沒有分子力場專用的優化代碼快,但此時可以設置柔性掃描。
(4)分子力場計算時,至少對于這個體系,并行計算會令耗時不降反升,所以這里靠%nprocs=1刻意要求不用并行計算。
(5)掃描的步長太小的話把分子拉直需要的步數太多,顯然不行,而步長太大則容易導致中途報錯(此時還沒充分拉直),根據我的經驗步長設0.5埃左右比較合適。由于事先往往不好估計拉直時兩個原子相距多遠,所以可以把掃描步數設得比較大(比如本例設50步),當分子已經被拉得很直后,繼續掃下一步時程序通常會報錯,此時用GaussView載入輸出文件查看掃描軌跡,若最后一幀結構就是想要的平直的結構的話就取最后一幀結構即可。如果步數一開始設小了,導致最后沒有完全拉直也沒關系,拿最后一步的結構繼續做拉長的掃描即可。
(6)當前體系是之前我用DFT優化過的結構,已經很合理了,所以沒有明確靠geom=connectivity指定原子間連接關系,而直接讓Gaussian根據當前結構猜連接關系并用于力場計算。如果初始結構沒有優化過,最好讓GaussView保存出帶有恰當連接關系的輸入文件以免自動判斷的連接關系有誤。
使用Gaussian對上面的輸入文件進行計算,算了1分鐘左右掃描到第31步后報錯,報錯前最后一步結構如下所示,可見是我們想要的平直的結構。
3.2 方法2:利用外電場加外力
Gaussian里可以加外電場,外電場會給帶電粒子施加額外的力,在《一篇文章深入揭示外電場對18碳環的超強調控作用》(http://www.shanxitv.org/570)里介紹的筆者的文章中有很多介紹和討論。因此,可以在UFF力場計算時,把25號原子坐標凍結住,而讓8號原子帶+1.0原子電荷,然后按照下圖所示向X軸正方向加足夠強度的電場,這樣8號原子就會受電場力往X正方向運動,從而實現拉直分子的目的。
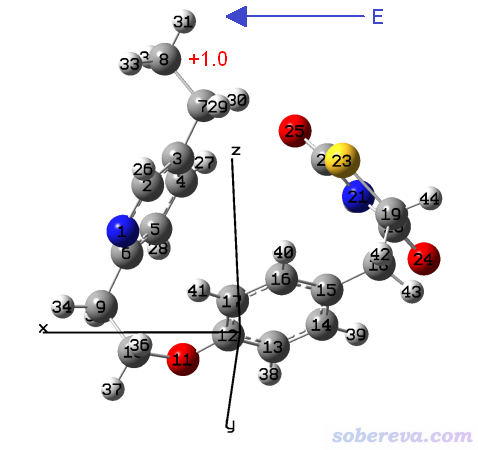
此例輸入文件如下
%nprocs=1
# UFF opt(loose,nomicro,modredundant) field=x-300 nosymm
Generated by Multiwfn
0 1
N 2.96734200 -1.21688000 2.24577900
C 2.32051000 -1.62618600 3.34057500
C 1.63462400 -2.84168400 3.45382100
C 1.64984500 -3.67567100 2.33272400
C 2.32098400 -3.26708600 1.18384000
C 2.96534100 -2.02795600 1.17397000
C 0.96378000 -3.24289700 4.74461900
C--1.0 1.95041300 -3.88295500 5.73536500
C 3.62426800 -1.48867000 -0.07067000
[略]
25 F
此例C--1.0代表對這個8號原子不手動指定原子類型(兩個橫杠之間沒寫內容),原子電荷為+1.0。沒設原子電荷的原子的原子電荷默認為0。Gaussian里電場方向和習俗相反,因此field=x后面是負號。電場強度用300(0.03 a.u.)一般就可以,如果發現最終結果怪異,如有些鍵被拉得太長,結構都扭曲了,應當適當減小電場以減小外力;如果分子拉得還不夠直,可以稍微增大電場來加大外力。nosymm必須寫,要不然結構被自動弄到標準朝向下后外電場相對于分子的方向就和期望不符了,關于nosymm更多信息見《談談Gaussian中的對稱性與nosymm關鍵詞的使用》(http://www.shanxitv.org/297)。nomicro還是要寫,要不然凍結設定不生效,其電場設置也不會對優化起作用。
用Gaussian計算上面的輸入文件,68步后收斂,耗時才十幾秒。最終結構如下,可見是我們想要的
3.3 方法3:令兩端的原子彼此間受到靜電斥力
這個做法超級簡單,而且超級快速(直接用分子力場的專用優化代碼),比前述兩種方法通常更值得推薦。原理很簡單,即給兩端的原子都設數值較大的相同符號的原子電荷,二者通過強烈的靜電斥力自然就會彼此遠離,從而把分子拉直。輸入文件如下,只有8號和25號原子設了原子電荷,都設為了+10.0。設多大合適應當看實際情況,比如如果分子特別長,那么原子電荷應設大一些,要不然靜電斥力不夠強。此例輸入文件如下
%nprocs=1
# UFF opt
Generated by Multiwfn
0 1
[略]
C 0.96378000 -3.24289700 4.74461900
C--10.0 1.95041300 -3.88295500 5.73536500
C 3.62426800 -1.48867000 -0.07067000
[略]
S -2.51218200 -3.05838400 3.26014600
O--10.0 -4.37962400 -3.91727700 -0.05061200
O -1.13156500 -5.32519500 2.86775900
[略]
用Gaussian計算,僅花了一秒鐘就算完了!結果如下所示,非常平直,特別理想!
網友還給我發過一個含有200多個原子的彎曲的很長的分子,如下所示
用這一節的做法也很順利地優化成了平直的構象,如下所示。計算僅僅花了5秒鐘。

4 總結
本文介紹了筆者想到的四種把分子從彎曲結構拉成平直的方法,思路各有不同。實際當中會遇上各種特殊情況,比如可能需要同時拉直兩條鏈(如磷脂的兩條尾巴),應當在理解這些方法思想的基礎上根據實際情況恰當選擇、靈活運用。
本文的做法適合一般中、小分子體系。如果是大分子體系,比如蛋白質,比較適合GROMACS等專門的基于分子力場計算的程序。在GROMACS里面可以用pull相關的選項做拉伸動力學(見比如《在Gromacs中模擬金納米線拉伸過程(含視頻)》http://www.shanxitv.org/153),也可以將體系一端的原子定義為凍結組(freezegrps)而另一端定義為受常外力的組(acc-grps,結合accelerate設置)從而拉開。
值得一提的是,本文介紹的雖然是把分子拉直的方法,但實際上也可以將這些方法應用到研究環狀、籠狀、簇狀等化學體系對外力造成的拉伸和壓縮問題的研究上。
補充:使用Avogadro程序拉直分子的做法
通過Avogadro可視化程序也可以拉直分子,此程序可以在http://avogadro.cc免費下載。在此程序中拉直分子比較直觀,直接按住鼠標拖拽即可,這和SAMSON可視化程序里的twist工具非常類似。啟動Avogadro后,先載入pdb之類的結構文件,點擊下圖上方箭頭所指的按鈕,然后再點擊下圖左側箭頭所指的Start按鈕,之后按住鼠標左鍵拖動特定原子即可,按照下圖的綠色箭頭分別把兩端的原子往兩頭拉就能最終拉直。
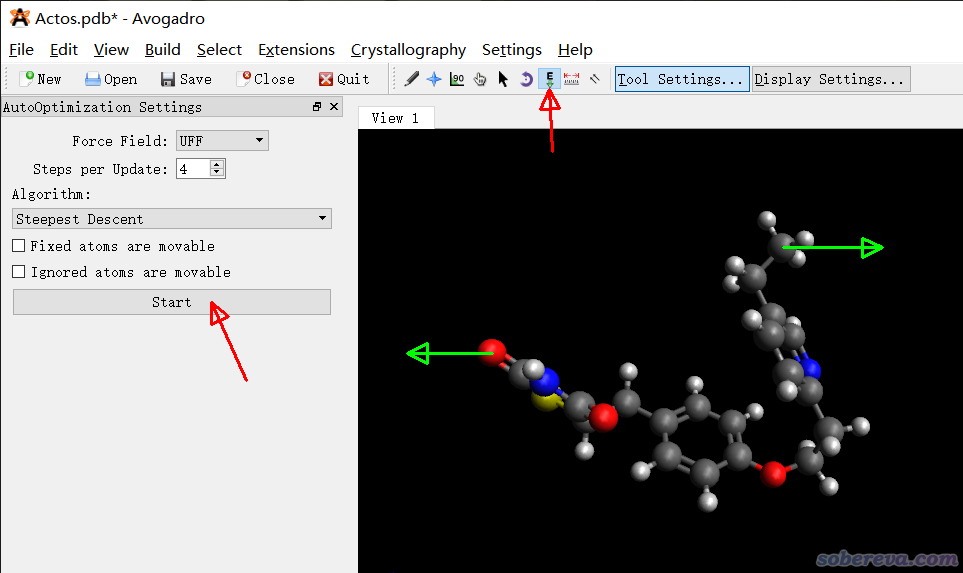
Avogadro的這個功能實際上是持續通過指定的力場優化體系幾何結構,因此當你拖動某個原子時,其它原子的位置會自發地弛豫來降低能量,因此鍵長、鍵角、二面角始終能保持比較合理的狀態。但我發現Avogadro的這個功能拉直小分子不錯,而拉直第3節最后那個特別長的聚合物則很難實現,而且特別卡頓。
]]>A simple example of performing ab initio dynamics (AIMD) using ORCA
Sobereva@北京科音
First release: 2020-Dec-13 Last update: 2025-May-9
1 前言
基于量子化學方法的動力學一般稱為從頭算動力學(Ab initio molecular dynamics, AIMD),相比于基于一般的經典力場的動力學,其關鍵優勢在于精度高、普適性強、能夠描述化學反應,代價是耗時相差N個數量級。ORCA量子化學程序有不錯的做BOMD形式的從頭算動力學的功能,使用很方便,而且本身ORCA做DFT的效率又高,是做孤立體系AIMD的首選程序之一。雖然有些特性不支持,比如沒法像Gaussian的BOMD那樣直接做準經典動力學,不能根據原子距離等標準判斷什么時候自動結束任務等等,但都不是大問題。對于跑跑普通的AIMD來說,筆者感覺ORCA明顯比Gaussian的ADMP或BOMD更好用(Gaussian的AIMD輸入文件較為抽象,手冊相應部分寫得很爛,而且連個像樣的熱浴都沒有),而且速度也明顯更快。
筆者發表的18碳環(cyclo[18]carbon)的研究論文中,其單體和二聚體做的分子動力學就是用ORCA跑的,分別見《揭示各種新奇的碳環體系的振動特征》(http://www.shanxitv.org/578)對中Chem. Asian J., 16, 56 (2021)和《全面探究18碳環獨特的分子間相互作用與pi-pi堆積特征》(http://www.shanxitv.org/572)中對Carbon, 171, 514 (2021)一文的介紹。在《18碳環等電子體B6N6C6獨特的芳香性:揭示碳原子橋聯硼-氮對電子離域的關鍵影響》(http://www.shanxitv.org/696)中介紹的筆者的Inorg. Chem., 62, 19986 (2023)文章中還用ORCA跑了B6C6N6的高溫動力學以證明其穩定性。在《18個氮原子組成的環狀分子長什么樣?一篇文章全面揭示18氮環的特征!》(http://www.shanxitv.org/696)中介紹的筆者的ChemPhysChem, 25, e202400377 (2024)文章中還用ORCA跑了18氮環的動力學以考察其熱分解行為。這些文章可以作為ORCA跑AIMD的典型范例,很推薦閱讀和引用。
筆者在北京科音高級量子化學培訓班(http://www.keinsci.com/workshop/KAQC_content.html)中會用多達兩百多頁的ppt專門深入詳細講AIMD的模擬,其中也包括ORCA做AIMD的各種細節、大量技巧以及諸多實例,并且此培訓中還會系統講授ORCA程序的使用,因此是使用ORCA專門做AIMD的讀者一定不可錯過的培訓。而本文只是舉一個簡單的例子,幫助讀者快速了解ORCA如何做最簡單的AIMD。注意ORCA只適合跑孤立體系的從頭算動力學,如果是做周期性體系的從頭算動力學,CP2K是最佳的選擇,在筆者講授的北京科音CP2K第一性原理計算培訓班(http://www.keinsci.com/workshop/KFP_content.html)中有極為全面、系統、詳細的講解并給了大量例子。
本文內容適用于Multiwfn最新版本、VMD 1.9.3、ORCA 4.2.x及以后版本的情況。
2021-Jul-7 針對ORCA 5.0的補充說明:對于2021-Jul-7及以后更新的Multiwfn,進入本文所述的Multiwfn產生輸入文件的界面后,可以通過選項-11選擇適合的ORCA版本,默認為ORCA 5.0及以后版本,而下文內容對應的是4.2.x版的情況。對于>=5.0版,Multiwfn自動設的熱浴是CSVR,比之前版本唯一支持的Berendsen熱浴更好,而且同樣普適。而且在run后面多加了CenterCOM,這是從5.0版本開始支持的消除整體質心運動的選項,而下文里提到的constraint add center這一行就沒有了。
2 實例:[Al(H2O)6]3+與NH3之間的質子轉移
ORCA的MD模塊的開發者網站上有個真空中[Al(H2O)6]3+與NH3之間的質子轉移的動畫,如下所示
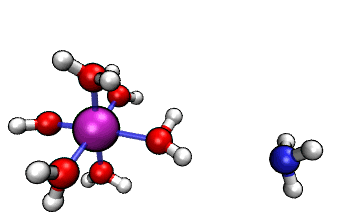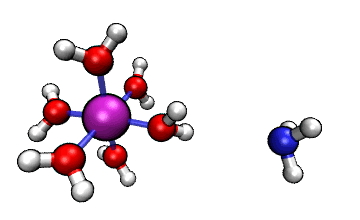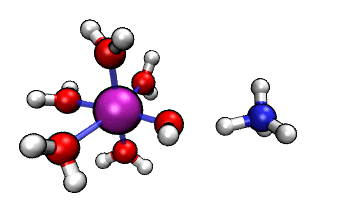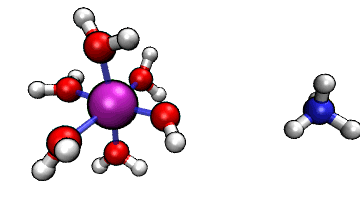
可見NH3向帶正電的[Al(H2O)6]3+逐漸靠近,水上的一個質子轉移到了氨氣分子上,然后由于靜電互斥,NH4+就逐漸遠離[Al(H2O)5(OH)]2+了。這里我們試圖重現這個過程。下面提到的文件都可以在http://www.shanxitv.org/attach/576/file.zip中獲得。
我們首先獲得[Al(H2O)6]3+的基本合理的結構。當然用ORCA優化也可以,這里筆者習慣性地用Gaussian來優化。在GaussView里搭建Al(H2O)6,保存為Al(H2O)6_optfreq.gjf,將關鍵詞改為# B3LYP/6-31G* opt freq,將電荷改為3,然后用Gaussian運行之,就得到了優化后的[Al(H2O)6]3+結構。再用GaussView打開輸出文件Al(H2O)6_optfreq.out,把一個氨氣分子畫在一個水的旁邊,如下所示,然后保存為complex.gjf。
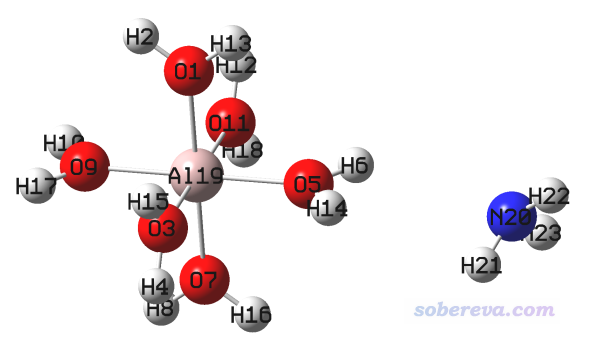
Multiwfn程序(http://www.shanxitv.org/multiwfn)有很便利的生成ORCA常見任務的輸入文件的功能,見《詳談Multiwfn產生ORCA量子化學程序的輸入文件的功能》(http://www.shanxitv.org/490),這里我們用Multiwfn生成ORCA的AIMD任務的標準輸入文件。
啟動Multiwfn,載入complex.gjf,然后輸入
oi // 生成ORCA輸入文件
0 // 選擇任務類型
6 // 分子動力學
1 // 計算級別用B97-3c
在當前目錄下就得到了ORCA的AIMD任務的標準輸入文件complex.inp。
在complex.inp里面將相應幾行改成下面這樣:
%maxcore 10000
%pal nprocs 10 end
timestep 1.0_fs
initvel 298.15_K
* xyz 3 1
現在的complex.inp的完整內容如下。以#為開頭的行代表后面的設置被注釋掉了,不會生效,想啟用可以去掉#。此文件里也有大量Multiwfn自動添加的注釋,只要一看注釋馬上就明白相應的行是什么含義,巨貼心,都省得查手冊了。
! B97-3c noautostart miniprint nopop
%maxcore 10000
%pal nprocs 10 end
%md
#restart ifexists # Continue MD by reading [basename].mdrestart if it exists. In this case "initvel" should be commented
#minimize # Do minimization prior to MD simulation
timestep 1.0_fs # This stepsize is safe at several hundreds of Kelvin
initvel 298.15_K no_overwrite # Assign velocity according to temperature for atoms whose velocities are not available
thermostat berendsen 298.15_K timecon 30.0_fs # Target temperature and coupling time constant
dump position stride 1 format xyz filename "pos.xyz" # Dump position every "stride" steps
#dump force stride 1 format xyz filename "force.xyz" # Dump force every "stride" steps
#dump velocity stride 1 format xyz filename "vel.xyz" # Dump velocity every "stride" steps
#dump gbw stride 20 filename "wfn" # Dump wavefunction to "wfn[step].gbw" files every "stride" steps
constraint add center 0..22 # Fix center of mass at initial position
run 2000 # Number of MD steps
end
* xyz 3 1
[坐標部分]
*
下面簡單說一下complex.inp里這些設置的含義、為什么這么設。
? B97-3c是一個又便宜又不錯的計算級別,在ORCA里還自動會啟用RIJ加速,速度很快,因此很適合跑AIMD,描述當前體系沒有問題。B97-3c的介紹見《詳談Multiwfn產生ORCA量子化學程序的輸入文件的功能》(http://www.shanxitv.org/490)的相應部分。但B97-3c也不是什么時候都能用,比如18碳環用純泛函描述皆失敗,見筆者在Carbon, 165, 468 (2020) 里的討論,顯然就不能用這個了,筆者跑涉及18碳環的AIMD的時候都用的是ωB97X-D3。
? %pal nprocs 10 end代表用10核。筆者當前任務實際上是在雙路E5-2696v3共36個物理核心的機子上跑的,但卻故意用了10核。這是因為根據筆者以前的測試,發現ORCA做DFT的AIMD的并行效率不理想,尤其是對于小體系,用核數太多甚至反倒速度更慢(大家可以對實際情況短暫跑比如5步實測一下設多少核速度最快)。一般來說就設10核就行了,當機子里有明顯更多核的時候,可以跑多個AIMD任務來充分利用計算資源,但應當對CPU內核進行綁定,否則AIMD計算速度可能顯著降低,見《通過設置CPU內核綁定降低ORCA同時做多任務的耗時》(http://www.shanxitv.org/553)。
? %maxcore 10000代表每個ORCA進程最多用10000MB。其實完全沒必要這么大,普通泛函下的AIMD不怎么耗內存,給1000都絕對夠了。
? restart ifexists這句被注釋掉了。如果你的AIMD想續跑,且當前目錄下有之前跑出來的與當前任務同名的后綴為.mdrestart的文件,可以取消注釋,任務就會延續之前AIMD最后的狀態續跑,新軌跡會在原有軌跡文件后面續寫。
? minimize這句被注釋掉了。如果取消注釋的話,動力學開始前會自動在當前級別下做幾何優化。
? timestep 1.0_fs代表動力學步長用1 fs。對于此例常溫下的模擬,1 fs步長是合適的,改大的話可能造成動力學不穩定,而改小的話跑同樣的時間長度需要更多步數導致需要更多耗時。如果追求絕對穩妥,或者是在明顯更高溫度下模擬,可以用0.5 fs。
? initvel 298.15_K代表根據298.15 K溫度通過Maxwell分布隨機初始化原子速度。no_overwrite代表如果之前已經有初速度信息了(比如可以是續跑時從之前的mdrestart文件里繼承來的),就不再產生新的初速度。
? thermostat berendsen 298.15_K timecon 30.0_fs代表用Berendsen熱浴控溫在298.15 K(此例筆者用的ORCA 4.2.1只能用這個熱浴,據悉從ORCA 5.0開始可以用更好的velocity rescale熱浴),時間常數為30 fs,通常這個時間常數是合適的。
? dump position stride 1 format xyz filename "pos.xyz"代表每一步都往當前目錄下的pos.xyz文件里寫入當前的坐標,因此pos.xyz是多幀xyz格式的軌跡文件。此格式的介紹見《談談記錄化學體系結構的xyz文件》(http://www.shanxitv.org/477)。
? dump force和dump velocity開頭的行都被注釋掉了,這兩行用于把模擬過程中的原子受力和原子速度分別保存到相應xyz文件里。dump gbw開頭的行也被注釋掉了,它可以在MD過程中每隔指定步數保存一次gbw文件,之后可以通過寫腳本調用Multiwfn對它們進行批量分析,從而考察動力學過程中電子結構變化,得到豐富的有化學意義的信息(如動力學中的電荷轉移情況、成鍵變化情況、電子定域性變化等等,在北京科音高級量子化學培訓班里筆者會給出很多這種例子)
? constraint add center 0..22代表將當前整個體系(0號到22號原子)的質心約束在初始位置。之所以這么做,是因為盡管ORCA在產生初速度時已經去掉了整體平移的速度分量,但實際模擬過程中由于數值問題,仍然往往會看到有明顯的整體漂移的現象,因而有礙觀察(在VMD里觀看軌跡時還得做一下align才能消掉)。因此直接把質心位置約束住就沒有這個問題了。
? run 2000代表總共跑2000步,當前步長是1 fs,即最多跑2 ps。當前這個模擬的目的是觀察到質子轉移,跑多長時間合適并不好預估,所以可以一次先跑2000步看看,若不夠到時候再續跑。值得一提的是,至少對于筆者現在用的ORCA 4.2.1,我發現如果一次跑的步數很多,達到2000步左右之后,之后每一步的耗時會有逐漸的上升趨勢,原因不明。因此我建議每次跑最多不宜超過3000步,如果需要跑更長的話,最好分多段來跑。
重要提示:如果你用的是ORCA較新版本,應去掉constraint add center 0..22,而把run 2000改為run 2000 CenterCOM,否則可能跑不了。
現在用ORCA運行complex.inp,模擬過程中可以看到每一步的實時情況:
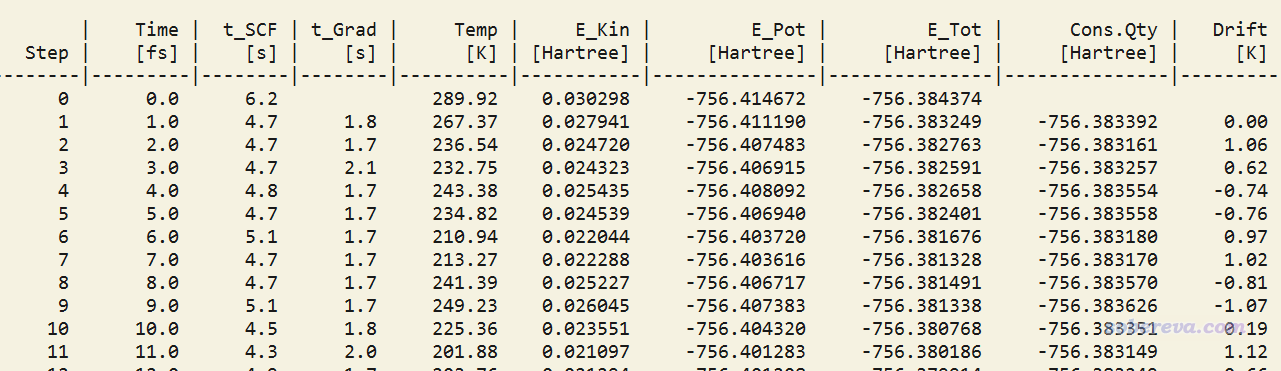
Step是當前的步數,Time是當前的時間,t_SCF和t_Grad分別是算這一步的SCF過程和計算受力的耗時,二者加和就是這一步的總耗時。可見每一步耗時約6~7秒,乘以要跑的步數就可以估計跑完整個任務的耗時。后面還可以看到每一步的溫度、動能、勢能等信息。由于當前體系原子數很少,溫度相對于熱浴的參考溫度波動大是很正常的事。而且由于用了熱浴,所以總能量E_tot也有明顯波動。
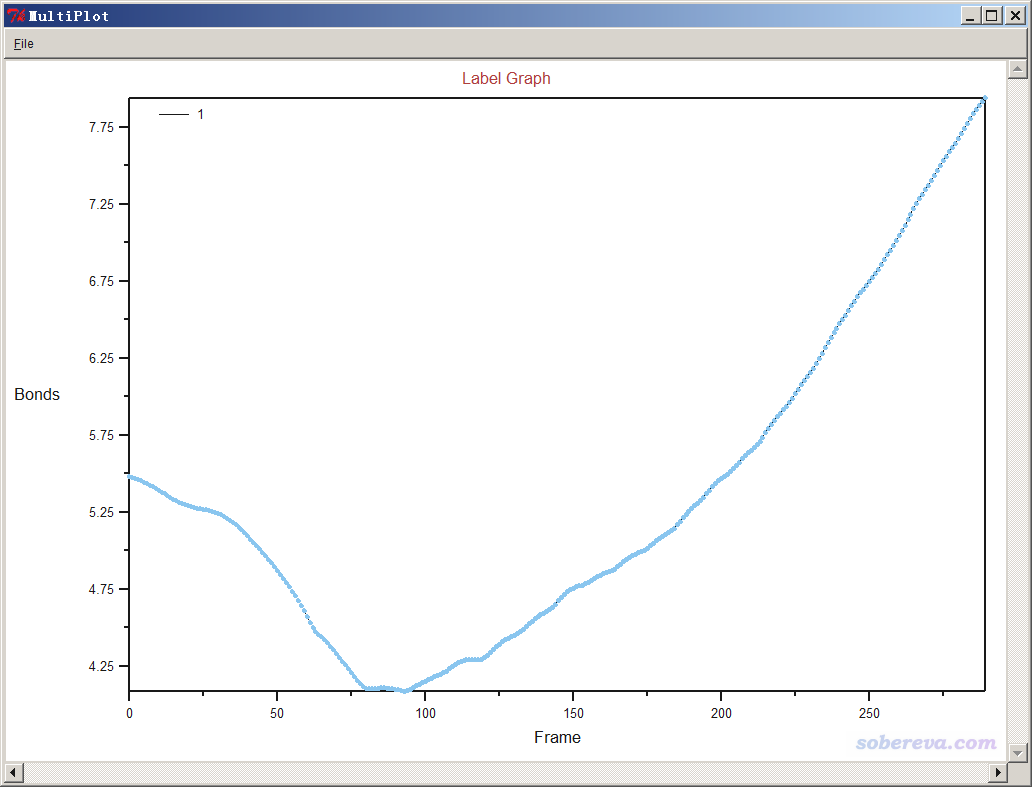
VMD是觀看動力學軌跡最好的程序,可以在http://www.ks.uiuc.edu/Research/vmd/免費下載。建議在模擬過程中,隔一陣子就用VMD把pos.xyz載入進去,看看當前的動態行為如何、跑成什么結構了。跑到289步的時候,筆者看了一眼pos.xyz,發現質子不僅已經完全轉移,而且NH4+都已經跑走一定距離了,所以就沒必要繼續跑了,故把ORCA直接殺掉了。
模擬過程中ORCA還產生其它一些文件。complex.md.log是ORCA的MD模塊輸出的信息,相當于整個輸出文件中的子集。complex.mdrestart是用來續跑的文件,每一步都會往里面寫入當前步的時間、坐標、速度、受力等信息。complex-md-ener.csv是把每一步的時間、耗時、溫度、能量等信息以csv格式保存的文件。還有其它一些零碎的文件,通常不是一般用戶關心的,這里就不說了,都可以放心刪掉,留著也沒用。
現在我們來看模擬軌跡。建議大家根據《VMD初始化文件(vmd.rc)我的推薦設置》(http://www.shanxitv.org/545)里說的修改VMD的初始化文件,從而添加自定義命令bt,這樣在播放軌跡的時候對每一幀會自動重新判斷成鍵關系。
啟動VMD,載入pos.xyz(在本文文件包里已提供,共290幀)。然后在文本窗口輸入bt,按回車,使得每幀都更新連接關系。然后再輸入bw,按回車使得背景變為白色。選Graphics - Representation,將Drawing Method改為CPK。然后點擊VMD界面右下角的三角播放動畫,會看到如下結果。如果想在VMD圖形窗口中顯示出幀號或時間,看《使VMD播放軌跡時同步顯示幀號》(http://www.shanxitv.org/13)。
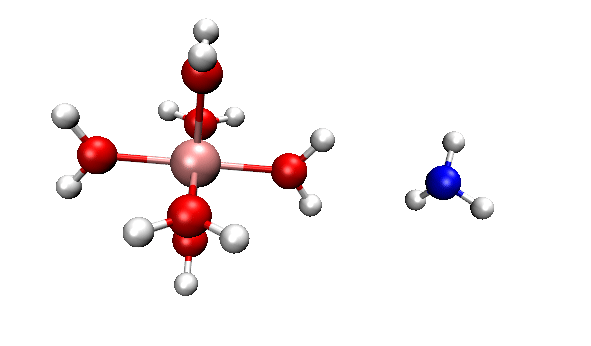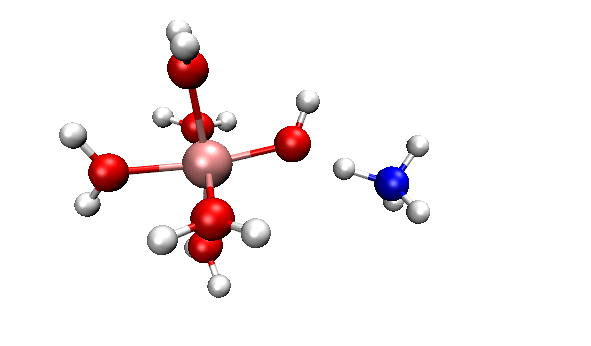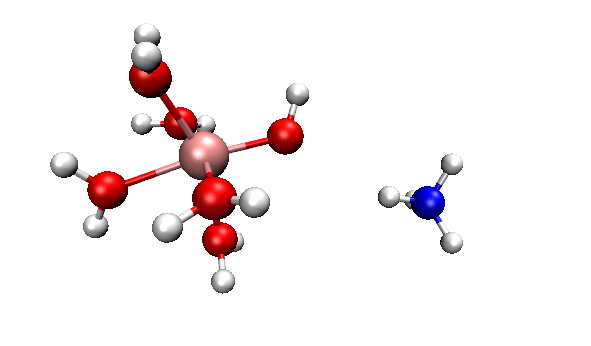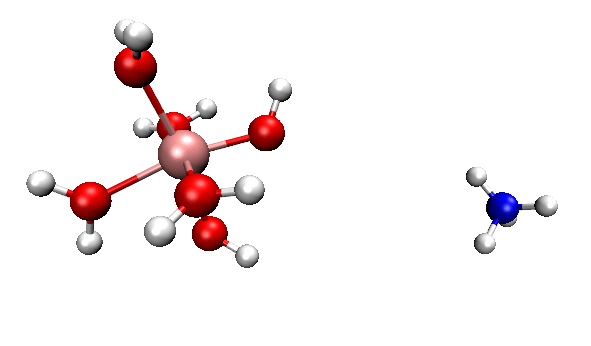
可見,我們跑出來的動力學現象和本文一開始的那個官方動畫幾乎完全一致。都是兩個反應物先接近,然后形成復合狀態時質子在二者之間震蕩,最后NH4+跑掉。
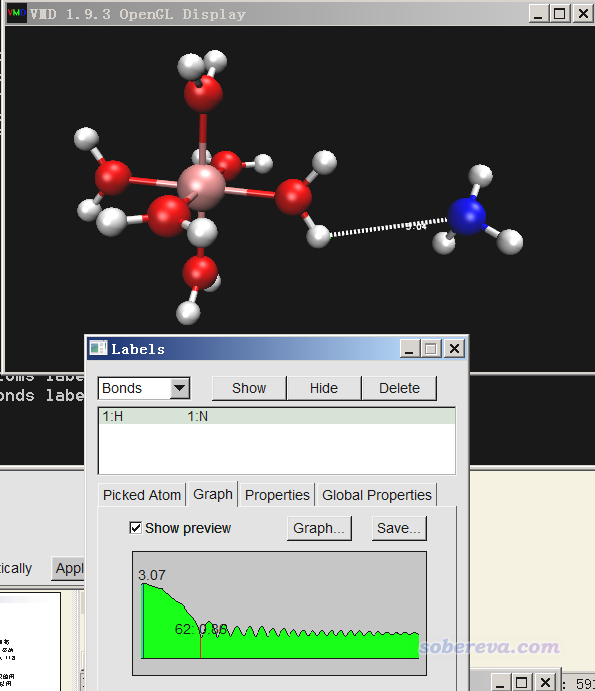
如果想把質子轉移情況通過距離隨時間的變化曲線方式呈獻給讀者,可以在點擊VMD的三維圖形窗口后,按鍵盤上的2鍵(之后按r可以恢復為默認的旋轉模式),然后點擊兩個原子正中心,二者之間就會增加Bond label(默認是以白色字顯示距離,在黑背景下才看得清楚),這里筆者把N和轉移過去的質子之間增加了Bond label。然后進入Graphics - Labels,切換到Bonds,選Graph,點擊Show preview復選框,然后點擊1:H 1:N這項,就會看到距離變化已經顯示在預覽窗口了,如下所示,其中紅色和藍色豎條標記的分別是最小距離和最大距離位置和數值。
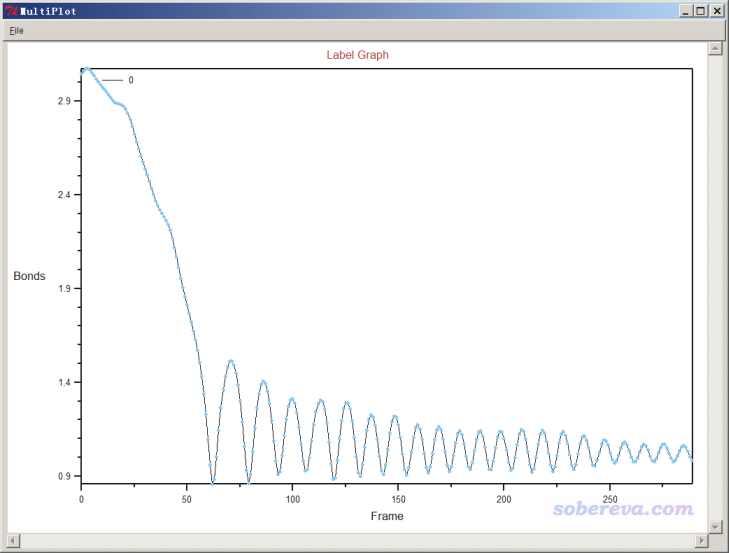
如果點擊Graph按鈕,就會把曲線顯示在大窗口中,如下所示。可見,在大約60幀,也即60 fs左右,N-H鍵就算是基本形成了,之后N-H鍵不斷振動。
以類似方式,我們可以標記Al與N的距離,隨時間變化如下所示。可見Al與N先接近,質子轉移完畢后,二者就逐漸遠離了。
在Labels窗口里還可以點擊save,把距離變化數據保存到文本文件里,之后可以導入Origin等程序里繪制曲線。
用VMD還可以測量角度、二面角的變化,分別是在圖形窗口里按3然后點三個原子、按4然后點四個原子進行標記,之后在Graphics - Labels里觀看。
3 總結&其它
通過上面的例子,可以看到ORCA做AIMD是相當容易的,只要把Multiwfn支持的含有結構信息的文件(如pdb/gjf/xyz/mol/mol2/fch等等,見Multiwfn手冊2.5節)載入Multiwfn,敲幾下鍵盤產生標準AIMD任務的輸入文件,然后根據實際情況稍微改幾個設定就可以跑了。
以幾十核的一般雙路服務器的運算能力,ORCA里用B97-3c跑幾十原子有機體系的幾十ps的動力學不是特別困難的事。不過,能跑的時間尺度仍遠遠比不上xtb跑半經驗層面DFT的GFN-xTB方法的動力學,xtb跑動力學的粗淺介紹和例子看此文的相應部分:《使用Molclus結合xtb做的動力學模擬對瑞德西韋(Remdesivir)做構象搜索》(http://bbs.keinsci.com/thread-16255-1-1.html)。因此,拿ORCA跑DFT的動力學之前,先拿xtb初步跑跑,找找感覺,大體摸索出自己期望的現象能出現的條件(如溫度、初始結構、反應物相對位置和碰撞方式等),然后再用DFT跑通常是比較好的做法,免得做昂貴的DFT的MD試來試去把時間都耽誤了。
本文只涉及了VMD一丁點皮毛,VMD對于做動力學的人是必須玩得非常溜的。筆者在北京科音分子動力學與GROMACS培訓班(http://www.keinsci.com/workshop/KGMX_content.html)里對VMD有非常深入全面的介紹,包括tcl腳本的編寫。利用VMD的tcl腳本可以對軌跡做千變萬化的分析,有些分析諸如質心距離變化、平面間夾角變化、某幾何變量分布統計、不同結構出現比率等,是必須靠寫腳本才能實現的。
光是分析分析動力學過程的能量、結構變化是很膚淺的。利用Multiwfn,可以對ORCA跑的動力學的全過程的電子結構做深入透徹的分析,從而考察化學鍵、弱相互作用、電荷分布等等在動力學過程中的變化,由此能夠從提供深入的視角,使研究文章信息更豐富、明顯更上檔次。非常建議詳細閱讀《詳談Multiwfn的命令行方式運行和批量運行的方法》(http://www.shanxitv.org/612),里面第4節專門講了怎么做這樣的分析,你會發現特別容易也特別有價值。
如果Multiwfn創建ORCA做動力學輸入文件的功能對你的實際研究產生了幫助,希望在寫文章的時候提及諸如The input file of ab-initio molecular dynamics was prepared with the help of Multiwfn program并引用Multiwfn原文,這是對Multiwfn此功能開發最好的支持。
]]>Reducing time-consuming of multitasking of ORCA by setting CPU core binding
文/Sobereva@北京科音 2020-Jun-1
1 前言
由于任何程序的并行效率都是隨著核數增加而降低,當機子核數比較多的時候,比如有好幾十核,而且又有許多任務要跑的時候,比起一個一個調用所有核心來跑,同時跑兩個或者多個任務但是每個都用較少的核心數(總和不超過物理核心數),總耗時通常會更低。對于某些程序跑某些任務,甚至并行核數較少的時候反倒比核數較多的時候速度還更快。因此在核數較多的機子上,同時跑多個任務是很常見的事情。
然而,同時跑多個任務涉及到資源爭搶問題,如果爭搶得比較厲害,跑多個任務的效率會大打折扣,甚至可能還不如一個一個用所有核來跑。
如今很多程序在并行計算的時候都支持線程或進程與CPU內核的綁定,從而避免操作系統自動調度導致任務在不同CPU核心上頻繁切換執行引起運行效率的下降,恰當綁定也可以有效降低CPU資源爭搶問題。ORCA自身沒有直接提供內核綁定的設置,但可以通過自行指定OpenMPI的運行選項來實現,本文就說說具體做法和實際效果。
2 CPU內核綁定的實現
2.1 在ORCA運行時使用內核綁定
ORCA在Linux下并行一般結合OpenMPI使用,本文也只說OpenMPI的情況。OpenMPI基于hwloc庫來獲取硬件信息,而hwloc庫又要利用libnuma。只有在編譯OpenMPI時提供了libnuma靜態庫,之后ORCA運行時才能借助OpenMPI的設置實現內核綁定。多數情況下系統里是沒有裝libnuma靜態庫的,對于CentOS 7.x,可以通過yum install numactl-devel命令來裝上,之后再按照《量子化學程序ORCA的安裝方法》(http://sobereva.com/451)里說的照常編譯OpenMPI就可以了。
ORCA運行命令中可以加入傳遞給OpenMPI的mpirun的選項,比如:
orca test.inp "--bind-to core"
此時ORCA利用mpirun來運行其支持并行的模塊的時候就會把--bind-to core選項傳遞給mpirun。OpenMPI 3.1.x版的mpirun支持的選項在這里都能看到:https://www.open-mpi.org/doc/v3.1/man1/mpirun.1.php,其中就包括了內核綁定的設置的說明。
2.2 內核綁定規則的設置
mpirun的選項中--bind-to [值]指定的是綁定的對象什么,而--map-by [值]是設定按照什么順序循環被綁定物。例如當前機子是雙路的,每個CPU有六個核,看以下兩種情況:
--bind-to core --map-by core:按照循環各個核的順序綁定,圖例如下。B是代表運行當前進程的CPU核心
進程0:[B . . . . .][. . . . . .]
進程1:[. B . . . .][. . . . . .]
進程2:[. . B . . .][. . . . . .]
進程3:[. . . B . .][. . . . . .]
...略
這表明進程0綁定了第一個CPU的第一個核心,只有這個核心被用來跑這個進程。
--bind-to socket --map-by socket:按照循環各個CPU的順序綁定(socket此處是指CPU插槽),圖例如下
進程0:[B B B B B B][. . . . . .]
進程1:[. . . . . .][B B B B B B]
進程2:[B B B B B B][. . . . . .]
進程3:[. . . . . .][B B B B B B]
...略
可見,此時進程0綁定了第一個CPU的所有核心,這些核心一起執行這個進程,而第二個CPU核心不會跑這個進程。
mpirun加上--report-bindings選項可以在開始并行執行時顯示當前是怎么綁定的,用以確認綁定方式合乎自己的要求。
如果想精細控制綁定規則,需要創建rankfile文件。比如rankfile文件是/sob/miku.txt,可以這么運行./violet.x程序:mpiexec -np 3 -rf /sob/miku.txt --report-bindings ./violet.x,此時一共3個進程會按照rankfile里指定的方式來綁定。
比如rankfile文件內容如下
rank 0=Saber110 slot=1:0,1
rank 1=Saber109 slot=0:*
rank 2=Saber108 slot=1:1-3
rank 3=Saber17 slot=0:1,1:0-2
rank 4=Saber109 slot=0:*,1:*
rank 5=Saber109 slot=0-2
就意味著(注意此處進程、CPU編號和核心編號都是從0開始的)
第0個進程與Saber110節點的第1號CPU的0、1兩個核心綁定
第1個進程與Saber109節點的第0號CPU的所有核心綁定
第2個進程與Saber108節點的第1號CPU的1、2、3核心綁定
第3個進程與Saber17節點的第0號CPU的1號核心,以及第1號CPU的0~2號核心綁定
第4個進程與Saber109節點的第0、1號CPU的所有核心綁定
第5個進程與Saber109節點的第0、1、2號邏輯核心綁定
如果比如當前是-np 3跑3個進程,則只有前3個規則生效,而rank 3、rank 4、rank 5的設置不會被利用。
2.3 讓每個ORCA任務獨占不同的CPU核心
如果只跑一個任務的話,設內核綁定沒什么用處。而跑多個任務時,給不同任務指定不同的CPU核心可用范圍,則有可能降低CPU資源爭搶來降低耗時。為此,需要設置不同rankfile。比如雙路共36核72線程機子,其邏輯核心的序號是下面的順序:
第一個CPU的18個物理核心:依次為0~17
第二個CPU的18個物理核心:依次為18~35
第一個CPU的18個虛擬核心:依次為36~53
第二個CPU的18個虛擬核心:依次為54~71
更確切來說,比如0和36號邏輯核心的任務其實都是第一個CPU的實際的0號核心做的,如果就是一個MPI進程要跑,在0還是36號上執行速度都一樣,但如果有兩個MPI進程,若在0和36號上面同時跑,速度就只有之前的約一半了。詳見《正確認識超線程(HT)技術對計算化學運算的影響》(http://sobereva.com/392)。下文就忽略掉由于超線程技術而多出來的36~71號核心了。
如果有兩個ORCA任務要跑,每個都設nprocs 18,那么默認的情況下,每個任務會同時在兩個CPU上跑,有可能存在資源爭搶。如果希望這兩個任務分別在第一和第二個CPU上跑,可以設置這以下兩個rankfile文件。
CPU1.txt,內容為
rank 0=2696v3 slot=0:0
rank 1=2696v3 slot=0:1
rank 2=2696v3 slot=0:2
rank 3=2696v3 slot=0:3
rank 4=2696v3 slot=0:4
rank 5=2696v3 slot=0:5
rank 6=2696v3 slot=0:6
rank 7=2696v3 slot=0:7
rank 8=2696v3 slot=0:8
rank 9=2696v3 slot=0:9
rank 10=2696v3 slot=0:10
rank 11=2696v3 slot=0:11
rank 12=2696v3 slot=0:12
rank 13=2696v3 slot=0:13
rank 14=2696v3 slot=0:14
rank 15=2696v3 slot=0:15
rank 16=2696v3 slot=0:16
rank 17=2696v3 slot=0:17
CPU2.txt,內容為
rank 0=2696v3 slot=1:0
rank 1=2696v3 slot=1:1
rank 2=2696v3 slot=1:2
rank 3=2696v3 slot=1:3
rank 4=2696v3 slot=1:4
rank 5=2696v3 slot=1:5
rank 6=2696v3 slot=1:6
rank 7=2696v3 slot=1:7
rank 8=2696v3 slot=1:8
rank 9=2696v3 slot=1:9
rank 10=2696v3 slot=1:10
rank 11=2696v3 slot=1:11
rank 12=2696v3 slot=1:12
rank 13=2696v3 slot=1:13
rank 14=2696v3 slot=1:14
rank 15=2696v3 slot=1:15
rank 16=2696v3 slot=1:16
rank 17=2696v3 slot=1:17
這里2696v3是當前的主機名,運行hostname命令時顯示什么就填什么(如果顯示的是IP號,這里就填IP號)。
假設這兩個rankfile文件都在/sob目錄下,然后執行以下兩條命令分別跑ORCA任務
orca MD1.inp "-rf /sob/CPU1.txt" > MD1.out
orca MD2.inp "-rf /sob/CPU2.txt" > MD2.out
則MD1和MD2任務就分別只在第一個和第二個CPU上跑了
如果你有四個任務要跑,每個任務給9個核,那么可以創建四個rankfile:
CPU1a.txt的內容
rank 0=2696v3 slot=0:0
rank 1=2696v3 slot=0:1
rank 2=2696v3 slot=0:2
rank 3=2696v3 slot=0:3
rank 4=2696v3 slot=0:4
rank 5=2696v3 slot=0:5
rank 6=2696v3 slot=0:6
rank 7=2696v3 slot=0:7
rank 8=2696v3 slot=0:8
CPU1b.txt的內容
rank 0=2696v3 slot=0:9
rank 1=2696v3 slot=0:10
rank 2=2696v3 slot=0:11
rank 3=2696v3 slot=0:12
rank 4=2696v3 slot=0:13
rank 5=2696v3 slot=0:14
rank 6=2696v3 slot=0:15
rank 7=2696v3 slot=0:16
rank 8=2696v3 slot=0:17
CPU2a.txt的內容
rank 0=2696v3 slot=1:0
rank 1=2696v3 slot=1:1
rank 2=2696v3 slot=1:2
rank 3=2696v3 slot=1:3
rank 4=2696v3 slot=1:4
rank 5=2696v3 slot=1:5
rank 6=2696v3 slot=1:6
rank 7=2696v3 slot=1:7
rank 8=2696v3 slot=1:8
CPU2b.txt的內容
rank 0=2696v3 slot=1:9
rank 1=2696v3 slot=1:10
rank 2=2696v3 slot=1:11
rank 3=2696v3 slot=1:12
rank 4=2696v3 slot=1:13
rank 5=2696v3 slot=1:14
rank 6=2696v3 slot=1:15
rank 7=2696v3 slot=1:16
rank 8=2696v3 slot=1:17
之后跑四個ORCA任務的時候分別指定這四個rankfile,則第一、二個任務就分別在第一個CPU的前9個、后9個核心上跑,第三、四個任務就分別在第二個CPU的前9個、后9個核心上跑,這盡可能減小了四個任務彼此的干擾(但干擾是不可能完全杜絕的)。
3 綁定不同CPU核心跑多任務對耗時的影響
為了測試綁定不同CPU核心對于跑多任務的耗時影響,我選了一個堿基對當測試體系,一共29個原子,在B3LYP-D3(BJ)/def2-QZVP結合RIJCOSX級別下,用筆者的XEON 2696v3雙路機子(36核72線程)進行測試。結果如下
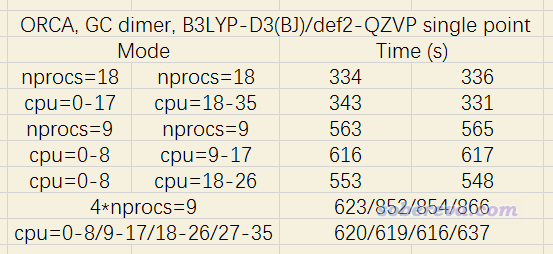
其中諸如nprocs=18代表不對內核進行綁定設置的情況。可見跑兩個18核任務的時候,是否綁定對耗時影響可以忽略。跑兩個9核的任務時,分別在兩個CPU上跑的耗時明顯比讓這倆任務分別占同一個CPU的前9個和后9個核心的耗時要低,顯然后者的情況會對第一個CPU有一定程度的資源爭搶。內核綁定的效果對于同時跑四個任務很明顯,直接跑四個9核的任務的耗時比讓這四個任務綁定不同核心(分別綁定0-8、9-17、18-26、27-35號核心)的耗時高了1/3有余!
這種綁定的做法對于不同類型任務產生的效果不一樣。筆者之前拿ORCA跑了一個環狀體系的AIMD模擬,哪怕只是同時跑兩條對應于不同溫度下的軌跡,每個都給9核,分別綁在兩個CPU上跑的速度都顯著快于不設綁定直接提交兩個任務的情況。還可以觀察到一個現象,如果以普通方式運行,當第二個AIMD任務交上去之后,就會發現ORCA顯示的第一個AIMD任務的每步的耗時增加了很多;而如果兩個任務在運行時分別綁在不同CPU的核心上,則提交第二個任務后,之前正在跑的第一個任務每步的耗時幾乎沒有變化。
總之,對于ORCA用戶,如果經常同時跑多任務的話,不妨留意一下內核綁定問題。對于跑AIMD,我個人總是將多個任務綁定不同內核來跑,哪怕只是同時跑兩個任務。
Gaussian從16版開始支持%cpu設置,可直接控制任務在哪些CPU核心上運行,這在《正確認識超線程(HT)技術對計算化學運算的影響》(http://sobereva.com/392)中我專門提過。筆者也測試了一下在Gaussian中把同時提交的任務指定在不同CPU核心上對速度的影響。還是用堿基對體系,在M06-2X/def2-TZVP下做單點計算,耗時如下

可見是否將不同任務綁在不同CPU核心上運行對耗時基本沒影響,所以Gaussian用戶不需要特意考慮內核綁定問題。如果兩個9線程的任務分別綁定在同一個CPU的前9個和后9個核來跑,速度則會明顯不如照常提交這兩個任務,這和測試ORCA時看到的情況是一致的。
]]>Example of performing counterpoise correction and calculation of intermolecular binding energy in ORCA
文/Sobereva@北京科音
First release: 2020-Mar-13 Last update: 2024-Dec-31
在《談談BSSE校正與Gaussian對它的處理》(http://www.shanxitv.org/46)一文中介紹過BSSE問題與counterpoise校正。Counterpoise校正在高精度弱相互作用計算時通常需要考慮。Gaussian有現成的counterpoise關鍵詞,因此做counterpoise校正很簡單。但是ORCA程序(至少對于撰文時最新的4.2.1版來說)沒有提供現成的counterpoise關鍵詞,只能通過定義鬼原子的方式手工實現,因此實現起來稍微麻煩。不熟悉ORCA者看http://www.shanxitv.org右側ORCA分類里我寫過的大量相關文章,在北京科音高級量子化學培訓班(http://www.keinsci.com/KAQC)里有非常詳細的ORCA使用全套講解。
之前在《詳談Multiwfn產生ORCA量子化學程序的輸入文件的功能》(http://www.shanxitv.org/490)中筆者專門介紹了Multiwfn產生ORCA量子化學程序的輸入文件的功能。為了使得用ORCA做counterpoise任務更方便,筆者2020-Mar-13給Multiwfn的這個功能增加了相應選項,在這里通過一個實例簡單演示一下。最新版Multiwfn可以在http://www.shanxitv.org/multiwfn免費下載,相關知識見《Multiwfn FAQ》(http://www.shanxitv.org/452)。
注意按照下文例子算出來的實際上是復合物結構中單體間相互作用能。而一般意義的結合能在計算時是要考慮單體形成復合物過程中形變造成的能量改變(變形能)。由于下面的例子里單體是剛性的,變形能可以忽略不計,因此下文仍稱作結合能。強烈建議讀者閱讀《談談分子間結合能的構成以及分解分析思想》(http://www.shanxitv.org/733)搞清楚邏輯關系。
本文用的示例體系是下圖這個體系,我們要在DLPNO-CCSD(T)/aug-cc-pVTZ下計算其結合能,并考慮counterpoise校正。
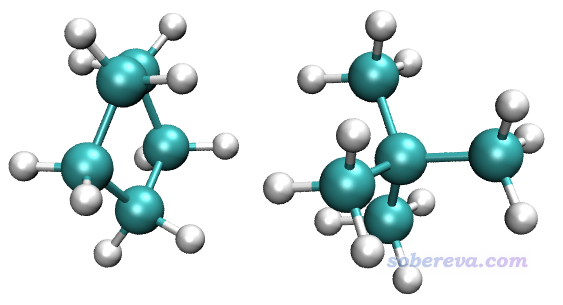
此體系的xyz格式的結構文件,后文中涉及的輸入輸出文件以及要用的shell腳本都可以在這里下載:http://www.shanxitv.org/attach/542/file.rar。并不是必須得用xyz格式,只要是Multiwfn支持的含有結構信息的格式(如pdb、mol2...)都可以作為輸入文件,見《詳談Multiwfn支持的輸入文件類型、產生方法以及相互轉換》(http://www.shanxitv.org/379)。
啟動Multiwfn,輸入dimer.xyz的路徑載入之。如果你還不知道兩個單體的原子序號范圍,可以進入Multiwfn的主功能0來查詢。進入主功能0后,選擇Tools - Select fragment,輸入其中一個片段中的任意一個原子的序號比如16,整個片段就被高亮,而且序號也顯示出來了,如下所示。
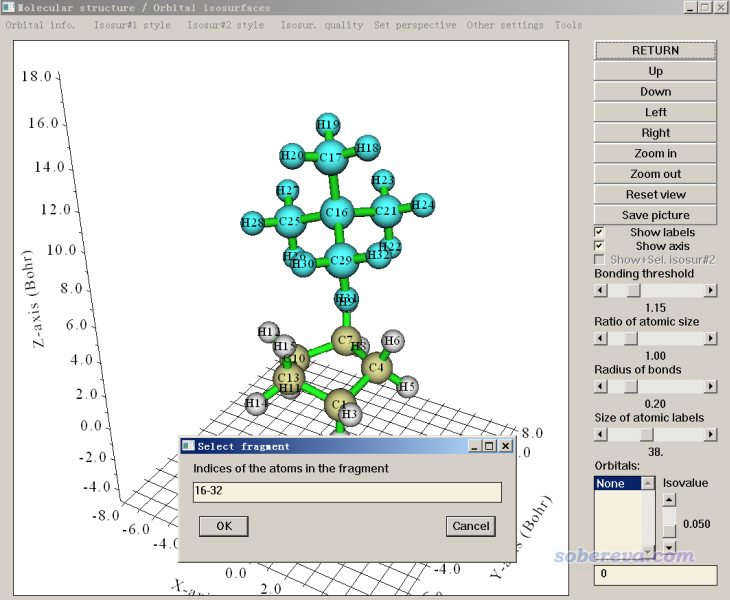
如此這般,把兩個片段序號范圍都拷出來備用,然后點Return退出主功能0,之后依次輸入以下內容:
oi //產生ORCA的輸入文件
[按回車] //輸出文件與輸入文件同名但用inp作為后綴
0 //選擇任務類型
7 //考慮counterpoise校正的結合能
1-15 //片段1的序號
16-32 //片段2的序號
-2 //加彌散函數
8 //用DLPNO-CCSD(T) with NormalPNO
在當前目錄下馬上就有了dimer.inp,內容如下
%pal nprocs 4 end
! DLPNO-CCSD(T) normalPNO RIJK aug-cc-pVTZ aug-cc-pVTZ/JK aug-cc-pVTZ/C tightSCF noautostart miniprint nopop
%maxcore 1000
* xyz 0 1
C 0.79991408 -1.02205164 0.68773696
H 0.85355588 -1.12205101 -0.39801435
H 1.49140210 -1.74416936 1.11972040
C 1.11688700 0.42495279 1.09966205
H 1.83814230 0.89014504 0.43045256
H 1.55556959 0.43982464 2.09708356
C -0.24455916 1.16568959 1.10297714
H -0.25807760 2.00086313 0.40532333
H -0.44880450 1.57699582 2.09098447
C -1.29871418 0.10381191 0.73930899
H -1.47356078 0.10524338 -0.33800545
H -2.25673428 0.27804118 1.22715843
C -0.64687993 -1.22006836 1.13630660
H -1.12443918 -2.08762702 0.68299327
H -0.68601864 -1.34528332 2.22022006
C 0.04984615 0.09420760 5.61627735
C -0.04649805 -0.05787837 7.13191782
H 0.94604832 -0.07334458 7.58427505
H -0.60542282 0.77000613 7.57035274
H -0.55366275 -0.98654445 7.39726741
C 0.76389939 1.40111272 5.28065247
H 0.84541894 1.53461185 4.20097059
H 0.22042700 2.25580115 5.68615385
H 1.77150393 1.41176313 5.69888547
C -1.35516567 0.11403225 5.01895782
H -1.31823408 0.23122219 3.93510886
H -1.93746520 0.94145581 5.42730374
H -1.88506873 -0.81375459 5.24028712
C 0.83774596 -1.07927730 5.03893917
H 0.34252564 -2.02626804 5.25918232
H 0.93258913 -0.99209454 3.95580439
H 1.84246405 -1.11668194 5.46268763
*
$new_job
! DLPNO-CCSD(T) normalPNO RIJK aug-cc-pVTZ aug-cc-pVTZ/JK aug-cc-pVTZ/C tightSCF noautostart miniprint nopop Pmodel
%maxcore 1000
* xyz 0 1
C 0.79991408 -1.02205164 0.68773696
H 0.85355588 -1.12205101 -0.39801435
H 1.49140210 -1.74416936 1.11972040
C 1.11688700 0.42495279 1.09966205
H 1.83814230 0.89014504 0.43045256
H 1.55556959 0.43982464 2.09708356
C -0.24455916 1.16568959 1.10297714
H -0.25807760 2.00086313 0.40532333
H -0.44880450 1.57699582 2.09098447
C -1.29871418 0.10381191 0.73930899
H -1.47356078 0.10524338 -0.33800545
H -2.25673428 0.27804118 1.22715843
C -0.64687993 -1.22006836 1.13630660
H -1.12443918 -2.08762702 0.68299327
H -0.68601864 -1.34528332 2.22022006
C: 0.04984615 0.09420760 5.61627735
C: -0.04649805 -0.05787837 7.13191782
H: 0.94604832 -0.07334458 7.58427505
H: -0.60542282 0.77000613 7.57035274
H: -0.55366275 -0.98654445 7.39726741
C: 0.76389939 1.40111272 5.28065247
H: 0.84541894 1.53461185 4.20097059
H: 0.22042700 2.25580115 5.68615385
H: 1.77150393 1.41176313 5.69888547
C: -1.35516567 0.11403225 5.01895782
H: -1.31823408 0.23122219 3.93510886
H: -1.93746520 0.94145581 5.42730374
H: -1.88506873 -0.81375459 5.24028712
C: 0.83774596 -1.07927730 5.03893917
H: 0.34252564 -2.02626804 5.25918232
H: 0.93258913 -0.99209454 3.95580439
H: 1.84246405 -1.11668194 5.46268763
*
$new_job
! DLPNO-CCSD(T) normalPNO RIJK aug-cc-pVTZ aug-cc-pVTZ/JK aug-cc-pVTZ/C tightSCF noautostart miniprint nopop Pmodel
%maxcore 1000
* xyz 0 1
C: 0.79991408 -1.02205164 0.68773696
H: 0.85355588 -1.12205101 -0.39801435
H: 1.49140210 -1.74416936 1.11972040
C: 1.11688700 0.42495279 1.09966205
H: 1.83814230 0.89014504 0.43045256
H: 1.55556959 0.43982464 2.09708356
C: -0.24455916 1.16568959 1.10297714
H: -0.25807760 2.00086313 0.40532333
H: -0.44880450 1.57699582 2.09098447
C: -1.29871418 0.10381191 0.73930899
H: -1.47356078 0.10524338 -0.33800545
H: -2.25673428 0.27804118 1.22715843
C: -0.64687993 -1.22006836 1.13630660
H: -1.12443918 -2.08762702 0.68299327
H: -0.68601864 -1.34528332 2.22022006
C 0.04984615 0.09420760 5.61627735
C -0.04649805 -0.05787837 7.13191782
H 0.94604832 -0.07334458 7.58427505
H -0.60542282 0.77000613 7.57035274
H -0.55366275 -0.98654445 7.39726741
C 0.76389939 1.40111272 5.28065247
H 0.84541894 1.53461185 4.20097059
H 0.22042700 2.25580115 5.68615385
H 1.77150393 1.41176313 5.69888547
C -1.35516567 0.11403225 5.01895782
H -1.31823408 0.23122219 3.93510886
H -1.93746520 0.94145581 5.42730374
H -1.88506873 -0.81375459 5.24028712
C 0.83774596 -1.07927730 5.03893917
H 0.34252564 -2.02626804 5.25918232
H 0.93258913 -0.99209454 3.95580439
H 1.84246405 -1.11668194 5.46268763
*
這個輸入文件包含了DLPNO-CCSD(T)/aug-cc-pVTZ with normalPNO級別下的三個單點任務。第一個子任務是對二聚體做計算,輸出的能量我們叫E_AB;第二個子任務是對第1個片段在二聚體基組下做計算,輸出的能量我們叫E_A(AB);第三個子任務是對第2個片段在二聚體基組下做計算,輸出的能量我們叫E_B(AB)。原子名后面帶冒號的說明這個原子是鬼原子。計算前別忘了把maxcore和nproc都設為恰當情況。其中nproc只要在開頭定義一次就夠了。
在筆者的2*Intel E5-2696v3(36核)機子上,通過36核并行,每個核給6000MB內存,花了41分鐘跑完。在輸出文件中搜索FINAL SINGLE POINT ENERGY,總共會搜索到三個:
FINAL SINGLE POINT ENERGY -393.613585294163
FINAL SINGLE POINT ENERGY -196.197449643018
FINAL SINGLE POINT ENERGY -197.412738820804
依次為E_AB、E_A(AB)、E_B(AB)。然后按照計算E_AB - E_A(AB) - E_B(AB)就得到考慮counterpoise校正的結合能了。如果不理解這點,看我的培訓班(http://www.keinsci.com/workshop/KBQC_content.html)里的一頁ppt

對于當前例子,counterpoise校正的結合能因此為627.51*(-393.613585294163+196.197449643018+197.412738820804)= -2.13 kcal/mol。
本文的這個體系其實是著名的S66弱相互作用測試集里的,用的結構也是其補充材料里的結構。在S66文中通過CCSD(T)/CBS計算出來的結果是-2.40 kcal/mol,可見本文的結果和金標準符合得非常好。
為了讓結合能計算更省事,我還專門寫了個腳本,是本文文件包里的getEbind.sh。此腳本在運行后會處理當前目錄下的所有out文件,自動提取整體和兩個片段的能量,并求差給出不同單位的結合能。例如當前目錄下有三個.out文件,都是上文的方法用Multiwfn產生的任務的輸出文件,getEbind.sh輸出的信息如下,可見提取結合能非常方便。
H2O_ext.out
Etot = -761.79953946 E1 = -685.35948656 E2 = -76.43844639 Hartree
-0.0016065 Hartree
-1.008 kcal/mol
-4.218 kJ/mol
H2O_int.out
Etot = -761.80467188 E1 = -685.35943188 E2 = -76.43928426 Hartree
-0.0059557 Hartree
-3.737 kcal/mol
-15.637 kJ/mol
H2.out
Etot = -686.52275741 E1 = -685.35832899 E2 = -1.16228297 Hartree
-0.0021455 Hartree
-1.346 kcal/mol
-5.633 kJ/mol
2022-Jan-9補充
從2022-Jan-9更新的Multiwfn開始,在產生ORCA輸入文件的界面里選擇任務類型時可以選擇-7,用法同前。此時產生的ORCA輸入文件會計算5個能量,依次為:整體的能量、整體基函數下片段1的能量、整體基函數下片段2的能量、片段1的能量、片段2的能量。執行本文文件包里的ORCA_CP.sh就可以對當前目錄下所有這種任務的輸出文件進行處理,此腳本對每個文件會輸出這些信息:(1)5個能量的具體數值 (2)原始的相互作用能 (3)BSSE校正后的相互作用能 (4)BSSE校正能 (5)BSSE校正后的整體的能量。這種任務所做的計算、腳本給出的信息,和Gaussian程序的counterpoise關鍵詞完全相同。
本文文件包里的waterdimer.out是一個例子文件,ORCA_CP.sh對它進行處理的輸出如下:
File: waterdimer.out
E(AB) = -152.054002577 Hartree
E(A)_AB = -76.022655799 E(A) = -76.022397044 Hartree
E(B)_AB = -76.024059371 E(B) = -76.022540157 Hartree
Raw interaction energy:
-0.0090654 Hartree
-5.689 kcal/mol
-23.801 kJ/mol
BSSE corrected interaction energy:
-0.0072874 Hartree
-4.573 kcal/mol
-19.133 kJ/mol
BSSE correction energy:
0.0017780 Hartree
1.116 kcal/mol
4.668 kJ/mol
BSSE corrected complex energy:
-152.0522246 Hartree
Calculation of metal surface adsorption problems based on cluster models using quantum chemistry programs
文/Sobereva@北京科音
First release: 2020-Mar-8 Last update: 2020-Mar-10
簡介:本文首先對量子化學程序基于簇模型計算表面反應或吸附問題做簡單介紹,然后基于ORCA量子化學程序,簡單演示如何計算苯分子在Ag(111)表面上的吸附能。本文目的是令讀者認識到量子化學程序研究表面問題的可行性,并了解實際計算中需要值得注意的地方。
筆者還另有一篇博文章《18碳環(cyclo[18]carbon)與石墨烯的相互作用:基于簇模型的研究一例》(http://www.shanxitv.org/615),也和本文主題密切相關,建議讀者一看。
1 關于簇模型
一說到基于量子力學方法的固體表面反應/吸附的計算,很多人就首先想到用第一性原理程序諸如Quantum Espresso、CP2K去做,甚至以為以計算孤立體系為主的Gaussian、ORCA等量子化學程序完全不適合,實際上這是不對的。很多固體表面問題的計算都可以通過構建簇模型(cluster model)來使用量子化學程序計算。所謂簇模型就是把反應/吸附位點附近區域的原子挖出來當做孤立體系計算。
用量子化學程序通過簇模型來算和使用第一性程序通過周期性方式來算這類問題各有利弊。用簇模型的優點在于:
? 整體來說,量子化學程序支持的理論方法遠比第一性原理程序豐富得多得多,選擇余地明顯更大,還可以做到更高精度(如雙雜化、DLPNO-CCSD(T)等)。用雜化泛函相對于純泛函的耗時增加程度遠低于基于平面波的第一性原理程序。
? 在計算設置上更方便,也不需要考慮k點、真空層、分子與相鄰鏡像作用、偶極校正之類亂七八糟的事。
? 支持的任務類型更豐富,關鍵詞寫著省事,可視化程序也多。例如Gaussian vs. VASP
? 結合強大的Multiwfn可以做非常豐富的波函數分析,使文章的分析討論部分明顯更充實、更上檔次。參考比如《Multiwfn支持的分析化學鍵的方法一覽》(http://www.shanxitv.org/471)、《Multiwfn支持的弱相互作用的分析方法概覽》(http://www.shanxitv.org/252)、《Multiwfn支持的電子激發分析方法一覽》(http://www.shanxitv.org/437)等大量文章,在《Multiwfn入門tips》(http://www.shanxitv.org/167)里有所有Multiwfn做分析的博文匯總。而使用第一性原理程序的話,能做的分析少得多,不過最新的Multiwfn結合CP2K也已經能對周期性體系做諸多波函數分析,比如《使用Multiwfn做IGMH分析非常清晰直觀地展現化學體系中的相互作用》(http://www.shanxitv.org/621)、《使用Multiwfn結合CP2K通過NCI和IGM方法圖形化考察固體和表面的弱相互作用》(http://www.shanxitv.org/588)、《使用Multiwfn考察周期性體系的芳香性》(http://www.shanxitv.org/722)、《使用Multiwfn對周期性體系做鍵級分析和NAdO分析考察成鍵特征》(http://www.shanxitv.org/719)、《使用Multiwfn結合CP2K做周期性體系的atom-in-molecules (AIM)拓撲分析》(http://www.shanxitv.org/717)、《使用Multiwfn結合CP2K對周期性體系做電荷分解分析(CDA)》(http://www.shanxitv.org/716)、《使用Multiwfn對周期性體系計算Hirshfeld(-I)、CM5和MBIS原子電荷》(http://www.shanxitv.org/712)、《使用Multiwfn結合CP2K計算晶體中原子的氧化態》(http://www.shanxitv.org/711)
用簇模型的弊端在于:
? 需要合理考慮邊界效應。邊界效應此處指的是使用有限的簇模型作為周期性體系的近似導致的對被研究問題的計算精度的影響。邊界效應取決于兩點:(1)簇的尺寸 (2)對邊界的處理。簇的尺寸越大、讓邊界原子所處的環境越像體相,則邊界效應越小。
簇的大小選取有很大任意性。固體部分截的原子太少的話邊界效應太強,結果不準;而截得大的話,則計算耗時太高。除非團簇截得極大,否則都要考慮怎么恰當處理邊界。比如有機體系(如石墨烯),邊界往往用氫飽和,見比如《18碳環(cyclo[18]carbon)與石墨烯的相互作用:基于簇模型的研究一例》(http://www.shanxitv.org/615);如果是無機晶體,有用贗氫、capped ECP、用大量背景電荷表現更長程區域原子的靜電勢等做法;如果是純金屬的話倒是不需要做特別的考慮。
? 對于原子致密排布的塊狀固體材料(不是那種稀疏的、有孔洞的諸如MOF等,或者單層材料),用簇模型的話耗時比起用第一性原理程序當周期性算往往更高。但這點具體看用什么程序、什么基組、數值方面的設定等等,不能一概而論。
? 用簇模型算過渡金屬及其化合物的晶體經常會在SCF收斂上遇到麻煩,有時需要大量折騰。但也不是絕對解決不了,可結合《解決SCF不收斂問題的方法》(http://www.shanxitv.org/61)里的說明恰當嘗試。但即便SCF收斂了,也很有可能收斂到無意義的不穩定的波函數。
總的來說,用第一性原理程序做周期性計算研究表面問題是主流的做法,通過北京科音CP2K第一性原理計算培訓班(http://www.keinsci.com/KFP)學習一遍可以非常容易地上手這類的計算(若事先不具備基本量子化學計算常識的話則應當同時參加北京科音初級量子化學培訓班:http://www.keinsci.com/KEQC)。但簇模型計算這類問題也絕非很稀罕,相關研究文章也有不少,如《18碳環(cyclo[18]carbon)與石墨烯的相互作用:基于簇模型的研究一例》(http://www.shanxitv.org/615)所介紹的研究。還有的研究將兩種方式相結合,取二者長處,比如ACS Catal., 8, 3825 (2018)研究石墨烯上催化二氧化硫與氧氣反應形成硫酸鹽,就先用了VASP做了周期性計算優化了極小點和過渡態,再用Gausisan基于VASP優化的結構用簇模型做了單點計算,將得到的波函數用Multiwfn分析了催化反應機理以及石墨烯表面與被催化物質的弱相互作用。
2 簇模型計算表面問題的實際例子:苯分子在Ag(111)表面物理吸附的結合能的計算
下面就通過一個簡單例子,說明如何基于簇模型計算物理吸附的結合能。
2.1 相關說明
本例涉及的各種文件都可以在這里下載:http://www.shanxitv.org/attach/540/file.rar。
這個體系在DFT-D3原文J. Chem. Phys., 132, 154104 (2010)中被研究過,苯與銀表面間距的剛性掃描圖如下所示
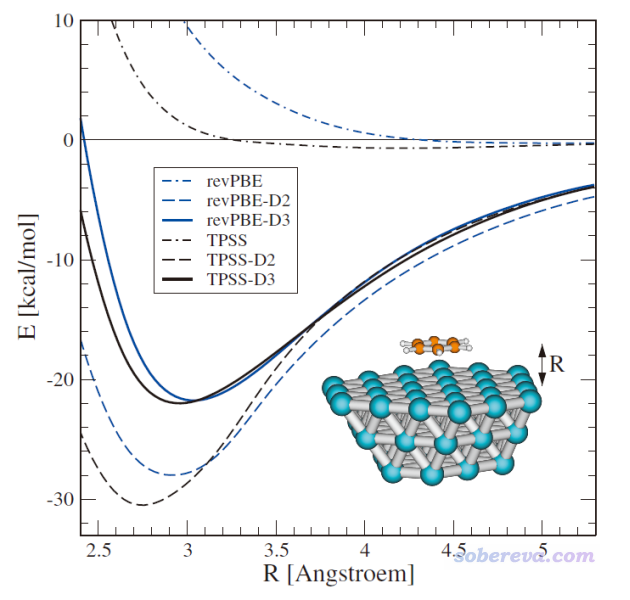
文中也說了,實驗測定的苯吸附到Ag(111)上的焓變是-13 kcal/mol。
下面我們來算苯分子在Ag(111)表面物理吸附對應的結合能。這里說的結合能是習俗上定義的,即只考慮電子能量的變化,而且片段能量用的是復合物中的結構算的。算這個問題分為以下幾步
(1)構建金屬簇
(2)把苯分子放進去得到復合物初猜結構
(3)優化復合物
(4)計算各個片段的單點能
(5)求差得到結合能
之后還可以再用Multiwfn做一些波函數分析。
嚴格來說,就算哪怕不計算熱力學量,優化完復合物也應當做一下振動分析來看看虛頻情況,如果苯分子有沿表面運動的虛頻模式,應當恰當消掉,基本策略參考《Gaussian中幾何優化收斂后Freq時出現NO或虛頻的原因和解決方法》(http://www.shanxitv.org/278)。但由于當前體系不小,做振動分析耗時很高,本文就忽略這點了。
本例使用ORCA程序,一方面是因為免費,另一方面是支持RIJCOSX,可以令這種大團簇體系在單點計算和優化時耗時降低極多,明顯快過Gaussian。不過ORCA做振動分析慢,也是本文沒做振動分析的原因。ORCA的基本常識看《量子化學程序ORCA的安裝方法》(http://www.shanxitv.org/451)和《基于ORCA量子化學程序對分子做優化、振動分析、觀看紅外光譜、觀看軌道的簡單演示》(https://www.bilibili.com/video/av59599938)。本文用的是ORCA 4.2.1。
2.2 建模
本文用的對應Ag(111)表面的團簇結構就按照上一節文獻里的圖來構建就行了,用什么程序隨意。比如可以去ICSD找到Ag的晶體結構,用VESTA、GaussView、Avogadro之類復制延展成足夠大的復晶胞,然后一點點刪原子就可以得到。你也可以用某貴得離譜的有切表面功能的可視化程序先載入自帶的庫里的Ag原胞,用切表面的選項切出(111)表面,然后再復制延展成面積足夠大、至少有三層原子厚度的表面,之后再一點點刪多余的原子。為了不讓邊界效應太顯著,對于簇模型研究表面問題,一般建議至少三層原子,而且固體團簇要比被吸附的分子明顯大一圈。像上面圖片里構造的Ag58團簇對于研究苯的吸附就是很得當的,不大不小。再大一圈的話耗時就忒高了,再小一圈的話邊界效應就較為顯著了。而且為了節約原子來降低耗時,可見上圖中的團簇是上面寬下面窄,比起上下一樣寬時原子數明顯更少。由于苯分子豎著看起來是近乎圓形的,因此團簇的表面最好也接近圓形。顯然,分子如果是長條形的,簇的表面也應當是長條形。
筆者建立好的簇如下所示,為了看得清楚每層用了不同顏色。在gview里可能默認沒有把Ag-Ag之間判斷為成鍵,導致構建簇模型的時候不好判斷原子位置,可以根據《談談原子間是否成鍵的判斷問題》(http://www.shanxitv.org/414)文中所述自定義成鍵判斷閾值,筆者設的是小于3埃就判斷成成鍵。
然后用把苯分子放在金屬簇的上方正中央位置。距離不要太遠,要不然優化到平衡位置需要多花很多步;也不要太近,本來過渡金屬團簇就是SCF難收斂的典型,如果被吸附分子放得太近導致電子結構變得更復雜的話會雪上加霜。本文把苯分子放在團簇表面上方3埃的位置。從DFT-D3原文里那張勢能面掃描圖來看,TPSS-D3的極小點位置也差不多就是3埃。
當前的結構不能直接就拿去優化,因為簇模型里邊界的原子沒有感受到真實體相環境的束縛,如果不把邊界的原子進行固定就優化的話,可能導致邊界的原子位置大幅改變,和真實情況明顯不符。因此我們把最靠邊的Ag都凍結,而中間的,即與苯分子密切接觸的Ag原子不設凍結,這是為了讓它們的位置自發地調整來響應相互作用造成的實際的結構變化。
下圖就是筆者在GaussView里最終構建的復合物初始結構,為了看得清楚給了兩個視角,黃色是被凍結的原子。在GaussView的Tools - Atom Selection界面里可以直接查詢到這些被選成黃色的原子的序號,即1-11,14,17,19-23,26,29,32,34-38,41,44,46-50,52,54-58。
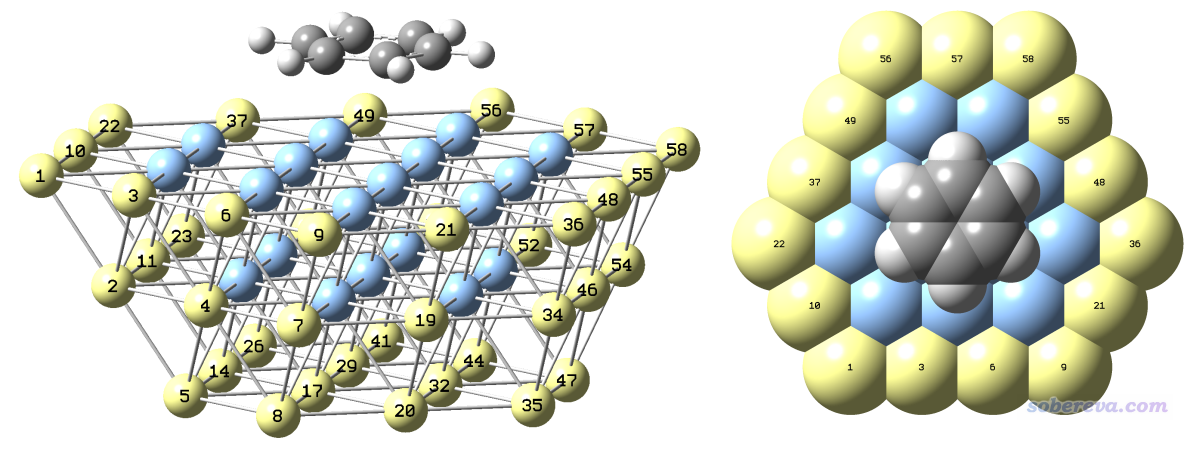
這個初始結構用GaussView保存出來的gjf文件是本文文件包里的Ag58_benzene_freeze.gjf。
值得一提的是,但凡若有可能,都最好讓簇模型處于閉殼層狀態,這樣算起來比開殼層的情況快得多。對于當前的體系,苯分子是閉殼層的,Ag是奇數個電子而且靠s電子成鍵(成鍵情況比較簡單),故只要Ag原子是偶數個,就可以讓整個體系是閉殼層。當前Ag是58個,所以整個體系是閉殼層。如果你用的銀團簇是比如Ag59或者Ag57,那么整體就是開殼層了,算起來就費勁多了。所以在設計金屬團簇的時候應當注意考慮這點。
2.3 幾何優化
下面用Multiwfn產生ORCA程序對上面的體系做限制性優化的輸入文件。相關信息看《詳談Multiwfn產生ORCA量子化學程序的輸入文件的功能》(http://www.shanxitv.org/490)。本文用的是2020-Mar-5更新的Multiwfn,可在官網http://www.shanxitv.org/multiwfn下載。啟動Multiwfn,然后輸入
Ag58_benzene_freeze.gjf //gjf文件的實際路徑,Multiwfn會從中讀取結構
oi //產生ORCA輸入文件
complex_opt.inp //產生的文件的名字
0 //設置任務類型
2 //優化
-3 //設置原子凍結
1-11,14,17,19-23,26,29,32,34-38,41,44,46-50,52,54-58 //被凍結的原子序號
3 //用RI-B3LYP-D3(BJ)/def2-TZVP(-f)
然后我們就在當前目錄下得到了complex_opt.inp文件。把其中的B3LYP改成PBE0,把def2-TZVP(-f)改為def2-SV(P),把CPU核數和內存分配量設為實際情況,其它內容不變。此時這個文件對應于PBE0-D3(BJ)/def2-SV(P)下進行限制性優化,是我們實際要用的。
這里解釋下為什么用這個級別做優化。前面我的博文里提到過,在ORCA里開RI時,純泛函的耗時遠低于雜化泛函,而且純泛函PBE對純金屬的描述也不錯,而之所以這里非要用雜化泛函PBE0,是因為用純泛函時SCF收斂難度比雜化泛函通常明顯更大,尤其是當前這種過渡金屬團簇類型的體系本來就是SCF難收斂的體系,嘗試過PBE就會發現收斂情況就像噩夢,所以用了雜化泛函。之所以把原本輸入文件里的B3LYP關鍵詞給改成了PBE0,這是因為J. Chem. Phys., 127, 024103 (2007)中專門說了B3LYP算金屬很爛,而量子化學領域里另一個很常用的雜化泛函PBE0則在這方面可靠得多,也有不少文章用PBE0算過Ag表面,如J. Phys. Chem. C, 117, 5075 (2013)。對于PBE0這種描述色散作用垃圾的泛函,在計算物理吸附時DFT-D校正顯然是需要的,見《談談“計算時是否需要加DFT-D3色散校正?”》(http://www.shanxitv.org/413)、《亂談DFT-D》(http://www.shanxitv.org/83),要不然結果就搞笑了。雖然目前ORCA也支持DFT-D4了,見《DFT-D4色散校正的簡介與使用》(http://www.shanxitv.org/464),但這里還是用D3,一方面是D3目前已經被極為廣泛地使用,比D4更放心一些,另一方面是D4比D3強的地方關鍵在于能考慮原子帶電狀態對色散校正的影響,而當前體系里金屬基本不帶電荷,因此用D4也不會帶來什么明顯好處(改進較大的是離子、過渡金屬配合物類型的體系)。
至于優化此體系用的基組的選擇,考慮到當前體系不僅原子數多,而且絕大多數都是重原子,本來耗時就肯定比較高,再加上幾何優化對基組不敏感(見《淺談為什么優化和振動分析不需要用大基組》http://www.shanxitv.org/387),所以基組用個便宜的2-zeta檔次的就夠了。lanl2DZ雖然便宜且也能基本滿足幾何優化目的,但沒有標配的輔助基組,如果用autoaux關鍵詞自動產生輔助基組的話不劃算。def2-SVP是ORCA用戶非常常用的中等偏小的基組,有標配的輔助基組,且對Ag是贗勢基組,耗時不會很高,用于優化比較合適。由于此基組對Ag有f極化函數,它會造成耗時增加不少,而對優化來說影響不大,因此我們實際用的是def2-SV(P),它把f極化函數給砍掉了,同時也砍掉了對氫的p極化,這對優化影響也甚微。
復合物優化任務的輸入文件是本文文件包里的complex_opt.inp,在雙路XEON E5-2696v3(共36核)服務器上花了10小時算完。產生的最終結構文件是本文文件包里的complex_opt.xyz。為了觀看優化過程的收斂趨勢,筆者用《OfakeG:使GaussView能夠可視化ORCA輸出文件的工具》(http://www.shanxitv.org/498)里的OfakeG工具將輸出文件轉成了贗Gaussian輸出文件,即本文文件包里的complex_opt_fake.out。用GaussView打開,收斂曲線如下所示,可見收斂還蠻順利的
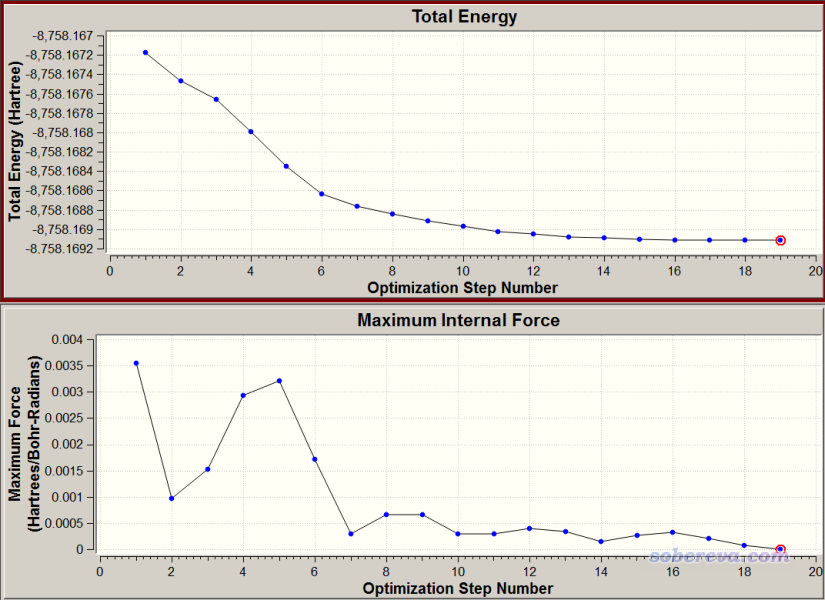
大家通過gview看結構變化也可以發現,由于復合物初始結構本來擺得就比較理想,所以優化過程中只是苯分子位置稍微調節了一下而已,整體沒有明顯變化,和苯接觸的銀原子也沒怎么動。
2.4 單點計算
現在產生單點任務的輸入文件。啟動Multiwfn,載入complex_opt.xyz,然后輸入
oi //產生ORCA輸入文件
complex_SP.inp //產生的文件的名字
4 //選擇計算級別
如果你對結合能計算精度要求不是特別高,或者計算條件不是很好,將產生的complex_SP.inp里的B3LYP改成PBE0就可以開始算了,此時對應于PBE0-D3(BJ)/def2-TZVP級別。
不過,有常識的人都知道,精確計算弱相互作用能需要考慮BSSE問題,見《談談BSSE校正與Gaussian對它的處理》(http://www.shanxitv.org/46)。在def2-TZVP下雖然BSSE問題已經不太大了,但對于弱相互作用能計算的影響還是不可忽視的,有四個解決辦法解決BSSE問題:
(1)做counterpoise校正。但這需要多花兩倍左右時間。若想要用這種做法,參看《在ORCA中做counterpoise校正并計算分子間結合能的例子》(http://www.shanxitv.org/542)。
(2)用gCP方法以免費方式近似解決BSSE問題。gCP校正的簡介見《大體系弱相互作用計算的解決之道》(http://www.shanxitv.org/214)。gCP對def2-TZVP有現成的參數,在ORCA里要用這種校正就寫上gCP(DFT/TZ)即可。而且gCP校正有解析梯度,完全可以用于前面的幾何優化過程中,在其輸入文件里寫上gCP(DFT/SV(P))即可。然而,為什么我們對此例不用gCP?這是因為gCP原本只對前四周期元素擬合了參數,對于之后的元素,會用同族的第四周期元素來湊合,比如Ag的gCP參數會用Cu的參數來湊合。這種湊合的做法,對于第四周期之后元素的原子數較少(比如過渡金屬配合物)的時候是可以接受的,但當前體系有一大堆Ag,其參數是影響結合能計算的重點,顯然這么湊合是不靠譜的近似。
(3)給基組加彌散函數,即改成ma-def2-TZVP,這樣可有效改進弱相互作用計算精度并減小BSSE問題。但這種做法我不推薦用于當前計算,因為此時沒有標配的輔助基組,用autoaux自動產生的不是特別劃算。而且本來當前體系SCF就比較難收斂,而眾所周知加彌散函數會導致SCF收斂難度暴增,對于當前這種原子堆得緊密的情況更是會導致線性依賴問題極度顯著,所以加彌散是餿主意。
(4)用更大的def2-QZVP基組。這個基組對于普通泛函的DFT計算而言已經基本達到了完備基組極限了。此時BSSE問題已可以忽略,而且比def2-TZVP時對電子結構描述又有少量改進,而且DFT-D3參數原本又是在def2-QZVP下搞的,因此在這個層面上顯得很完美。def2-QZVP的確相當貴,但在ORCA中利用RIJCOSX技術,用這么大的基組對當前體系進行單點計算,耗時依然是完全可接受的。因此,本例使用PBE0-D3(BJ)/def2-QZVP來做單點計算。
還有需要注意的是,通常基組越大SCF越不容易收斂。筆者嘗試過,在優化后的復合物結構下用def2-TZVP可以直接收斂(雖然SCF花了多達46圈),而用def2-QZVP則迭代幾十次之后依然沒有收斂的跡象,即便讀def2-TZVP收斂的波函數當初猜也不行。經嘗試,我發現把incremental Fock加速技術關閉后可以直接解決,即在輸入文件里加上%scf DirectResetFreq=1 end。
(補充:如果你是用ORCA 5.0及以后版本,請忽略下面grid4 gridx4部分的說明,輸入文件里也不要加上grid4 gridx4,否則會報錯)
另外,建議在關鍵詞里加上grid4 gridx4關鍵詞,使用比默認更好的交換相關泛函積分格點和COSX格點。之所以這樣,是因為當前我們希望算的結果比較準,故提升積分格點精度有好處。而且,當前體系大量原子致密堆積,默認積分格點下的積分精度差點意思。比如在def2-TZVP下用默認格點,在SCF收斂后自動做Final grid步驟(提升積分格點精度算最終能量)的時候提示如下
Change in XC energy ... -0.003667837
Integrated number of electrons ... 1144.003054396
Previous integrated no of electrons ... 1143.933959260
可見迭代過程用的積分格點對電子數的積分值1143.9339和整數相差不小。即便在final grid時,積分值1144.0030偏離整數仍不可忽略。反之,如果用grid4 gridx4,則輸出變為
Change in XC energy ... 0.001404285
Integrated number of electrons ... 1144.000217645
Previous integrated no of electrons ... 1144.003114364
可見積分精度改進不小,明顯更接近于整數。提升積分格點精度還有一個好處是對于某些體系可能令SCF收斂更容易。當前這種特征的體系結合大基組本來SCF就很難收斂,顯然多花點耗時來提升SCF收斂的可能性是劃算的。總之,對本文這種類型的體系在算單點的時候,都建議用grid4 gridx4,一舉多得,而對耗時的增加完全可以接受,而且還可能因為達到收斂限所需步數變少而反倒令耗時更低(在def2-TZVP下確實如此,默認格點下用了46輪收斂,grid4 gridx4下用了37輪,導致后者反倒耗時比前者還低了幾分鐘)。
考慮到上面提到的問題,最終我們對復合物用的單點任務的輸入文件的關鍵詞部分如下
! PBE0 D3 grid4 gridx4 def2-QZVP def2/J RIJCOSX noautostart miniprint nopop
%scf DirectResetFreq=1 end
計算用了40輪收斂,在2*E5-2696 v3 36核機子上花了5個小時。注意這類體系本身就是比較難收斂的,所以切勿看到跑了比如30輪左右還沒收斂,而且收斂情況還有些波動就把任務給斷了,一定要沉得住氣。
復合物算完后,我們把complex_SP.inp復制為benene_SP.inp并把其中銀原子都去掉,就成了苯分子的單點任務文件,計算之。再把complex_SP.inp復制為Ag58_SP.inp并把其中苯分子部分去掉,就成了銀團簇的單點任務文件,計算之。算完的同名out文件在本文文件包里都提供了。
2.5 計算結合能
從單點任務輸出文件中提取單點能,按照E_bind = E(Ag58_Benzene) - E(benzene) - E(Ag58)即可計算出結合能,即627.51*(-8760.730415916696+232.068063781533+8528.63
2371853557)=-18.8 kcal/mol。
前面提到,苯吸附過程的實驗焓變是-13 kcal/mol,我們算的和實驗定性吻合(比DFT-D3原文里勢能面掃描圖體現的結果還更好)。由于這個結合能已經算是很大了(苯二聚體結合能才-2.8 kcal/mol),而且這又不是容易算的體系,能算到這種精度就已經不容易了。我們的計算結果和實驗值的差異來自于幾個方面:
(1)邊界效應仍不可忽略,用更大的簇可能能改進結果
(2)PBE0-D3(BJ)理論方法的誤差。可嘗試其它泛函或結合其它的色散校正方式
(3)實驗數據對應焓變,本身和我們算的結合能就缺乏可比性。尤其是,形成復合物后,被吸附物被基底束縛,從而會誕生新的振動模式,這會對復合物的焓產生明顯貢獻(使之變得更正),忽略了這點會導致算出來的結合能偏大(即overbinding、結合能過負)
(4)忽略了從無限遠離到被吸附狀態過程中的分子和金屬表面結構變化對能量的影響。但由于當前的體系比較剛,這基本可以忽略
(5)實驗數據本身就有誤差
有一點非常值得一提,也是絕大多數人都忽略的,也就是DFT-D3的三體校正項,這在《DFT-D色散校正的使用》(http://www.shanxitv.org/210)中專門說過。平時我們用Gaussian、ORCA做計算的時候加D3校正都沒考慮三體校正項,因為這項對結合能的影響通常很小。然而,對于原子數很多,尤其是當前體系這樣原子還是緊密堆積的情況,三體校正項對結合能的影響可能是不容忽視的。
如上面的博文所述,ORCA里寫上ABC關鍵詞就可以在計算D3校正時也考慮三體校正項,但我們前面計算的時候沒寫這個。為了快速得到考慮三體校正項之后的結果,我們用Grimme獨立的dftd3程序來算。把分子坐標弄成xyz格式,運行dftd3 mol.xyz -func pbe0 -bj -abc,輸出的就是mol.xyz里的體系在PBE0泛函下的帶三體校正項的DFT-D3(BJ)色散修正能了。下面的表格里把ORCA輸出文件里的PBE0能量、dftd3給出的帶和不帶三體校正項(ABC)的DFT-D3(BJ)色散校正能都匯總到了一起
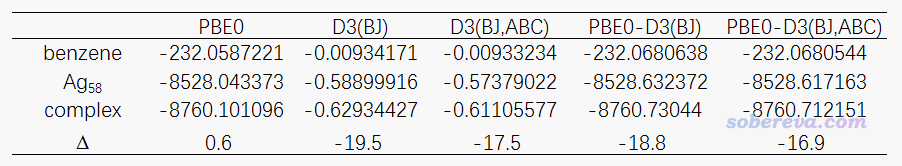
由上表可見,如果不考慮色散校正,結合能是正值,這是因為PBE0幾乎完全不能描述色散作用(這是苯與Ag表面吸引作用的最主要來源),考慮DFT-D3(BJ)校正后才定性正確。如果再把三體校正項考慮進去,算出來的結合能-16.9 kcal/mol比不考慮三體校正項時明顯更接近實驗的吸附過程的焓變,改進了約2 kcal/mol。如果再考慮到把焓的熱校正考慮進去還能進一步改進結果,且實驗數據本來就有誤差,可以認為當前計算模型的精度已經相當不錯了。
我建議大家用ORCA基于簇模型計算表面問題時都帶著ABC關鍵詞。幾何優化、振動分析帶不帶無所謂(PS:三體校正項沒有解析Hessian),關鍵是單點任務時應當帶上,反正對耗時沒有增加,還有可能對結果有不可忽視的改進。
2.6 弱相互作用分析
順帶一提,用簇模型算完之后可以結合Multiwfn做各種各樣的波函數分析。比如《使用Multiwfn圖形化研究弱相互作用》(http://www.shanxitv.org/68)和《使用Multiwfn做NCI分析展現分子內和分子間弱相互作用》(https://www.bilibili.com/video/av71561024)中介紹的NCI分析是非常流行的圖形化考察弱相互作用的方法,這里我們也將之用于苯與銀表面的物理吸附問題上。
是使用單點計算產生的波函數還是幾何優化產生的波函數來做NCI分析?我建議用后者,因為后者是在def2-SV(P)下產生的,前者是在def2-QZVP下產生的,用后者耗時顯然低得多得多,而且NCI分析對于波函數質量并不敏感,基組基本像樣即可。故基于優化任務產生的gbw文件,我們就可以做分析了。如《詳談Multiwfn支持的輸入文件類型、產生方法以及相互轉換》(http://www.shanxitv.org/379)所述,要么用orca_2mkl將gbw轉成molden輸入文件再載入Multiwfn,要么通過設置Multiwfn的settings.ini里的orca_2mklpath使得multiwfn能直接載入gbw文件了。對當前體系做NCI分析值得注意的一點是,為了節約時間,在設置格點時,應當讓空間范圍只涵蓋苯與金屬作用的區域,而不要把整個體系都納入。具體來說,在NCI分析功能設置格點的步驟中應選擇選項10 Set box of grid data visually using a GUI window,然后把盒子范圍調整成下面這樣
雖然當前體系很大,但由于盒子小,在基本能保證圖像精細的0.25 Bohr格點間距下,哪怕用4核機子也能很快就算出來結果。如果想要讓圖像更光滑,可在上圖的界面的右下角把格點間距再改小點,比如0.15 Bohr。下圖是0.19 Bohr時用VMD繪制出來的結果,色彩刻度用的-0.04~0.03 a.u.
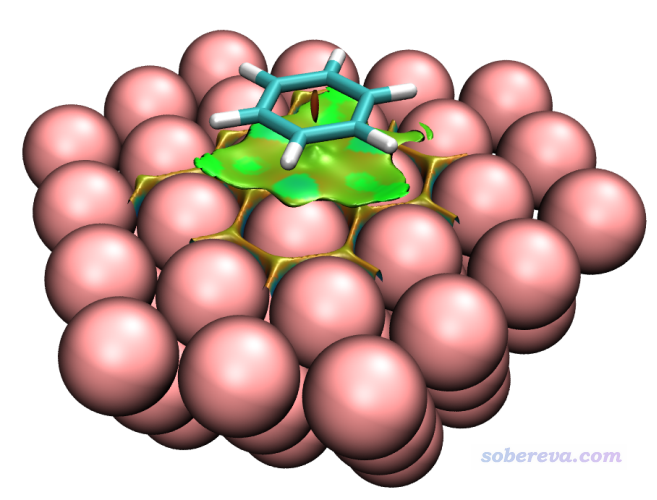
可見苯和銀表面之間有大面積的綠色區域,直觀展現出了苯的pi電子與銀表面電子海洋的大范圍色散相互作用,這有點類似于pi-pi堆積的景象(當然,銀沒有pi電子)。另外,在銀與銀之間也有等值面出現,我們不用管它,這體現的是Ag-Ag之間的作用,如果想屏蔽掉也可以,做法參看《用Multiwfn+VMD做RDG分析時的一些要點和常見問題》(http://www.shanxitv.org/291)。
3 總結&其它
本文通過苯在Ag(111)表面吸附的一個簡單例子,示例了怎么用簇模型研究表面問題,并且針對相關知識、涉及到的一些細節和要點做了討論,希望讀者舉一反三。本文的情況其實還算比較簡單的,不牽扯對邊界進行特殊處理的問題。
像當前這種不易SCF收斂的體系,可能光是在優化階段一開始就已經出現SCF不收斂了(這和復合物初始結構也有關。比如把苯放到距離銀表面2.7埃的距離,SCF就比3.0埃的時候難收斂,因為其更偏離平衡距離)。這種情況可以適度降低SCF收斂限。ORCA對優化默認用tightSCF收斂限比較嚴,如果發現SCF差一點就能收斂,但死活就是達不到收斂限,不妨用scfconv7關鍵詞,即把SCF收斂閾值設成單點任務默認閾值與tightSCF之間,這樣優化出來的結果一般也足夠精確。如果scfconv7都收斂不了,但是用scfconv6能收斂,在優化完了之后建議再用默認的tightSCF進一步優化確保結構準確。如果是初始結構太離譜導致SCF難收斂,自己又不知道大概怎么擺合適,用xtb程序做個預優化也可以,相關信息見《將Gaussian與Grimme的xtb程序聯用搜索過渡態、產生IRC、做振動分析》(http://www.shanxitv.org/421)。
對于單元素團簇類型體系,SCF收斂難度通常是:稀土>d族>ds族>主族。本文的Ag團簇的情況雖然已經不是很容易SCF收斂了,但還不是真正難搞的。諸如Fe團簇等,收斂難度遠高于Ag團簇,而且還有較高可能性出現波函數不穩定等情況,如果是初學者沒經驗的話不建議輕易去用簇模型做這些金屬的表面問題的計算。
值得一提的是,當前體系是C3v對稱性的。如果你用Gaussian來算的話,且在計算之前用gview對稱化為C3v,計算會快非常多,收斂也會比較容易。
筆者講授的北京科音中級量子化學培訓班(http://www.keinsci.com/workshop/KBQC_content.html)里的過渡態搜索部分有個實例也是基于簇模型的,如下,大家作為練習可以自行做一下。除了要凍結邊緣原子外,搜索過渡態和走IRC的過程和普通分子體系無異。
簇模型不僅限于研究表面問題。比如計算酶催化反應,整個蛋白酶動輒幾千原子,直接用量子化學算算不動,此時可以把反應位點區域挖出來,即保留底物以及周圍一圈跟它發生反應或者有顯著弱相互作用的殘基的側鏈和alpha碳,并且把alpha碳凍結并加氫飽和。這種研究方法可以參看這篇綜述:J. Am. Chem. Soc., 139, 6780 (2017)。筆者在北京科音高級量子化學培訓班里會講這種計算的具體細節。雖然也有很多人拿ONIOM(QM:MM)算這種問題,但補力場參數相當麻煩,好多任務和分析都做不了、容易出現亂七八糟問題,非常折騰,從結果上看一般也不比用簇模型有多大好處,所以沒有特殊情況的話我很不建議用ONIOM(QM:MM)。筆者專門有一篇文章說這事:《要善用簇模型,不要盲目用ONIOM算蛋白質-小分子相互作用問題》(http://www.shanxitv.org/597)。
]]>The method of using the converged wavefunction of Gaussian or other program as the initial guess wavefunction of ORCA
文/Sobereva@北京科音
First release: 2019-Oct-11 Last update: 2021-Oct-1
1 說明
ORCA是非常強大的量子化學程序,筆者之前也寫過不少相關文章,開發了不少輔助工具,見www.shanxitv.org右側的ORCA分類。ORCA相對于最常用的量子化學程序Gaussian來說,有一個缺點是SCF收斂做得不夠好,很多Gaussian能收斂的情況ORCA難以收斂,而且Gaussian的SCF不收斂的解決方案比較成熟,見《解決SCF不收斂問題的方法》(http://www.shanxitv.org/61)。另外,ORCA在產生初猜波函數方面也沒Gaussian靈活,比如Gaussian用戶可以用GaussView構建片段組合波函數作為初猜,而且利用stable=opt關鍵詞還可以自動優化出穩定的波函數,這對于雙自由基、反鐵磁性耦合體系的研究十分重要,參看《談談片段組合波函數與自旋極化單重態》(http://www.shanxitv.org/82)。雖說ORCA也不是沒法算這類自旋極化單重態體系,利用%SCF FlipSpin也可以實現,但往往明顯不如Gaussian方便。
顯然,如果能讓其它量子化學程序,特別是波函數初猜以及SCF迭代方面做得很好的Gaussian產生的波函數作為ORCA的初猜波函數,就能令ORCA取長補短,從很多惱人的問題中解脫。
Multiwfn(主頁&下載地址:http://www.shanxitv.org/multiwfn)程序支持載入.fch、.molden、GAMESS-US/Firefly輸出文件這些含有基組定義以及軌道信息的格式(在Multiwfn里稱為“基函數信息”),并且可以導出各種含有波函數信息的格式,支持的文件格式詳見《詳談Multiwfn支持的輸入文件類型、產生方法以及相互轉換》(http://www.shanxitv.org/379)。Multiwfn支持絕大多數主流量化程序產生的波函數,并可以作為波函數文件格式的轉換器來用。從2019-Oct-10更新的Multiwfn開始,Multiwfn的導出文件功能更是新增了產生.mkl文件的功能。.mkl文件是老版本Molekel的輸入文件,可以被ORCA自帶的orca_2mkl工具轉換成.gbw文件,ORCA計算時可以從.gbw文件中讀取軌道作為初猜波函數。顯然,利用Multiwfn可以直接實現將其它量化程序產生的波函數作為ORCA的初猜波函數的目的。甚至可以說,只要存在一種量化程序有辦法得到當前體系當前級別下收斂的波函數,用ORCA計算也一定能收斂。
有一點要提及的是ORCA是基于球諧型高斯函數做的計算,因此用Multiwfn實現這個目的,其它程序計算時也應當用球諧型高斯函數。不了解這方面的話看《談談5d、6d型d殼層基函數與它們在Gaussian中的標識》(http://www.shanxitv.org/51)。
如果本文介紹的Multiwfn的功能給你的研究帶來了幫助,請在論文中引用Multiwfn剛啟動時提示的Multiwfn程序的原文,這是對Multiwfn開發和維護最好的支持!
2 例子:丁烷雙自由基
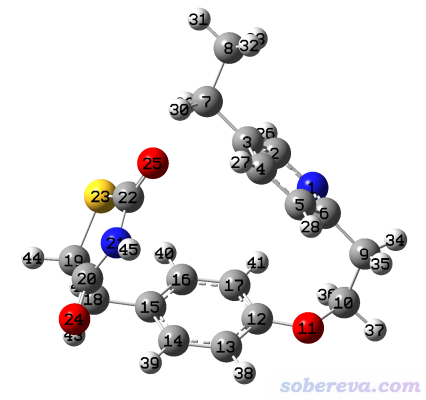
這里舉個例子,計算丁烷雙自由基C4H8,用以說明如何將Gaussian收斂的波函數作為ORCA的初猜波函數,請大家舉一反三。本例涉及的文件都在http://www.shanxitv.org/attach/517/file.rar里。
本例使用2020-Jul-1更新的Multiwfn,Gaussian使用G16 A.03版,ORCA使用4.2版。
首先用Gaussian運行C4H8.gjf,內容如下。任務是對C4H8在B3LYP/def2-SVP級別下產生最穩定波函數,對此體系也是對稱破缺波函數。使用def2-SVP的時候Gaussian默認用的是球諧型基函數。
%chk=C:\C4H8.chk
# B3LYP/def2SVP stable=opt
[空行]
ub3lyp/6-31g(d) opted
[空行]
0 1
C -0.74400100 1.78566400 0.00000000
H -0.60282700 2.33865300 0.92499500
H -0.60282700 2.33865300 -0.92499500
C -0.74400100 0.30988100 0.00000000
H -1.25452600 -0.08746700 0.88463900
H -1.25452600 -0.08746700 -0.88463900
C 0.74400100 -0.30988100 0.00000000
H 1.25452600 0.08746700 -0.88463900
H 1.25452600 0.08746700 0.88463900
C 0.74400100 -1.78566400 0.00000000
H 0.60282700 -2.33865300 -0.92499500
H 0.60282700 -2.33865300 0.92499500
然后用formchk將chk轉換為fch,載入Multiwfn后依次輸入
100 //其它功能 Part 1
2 //導出文件
9 //導出mkl文件
C4H8.mkl
y //表明產生的mkl文件是給ORCA當初猜用,程序會做特殊處理
現在當前目錄下就有了C4H8.mkl。
在當前目錄下運行orca_2mkl C4H8 -gbw,就把C4H8.mkl轉換為了C4H8.gbw。下面,我們將C4H8.gbw里的軌道作為初猜波函數,用ORCA也在B3LYP/def2-SVP下跑一下這個雙自由基的單點。輸入文件如下,將之命名為C4H8.inp,把它和C4H8.gbw都放在當前目錄下,然后進行計算。運行時ORCA會自動從C4H8.gbw中讀取軌道作為初猜波函數。由于C4H8.gbw里的波函數是非限制性波函數,因此寫了UKS關鍵詞。由于ORCA的B3LYP和Gaussian的定義不同,所以加了/G來和Gaussian一致。此文件里的結構和上面Gaussian輸入文件里的結構精確一致。
! UKS B3LYP/G def2-SVP miniprint nopop
* xyz 0 1
C -0.74400100 1.78566400 0.00000000
H -0.60282700 2.33865300 0.92499500
H -0.60282700 2.33865300 -0.92499500
C -0.74400100 0.30988100 0.00000000
H -1.25452600 -0.08746700 0.88463900
H -1.25452600 -0.08746700 -0.88463900
C 0.74400100 -0.30988100 0.00000000
H 1.25452600 0.08746700 -0.88463900
H 1.25452600 0.08746700 0.88463900
C 0.74400100 -1.78566400 0.00000000
H 0.60282700 -2.33865300 -0.92499500
H 0.60282700 -2.33865300 0.92499500
*
迭代過程信息如下
ITER Energy Delta-E Max-DP RMS-DP [F,P] Damp
*** Starting incremental Fock matrix formation ***
0 -157.0020361024 0.000000000000 0.00099750 0.00002892 0.0004319 0.7000
1 -157.0020389471 -0.000002844760 0.00101224 0.00002810 0.0003439 0.7000
***Turning on DIIS***
2 -157.0020411727 -0.000002225540 0.00299633 0.00007975 0.0002632 0.0000
3 -157.0020467355 -0.000005562801 0.00029108 0.00000721 0.0000572 0.0000
4 -157.0020467759 -0.000000040442 0.00006147 0.00000180 0.0000542 0.0000
**** Energy Check signals convergence ****
*****************************************************
* SUCCESS *
* SCF CONVERGED AFTER 5 CYCLES *
*****************************************************
可見由于用了Gaussian收斂的波函數當做初猜,收斂非常快和順利,第一輪迭代時的能量和密度矩陣變化就已經極小了,之后很快就收斂了。之所以不是一輪就收斂,是因為Gaussian和ORCA用的DFT積分格點有異。如果二者都用的是HF方法的話,ORCA里僅僅一輪就能收斂。
如果gbw和輸入文件不同名,為了能從gbw中讀取初猜波函數,應當寫上moread關鍵詞,然后加上一行諸如%moinp "/sob/Azusa_Nakano.gbw"來指明讀取的gbw文件位置。
為了讓Gaussian收斂的波函數放到ORCA里能夠收斂的概率盡可能高,有以下一些注意事項和建議
(1)確保Gaussian里用的基組和ORCA里精確一致。比如Gaussian里用6-31G系列基組時,默認是用笛卡爾型d基函數,而ORCA總是用球諧型基函數,因此Gaussian計算時要寫5d關鍵詞(不過ORCA用戶極少會用Pople系列基組,所以這無所謂)
(2)讓Gaussian計算時實際坐標與ORCA一致。Gaussian在計算時會自動將輸入朝向擺到標準朝向,因此chk文件最后轉出來的gbw里的笛卡爾坐標(標準朝向的)可能和ORCA計算時的不一樣,因此要么ORCA輸入文件里的坐標就用標準朝向的,要么Gaussian計算時候加上nosymm關鍵詞避免擺到標準朝向下,詳見《談談Gaussian中的對稱性與nosymm關鍵詞的使用》(http://www.shanxitv.org/297)。
(3)Gaussian為了節約電子積分計算的耗時,廣義收縮基組如ANO,以及部分廣義收縮基組,如Dunning相關一致性基組、Cl等一部分元素的6-311G系列基組,在計算的時候會自動把某些指數重復的primitive GTF給去掉。程序還會把收縮系數很小的primitive GTF給去掉。由于ORCA不自動做這種處理,可能導致個別情況下Gaussian里收斂的波函數拿到ORCA里還是難收斂或沒法收斂。為避免這個問題,可以在Gaussian計算時加上int=NoBasisTransform關鍵詞避免去掉任何primitive GTF,即基組原始怎么定義的就怎么用。如果你不清楚什么是(部分)廣義收縮基組的話,強烈建議總是帶上這個關鍵詞!
(4)建議帶上IOp(3/32=2)關鍵詞(尤其是帶彌散函數時)避免Gaussian自動去除線性依賴的基函數,以確保和ORCA計算使用的基函數的一致性。
(5)對于DFT計算,如果上面問題都已經考慮了,但放到ORCA里還是不能收斂,可以讓Gaussian和ORCA在計算時都用更好的DFT積分格點精度,并且在ORCA里不開RI。
順帶一提,利用Multiwfn還能將其它量化程序產生的波函數作為GAMESS-US的初猜波函數,因為如果載入的波函數文件含有基函數信息,Multiwfn產生的GAMESS-US輸入文件里可以直接帶初猜信息。Multiwfn也可以將其它量化程序產生的波函數當Gaussian的初猜,因為Multiwfn可以導出fch格式,用Gaussian的unfchk轉成chk后,就可以用guess=read來從中讀取波函數當初猜了。若有不清楚的,參看前述的《詳談Multiwfn支持的輸入文件類型、產生方法以及相互轉換》。
3 chk到gbw的批量轉換腳本
為了讓大家可以更方便地把chk轉換成gbw,筆者寫了一個批量自動轉換的bash小腳本chk2gbw.sh,可以在這里下載:http://www.shanxitv.org/attach/517/chk2gbw.sh。運行前需要先把Gaussian、Multiwfn、ORCA都安裝好,運行過程中會自動調用formchk、Multiwfn和orca_2mkl工具。
運行此腳本后,腳本會對當前目錄下每個chk文件進行轉換。比如有個文件叫popipa.chk,運行此腳本后會得到popipa_Gau.gbw,還會得到popipa.inp,這是ORCA輸入文件,打開一看就會看到里面已經寫了moread關鍵詞和%moinp "popipa_Gau.gbw",坐標和chk里的直接對應,因此用戶現在只需要把定義計算級別的關鍵詞改成實際情況即可開始ORCA計算。可見此腳本方便至極!
運行時輸出信息示例:
Converting B2H6.chk to B2H6.gbw ... (1 of 3)
Converting C2H5F.chk to C2H5F.gbw ... (2 of 3)
Converting CCl4.chk to CCl4.gbw ... (3 of 3)
4 gbw到chk的批量轉換腳本
從2020年8月21日更新的Multiwfn開始,在其examples\scripts目錄下提供了gbw2chk.sh文件,只要運行,就會把當前目錄下的所有gbw文件轉化為同名的chk文件,以便通過Gaussian使用guess=read關鍵詞讀取其中的波函數當初猜。運行前需要先把Gaussian、Multiwfn、ORCA都安裝好。注:如果波函數是UHF/UKS計算得到的,必須用2021年10月1日及以后更新的Multiwfn。
]]>OfakeG: A tool that enables GaussView to visualize ORCA output files
文/Sobereva@北京科音
First release: 2019-Jul-17 Last update: 2024-Oct-30
1 前言
量子化學程序ORCA用的人越來越多,功能很強大而且免費,用戶數在所有量化程序中已經是第二高(雖然跟Gaussian比還遙不可及)。但至少在筆者撰寫此文時,相對于用戶數占絕對主導地位的Gaussian程序而言,仍有一個不足之處是沒有像GaussView那樣的很理想的圖形界面。雖然也有Avogadro、Chemcraft、Gabedit等程序能支持ORCA,但都沒GaussView用著舒服。
在產生輸入文件方面,Multiwfn已經提供了產生ORCA常見任務的輸入文件的功能,見《詳談Multiwfn產生ORCA量子化學程序的輸入文件的功能》(http://www.shanxitv.org/490),用戶只需要用GaussView畫好結構,保存為gjf/mol/mol2/pdb格式,就可以用Multiwfn很方便地得到ORCA輸入文件,所以在建模、產生輸入文件方面,對ORCA用戶沒什么困難的。在觀看ORCA產生的軌道、做波函數分析方面,Multiwfn也都提供了極其豐富的功能,相關信息見《使用Multiwfn觀看分子軌道》(http://www.shanxitv.org/269)、《Multiwfn FAQ》(http://www.shanxitv.org/452)等文章,因此ORCA用戶在后處理分析方面也沒任何壓力。另外,Multiwfn可以基于ORCA的輸出文件繪制各類光譜圖,所以ORCA用戶在光譜研究方面也已經很方便了,相關信息見《Simulating UV-Vis and ECD spectra using ORCA and Multiwfn》(http://www.shanxitv.org/485)、《Simulating UV-Vis and ECD spectra using ORCA and Multiwfn》(http://www.shanxitv.org/485)。筆者在北京科音高級量子化學培訓班(http://www.keinsci.com/workshop/KAQC_content.html)里還對ORCA的各方面使用做了相當全面系統的講解,可以很快上手并用得游刃有余。
雖然Multiwfn已經解決了ORCA用戶在使用方面的大量障礙,但幾何優化軌跡/收斂情況的考察以及振動模式的觀看不屬于Multiwfn的范疇,而目前卻沒有理想的解決辦法。雖然Chemcraft也能觀看ORCA的優化軌跡,但終究沒有常用的GaussView用著舒服,而且還是收費程序;Avogadro雖然也觀看ORCA的振動分析對應的振動動畫,但顯示效果不理想,在Windows下容易崩潰、出現異常(至少在筆者的Win7-64bit機子上是如此)。盡管也可以讓Gaussian與ORCA掛接,這樣可以使ORCA做計算但是輸出的是Gaussian格式的信息,從而等效實現讓GaussView觀看ORCA計算的結果的目的,見《將Gaussian與ORCA聯用搜索過渡態、產生IRC、做振動分析》(http://www.shanxitv.org/422),但這樣做稍顯麻煩、在Windows下也沒法用。
顯然,如果能開發個程序把ORCA的優化、振動分析、優化+振動分析的輸出文件“偽造”成Gaussian的,這樣就可以令GaussView直接支持讀取ORCA的輸出文件了,使得ORCA對于常見問題的研究用起來方便得多,也明顯便于Gaussian用戶同時掌握ORCA程序。筆者開發的OfakeG程序就是實現這個目的,下面介紹一下。如果你想一睹為快這個程序的實際效果,可以看這個視頻:《基于ORCA量子化學程序對分子做優化、振動分析、觀看紅外光譜、觀看軌道的簡單演示》(https://www.bilibili.com/video/av59599938),其中用到了此程序。
OfakeG的學術合理性聲明:本程序的開發靈感來自于Grimme的xtb程序。xtb程序做振動分析的時候會自動輸出一個偽造Gaussian的振動分析輸出文件,目的是為了讓用戶看振動模式方便;xtb程序為了兼容GSM也官方支持偽造ORCA的輸出文件。大牛Grimme直接用的就是fake這個詞。顯然令A程序輸出B程序的格式在學術界是非常正常的事情。Gaussian的輸出格式是公開的而非加密的,GaussView能讀入的格式也相當于是公開的,本文的OfakeG程序亦沒有對GaussView本身做任何篡改,明顯從各個角度上本程序的開發是完全學術正當的。本程序所做的事僅僅是將ORCA的輸出信息轉化成Gaussian的格式而已,文中所謂“偽造”只不過是常規的文件格式轉換而已,和數據層面的“造假”有天壤之別,轉換出的文件里也根本沒有任何文字體現這文件是靠Gaussian程序算出來的。此程序的開發目的是給廣大科研工作者提供個便利,開發/使用此程序不涉及任何侵權和學術不端(除非你利用OfakeG之后,把ORCA算的結果說成是Gaussian算的)。此程序愿意用就用,不愛用的、缺乏對計算化學領域程序狀況基本認識的、懷有惡意的、有特殊利益驅動的人以及杠精,不要強詞奪理在學術合理性上亂噴此程序。
2 OfakeG程序
2.1 介紹+使用方法
OfakeG程序可以在官方頁面下載:http://www.shanxitv.org/soft/OfakeG
其中帶.exe后綴的是Windows版,不帶后綴的是Linux版。
此程序目前支持處理ORCA的opt、freq和opt freq任務的輸出文件(不支持單點任務文件,因為根本沒有任何轉換的意義!)。此程序對ORCA 4.x、5.x、6.x版經測試可用,對于其它版本可能兼容也可能不兼容,請讀者自行嘗試。等ORCA以后出新版本,并且筆者發現和OfakeG不兼容時,預計筆者會更新此程序并更新本文。
OfakeG使用非常簡單。啟動此程序后,把上述任務的ORCA輸出文件路徑輸入進去(對于Windows也可以直接把文件拖進去,路徑會直接顯示出來),一按回車,就會在當前目錄下產生偽造的Gaussian輸出文件。如果輸入文件名字是yuri.out,則輸出文件將是yuri_fake.out。這個輸出文件可以直接載入GaussView,對opt或opt freq任務可以播放優化過程的動畫、用results - Optimization觀看優化過程的收斂情況,對freq或opt freq任務可以用results - Vibrations觀看振動模式。
在Windows下還有更省事的運行方式,即可以直接將ORCA輸出文件拖到OfakeG.exe圖標上,此時在ORCA輸出文件的目錄下會出現文件名帶_fake的偽造的Gaussian輸出文件。
OfakeG也可以通過命令行方式使用,比如在Linux下可以在OfakeG所在目錄下運行./OfakeG Aika.out,將在當前目錄下得到Aika_fake.out。顯然,你也可以自寫shell腳本用這個程序大批量轉換ORCA輸出文件。
OfakeG文件包里的.out文件是一些ORCA的示例輸出文件。如果你的輸出文件轉換不成功,請嘗試通過對照這些示例文件搞清楚是怎么回事。目前OfakeG名義上只支持HF/DFT的輸出文件,其它理論方法不一定能支持。對于加了亂七八糟復雜關鍵詞的情況,OfakeG也不一定能處理。
如果OfakeG處理你的文件時崩潰,且得到的_fake后綴的文件里只有幾行信息,很有可能是因為你的ORCA輸出文件的編碼是UTF16造成的,OfakeG是處理不了前者的情況的,是什么編碼和你用的終端有關系。比如Windows的cmd終端重定向輸出的文件是ASCII編碼的,而PowerShell是UTF16編碼的。對于UTF16編碼的輸出文件,你可以用比如Ultraedit打開,選另存為,把編碼改成Unicode或UTF8,之后再用OFakeG處理。如果你平時習慣用PowerShell且希望重定向出的文件直接就ASCII編碼,可以用諸如這樣的命令運行test.inp得到test.out:D:\study\orca\orca test.inp | out-file test.out -encoding ascii
如果是Win10,還可以直接指定默認的重定向的編碼,詳見https://stackoverflow.com/questions/40098771/changing-powershells-default-output-encoding-to-utf-8。
如果你怎么也搞不清楚為什么你的ORCA輸出文件無法轉化成功,或者可判定OfakeG程序有bug,請在http://bbs.keinsci.com/thread-13952-1-1.html貼子里發回帖,把文件壓縮后上傳。
2.2 OfakeG的幾個細節
以下內容建議留意一下,以更好地理解OfakeG的細節,但初學者不看也可以。
OfakeG給出的是簡化到不能再簡化的能令GaussView正常讀取的偽造的Gaussian輸出文件,因此如果你寫類似工具把其它程序的輸出文件也偽造成類似格式,就也可以令GaussView讀取。
GaussView要求輸出文件里必須有basis functions、alpha electrons、beta electrons信息,但ORCA輸出文件里不直接體現,而且這仨對于觀看優化和振動分析沒有意義,因此在偽造的輸出文件開頭有這仨信息,但數值都為0。
OfakeG從ORCA輸出文件里讀能量的時候讀的是FINAL SINGLE POINT ENERGY,即當前計算級別下的最終能量。而產生偽造的Gaussian輸出文件時,為了省事和統一,是以SCF Done標簽來輸出的。
Gaussian做優化任務的時候,對每一步,輸出次序是[結構i]-[結構i的能量]-[結構i的受力]-[結構i的收斂情況],所以結構、能量、收斂信息都是一一對應的。而對于ORCA,輸出也是這樣的順序,但最后在第i步時發現已經滿足收斂限了,之后還會根據第i步的信息再預測出第i+1步的結構,并且計算這個結構下的能量(也順帶得到波函數),而這個i+1結構就不再計算受力了,也因此對這個結構也不再輸出收斂判斷信息。所以OfakeG產生的偽造的Gaussian輸出文件中,第1步到第i+1步的結構、能量、收斂情況都會給出,但最后一次輸出的收斂情況信息里當前值全都被設成0來占位。
優化過程中除了像Gaussian一樣用受力/位移的最大/RMS值作為判斷標準外,ORCA還用能量變化作為判斷標準。為了體現這點,在偽造的Gaussian輸出文件中也在收斂判斷部分添加了這項,但這項不會被GaussView所讀取,大家可以自行考察。
對于振動分析,由于ORCA不會給出約化質量和振動模式的力常數,所以偽造的Gaussian輸出文件里也沒這項,這不影響一般的分析。由于ORCA不給出振動模式的不可約表示,所以OfakeG把不可約表示都一律輸出為A。
OfakeG把ORCA振動分析輸出的熱力學數據也都轉化為了Gaussian的輸出形式,對于用慣了Gaussian的人來說讀起來方便不少,并且還順帶多顯示了一項Electronic energy=,后面是振動分析對應的結構的電子能量。
OfakeG以后版本也有可能支持處理ORCA的IRC任務的輸出文件,但目前沒有打算支持。因為筆者撰文時最新的ORCA 4.1.2版的IRC功能非常弱、速度慢,甚至就連反應坐標都不給出來,原理上沒法轉換成Gaussian的格式。另外OfakeG也不會去支持轉換ORCA的TDDFT等電子激發任務的輸出文件,因為做這個轉換沒有任何實際意義。Multiwfn直接就能基于ORCA的TDDFT輸出信息繪制各種電子光譜和做電子激發分析(后者我都有現成的例子,見http://www.shanxitv.org/485。Multiwfn繪制光譜的更詳細介紹見http://www.shanxitv.org/224),而且ORCA目前版本給出的是TDDFT組態函數的貢獻而不是系數,原理上也不可能轉換為Gaussian形式的輸出。
OfakeG是100%純Fortran寫的,沒有利用任何庫和其它任何編程語言(或許有的人能猜到我為什么刻意彰顯這點)。
]]>
詳談Multiwfn產生ORCA量子化學程序的輸入文件的功能
On the function of generating ORCA input file in Multiwfn
文/Sobereva@北京科音
First release: 2019-Jun-23 Last update: 2025-Jun-17
1 簡介
ORCA量子化學程序如今的用戶越來越多,用戶數僅次于Gaussian,不僅免費,而且有很多優點,例如支持很多Gaussian不支持的較重要方法,如wB97M-V和DLPNO-CCSD(T),并且由于充分利用了RI技術,使得DFT、TDDFT的效率很高,這點在《大體系弱相互作用計算的解決之道》(http://www.shanxitv.org/214)和《使用ORCA在TDDFT下計算旋軌耦合矩陣元和繪制旋軌耦合校正的UV-Vis光譜》(http://www.shanxitv.org/462)我都已經說過。ORCA的安裝方法見《量子化學程序ORCA的安裝方法》(http://www.shanxitv.org/451)。
最強大的波函數分析程序Multiwfn也可以產生ORCA常見任務的輸入文件,這個功能實用性極高。產生的關鍵詞對于ORCA最新版都是合適的。隨著ORCA的更新、用法的變動,Multiwfn也會相應地更新。最新版Multiwfn可以從主頁http://www.shanxitv.org/multiwfn免費獲取。
Multiwfn的這個功能并不是那種類似GaussView產生Gaussian輸入文件的界面那樣可以充分指定各種各樣的選項,我覺得這種創建輸入文件的工具對于ORCA用戶而言并沒什么實際意義。實際上有很大一批人用ORCA只是沖著ORCA的那些長處、特色功能去的,把ORCA作為Gaussian的補充,特別常用的任務類型就那么些,他們沒精力也不需要把ORCA的所有細節都摸清楚。Multiwfn的這個產生ORCA輸入文件的功能更像是把當前載入的體系的坐標套入特定類型任務的關鍵詞模板里,之后用戶只需把內存使用量、核數等參數簡單地改一下,馬上就可以跑,這顯著拉低了ORCA的使用門檻,也使得ORCA用起來方便不少。
如果你的研究中使用了Multiwfn創建了ORCA輸入文件并給你帶來了便利,希望在寫文章的時候提及諸如The input files of ORCA program were prepared with the help of Multiwfn code并引用Multiwfn程序在剛啟動時顯示的Multiwfn原文,這是對Multiwfn此功能開發最好的支持。
特別注意:本文介紹的功能僅僅能讓你用ORCA做一些最最簡單、最最常見的計算,以及在使用上提供便利。而正經用ORCA做計算的話是必須要花時間專門具體學習ORCA的用法和各種相關理論背景知識的,而絕對不能稀里糊涂當黑箱用!非常推薦參加北京科音高級量子化學培訓班(http://www.keinsci.com/KAQC),里面專門有一節用300多頁幻燈片對ORCA的使用進行極度系統的講解并給出大量使用經驗,并且在不同小節中還對于以下提到的RI/密度擬合、COSX、DLPNO、F12顯式相關、STEOM、3c系列方法、sTD-DFT等關鍵知識有特別詳細的介紹,具備了這些知識才可能把ORCA用得得心應手、游刃有余!
2 產生ORCA輸入文件的界面
可以用任意Multiwfn支持的含有結構信息的文件作為Multiwfn的這個功能的輸入文件,比如fch、molden、wfn、mol、mol2、pdb、xyz、gjf等。然后進入主功能100的子功能2中的選項12(更快速進入的方法是在主菜單里直接輸入oi),輸入要產生的ORCA輸入文件的文件名,之后就會看到以下界面。注意隨著Multiwfn的版本更新,這個界面也可能會有所變化,屆時筆者也會相應地更新本文。
-100 Use template input file provided by user to generate new input file
-11 Choose ORCA version compatibility, current: >ORCA 5.0
-10 Set computational resources, core: 8 memory/core: 1000 MB
-2 Toggle adding diffuse functions, current: No
-1 Toggle employing implicit solvation model, current: No
0 Select task, current: Single point
1 B97-3c 1b r2SCAN-3c
2 RI-BLYP-D3(BJ)/def2-TZVP
3 RI-B3LYP-D3(BJ)/def2-TZVP(-f) 4 RI-B3LYP-D3(BJ)/def2-TZVP
5 RI-wB97M-V/def2-TZVP
6 RI-PWPB95-D4/def2-TZVPP 7 RI-PWPB95-D4/def2-QZVPP
6b RI-wB97X-2-D3(BJ)/def2-TZVPP 7b RI-wB97X-2-D3(BJ)/def2-QZVPP
6c RI-revDSD-PBEP86-D4/def2-TZVPP 7c RI-revDSD-PBEP86-D4/def2-QZVPP
8 DLPNO-CCSD(T)/def2-TZVPP with normalPNO and RIJCOSX
9 DLPNO-CCSD(T)/def2-TZVPP with tightPNO and RIJCOSX
10 CCSD(T)/cc-pVTZ
11 CCSD(T)-F12/cc-pVDZ-F12 with RI
12 Approximated CCSD(T)/CBS with help of MP2 (cc-pVTZ->QZ extrapolation)
13 DLPNO-CCSD(T)/CBS with RIJCOSX & tightPNO (def2-TZVPP->QZVPP extrapolation)
14 CCSD(T)/CBS (cc-pVTZ->QZ extrapolation)
20 sTD-DFT based on RI-wB97X-D3/def2-SV(P) orbitals
22 TDDFT RI-PBE0/def2-SV(P)
23 TDDFT RI-RSX-QIDH/def2-TZVP 231 TDDFT RI-DSD-PBEP86/def2-TZVP
24 EOM-CCSD/cc-pVTZ 25 STEOM-DLPNO-CCSD/def2-TZVP(-f)
上述選項里,從1開始都是最常用的ORCA計算級別,20之前的都是基態計算任務,從20開始的都是激發態計算任務,都是序號越大精度越高但耗時也越高。選了之后就會生成ORCA輸入文件,關鍵詞是相應級別計算時最恰當的。之后用戶可以再根據實際需要把輸入文件里的基組、核數(%pal nprocs后面的值)、內存使用量(%maxcore后面的值)改成實際情況。
由于ORCA不同大版本的情況不同,某些關鍵詞寫法相差甚大,為了兼容性考慮,界面里專門留了個選項-11可以用來切換ORCA版本。默認對應的是最新的ORCA版本。
選擇計算級別前,可以先用選項0選擇任務類型。默認是單點,也可以選優化極小點、振動分析、優化極小點+振動分析、優化過渡態+振動分析、分子動力學、考慮counterpoise校正算結合能。對于振動分析任務,關鍵詞會自動加上tightSCF來使用更嚴格的SCF收斂限(而對于優化任務ORCA默認就是用tightSCF)。分子動力學任務默認設置對應的是控溫在298.15 K下用0.5 fs步長模擬500步。
默認不使用溶劑,如果想用SMD溶劑模型表現溶劑環境,可以選擇選項-1,然后輸入溶劑名,之后導出的輸入文件里就帶著溶劑模型關鍵詞了。ORCA直接支持的SMD溶劑名詳見手冊(一搜1-HEXANOL就可以跳到對應的頁)。若在這個選項里輸入c,也可以用CPCM溶劑模型、自定義溶劑。
如《談談彌散函數和“月份”基組》(http://www.shanxitv.org/119)中所述,很多情況用彌散函數是必須的,比如陰離子體系的單點等。如果你想把基組改成帶彌散函數的版本,就選擇一次選項-2將其狀態切換為Yes。此時cc-pVnZ系列基組都會被改為aug-cc-pVnZ,def2系列基組都會被改為ma-def2系列,不熟悉此基組的話見《給ahlrichs的def2系列基組加彌散的方法》(http://www.shanxitv.org/340)。與此同時,輔助基組也會改成恰當的。對于ma-def2系列,由于沒有標配的輔助基組,所以會自動用autoaux關鍵詞來自動構建。僅B97-3c、r2SCAN-3c和CCSD(T)-F12/cc-pVDZ with RI沒法加彌散函數。
如果你載入進Multiwfn的文件是比如fch、wfn、molden這樣帶有波函數信息的,那么原本波函數對應的電荷和自旋多重度是多少,則產生的ORCA輸入文件里的值也是多少。如果載入的是諸如xyz、pdb這樣只含有結構信息的文件,那么別忘了需要根據實際情況自己改一下電荷和自旋多重度。
注意有些方法和任務類型之間不兼容,比如對于ORCA 5.0版而言,PWPB95-D3(BJ)沒有解析梯度而無法直接用于幾何優化任務。也有些方法本身可以用于優化,比如TDDFT,但使用了SMD溶劑模型后就不再支持。這種情況請大家自行手動調整輸入文件里的關鍵詞,比如需要改用數值梯度、數值Hessian的自己分別寫上numgrad、numfreq
輸入文件里的%maxcore控制ORCA并行計算時每個進程用的內存量(MB)的上限。因此比如你想8核并行,機子有32GB物理內存,若扣除操作系統、后臺任務占的內存,假設剩30GB(約30000MB),那么maxcore不應超過30000/8=3750,但鑒于ORCA計算時進程實際使用的內存量上限往往會超過maxcore,因此為保險起見此時maxcore建議設為3000。計算使用的CPU核數、maxcore設置既可以自己編輯Multiwfn產生的ORCA輸入文件,也可以直接在當前界面里用選項-10修改,默認的核數與Multiwfn的settings.ini里的nthreads參數等同。
Multiwfn產生的ORCA輸入文件里都帶著noautostart miniprint關鍵詞。ORCA會自動試圖從當前目錄下與當前任務同名的gbw里讀取結構和波函數作為初猜,noautostart代表要求不這樣干。miniprint代表避免輸出一些對普通用戶沒什么意義的中間信息,且也不自動做布居分析,因為ORCA默認做的布居分析的輸出會導致輸出文件的信息量巨大,而這些信息沒太大意義而且格式還不好讀。真想要做布居分析討論電子結構,隨時可以用Multiwfn基于ORCA產生的molden文件來實現,又快又方便信息又容易讀,見《詳談Multiwfn支持的輸入文件類型、產生方法以及相互轉換》(http://www.shanxitv.org/379)以及Multiwfn手冊4.7節的布居分析例子。
此界面里有個選項-100,選了之后用戶要提供一個ORCA模板文件,其中包含實際計算要用的所有設置,只不過坐標部分寫成[geometry],Multiwfn會將這部分替換為當前體系的坐標來產生ORCA輸入文件。通過這個功能可以完全自定義地創建ORCA輸入文件,還可以通過自寫簡單的腳本實現諸如把一大批xyz文件轉化為帶有特殊關鍵詞的ORCA輸入文件。一個模板文件的例子如下,用來產生DSD-BLYP雙雜化泛函級別的自然軌道(算完后產生的以.mp2nat為后綴的文件可自行改成.gbw后綴,然后就可以用orca_2mkl轉成.molden輸入文件,之后可以載入Multiwfn做波函數分析)。
! DSD-BLYP def2-TZVP tightscf miniprint
%maxcore 6000
%pal nprocs 10 end
%mp2
density relaxed
NatOrbs true
end
* xyz 0 1
[geometry]
*
3 使用Multiwfn產生ORCA輸入文件的簡單例子
此例我們要用ORCA對甲醇在B97-3c級別下優化,之后用wB97M-V/def2-TZVP算單點,算單點時使用SMD模型表現乙醇環境。
首先用GaussView(或其它建模程序)畫個甲醇,保存為gjf或mol或mol2或pdb文件,然后載入啟動Multiwfn,載入此文件,依次輸入
100 //其它功能,Part 1
2 //導出文件、產生量化程序輸入文件
12 //產生ORCA輸入文件
opt.inp //輸出的文件名
0 //選擇任務類型
2 //優化
1 //B97-3c
現在當前目錄下出現了opt.inp,恰當設置里面的maxcore和nprocs,然后用ORCA運行之。
算完后當前目錄下出現了opt.xyz,此為優化后的結構文件,將之載入Multiwfn,依次輸入
100 //其它功能,Part 1
2 //導出文件、產生量化程序輸入文件
12 //產生ORCA輸入文件
SP.inp //文件名
-1 //啟用SMD溶劑模型
ethanol //溶劑名
5 //RI-wB97M-V/def2-TZVP
然后將當前目錄下得到的SP.inp再次用ORCA運算即可。
在《Simulating UV-Vis and ECD spectra using ORCA and Multiwfn》(http://www.shanxitv.org/485)一文中,筆者還演示了使用了Multiwfn創建ORCA輸入文件,然后通過ORCA做TDDFT計算,最后用Multiwfn產生電子光譜的全過程。在《基于ORCA量子化學程序對分子做優化、振動分析、觀看紅外光譜、觀看軌道的簡單演示》(https://www.bilibili.com/video/av59599938)視頻中,筆者演示了利用ORCA結合Multiwfn等程序,對有機小分子做一系列最常見的計算任務的基本操作。
4 計算級別簡介和關鍵詞解讀
下面解釋一下前述計算級別的特點以及解讀一下相關的關鍵詞,以便于讀者準確地認識這些計算級別的特點、什么時候適合用什么級別、為什么筆者令Multiwfn支持這些級別,以及理解為什么關鍵詞是那樣定義的。
注意采用的基組沒有一個是Pople系列基組,因為如《談談量子化學中基組的選擇》(http://www.shanxitv.org/336)提到的,這套基組對于較好精度的計算沒有一個是劃算的,而對于便宜的級別,這套基組又沒有標配的給RI用的輔助基組,因此Pople系列基組在ORCA中幾乎是擺設。DFT相關的計算筆者配的都是def2系列,這非常適合DFT,而對于后HF類型計算用的都是Dunning相關一致性基組,但實際上改用檔次相當的def2系列基組也完全可以。
從ORCA 5.0開始,對于雜化泛函默認就會用RIJCOSX,因此下面關鍵詞里面RIJCOSX對于ORCA >=5.0版做雜化泛函計算其實是多余的,但為了與老版本的兼容性考慮還是留著了。
4.1 基態計算
? B97-3c。關鍵詞:B97-3c
這是Grimme提出來的一個又便宜又快的組合式方法,用的是純泛函,基組是方法直接內定的,還帶了DFT-D3、SRB校正項。對于主族和過渡金屬體系都適用。在ORCA里的耗時僅略高于RI-BLYP/def2-SVP一點點,但結果肯定整體更好。更詳細介紹見《盤點Grimme迄今對理論化學的貢獻》(http://www.shanxitv.org/388)。我建議使用此方法去優化、計算大體系能量,或者粗略計算小體系用于初步篩選的目的,比如筆者在《gentor:掃描方式做分子構象搜索的便捷工具》(http://bbs.keinsci.com/thread-2388-1-1.html)的實例中就是這樣做的。根據筆者自己的一些研究,用Intel 36核機子用B97-3c在真空下優化一個160原子的弱相互作用顯著的有機體系,用了75步收斂,總共耗時才不到三個小時,比起在Gaussian下用算這種體系常用的B3LYP-D3(BJ)/6-311G*耗時低得多得多。之后在同樣條件下用B97-3c對這個體系做振動分析,耗時三個半小時,也完全可以接受。使用B97-3c時不需要寫與RI相關的關鍵詞和指定輔助基組,因為默認就會用RIJ和恰當的輔助基組加速計算。
? r2SCAN-3c。關鍵詞:r2SCAN-3c
這是B97-3c的后繼者,基于2020年末提出的r2SCAN泛函弄的-3c方法。耗時比之高了百分之幾十,但精度全面提升了不少。因此如果不是特別窮的話,建議總是用r2SCAN-3c代替B97-3c。
? RI-BLYP-D3(BJ)/def2-TZVP。關鍵詞:BLYP D3 def2-TZVP def2/J
BLYP泛函平時用得不多,畢竟算有機體系精度遠不如雜化泛函,但是BLYP這樣的純泛函在ORCA下利用RIJ方式加速庫侖部分的計算后(記為RI-BLYP),對于稍大點的體系,速度能比Gaussian快一個數量級以上(振動分析除外)!。因此以BLYP為典型代表的純泛函還是很有存在意義的。關鍵詞里D3代表加上DFT-D3(BJ)色散校正,詳見《DFT-D色散校正的使用》(http://www.shanxitv.org/210)。對于算弱相互作用,根據GMTKN55測試集的測試,BLYP加上DFT-D3(BJ)后,在所有的“純泛函-D3”里是頂尖的。def2-TZVP雖然是一個中等偏上的基組,在Gaussian里算是偏貴的,但是在ORCA里利用RIJ加速后其實挺便宜的。使用RIJ技術加速需要指定輔助基組,關鍵詞中def2/J就代表使用對def2系列基組通用的用于RIJ目的的輔助基組。不需要特意寫RIJ關鍵詞,因為對純泛函默認會使用RIJ。注意BLYP-D3(BJ)/def2-TZVP的耗時比B97-3c稍高,但精度未必比B97-3c好。特別是從ORCA 5.0開始支持了r2SCAN-3c后,RI-BLYP-D3(BJ)/def2-TZVP這個組合其實就沒什么任何使用價值了。
? RI-B3LYP-D3(BJ)/def2-TZVP(-f)。關鍵詞:B3LYP D3 def2-TZVP(-f) def2/J RIJCOSX
由于B3LYP便宜、穩健、被支持廣泛,雖然算熱化學方面的精度已經嚴重落伍,但即便到現在還是被使用得最多的泛函,結合DFT-D色散校正后又令它的生命周期進一步延長,相關信息看《簡談量子化學計算中DFT泛函的選擇》(http://www.shanxitv.org/272)。對于B3LYP這樣的雜化泛函,ORCA里可以用RIJK或RIJCOSX來加速,后者在ORCA里能用的特征更多,而且對于二、三十個原子以上的情況,通常RIJCOSX比RIJK明顯更快,RIJCOSX關鍵詞代表啟用之。def2-TZVP(-f)是在def2-TZVP基組的基礎之上把f極化函數閹割掉的結果,這可以顯著節約時間,而對精度損失不算特別嚴重,此時尺寸與6-311G(2d,p)相仿佛。雜化泛函結合RIJCOSX的耗時顯著高于純泛函結合RIJ,因此RI-B3LYP即便結合def2-TZVP(-f),耗時也比RI-BLYP/def2-TZVP高得多,且速度遠勝于在Gaussian里的計算速度(Gaussian對雜化泛函不支持RI)。對于主族體系各方面計算以及弱相互作用計算而言,B3LYP-D3(BJ)比上述的BLYP-D3(BJ)好是肯定的,因此是否適合使用B3LYP-D3(BJ)應當自行掂量。
? RI-B3LYP-D3(BJ)/def2-TZVP。關鍵詞:B3LYP D3 def2-TZVP def2/J RIJCOSX
同上,只不過基組變成了完整的def2-TZVP
? RI-wB97M-V/def2-TZVP。關鍵詞:wB97M-V def2-TZVP def2/J RIJCOSX strongSCF
根據Phys. Chem. Chem. Phys., 19, 32184 (2017)、Mol. Phys., 115, 2315 (2017)、J. Chem. Theory Comput., 15, 3610 (2019)等測試,ωB97M-V在除了雙雜化泛函以外的泛函中,無論算主族還是含過渡金屬的體系,無論算反應能、勢壘還是弱相互作用,性能都是頂級的,在《簡談量子化學計算中DFT泛函的選擇》我也提到了。雖然從ORCA 5.0開始支持了ωB97M-V的解析梯度,但沒有解析Hessian,因此不便于做振動分析檢驗虛頻情況,所以我不建議用這個泛函做優化,而且本身這個泛函也比B3LYP等更貴一些。另外,眾所周知,幾何優化耗時遠高于單點計算,而幾何優化用的級別可以比單點要低,因為幾何優化結果對計算級別敏感度遠低于能量計算,這點在《淺談為什么優化和振動分析不需要用大基組》(http://www.shanxitv.org/387)專門提了,因此推薦大家用相對便宜的B97-3c或比它更好也更貴的RI-B3LYP-D3(BJ)/def2-TZVP(-f)來優化結構,最后用RI-wB97M-V/def2-TZVP來算單點。對于ωB97M-V這樣的非雙雜化泛函而言,對于一般問題用def2-TZVP就已經夠用了,要求更高的話也可以考慮def2-QZVPP。如果你要算弱相互作用能的話也推薦用def2-QZVPP,若結合counterpoise校正還能更好一點。由于ORCA的默認的SCF收斂限相對于wB97M-V的精度水平來說過于寬松了些,因此同時用了strongSCF來讓能量收斂到更高精度。
值得一提的是雖然M06-2X很流行,很適合算主族體系,但在上面只字未提。這是因為M06-2X做幾何優化方面比B3LYP-D3(BJ)優勢并不明顯,而耗時明顯更高,對積分格點要求也高,還更難收斂。雖然M06-2X用于算主族體系的能量很不錯,但是和ωB97M-V比又明顯遜色,在速度上也沒優勢,故M06-2X在當前的ORCA中沒多大用武之地。
? RI-PWPB95-D4/def2-TZVPP。關鍵詞:PWPB95 D4 def2-TZVPP def2/J def2-TZVPP/C RIJCOSX tightSCF
PWPPWPB95-D4在《簡談量子化學計算中DFT泛函的選擇》中專門說了,是很穩健且精度很好的雙雜化泛函,精度介于ωB97M-V和CCSD(T)檔次之間。當前關鍵詞令PWPB95-D4泛函的雜化泛函部分做SCF的過程中通過RIJCOSX加速來降低耗時,而在之后計算類似MP2部分的時候RI來顯著降低耗時(只要用了RIJCOSX關鍵詞,默認就會在MP2部分也用RI),這一步使用def2-TZVPP/C輔助基組。為了讓SCF部分盡量準確從而得到數值層面較準確的PWPB95-D4的結果,因此用了tightSCF。筆者用Intel 36核服務器做過測試,對于168個原子的有機體系的單點,這樣的計算級別耗時不到三個小時,耗硬盤也不多,而如果用Gaussian的話根本沒戲,差不多80個原子就封頂了,還特別耗硬盤。對于66個原子的有機體系的單點,ORCA下用這個級別僅需六分鐘就能算完。總的來說,RI-雙雜化/def2-TZVPP用Intel三、四十核的服務器跑200原子以內的單點毫無壓力。另外,如果你希望耗時更低的話,可以手動加上float關鍵詞,代表通過單精度變量而非默認的雙精度變量儲存中間數據,耗時能節約一小半,而且還能省一倍硬盤。
? RI-PWPB95-D4/def2-QZVPP。關鍵詞:PWPB95 D4 def2-QZVPP def2/J def2-QZVPP/C RIJCOSX tightSCF
我之前在《談談量子化學中基組的選擇》(http://www.shanxitv.org/336)中說過,后HF、雙雜化泛函對基組的要求顯著高于普通泛函,因此為了充分發揮PWPB95-D4的潛力,如果你能接受更大計算量的話,推薦結合def2-QZVPP。對于66個原子的有機體系的單點,在Intel 36核服務器下ORCA下用這個級別用了30分鐘算完。
? RI-wB97X-2-D3(BJ)/def2-TZVPP(RI-wB97X-2-D3(BJ)/def2-QZVPP與之類似)
關鍵詞:wB97X-2 D3 def2-TZVPP def2/J def2-TZVPP/C RIJCOSX tightSCF
%method
D3S6 0.547
D3A1 3.520
D3S8 0.0
D3A2 7.795
end
ωB97X-2-D3(BJ)在PCCP, 20, 23175 (2018)的雙雜化泛函橫測當中表現出眾(不過在算分子間弱相互作用的測試上表現一般)。ωB97X-2從ORCA 5.0開始支持,但支持的只是2009年當時提出的原版,其搭配的DFT-D3(BJ)色散校正參數在PCCP, 20, 23175 (2018)的補充材料里才給出,沒有在ORCA里內置。因此Multiwfn產生的輸入文件里如上所示自動用%method ... end字段補充了色散校正參數。
? RI-revDSD-PBEP86-D4/def2-TZVPP(RI-revDSD-PBEP86-D4/def2-QZVPP與之類似)
關鍵詞:revDSD-PBEP86-D4/2021 def2-TZVPP def2/J def2-TZVPP/C RIJCOSX tightSCF
revDSD-PBEP86-D4在計算有機反應能以及弱相互作用能方面在現有的雙雜化泛函里都是頂級的,建議對這類問題使用。
? DLPNO-CCSD(T)/def2-TZVPP with normalPNO and RIJCOSX。關鍵詞:DLPNO-CCSD(T) normalPNO RIJCOSX def2-TZVPP def2/J def2-TZVPP/C tightSCF
CCSD(T)普遍被認為是計算靜態相關不是特別強的體系的金標準。DLPNO-CCSD(T)是ORCA中的黑科技,是一種對CCSD(T)的數值近似,可以把原本最多只能算得動不超過30原子的CCSD(T)的方法擴展到好幾十甚至上百原子,精度和耗時通過此方法中的一些閾值參數來控制。ORCA里有LoosePNO、NormalPNO、TightPNO三種標準,越往后越貴,但結果也越接近CCSD(T)。LoosePNO就太爛了,不建議用,而NormalPNO較有實用價值。用NormalPNO的時候精度就已經顯著超過revDSD-PBEP86-D4了,相對于CCSD(T)的誤差通常在1 kcal/mol以內。DLPNO-CCSD(T)的HF計算部分可以用RIJCOSX方法來加速,這是為什么關鍵詞里寫了RIJCOSX,并且指定了def2/J輔助基組。def2-TZVPP/C輔助基組是用于DLPNO-CCSD(T)的電子相關部分計算的。順帶一提,在北京科音高級量子化學培訓班(http://www.keinsci.com/workshop/KAQC_content.html)中專門有一節“低標度耦合簇方法”很詳細講授DLPNO相關方法以及LNO-CCSD(T)等其它低標度方法。
? DLPNO-CCSD(T)/def2-TZVPP with tightPNO and RIJCOSX。關鍵詞:DLPNO-CCSD(T) tightPNO RIJCOSX def2-TZVPP def2/J def2-TZVPP/C tightSCF
DLPNO-CCSD(T)結合tightPNO的時候,根據J. Chem. Theory Comput., 11, 4054 (2015)的測試(注意此文里有很多在耗時方面的嚴重誤導性說法),與CCSD(T)的誤差在1 kJ/mol的程度,幾乎可認為沒有差別。tightPNO的耗時比NormalPNO通常高幾倍。在筆者的Intel 36核機子上,用當前級別計算66個原子的有機體系耗時約8小時,耗硬盤最多時候為120GB。如果你還想要更好的精度,建議將基組提升至def2-QZVPP,但也會貴非常多。如果不用RIJCOSX,即去掉RIJCOSX def2/J關鍵詞,精度會有很輕微改進,但對較大的體系,SCF部分的耗時會增加許多。
? CCSD(T)/cc-pVTZ。關鍵詞:CCSD(T) cc-pVTZ tightSCF
這就是原版的CCSD(T)/cc-pVTZ,沒什么好說的。
注:上述幾個計算級別中,如果有cc-pVTZ不支持的元素,把cc-pVTZ替換為質量差不多的def2-TZVPP。如果提示某些元素沒有對應的輔助基組,把cc-pVTZ cc-pVTZ/JK cc-pVTZ/C替換為def2-TZVPP def2/JK def2-TZVPP/C。
? CCSD(T)-F12/cc-pVDZ-F12 with RI。關鍵詞:CCSD(T)-F12/RI cc-pVDZ-F12 cc-pVDZ-F12-CABS cc-pVTZ/C tightSCF
F12是顯式相關方法,可以結合到CCSD(T)、MP2等方法上。CCSD(T)-F12結合cc-pVDZ-F12基組時,耗時顯著低于CCSD(T)/cc-pVQZ,但精度則與之接近。因此如果你需要CCSD(T)/cc-pVQZ檔次的數據但算得很吃力的話,當前級別是個很好的選擇。計算過程中利用RI可以進一步顯著節約時間,所以寫了/RI。順帶一提,北京科音高級量子化學培訓班(http://www.keinsci.com/workshop/KAQC_content.html)專門有一節“顯式相關計算”用30多頁幻燈片非常完整深入講F12這類計算。
? Approximated CCSD(T)/CBS with help of MP2 (cc-pVTZ->QZ extrapolation)。關鍵詞:ExtrapolateEP2(3/4,cc,MP2) tightSCF
我在《談談能量的基組外推》(http://www.shanxitv.org/172)中專門介紹過基組外推的概念,這也叫CBS外推,即假定外推到了完備基組極限(CBS)。CCSD(T)檔次下最常用的外推就是基于cc-pVTZ和cc-pVQZ來外推,可以免費地得到更高一個檔次基組的結果,即大約cc-pV5Z的結果,這種計算方式常見于各種高精度小體系的研究文章當中。直接利用CCSD(T)結合cc-pVTZ和cc-pVQZ能量進行外推的話,后者耗時非常高,往往很難承受,因此可以借助MP2來進行“近似的外推”,即利用MP2/cc-pVTZ和QZ先外推出MP2/CBS相關能,然后CCSD(T)/CBS的相關能就近似可以估計為CCSD(T)/cc-pVTZ + MP2/CBS - MP2/cc-pVTZ,這就避免了直接做非常昂貴的CCSD(T)/cc-pVQZ計算了。這個近似精度很好,一般也就帶來0.1 kcal/mol左右程度的誤差,筆者在《各種后HF方法精度簡單橫測》(http://www.shanxitv.org/378)中專門做過測試,有興趣可以看看。ORCA提供了ExtrapolateEP2關鍵詞,可以直接實現上述CCSD(T)結合MP2的CBS外推。
其實如果將這里的CCSD(T)改為tightPNO的DLPNO-CCSD(T),能以相近的精度算明顯更大體系,但可惜這沒法在目前的ORCA里通過一套關鍵詞直接實現。
? DLPNO-CCSD(T)/CBS with RIJCOSX & tightPNO (def2-TZVPP->QZVPP extrapolation)。關鍵詞DLPNO-CCSD(T) tightPNO Extrapolate(3/4,def2) def2-QZVPP/C RIJCOSX tightSCF
? CCSD(T)/CBS (cc-pVTZ->QZ extrapolation)。關鍵詞:CCSD(T) Extrapolate(3/4,cc) tightSCF
ORCA里可以用Extrapolate關鍵詞對任意后HF方法進行外推,以上兩個級別就用了這點。一個是直接用CCSD(T),算十個原子都已經極度吃力;另一個是便宜得多但結果也糙一些的DLPNO-CCSD(T) with tightPNO,同時為了節約SCF時間而用了RIJCOSX,之所以改用了def2,是因為輔助基組層面的原因
4.2 激發態計算
? sTD-DFT based on RI-wB97X-D3/def2-SV(P) orbitals
關鍵詞:wB97X-D3 def2-SV(P) def2/J RIJCOSX
%tddft
Mode sTDDFT
Ethresh 7.0
PThresh 1e-4
PTLimit 30
maxcore 6000
end
對于好幾百個原子的很大體系的電子光譜計算,往往要算幾百個態才夠覆蓋感興趣的波長范圍。用wB97X-D3/def2-SV(P)對這樣大小的體系算單點能往往算得動,但是用TDDFT算這么多態往往太困難。此時可以用Grimme提出的sTD-DFT方法,此方法基于DFT計算求解的軌道,通過對TDDFT矩陣元進行高度近似,只需要非常少的計算量就可以算出大量激發態(sTDDFT與TDDFT的耗時關系有點像半經驗方法與DFT的耗時關系)。但代價是精度有所降低,不過對于很大體系的粗放式研究來說夠用了。sTD-DFT功能已被內置于ORCA中,上面的關鍵詞就是先做RI-wB97X-D3/def2-SV(P)單點計算,再基于其軌道做sTD-DFT,可以算出激發能為7 eV以內的所有態(這是一般感興趣的電子光譜能量范圍)。用wB97X-D3泛函是因為根據Phys.Chem.Chem.Phys.,16,14408(2014)的測試,將它結合sTD-DFT來用對于大體系(通常有顯著電子共軛)結果較理想。PThresh和PTLimit用于控制納入考慮的組態函數的范圍,不寫它們的話會考慮所有組態函數,這對于大體系可能耗時較高,當前的PThresh和PTLimit設置可以避免這個問題,對精度的犧牲可忽略不計。注意sTD-DFT目前只能對單個結構做電子激發計算,而不能用于激發態優化、振動分析等目的。順帶一提,北京科音高級量子化學培訓班(http://www.keinsci.com/workshop/KAQC_content.html)專門有一節“使用sTDA方法快速計算大體系電子光譜”用約40頁幻燈片非常全面完整詳細講這類計算。
? TDDFT RI-PBE0/def2-SV(P)
關鍵詞:PBE0 def2-SV(P) def2/J RIJCOSX tightSCF
%tddft
nroots 10
TDA false
end
這是用PBE0結合大小與6-31G*差不多的def2-SV(P)做TDDFT計算。PBE0是TDDFT計算激發態常用的泛函,但碰見大共軛體系建議改為CAM-B3LYP等,詳見《亂談激發態的計算方法》(http://www.shanxitv.org/265)。def2-SV(P)對于計算中、大體系的電子光譜是很適合的,既不貴,精度也基本夠用,如果有余力且希望結果更好建議把def2-SV(P)改為def2-TZVP(-f)或def-TZVP(關鍵詞寫為TZVP)。關鍵詞里nroots 10代表計算10個激發態,當然需要根據實際情況進行調節,同Gaussian的TDDFT計算中的nstates,詳見《Gaussian中用TDDFT計算激發態和吸收、熒光、磷光光譜的方法》(http://www.shanxitv.org/314)。用tightSCF是為了降低數值層面的誤差。
? TDDFT RI-DSD-PBEP86/def2-TZVP
關鍵詞:DSD-PBEP86 def2-TZVP def2/J def2-TZVP/C RIJCOSX tightSCF
(用TDDFT RI-RSX-QIDH/def2-TZVP則把上面的DSD-PBEP86改為RSX-QIDH)
%tddft
nroots 10
TDA false
end
ORCA是為數不多的支持雙雜化泛函下做TDDFT的程序,此級別精度比普通泛函做TDDFT更高,但代價是耗時也明顯更高。計算時應當同時指定/C輔助基組。根據JCTC, 17, 4211 (2021)的測試,在ORCA能用的雙雜化泛函中DSD-PBEP86算局域激發和分子內電荷轉移激發最好,在JCTC, 18, 1646 (2022)的圖5和表2中體現RSX-QIDH是算分子間CT激發的首選,所以Multiwfn給了兩個選項。用上述關鍵詞用對一個47原子的有機體系做TDDFT計算,筆者在Intel 2*2696v3 36核機子上用不到20分鐘算完,可見耗時毫不夸張,算稍大一些的體系也能算得動。
? EOM-CCSD/cc-pVTZ
關鍵詞:EOM-CCSD cc-pVTZ tightSCF
%mdci
nroots 3
end
EOM-CCSD計算激發態的精度算是很不錯了,比TD-雙雜化精度更高,也更穩健和普適,但耗時頗高,而且隨算的態數增加總耗時迅速增加,因此主要適合的是小體系的低階激發態的較高精度的研究。RI在EOM-CCSD計算時派不上用場,和Gaussian相比也沒有顯著優勢。
? STEOM-DLPNO-CCSD/def2-TZVP(-f)
關鍵詞:STEOM-DLPNO-CCSD RIJCOSX def2-TZVP(-f) def2/J def2-TZVP/C tightSCF
%mdci
nroots 3
end
STEOM-CCSD方法的初衷是降低EOM-CCSD的耗時,但不應將STEOM-CCSD視為是EOM-CCSD的近似,而應當視為兩種不同方法。二者精度差不多,而STEOM-CCSD對CT激發的結果往往比EOM-CCSD更好。EOM-CCSD明顯算不動的體系STEOM-CCSD照樣算不動,結合像樣的基組時一般頂多也就用于二十多個原子。DLPNO技術與STEOM-CCSD的結合誕生的STEOM-DLPNO-CCSD則可以用于大得多的體系,在def2-TZVP(-f)這樣的基組下甚至對超過60個原子的體系也能算(但注意超級耗硬盤,沒有好幾個TB的剩余空間就別指望了)。
Written by Tian Lu (sobereva@sina.com), 2019-May-20
Beijing Kein Research Center for Natural Sciences (http://www.keinsci.com)
1 Introduction
In this tutorial, I will show how to very easily using ORCA and Multiwfn to simulate UV-Vis and electronic circular dichroism (ECD) spectra for a typical organic system alanine in water environment. The ORCA is a free quantum chemistry program, it can be downloaded via https://orcaforum.kofo.mpg.de/app.php/portal. Although Multiwfn is a code aiming for wavefunction analysis, it also has a powerful module used to plot various kinds of spectra based on output file of Gaussian, ORCA, xtb, sTDA or plain text file. Multiwfn can be freely obtained at http://www.shanxitv.org/multiwfn. All the ORCA and Multiwfn supports Windows, Linux and MacOS platforms.
In this tutorial, I assume that you are using Windows platform. The version of ORCA is 4.1.1, the version of Multiwfn is 3.7(dev) updated on 2019-Jul-14 (do not use older version). The overall computational cost should be less than 10 minutes for an ordinary Intel quad-core CPU.
All files involved in this tutorial can be downloaded at http://www.shanxitv.org/attach/485/file.rar.
2 Optimization of ground state
Both UV-Vis and ECD are absorption spectra, commonly it is assume when a molecule undergo electronic excitation, the geometry is at minimum point of potential energy surface of ground state. Therefore, the ground state geometry should be optimized first. Since the conformational space of alanine is not complicated, construction of the alanine structure is relatively arbitrary. It is important to note that under water environment the alanine is a zwitterion. The initial geometry (initgeom.xyz) I built is shown below:
Now we generate input file of optimization task of ORCA. The easiest way in my opinion is using Multiwfn to do this. Double click icon of Multiwfn.exe to boot up Multiwfn, then input below commands. The texts behind // are comment.
initgeom.xyz //Other formats are also supported, e.g. mol, mol2, pdb, gjf, fch, molden...
100 //Other functions (Part 1)
2 //Exporting files or generating input file of mainstream quantum chemistry codes
12 //Generate ORCA input file
S0_opt.inp //The path of the input file to be generated
-1 //Enable using SMD solvation model
[Press ENTER button to use default solvent (water)]
0 //Select type of task
2 //Optimization
1 //B97-3c. This is an economical level but able to provide satisfactory geometry
Now S0_opt.inp has been generated in current folder. Open it by text editor, change the value behind "nprocs" to the actual number of physical CPU cores of your machine, also properly set the value behind "maxcore", which controls the maximal amount of memory can be utilized per CPU core (in MB).
Move the S0_opt.inp to an empty folder, open console window of your system and enter this folder, then input command such as D:\study\orca411\orca S0_opt.inp > S0_opt.out to conduct the calculation, where D:\study\orca411\orca is the absolute path of ORCA executable file in my machine.
After calculation, from the output file S0_opt.out you can find the optimization converged after 11 steps without any error. The S0_opt.xyz in current folder is the optimized geometry, while S0_opt.trj contains every structure of optimization trajectory (If you want to visualize the trajectory, you can manually change the suffix to .xyz, and then drag this file into VMD, which is an excellent visualization program and can be freely obtained via http://www.ks.uiuc.edu/Research/vmd/).
Use Multiwfn to load the S0_opt.xyz, and then enter main function 0 to visualize the geometry, it can be seen that the optimized geometry fully meets our expectation, as shown below
3 Calculation of excited states
Next, we perform excited state calculation using the popular TDDFT method. According to many benchmark articles, for TDDFT calculation of such a small molecule, PBE0 functional is very suitable (however, for large conjugated systems such as most organic dyes, in particular the states with strong charge-transfer character, CAM-B3LYP and wB97XD often work much better)
Return to main menu of Multiwfn, then input
100 //Other functions (Part 1)
2 //Exporting files or generating input file of quantum chemistry codes
12 //Generate ORCA input file
TDDFT.inp //The path of the input file to be generated
-1 //Enable using SMD solvation model
[Press ENTER button to use default solvent (water)]
22 //TDDFT task at PBE0/def2-SV(P) level with RI acceleration technique
The TDDFT.inp has been generated in current folder, do not forget to properly change the "nprocs" and "maxcore" parameters. Note that in this file the "nroots" is set to 10, namely ten lowest excited states will be calculate. For plotting absorption spectra purpose of small systems like alanine, this setting is adequate, however, if your system consists of much more atoms, "nroots" should also be accordingly increased, otherwise the finally simulated spectra cannot fully cover wavelength range of common interest (e.g. >250nm). In addition, since this system is fairly small, in order to reach higher accuracy, we can use basis set better than the def2-SV(P), therefore we replace the "def2-SV(P)" keyword with "def2-TZVP(-f)", which is a good basis set with size similar to 6-311(2d,p).
Run command like this: D:\study\orca411\orca TDDFT.inp > TDDFT.out
4 Simulating UV-Vis spectrum
From the TDDFT output file, you can find excitation energies and oscillator strengths under "ABSORPTION SPECTRUM VIA TRANSITION ELECTRIC DIPOLE MOMENTS". Based on these data, Multiwfn is able to plot theoretical UV-Vis spectrum. Now we do this.
Boot up Multiwfn and input
TDDFT.out
11 //Plot spectrum
3 //UV-Vis
0 //Show the spectrum on the screen
You can use abundant options in the interface to gradually adjust plotting parameters and then replot the graph until you are satisfied. Please check Section 3.13 of Multiwfn manual on the detailed introduction of the spectrum plotting module and related theoretical background. Section 4.11 provided examples of plotting other kind of spectra.
NOTE: If Multiwfn crashes before entering the plotting interface, it is possible that your ORCA output file is in Unicode encoding, while Multiwfn only supports parsing text file in ASCII encoding. Please check #6 in this post on how to solve this problem: http://www.shanxitv.org/wfnbbs/viewtopic.php?id=213.
5 Simulating ECD spectrum
Multiwfn is also able to simulate ECD spectrum based on excitation energies and rotatory strengths in the ORCA TDDFT output file. Now we do this.
Input below commands in the Multiwfn window:
-3 //Return to main menu
11 //Plot spectrum
4 //ECD
0 //Show the spectrum on the screen
6 Some worth noting points
There are several points I want to mention.
For most neutral system, geometry optimization can be carried out without solvation model to decrease time cost. However, for systems whose local region shows evident ionic character, such as the alanine in water environment, the solvation model should also be applied in optimization stage. For calculation of excited states, solvation model should always be employed to mimic real environment because its impact on electronic excitation is large, in particular for polar solvents.
ECD spectrum is very sensitive to conformation. The alanine under our present study does not have more than one accessible conformation, therefore we can safely use single structure to simulate the spectra. However, if there may be multiple thermally accessible conformations under present condition (this is usually true for flexible molecules containing rotable bonds at room temperature), commonly you should optimize structure and calculate free energy for each one, then use Boltzmann relationship to estimate occurrence percentage, then perform electron excitation calculation for all conformations having probability higher than e.g. 5%, and finally use Multiwfn to plot conformationally averaged ECD spectrum, as illustrated in Section 4.11.4 of Multiwfn manual.
BTW 1: To obtain all possible conformations of a highly flexible molecule, commonly you need to use conformational search software, there are many choice. Among which, one of best choice is the Molclus code developed by me, it is free and very flexible, the official site is http://www.keinsci.com/research/molclus.html (English version of manual will be available later).
BTW 2: To characterize and understand nature of electron excitations, Multiwfn provide numerous functions, see Section 4.A.12 of the manual for an overview.
BTW 3: There is a video tutorial illustrating how to use ORCA quantum chemistry program in combination with Multiwfn, OfakeG and GaussView to realize very common calculation tasks and analyses for a simple organic molecule methanol, taking a look at it is suggested: https://youtu.be/tiTmTbtbtig.
]]>