詳談使用Gaussian做勢能面掃描
詳談使用Gaussian做勢能面掃描
文/Sobereva@北京科音 2019-Apr-1
目錄
1 基本知識
2 用Gaussian做剛性掃描的方法和實例
2.1 基礎知識
2.2 剛性掃描實例1:掃描乙醇的羥基的二面角
2.3 剛性掃描實例2:掃描Li+...苯之間的距離
2.4 剛性掃描實例3:掃描氟化氫鍵長獲得解離曲線
2.5 剛性掃描實例4:同時掃描水分子的鍵長與鍵角
3 使用dimerscan結合Gaussian做二聚體的剛性掃描
3.1 實例:苯酚二聚體的氫鍵距離掃描
3.2 實例:主-客體復合物的接觸距離掃描
4 使用gentor結合Gaussian做二面角的剛性掃描
5 用Gaussian做柔性掃描的方法和實例
5.1 基本用法
5.2 柔性掃描的一些相關問題
5.3 柔性掃描實例1:柔性掃描C2H4ClBr的二面角
5.4 柔性掃描實例2:乙酰和甲胺封閉的丙氨酸(ACE-ALA-NME)的構象搜索
5.5 柔性掃描實例3:輔助尋找乙醇脫水的過渡態
5.6 柔性掃描實例4:把環丁烷拉成丁烷雙自由基
6 通過廣義化內坐標(GIC)進行柔性掃描
6.1 GIC掃描實例1:水+氮氣二聚體的幾何中心距離掃描
6.2 GIC掃描實例2:令1,3-丁二烯兩個亞甲基同步旋轉的掃描
7 總結&其它
本文將介紹勢能面掃描的概念,通過最常用的量子化學程序Gaussian演示怎么實現各種類型的勢能面掃描,并且介紹利用筆者開發的gentor和dimerscan程序結合Gaussian來實現一些只靠Gaussian實現不了的特殊的掃描。相信筆者讀完本文后會對勢能面掃描有十分全面的了解。在北京科音(http://www.keinsci.com)開辦的初級/基礎量子化學培訓班里會對這個主題講得更充分并給出更多例子。
1 基本知識
勢能面掃描用來考察體系能量隨著一個或多個幾何變量的改變而發生的改變。勢能面掃描有很多實際用處,比如
? 構造完整勢能面或者勢能面的子空間。之后基于此可以跑量子動力學、計算反應速率常數時考慮多維隧道效應等等
? 產生力場參數。一般通過勢能面擬合實現
? 幫助確定搜索過渡態適合的初猜結構
? 求解振動問題(振動能級、振動平均結構等)。相關信息參考《Molcas的計算雙原子分子光譜常數的模塊vibrot使用簡介》(http://www.shanxitv.org/372)、《談談溫度、壓力、同位素設定對量子化學計算結果產生的影響(http://www.shanxitv.org/423)。
? 尋找能量較低或最低的構象。參考《gentor:掃描方式做分子構象搜索的便捷工具》(http://bbs.keinsci.com/thread-2388-1-1.html)
? 幫助弄清楚反應機理、鍵的解離等化學上感興趣的過程
? 考察體系電子結構隨幾何結構特定方式的改變而發生的變化。見比如《制作動畫分析電子結構特征》(http://www.shanxitv.org/86)。
掃描分為剛性掃描(rigid scan)和柔性掃描(relaxed scan)。剛性掃描指的是讓程序按照你指定的掃描設定依次改變體系幾何結構,結構每變一次就算一次單點能,而不被掃描的那些幾何變量還是保持在初始結構的值。而在柔性掃描中,每次按照你的要求而改變特定幾何參數時,做的不是單點計算而是限制性優化,即沒有被掃描的變量都會被優化以使得體系能量盡可能低,相當于允許這些變量自發地弛豫(relax)。我們平時說掃描的時候一般默認指前者。因此如果你想告訴別人你做的是柔性掃描的話,那么絕對不要簡單地說成“掃描”或scan,免得造成誤會。
下面,筆者就結合最常用的量子化學程序Gaussian對勢能面掃描的操作進行介紹,并且舉各種勢能面掃描的例子。所有計算使用Gaussian16 A.03版完成,GaussView用的是6.0.16版,同時會利用到的xyz2QC和gentor程序來自于molclus 1.8版程序包(可在此免費獲取http://www.keinsci.com/research/molclus.html)。
本文例子的輸入文件都可以在此文件包里找到:http://www.shanxitv.org/attach/474/file.rar。文件名在例子中都提到了。輸出文件也給了。
2 用Gaussian做剛性掃描的方法和實例
2.1 基礎知識
在Gaussian里做剛性掃描很簡單,關鍵詞里寫上scan,然后對要掃的變量進行定義即可。鍵長、鍵角、二面角都可以掃描,被掃描的幾何坐標必須以變量形式表達。比如某個鍵長變量原本定義是B1= 1.3,那么把這個變量設置改為B1= 1.3 10 0.1,就代表以1.3埃為初始值掃描10步,每步增加0.1埃,最終掃到1.3+10*0.1=2.3埃。由于初始結構也會被計算一次,所以總共會計算11個單點能。算完之后,可以從輸出文件末尾讀取信息匯總,也可以把輸出文件用GaussView打開觀看掃描過程每一步的結構變化,還可以在Result - Scan里圖形化顯示能量在掃描過程中的變化。
還值得一提的是,掃描的每一步用的初猜波函數會自動用上一步收斂的波函數,主要考慮是相鄰兩步之間結構變化不是特別大,所以這樣獲得初猜波函數比重新產生初猜波函數通常更容易令SCF收斂,從而節約時間,通常還能使得上一步的電子結構能夠承接下去。如果你做掃描的時候,發現一開始能收斂,但是掃描中途有的點SCF不收斂,或者收斂到奇怪的情況(體現在能量出現突然波動),不妨將掃描步長改小試試。
下面我們來看一些例子,被考察的問題由簡單到復雜。
2.2 剛性掃描實例1:掃描甲醇的羥基的二面角
這個簡單例子的目的是考察一下甲醇的羥基二面角旋轉過程的能量變化。做掃描之前,我們先在B3LYP/6-31G*下對甲醇做一下幾何優化,優化后是下圖的結構,兩張圖是不同的視角。可見當前結構是Cs點群:
用GaussView把優化后的結構保存為methanol.gjf文件,在保存的界面里記得把Write Cartesians復選框取消掉,以使得保存出的坐標是以內坐標方式記錄的。
我們打算在B3LYP/6-31G*下做掃描,想讓C1-O5鍵轉180度,使得上圖的右圖中H6從H2的反方向轉到與H2重合。因此,我們得從methanol.gjf里去看內坐標的定義,看讓哪個二面角變量的改變可以使得H6繞著C1-O5軸旋轉。在gjf里可見
C
H 1 B1
H 1 B2 2 A1
H 1 B3 2 A2 3 D1
O 1 B4 2 A3 3 D2
H 5 B5 1 A4 2 D3
顯然,D3對應H6-O5-C1-H2二面角,因此如果讓D3變化,即繞著C1-O5轉,就可以達到目的了。我們讓這個變量每10度變化一次,因此總步數應當是180/10=18步。遂把gjf文件改成下面這樣,對應methanol_scan.gjf:
# b3lyp/6-31g(d) scan nosymm
[空行]
Title Card Required
[空行]
0 1
C
H 1 B1
H 1 B2 2 A1
H 1 B3 2 A2 3 D1 0
O 1 B4 2 A3 3 D2 0
H 5 B5 1 A4 2 D3 0
[空行]
B1 1.09337920
B2 1.10125884
B3 1.10125884
B4 1.41863900
B5 0.96872260
A1 108.05956655
A2 108.05956655
A3 106.69693538
A4 107.66742719
D1 117.09085412
D2 -121.45457294
D3 180.00000000 18 10.
注意,凡是程序需要讀入浮點數的地方,絕對不能寫成整數,因此此例步長必須寫10.或者10.0而不能寫10。另外,做剛性掃描的過程中,如果涉及到點群的改變,不加nosymm關鍵詞的話有時會出問題,而且看到的掃描軌跡可能不連續。一開始結構是Cs點群,二面角改變一下就成C1點群了,所以我們這里用了nosymm。關于nosymm的更多說明看《談談Gaussian中的對稱性與nosymm關鍵詞的使用》(http://www.shanxitv.org/297)。
這個體系小、計算級別低,很快就算完了。計算過程中途輸出的信息不用管,直接把輸出文件拉到末尾,可以看到下面的信息,是掃描過程的匯總。D3是指被掃描的那個變量,當前用的B3LYP屬于SCF一類方法,所以每個點的能量顯示在SCF那一列下面,單位是Hartree。
Summary of the potential surface scan:
N D3 SCF
---- --------- -----------
1 180.0000 -115.71441
2 190.0000 -115.71425
3 200.0000 -115.71382
4 210.0000 -115.71320
...略
把methanol_scan.out拖到GaussView里,在窗口左上角可以看到幀號。當前掃描18步,初始結構也算一個結構,因此一共19幀,第一幀對應輸入文件里的結構。選擇Results - Scan,可以看到掃描過程的能量變化,如下圖所示。點擊其中一個點,圖形窗口就會切換到相應的幀。在窗口下方可以看到這個點對應的能量和被掃描的坐標的當前值。
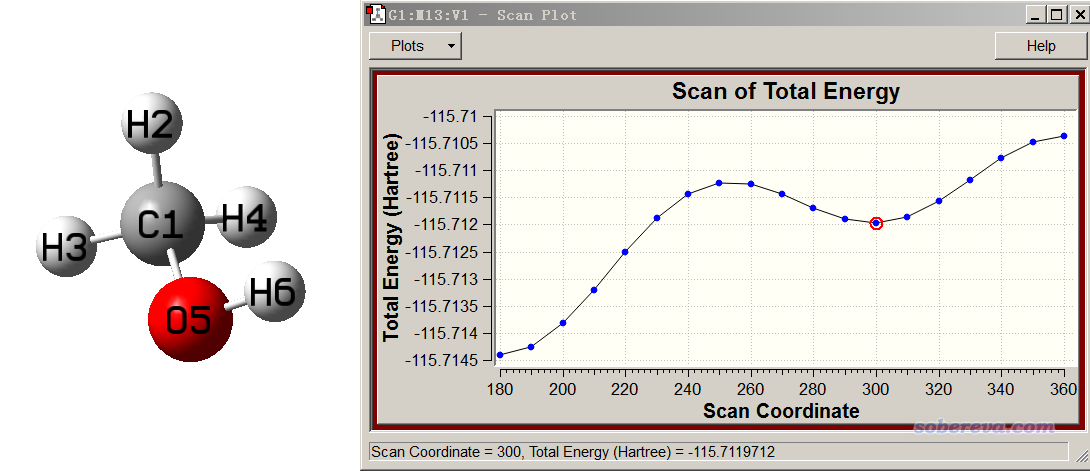
從掃描曲線上,可以看到有兩個極小點(對于這條曲線而非整個勢能面而言),一個是D3=180度,正好對應初始結構,這顯然是能量最低的結構。另一個局部極小點在D3大約300度的位置,結構如上圖左邊所示。而當H6轉到與H2沿著C-O鍵軸看正好重合的時候,能量達到了全局最大。
通過這個圖中的極大點和相鄰極小點位置,我們可以粗略估計旋轉勢壘。但是要想精確計算的話,應當用這個圖中極小點和極大點的坐標分別作為初猜結構去準確優化極小點和過渡態結構(找過渡態參考《簡談Gaussian里找過渡態的關鍵詞opt=TS和QST2、3》http://www.shanxitv.org/460),然后再用更高級別算單點能再求差。
做剛性掃描比較煩人的一個情況是想掃描的那個坐標在GaussView直接保存的gjf文件沒有恰好對應的變量。此時要么想辦法修改體系坐標書寫方式使得被掃描的坐標能通過某個幾何變量表示;要么手動在GaussView里每修改一次坐標就保存一個輸入文件,掃多少步就產生多少個輸入文件,然后批量運行、批量提取單點能。Linux下批量執行的方法看《使用Gaussian時的幾個實用腳本和命令》(http://www.shanxitv.org/258)。
2.3 剛性掃描實例2:掃描Li+...苯之間的距離
這個例子中我們要掃描Li+與苯的苯環中心在垂直于苯環方向的距離。這個目的有兩種實現方式,第一種是基于笛卡爾坐標來設定掃描方式,下面說一下。我們先在GaussView里畫一個苯,做對稱化成為D6h點群,用便宜的B3LYP/6-31G*優化一下,然后以笛卡爾坐標方式保存為gjf文件。此文件里,苯正好在Z=0的XY笛卡爾平面上,苯環中心就是(0,0,0)位置。假設我們想掃描Li+,使之從距離苯環中心5埃處逐漸接近苯環中心,共10步,每一步移動0.3埃,最終移動到相距2埃的位置,我們應當把輸入文件寫成這樣(Ben_Li_Cart.gjf):
# M062X/6-311g(d) scan pop=always
[空行]
Title Card Required
[空行]
1 1
C 0.00000000 1.39650157 0.00000000
C 1.20940584 0.69825078 -0.00000000
C 1.20940584 -0.69825078 -0.00000000
C 0.00000000 -1.39650157 0.00000000
C -1.20940584 -0.69825078 0.00000000
C -1.20940584 0.69825078 0.00000000
H 0.00000000 2.48320512 -0.00000000
H 2.15051871 1.24160256 -0.00000000
H 2.15051871 -1.24160256 -0.00000000
H -0.00000000 -2.48320512 0.00000000
H -2.15051871 -1.24160256 0.00000000
H -2.15051871 1.24160256 0.00000000
Li 0. 0. Z
[空行]
Z= 5.0 10 -0.3
可見我們手動添加了一個Li原子,X和Y都為0,Z坐標以變量表示,初值為5.0,最后一步距離苯環中心將是5.0+10*(-0.3)=2.0埃。此例為了讓結果達到基本定量合理,用了算弱相互作用不錯的M06-2X結合比前例稍微大一些的基組。由于掃描過程始終是C6v點群,所以此例我們不用寫nosymm關鍵詞,如果寫了這個關鍵詞的話由于沒法利用對稱性加速計算會多花很多時間。
有量子化學常識的人都知道Gaussian里只能設定體系總電荷,沒法直接指定Li帶的電荷。但Li在實際體系中到底帶多少電荷?隨著掃描的進行其電荷量如何變化?想了解這個問題,最簡單的做法就是在掃描的時候像本例這樣同時寫pop=always關鍵詞,這樣掃描的每一步都會做一次布居分析從而得到原子電荷。
執行上面這個任務,把輸出文件用GaussView打開,掃描曲線如下。圖應該從右往左看
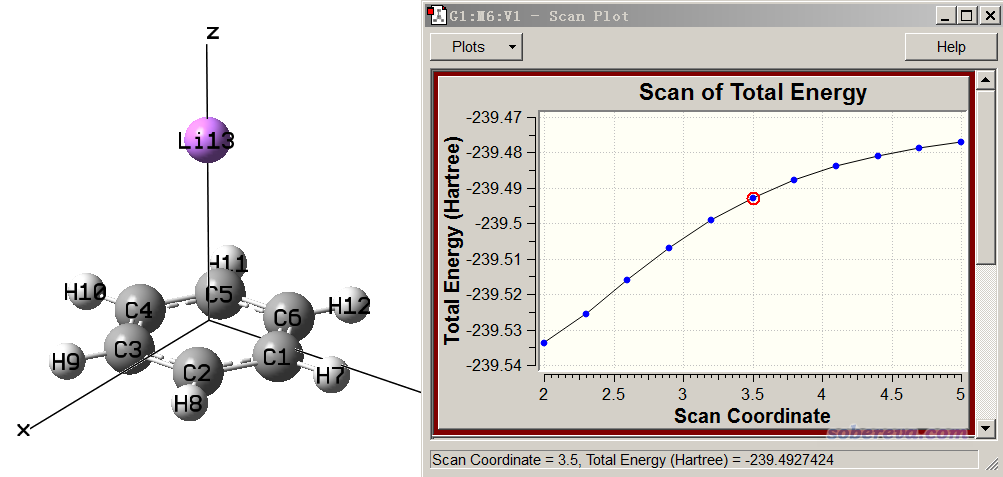
可見隨著Li逐漸接近苯環中心,體系能量顯著下降。我們可以直接在GaussView里觀看原子電荷的變化,做法是在Scan界面里選擇Plots,選Plot Molecular Property,再選Atomic Charge,然后輸入13,點OK。之后在能量變化圖的下方就可以看到序號為13的Li原子的Mulliken電荷隨掃描的變化了,如下所示
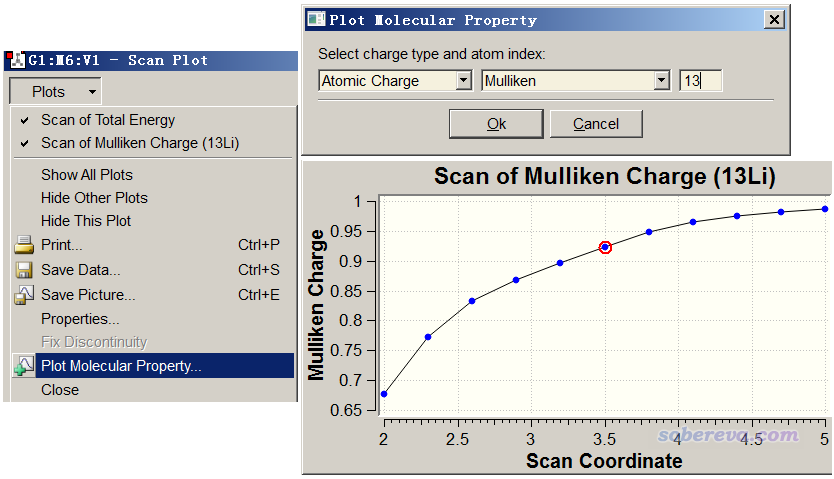
由圖可見,當Li距離苯很遠的時候,Li帶的原子電荷接近1.0,是個典型的陽離子。而隨著距離接近,苯上的電子就逐漸往Li+上轉移,導致在距離較近的時候Li帶的原子電荷明顯達不到1.0了。注意,Mulliken電荷只是一個比較low的原子電荷模型,存在容易低估離子性、怕彌散函數等問題,相關討論參見筆者的《原子電荷計算方法的對比》(http://www.whxb.pku.edu.cn/CN/abstract/abstract27818.shtml)一文,在實際研究中用ADCH或NPA電荷說事更好。如果想對電荷轉移特征了解更多,還可以用Multiwfn做更細致的布居分析,看Multiwfn手冊4.7.0節的例子,以及繪制密度差圖,看《使用Multiwfn作電子密度差圖》(http://www.shanxitv.org/113)。
Plot Molecular Property界面里還可以繪制別的,比如繪制幾何變量隨掃描的改變。使用pop=always后,還可以在這個界面里選擇繪制偶極矩矢量或其總大小。而如果不用pop=always的話,程序只會在初始結構計算時做布居分析。
這個例子也可以在內坐標下實現掃描,但是需要用到虛原子。做法是在GaussView里顯示出苯之后,在苯環中心添加一個虛原子X(把苯環上的六個碳都選中成為黃色,在Builder窗口里選X原子,然后點右鍵選Builder - Place Fragment at Centroid...),然后再在虛原子上加個氫,把X-H的鍵軸調成與苯環垂直,然后把氫替換成Li原子。之后保存內坐標形式的gjf文件,會發現其中有一個變量正好對應虛原子和Li原子之間的距離,因此對這個距離掃描即可。輸入文件是Ben_Li_Zmat.gjf。
實際研究中,可能環是斜著的,要掃描某個原子與它的環中心的距離。此時既可以用上面提的內坐標方式掃描,也可以用笛卡爾坐標方式掃描。對于后者,由于Gaussian里在剛性掃描的時候沒法讓某個原子按照某個自定義矢量來移動,因此需要先旋轉體系,讓環平面恰好平行于某個笛卡爾平面,比如平行于XY,這樣才能通過掃描Z坐標實現目的。按照《調節平面分子間距的方法》(http://www.shanxitv.org/178)里的“方法二”,借助筆者自寫的VMD程序的腳本,可以輕易讓某個環平面恰好平行XY平面。
值得一提的是,在掃描過程中如果能進行深入細致的電子結構分析,可以獲得遠比結構+能量豐富得多的信息,讓研究文章充實起來。想實現這點,可以用《產生Gaussian的IRC和SCAN任務每個點的波函數文件的工具》(http://www.shanxitv.org/199)一文介紹的筆者寫的SCANsplit工具載入剛性掃描的輸出文件,將掃描過程中每個結構轉化為單點任務輸入文件,批量執行后就有了記錄每個點波函數的fch或wfn文件,之后再通過腳本和一些Linux下的命令就可以自動調用Multiwfn考察電荷分布、成鍵等電子結構方面的信息隨掃描過程的變化,詳見《通過鍵級曲線和ELF/LOL/RDG等值面動畫研究化學反應過程》(http://www.shanxitv.org/200),可以討論的手段見《Multiwfn支持的分析化學鍵的方法一覽》(http://www.shanxitv.org/471)。
2.4 剛性掃描實例3:掃描氟化氫鍵長獲得解離曲線
這一節我們通過DFT方法研究氟化氫分子的H-F鍵斷裂過程的勢能曲線。這個曲線我之前在《淺談為什么優化和振動分析不需要用大基組》(http://www.shanxitv.org/387)給出過,當時對不同級別掃描出的曲線進行了對比。H-F鍵的斷裂過程中體系會由閉殼層分子逐漸變成H自由基和F自由基。當鍵長在平衡距離附近的時候,體系是閉殼層狀態,計算沒有什么特殊的,而距離拉遠后,超過所謂的“不穩定點”對應的鍵長,整個體系就成了雙自由基狀態,屬于自旋極化單重態,這時對于DFT計算而言需要做對稱破缺計算。如果對這點不懂,一定要看《談談片段組合波函數與自旋極化單重態》(http://www.shanxitv.org/82)。因此,掃描這種共價鍵均裂解離曲線不是看起來那么簡單。而如果你掃描的是NaCl這樣的離子鍵異裂成Na+和Cl-的解離曲線,那就不需要考慮這些問題了,因為解離后變成的Na+和Cl-都是閉殼層狀態。
對于這個體系的掃描,我的建議是這樣:
(1)先在H和F距離很遠、鍵完全斷裂的情況下產生對稱破缺波函數,對應雙自由基狀態
(2)用非限制性開殼層形式(U)做掃描,讓H-F距離逐漸減小,第一步的初猜波函數直接讀取(1)收斂的波函數。由于之后每一步都會用上一步收斂的波函數做初猜,所以一開始的波函數的對稱破缺狀態會一直維持下去。當距離減小到已經小于不穩定點的時候,由于此時基態波函數是閉殼層波函數,因此從此開始每一步的結果和用限制性閉殼層(R)形式計算的結果將會完全一樣。
第1步的輸入文件是下面這樣(HF_broken.gjf),對應鍵長4埃。用U明確指定做非限制性計算,用guess=mix是為了打破初猜波函數的對稱性,但是直接用guess=mix未必能收斂到真正的對應當前結構下基態的對稱破缺波函數,因此又加上了stable=opt來對收斂的波函數進行檢測,如果發現不穩定,會自動嘗試優化出最穩定的波函數。
%chk=C:\HF.chk
# UB3LYP/def2TZVP guess=mix stable=opt
[空行]
Title Card Required
[空行]
0 1
F
H 1 B1
[空行]
B1 4.0
第2步的輸入文件如下(HF_scan.gjf),從4.0埃掃到4.0-0.06*55=0.7埃。涉及對稱破缺的計算加上nosymm會比較穩妥,所以這里用了nosymm。
%oldchk=C:\HF.chk
# UB3LYP/def2TZVP scan guess=read nosymm
[空行]
Title Card Required
[空行]
0 1
F
H 1 B1
[空行]
B1 4.0 55 -0.06
最終掃出來的曲線如下,非常理想。如果掃描出的這種曲線有一些明顯的突躍,通常是那個點恰好沒有收斂到與周圍的點特征基本相同的波函數所致,此時可以嘗試把掃描步長減小再試,這樣有助于保持波函數的連續性,畢竟每個點的初猜自動用上一個點的收斂的波函數,二者結構相差越小則波函數的狀態越容易繼承下去。
掃描共價鍵的解離曲線還有個做法,只需要一步,就是使用比如# UM062X/TZVP guess(always,mix) scan nosymm這種關鍵詞,此時讓鍵長從大變小來掃描,還是讓鍵長從小變大來掃描,其實都一樣,因為這里用了always,代表每一步重新產生初猜波函數;而且由于用了mix來使得每一步都試圖得到對稱破缺態,如果對稱破缺態更穩定,那么這個點就收斂到對稱破缺態,而如果閉殼層態更穩定,那么就自動收斂到對應閉殼層的波函數。大家可以嘗試對乙烷的C-C鍵用這個關鍵詞掃描,會發現可以得到正確的解離曲線,掃到最后相當于兩個甲基自由基。但是這套關鍵詞用于掃描上面的氟化氫體系的話會發現行不通,會掃得亂七八糟,因為對這個體系的很多點,光靠guess=mix產生的初猜波函數收斂不到實際基態波函數上。不信的話可以看一下HF_scan_mix_always.out的曲線,就是以這種方式算的,其最后一個點SCF沒有收斂,只看其它點的話,會看到能量曲線不合理,因為拉遠后能量變化沒有趨于水平。雖然從輸出文件中看到超過不穩定點的結構的<S**2>確實不為0,因此確實得到了對稱破缺態,但是和之前我們掃描出來的輸出文件里相應的點<S**2>不符(明顯偏小)。有興趣的話讀者可以對諸如鍵長為3.5埃的時候用UB3LYP/def2TZVP guess(mix,always) nosymm產生fch文件,用Multiwfn看看自旋密度分布了解是怎么回事(參見《談談自旋密度、自旋布居以及在Multiwfn中的繪制和計算》http://www.shanxitv.org/353),你會發現,并不是如期望的在H和F上各有單電子且自旋相反,而是alpha和beta單電子同時出現在了F上面!
可以計算共價鍵解離曲線的方法極多。使用恰當的泛函,通過對稱破缺方式計算通常就可以給出不錯的解離曲線。但如果要求更高,可以用UCCSD(T)。而對于牽扯多重鍵斷裂的問題,情況復雜、靜態相關很強,通常需要考慮用較復雜但是普適性強的多參考方法如CASPT2以確保得到靠譜的結果。
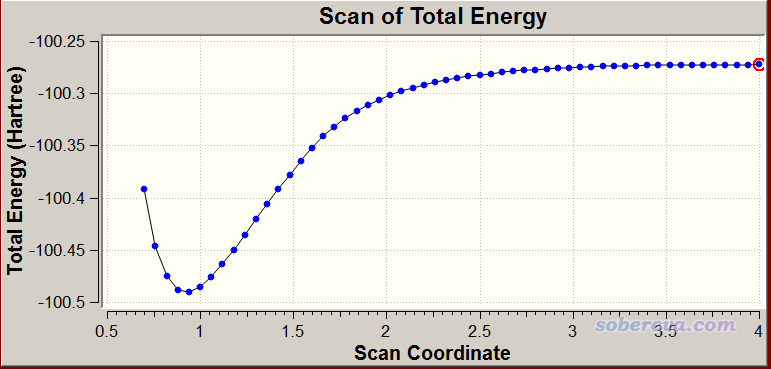
2.5 剛性掃描實例4:同時掃描水分子的鍵長與鍵角
這個例子演示二維掃描。對水分子,令兩個O-H鍵長同時從0.92掃到1.02埃,每步0.01埃,共10步。與此同時,讓鍵角從95度掃到115度,每步2埃,共10步。因此掃描任務中要算的單點能的次數是(10+1)*(10+1)=121個。顯然,隨著被掃描的變量數增加,剛性掃描的耗時呈幾何式增加。對某個剛性掃描任務要想估計能不能掃得動,你可以算一個單點看看耗時,然后乘以要算的點數。
當前這個任務的輸入文件如下(H2O_2Dscan.gjf)
# B3LYP/def2SVP scan
[空行]
Title Card Required
[空行]
0 1
O
H 1 B1
H 1 B1 2 A1
[空行]
B1 0.92 10 0.01
A1 95.0 10 2.0
算完之后,可以把輸出文件末尾的匯總數據用Sigmaplot、Surfer、Origin之類繪制成曲面圖或者填色圖或者等值線圖便于考察。如果將輸出文件載入GaussView,也可以看到有121幀,并可以觀看掃描軌跡。如果進入Results - Scan,會看到下圖,曲面圖的縱坐標對應電子能量,在下方還有投影的等值線圖。
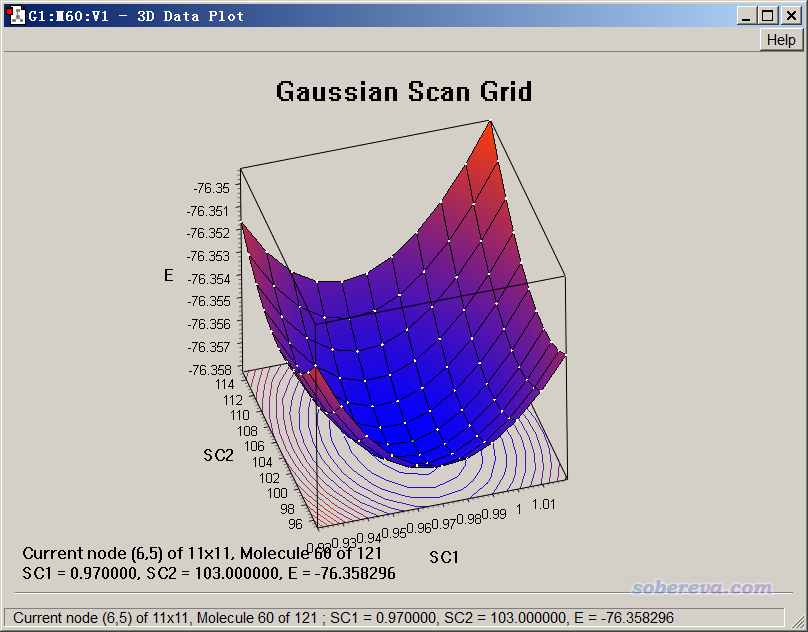
點擊曲面上的小白點使之成為綠色,可以使狀態欄下方顯示相應的點對應的幾何變量的數值和能量,圖形窗口里的結構也會切換到對應的幀。由上圖可見,所有掃描的點里能量最低的是B1=0.97埃、A1=103度的那個點,比起其它點更接近于水分子的勢能面最低點。
類似地,我們還可以用Gaussian同時掃描更多的坐標,但超過兩個坐標時,結果就沒法直接用GaussView繪圖直觀展現了。
3 使用dimerscan結合Gaussian做二聚體的剛性掃描
借助筆者寫的dimerscan和xyz2QC程序,我們可以實現很多沒法直接用Gaussian的scan設定實現的二聚體的剛性掃描。下面通過兩個例子來體現這點。dimerscan可以從《考察SAPT能量分解的能量項隨分子二聚體間距變化的簡單方法》(http://www.shanxitv.org/469)頁面里下載,xyz2QC是筆者開發的團簇構型與分子構象搜索molclus程序包中帶的工具,可以在molclus主頁下載:http://www.keinsci.com/research/molclus.html。
3.1 實例:苯酚二聚體的氫鍵距離掃描
經常研究分子間相互作用的人,往往會遇到一個問題就是想對二聚體之間做剛性掃描,但是GaussView產生的gjf文件里沒有正好對應于想掃描的坐標的幾何變量。比如下面的苯酚二聚體
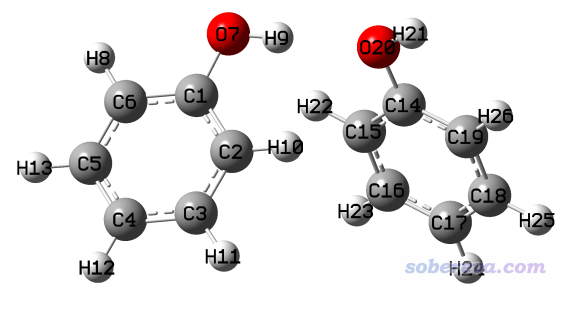
我們想考察體系能量隨氫鍵距離變化而發生的變化,從而得到不同距離下兩個單體間的弱相互作用能,因此我們應該掃描H9-O20或者O7-O20鍵長。但不幸的是,GaussView里保存的內坐標形式的輸入文件中(phenoldimer.gjf),O20的鍵長項是參考C14來定義的,因此只使用Gaussian的話沒法達到我們的掃描目的。雖然如后文所述,用柔性掃描的話想怎么定義都可以,但是柔性掃描不僅昂貴,而且掃描過程中其它坐標都可能顯著變化,和我們期望的研究目的不符。像這種情況,我們要掃描氫鍵距離,就得用dimerscan結合xyz2QC程序來實現。下面提到的文件都在本文文件包里rigid\phenoldimer目錄下。
我們先用GaussView打開phenoldimer.gjf,保存成笛卡爾坐標形式的gjf。將gjf里原子坐標以外的部分都刪掉,在坐標前面插入兩行,第一行是兩個單體各自的原子數,第二行是計算兩個單體時分別用的電荷和自旋多重度(對當前研究無影響),然后把這個文件保存為dimer.txt。此文件當前內容的形式如下
13 13
0 1 0 1
[苯酚單體1的坐標]
[苯酚單體2的坐標]
假設我們想掃描H9-O20,從1.7埃掃20步,每步增加0.1埃。將dimer.txt放到dimerscan.rar解壓后的目錄中,啟動dimerscan,輸入dimer.txt的路徑,然后依次輸入
9,20
1.7 //初始距離
20 //掃20步
0.1 //每步伸長0.1埃
最后按一次回車退出。此時當前目錄下出現了一堆.inp文件,不用管,那是給PSI4程序做SAPT用的。當前目錄下還出現了scan.xyz,里面每一幀對應掃描過程的一個結構。如果想檢驗下一生成的對不對,可以把這個文件拖到VMD程序(http://www.ks.uiuc.edu/Research/vmd/)里播放動畫確認一下,由動畫可見確實生成得沒錯:
把molclus程序包中的template.gjf里的關鍵詞改為要計算每個點用的級別,比如我們想用比較便宜的PM6-D3方法實現苯酚二聚體掃描,就把這個文件里的關鍵詞設為# PM6D3。然后啟動molclus程序包中的xyz2QC程序,選擇1 Generate multi-step Gaussian input file,輸入scan.xyz的路徑,然后直接按回車代表考慮所有幀,再按回車退出。當前目錄下馬上出現了Gaussian.gjf文件,此文件是Gaussian的多步任務文件,每一步是對每個結構算一次單點。
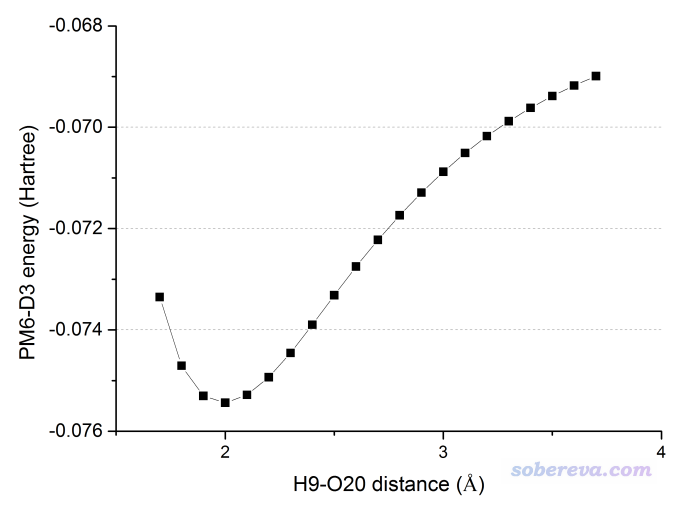
用Gaussian運行Gaussian.gjf,用ultraedit或者Linux下的grep命令將里面所有帶有E(RPM6D3)字樣的行都提取到一個文本文件里,會發現一共有21行(初始結構+掃描的20個結構)。然后把多余的列都刪掉只保留能量的列,導入到Origin里,把掃描的距離信息也插入到里面作為一列,然后用Origin作圖,如下所示(原始文件是phenoldimer.opj),完全與我們預期的相符。
注:當二聚體結構中不同單體的原子順序存在相互交錯的時候,需要先處理一下,確保單體里的原子序號是連著的,否則dimerscan沒法用。處理過程是用GaussView打開二聚體結構文件,在其中一個單體的原子上點右鍵選Select Fragment of ...使這個單體變成黃色,然后按Ctrl+X復制到剪切板,創建一個新窗口,再按Ctrl+Shift+V將之粘貼進去,保存成笛卡爾坐標形式的輸入文件1.gjf。之前的窗口還剩另一個單體,保存為2.gjf。最后將2.gjf里的坐標拼接到1.gjf的坐標后頭去。
3.2 實例:主-客體復合物的接觸距離掃描
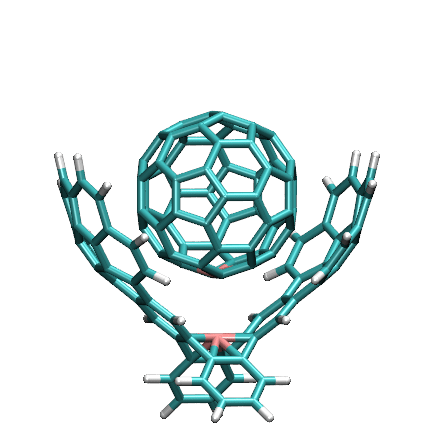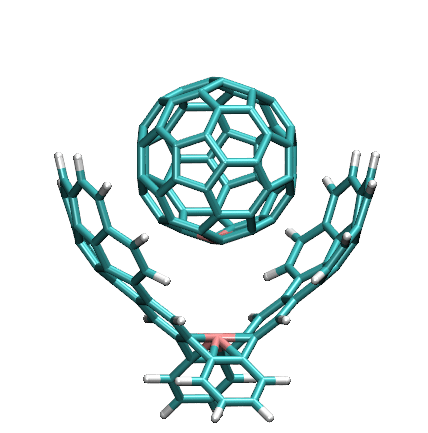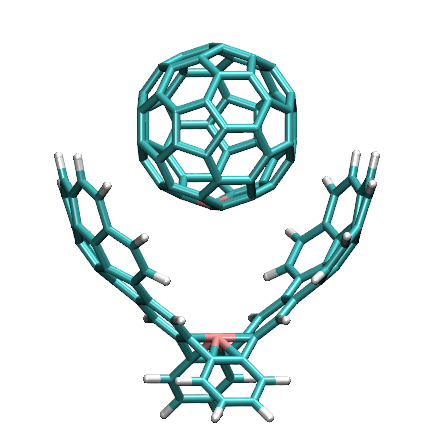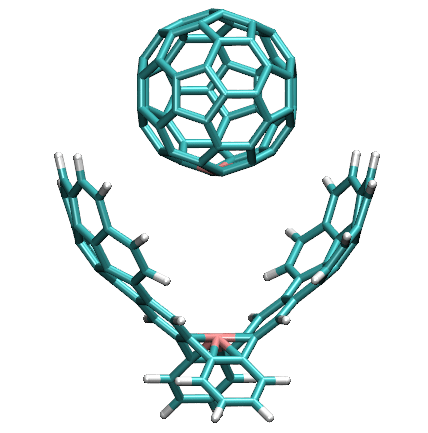
此例我們想對下圖所示的主-客體復合物在紅線方向上進行掃描,看這個富勒烯脫離主體分子過程的能量變化曲線。復合物結構是本文文件包里host-guest目錄下的host-guest.gjf。
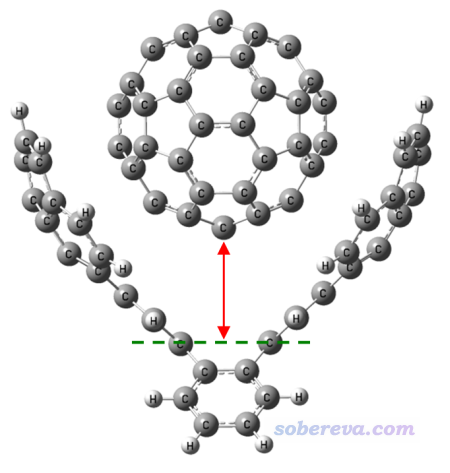
做這個體系的剛性掃描也沒法直接用Gaussian實現,不僅被掃描的兩端恰好沒有原子出現,而且即便有原子,一般也不會恰好有對應的變量可供掃描。對這種情況,我們還是可以用dimerscan+xyz2QC的組合來實現,只不過用之前我們先得在掃描的兩端位置添加虛原子。
我們先在GaussView里將客體分子剪切并粘貼到新窗口里,然后將主體和客體分子分別保存成host.gjf和guest.gjf。然后打開主體分子,在掃描的位點增加一個虛原子,然后保存gjf。對客體分子也這樣增加虛原子并保存。虛原子位置如下所示
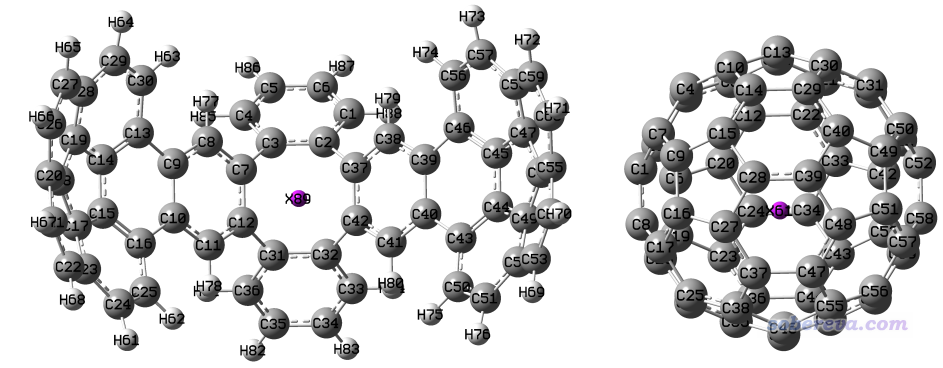
然后把host.gjf和guest.gjf里的坐標合并在一起,改成下面的格式,并保存為dimer.txt。目前包含虛原子X在內主體和客體分子分別有89和61個原子。
89 61
0 1 0 1
[主體分子的坐標]
[客體分子的坐標]
將dimer.txt拷到dimerscan目錄下,啟動dimerscan程序并輸入
dimer.txt
89,150 //新添加的兩個虛原子在整體中的序號,都是每個單體最后一個原子,因此第二個虛原子序號是89+61=150
2.8 //初始距離
30 //30步
0.15 //每步增加0.15埃
用VMD播放一下新產生的scan.xyz,會看到下面的動畫
還是按照上一節的方法,通過xyz2QC程序將scan.xyz轉換成多任務的Gaussian輸入文件Gaussian.gjf,使其中每個子任務都對應于用PM6-D3計算單點。用Gaussian執行此任務,然后提取SCF Done的數據進行作圖,會看到下圖的結果:
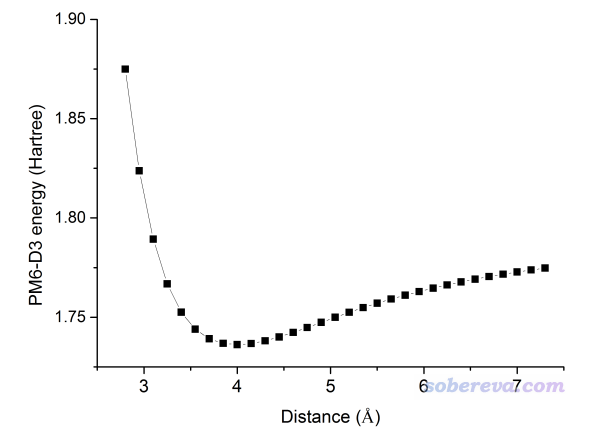
值得一提的是,一般繪制這種圖都應該用極小點結構作為縱軸零點,令每個點的能量都減去這個能量,使得圖上縱坐標體現的是相對于極小點的能量變化,此時縱坐標才有化學意義。
4 使用gentor結合Gaussian做二面角的剛性掃描
往往我們想剛性掃描二面角,并且掃描的時候讓整個基團連帶地轉動。比如下面這個體系,我們想看看繞著C1-C4鍵軸轉動時勢能曲線是什么樣。文件包里的C2H4ClBr.gjf是保存出來的基于內坐標的結構。
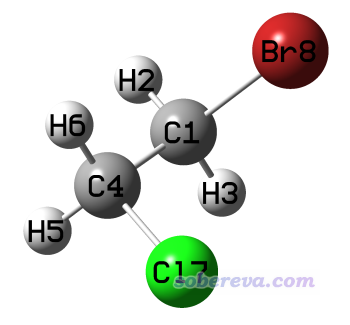
但我們的目的沒法簡單地達到。比如雖然gjf文件里可以發現D5對應Br8-C1-C4-H6二面角,但是如果掃這個角的話,發現Br8和H6雖然都轉動了,而其它原子并沒有跟著它倆的轉動而轉動,因此隨著掃描的進行結構愈發扭曲,完全違背我們的意愿。
對于這種繞著鍵旋轉的剛性掃描,需要借助筆者開發的molclus程序包里的gentor子程序實現。它可以按照用戶的要求對某個鍵依次進行旋轉,并將產生的結構依次寫入到traj.xyz文件里,之后再用xyz2QC轉化成Gaussian輸入文件,計算后提取所有能量就可以作圖了。
使用gentor前需要先把當前結構轉化成.xyz文件。最簡單的方法就是直接把C2H4ClBr.gjf載入GaussView,保存成笛卡爾坐標的形式的gjf,然后手動把此文件改寫成下面的樣子,第一行是原子數,第二行是注釋(內容隨意),然后把此文件保存為mol.xyz
8
niconiconi
C 0.00000000 0.00000000 0.00000000
H 0.00000000 0.00000000 1.07000000
H 1.00880579 0.00000000 -0.35666635
C -0.72596336 1.25740469 -0.51333288
H -0.22269110 2.13105477 -0.15506960
H -1.73533208 1.25642706 -0.15826411
Cl -0.72317775 1.25901418 -2.27332994
Br -0.90038238 -1.55950848 -0.63666702
把mol.xyz放到molclus目錄下的gentor目錄下,并把自帶的gentor.ini改寫為下面這樣:
1-4
e10
這代表從初始結構開始,繞著1-4鍵旋轉360度,每10度產生一個結構。其中第一個結構對應旋轉0度,最后一個結構對應旋轉350度,因此共36個結構。gentor提供了很豐富的選項來控制怎么旋轉,用法詳細介紹見《gentor:掃描方式做分子構象搜索的便捷工具》(http://bbs.keinsci.com/thread-2388-1-1.html),靠gentor甚至可以實現多個二面角同步變化的掃描。
運行gentor,從屏幕的提示中會看到成功產生了36個構象(倘若有嚴重不合理接觸的,會被自動剔除),這些結構輸出在了當前目錄下的traj.xyz中。將此文件載入VMD播放一下,會看到下面的動畫
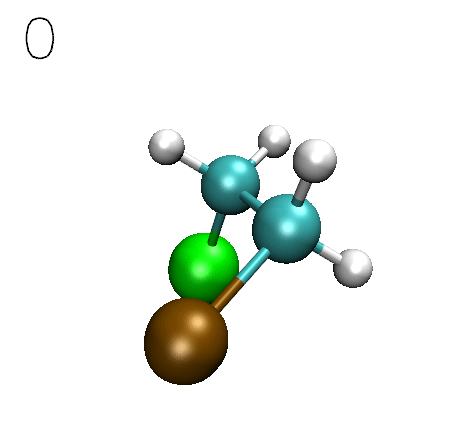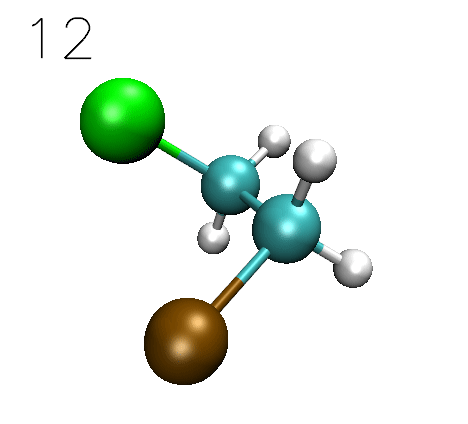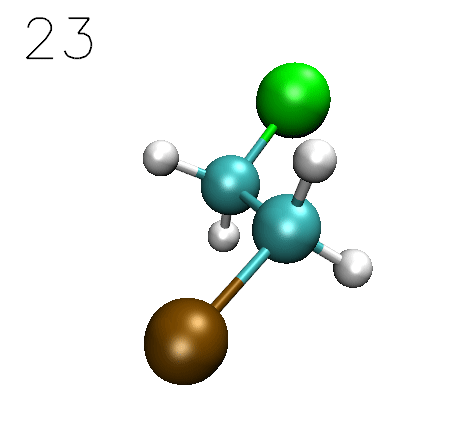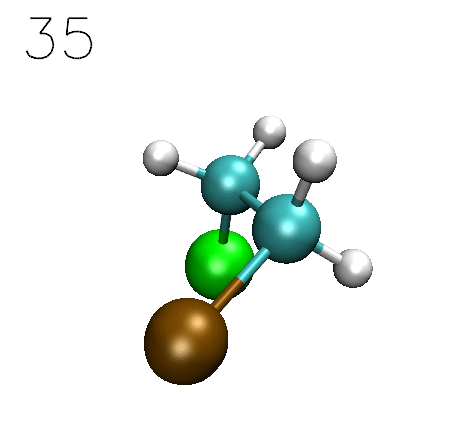
還是按之前的做法,用xyz2QC工具將traj.xyz轉化成含多任務的單一Gaussian輸入文件Gaussian.gjf,為省時間計算級別還是用粗糙的PM6-D3。按照之前的步驟把能量取出來,并且把每個點對應的Cl7-C4-C1-Br8二面角數值附上去,用Origin作圖,如下所示。(第一個結構的這個二面角數值-60.11104從GaussView里直接讀,之后各個結構的二面角數值利用excel產生等差數列就得到了)
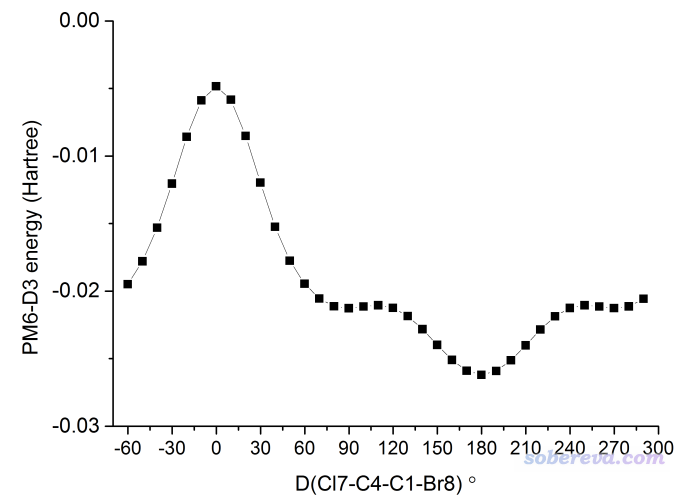
可見圖像首尾相接,因為正好轉了360度。圖中0度的時候正好是Cl和Br處于重疊構型,由于位阻大,所以能量很高。當轉到180度時,正好Cl在Br反方向,位阻最小,因此能量最低。
我們當前得到的這個二面角的剛性掃描圖其實實際意義并不太大。因為在現實中,隨著二面角旋轉,其它變量肯定會自發地變化,比如當Cl7-C4-C1-Br8正好處于0度時,中間的C-C鍵肯定會自發地輕微變長以讓Cl和Br的位阻適當減小,C-C-Br和C-C-Cl鍵角也肯定在二面角旋轉過程中出現可查覺的變化。如果忽略了這些效應,必定得到的二面角旋轉曲線不夠真實,并可能明顯高于實際旋轉勢壘。想較真實地考慮這些效應,就得用下文介紹的柔性掃描了。
5 用Gaussian做柔性掃描的方法和實例
5.1 基本用法
前面說了,柔性掃描就相當于對每一個掃描點結構中的非掃描變量做限制性優化。在Gaussian下做柔性掃描一般是在冗余內坐標下做,但其實也可以在內坐標下做,而不能在笛卡爾坐標下做。注意這里說的在什么坐標下做指的是限制性優化過程用的坐標,和輸入文件里把體系結構寫成什么坐標沒有任何關系(通常我們在冗余內坐標下做柔性掃描都是用比較方便的笛卡爾坐標形式)。
在冗余內坐標下做柔性掃描需要寫opt=modredundant關鍵詞,然后在坐標末尾空一行定義被掃描的變量,格式為:
原子1 原子2 [原子3] [原子4] S 步數 步長
寫兩個、三個、四個原子序號的話分別對應掃鍵長、鍵角、二面角。初值就是輸入文件里的結構的相應變量的數值,所以要改初值的話應該在GaussView里修改幾何坐標后再保存gjf。(在大約G09 C.01及之前版本,還可以在S后頭直接設定初值,但是對于之后的版本都沒法設初值了)
相對于剛性掃描,做柔性掃描的一個便利之處是不用考慮輸入文件里有沒有現成的對應于被掃描變量的坐標,想掃什么直接定義即可。柔性掃描也是可以同時掃多個,只要寫多行這種設定即可,對于二維柔性掃描也是可以用GaussView顯示出曲面圖。柔性掃描的輸出文件末尾不像剛性掃描那樣有匯總信息,但可以在GaussView里顯示出曲線或者曲面圖之后,在圖上點右鍵選導出數據,然后再用第三方程序繪圖。
做柔性掃描的時候也可以同時進行凍結設定,比如寫
8 F
10 F
就代表掃描過程中8、10號原子位置不允許改變(但使用nosymm的時候從動畫上才會看到完全沒動)。這點在《在Gaussian中做限制性優化的方法》(http://www.shanxitv.org/404)文中也介紹過。
柔性掃描時不能像剛性掃描時那樣允許虛原子出現在掃描設定中。
掃描某個原子時,它所在的片段上其它原子也會連帶地一起運動,這點比起剛性掃描好。比如之前需要借助gentor才能實現在剛性掃描過程中讓整個基團轉起來,而在柔性掃描時只需要對一個二面角掃描即可實現。
5.2 柔性掃描的一些相關問題
由于幾何優化比單點能的計算昂貴起碼一個數量級,因此,在同樣的掃描設定下,做柔性掃描比做剛性掃描也昂貴至少一個數量級,所以沒事別瞎用昂貴的柔性掃描。不得不用昂貴的柔性掃描的時候,如果對精度要求不是很高,可以用一些便宜的計算級別,比如PM7這樣的半經驗方法,比起用常用的DFT能節約兩個數量級的時間。另外,要注意柔性掃描是注定不可能用來得到極小點和過渡態的精確結構的,且不說別的,起碼被優化的變量一般不可能有某個點的數值恰好等于極小點或過渡態結構的數值。另外,柔性掃描是無法替代IRC的用處的,柔性掃描的曲線不可能與IRC正好相同,畢竟算法上相差很大,至多是對于極個別反應,其反應坐標基本上可以通過某個幾何變量表現(比如激發態質子轉移),那么掃描這個變量得到的曲線和IRC才有相似性。
由于柔性掃描過程本質上相當于一大批限制性優化,而幾何優化過程中Gaussian要求不能中途出現三個原子排成直線的情況,因此柔性掃描任務中途有可能因為出現三個原子排成直線而報錯。這也沒有什么簡單而且一定奏效的做法可以解決。碰上這種情況可以考慮調整掃描設定試圖避開出現這種現象的可能;或者對柔性掃描中失敗的點手動做限制性優化,然后遇到三個原子排成直線時,取最后的結構沿用之前的設定進一步優化(由于此時初始結構里就已經有三個原子排成直線,Gaussian會做一些恰當處理使得優化往往可以繼續進行)。
由于限制性優化時需要計算原子受力(梯度的負矢量),Gaussian只能對那些有解析梯度的方法做柔性掃描。諸如CCSD(T)那些只能算能量而連一階解析導數都沒有的方法,拿它們做柔性掃描想都別想;而且就算真的能做一般也根本算不動,因為柔性掃描這種任務本身就極其昂貴,其中要計算受力的次數巨多,而CCSD(T)就連算個單點能都很昂貴,更別提計算梯度了。之前看有的初學者居然試圖拿CCSD(T)/aug-cc-pVQZ去做柔性掃描,而且還打算掃62步,真是都不知該從哪吐槽。
做剛性掃描的時候,對計算級別的要求和單點一樣,也就是在計算條件允許范圍內,盡量用比較好的級別(但也別過度浪費),因為能量對計算級別敏感。而大家知道做幾何優化不需要太好級別就可以得到不錯結果,見《淺談為什么優化和振動分析不需要用大基組》(http://www.shanxitv.org/387),類似地,做柔性掃描的過程也完全不需要太好的級別。如果你想讓掃描曲線更準確,那么之后對柔性掃描產生的每個點再用較好級別計算單點能就行了。別直接直接用昂貴的級別做柔性掃描,否則純粹是在浪費計算資源。
經常有人問為什么他得到的柔性掃描曲線里出現了能量的突躍,問是否正常、結果能不能用。突躍是怎么造成的,通過下面這個勢能面圖就可以秒懂,越紅的地方勢能越高,越藍的地方勢能越低
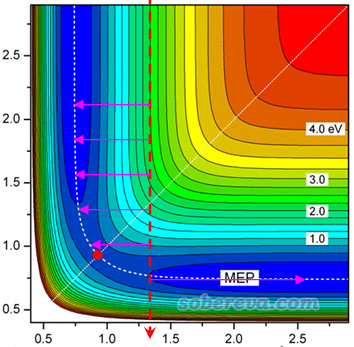
這張圖里面縱坐標是被掃描的變量值,按照箭頭方向從上到下掃描,橫坐標是不被掃描的變量。可見,由于掃描的每一步都是限制性優化,沒有被掃描的變量會被自動優化到與其初始坐標值最近的極小點去,相當于在水平方向上按照粉色箭頭的方向優化。圖中紅色圓球對應過渡態位置,當被掃描變量越過這個紅色圓球到達其下方之后,粉色箭頭的末端就會從圖的左側突然轉變到圖的右端去。由于紅色圓球附近的兩個掃描點在限制性優化后對應的結構發生了巨大變化,因此體系能量也會發生一個突躍,在曲線圖上就像一個斷崖一樣。而斷崖處的那個極大點,如果你觀看對應的結構發現與過渡態比較像,那么可以用這個點作為過渡態的初猜來用opt=TS找過渡態,本文5.5節會給出例子。
Gaussian的柔性掃描和上圖示意的還有所不同。對于Gaussian的柔性掃描的每一步,不被掃描的變量的初值并不是輸入文件里的這個變量值,而是上一步的這個變量被優化后的值。由于這點,柔性掃描過程存在“滯后性”。有人問為什么通過柔性掃描對某個二面角旋轉360度,得到的曲線的末端和始端能量、結構并不重合,其實就是這個原因。
柔性掃描和普通幾何優化一樣可能遇到難收斂之類情況,碰到這種問題時《量子化學計算中幫助幾何優化收斂的常用方法》(http://www.shanxitv.org/164)中提到的一些解決辦法也是可以嘗試的,比如改成gdiis算法、用recalc/calcall、放寬收斂限、恰當增大maxcyc等。
5.3 柔性掃描實例1:柔性掃描C2H4ClBr的二面角
我們之前將gentor與Gaussian結合對C2H4ClBr的C-C鍵做了剛性掃描,這回我們做一下柔性掃描。我們想讓Cl7-C4-C1-Br8的二面角從0度開始掃一圈,每10度掃一步,因此我們應當在GaussView里先把這個二面角設為0,然后保存gjf并改成下面這樣(C2H4ClBr_relax.gjf)
# PM7 opt=modredundant nosymm
[空行]
Title Card Required
[空行]
0 1
C 0.00000000 0.00000000 0.00000000
H 0.00000000 0.00000000 1.07000000
H 1.00880579 0.00000000 -0.35666635
C -0.72596336 1.25740469 -0.51333288
H -0.05342615 1.83983916 -1.10777699
H -1.06223201 1.83983874 0.31888943
Cl -2.10874985 0.77839276 -1.49111051
Br -0.90038252 -1.55950848 -0.63666682
[空行]
7 4 1 8 S 36 10.
這里為了省時間,用了從G16開始支持的PM7半經驗方法。nosymm加不加不影響掃描結果,這里之所以寫了nosymm是為了避免Gaussian每一步自動把結構調整到標準朝向下從而令掃描軌跡從視覺上看起來別扭、不連續。對于打算觀看柔性掃描軌跡的情況,一般建議在柔性掃描時加上nosymm。
把輸出文件載入進GaussView,在Results - Scan里觀看掃描曲線,如下所示
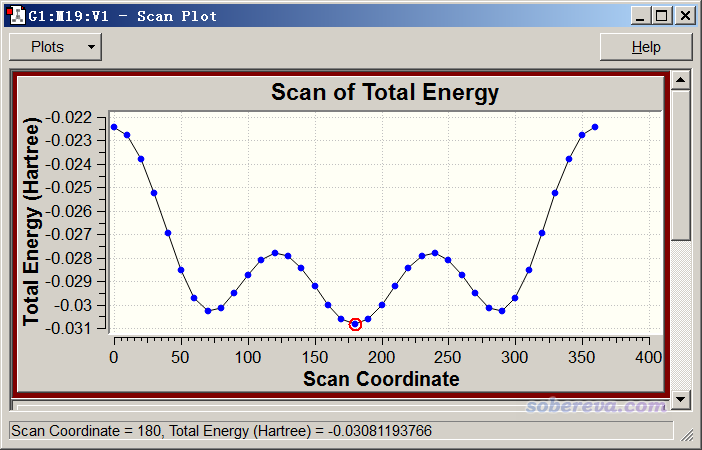
我們還可以考察一下中間的C-C鍵在掃描過程中的變化。選左上角的Plots - Plot Molecular Property,選Bond,然后輸入中間兩個碳的序號,即1和4,然后把窗口下拉框拉到最下面,看到下圖
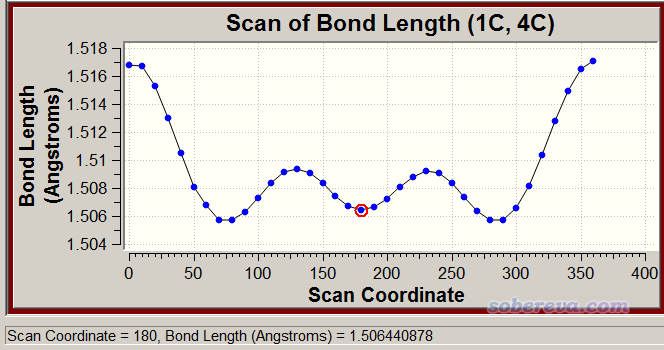
我們可以發現掃描過程中能量變化和C-C鍵鍵長變化有一定正相關性。能量越高,即兩邊基團間的位阻越大的結構下,中間的C-C鍵傾向于被撐得越長。
5.4 柔性掃描實例2:乙酰和甲胺封閉的丙氨酸(ACE-ALA-NME)的構象搜索
ACE-ALA-NME是對丙氨酸兩邊用乙酰和甲胺封閉后的模型結構,這里我們通過二維柔性掃描,試圖尋找這個體系的能量最低構象。體系結構如下所示
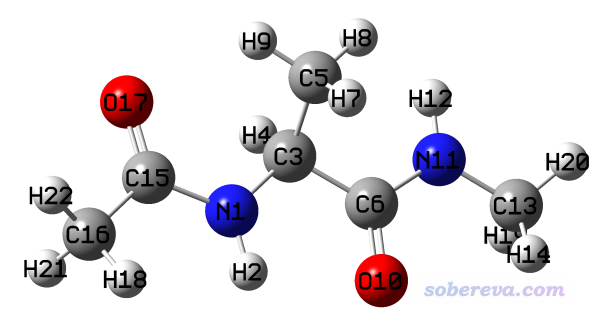
是否對這個體系里的甲基繞著C-C鍵做掃描意義不大,因為甲基非常小而且三個氫化學等價,其旋轉對勢能基本沒什么影響。此體系里的N1-C15和C6-N11都在肽鍵中,肽鍵由于存在局部pi共軛,旋轉勢壘很高,所以也沒必要考慮對其進行掃描。想通過掃描來找這個體系的能量最低構象只需要繞著容易旋轉的N1-C3和C3-C6鍵掃描就行了。對這兩個二面角做柔性掃描的輸入文件如下(ACE-ALA-NME.gjf):
# PM7 opt=modredundant
...略
5 3 1 15 S 6 60.
5 3 6 10 S 6 60.
將輸出文件弄到GaussView里查看,繪制曲面圖,如下所示。為了看得清楚把兩個視角都展示了出來,能量最低點用紅圈標了出來
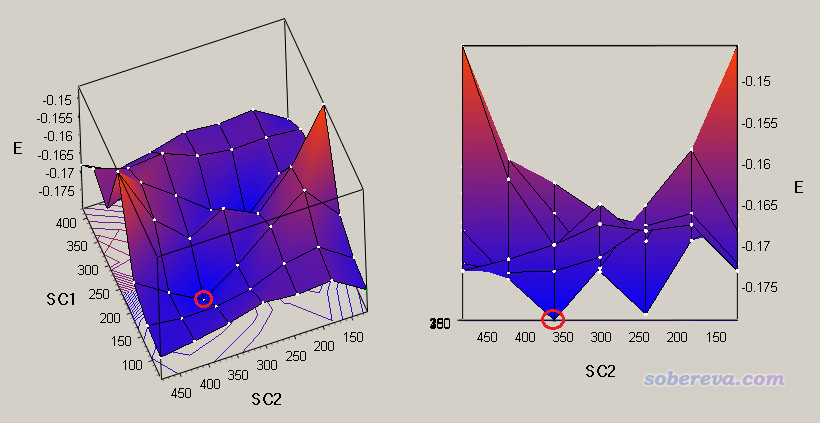
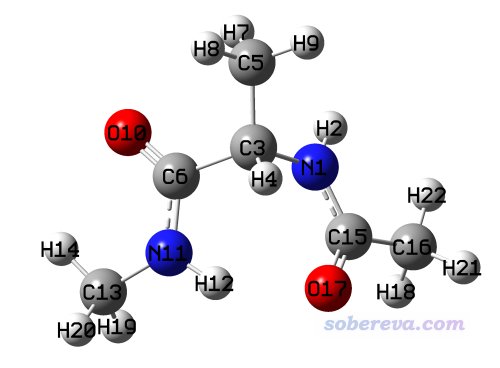
由于掃描的時候坐標是以固定步長變化的,因此上圖中能量最低點肯定還不是真正的能量最低構象,應當用這個點作為初始結構進一步做幾何優化。我們在GaussView中切換到這個點,保存文件成ACE-ALA-NME_opt.gjf,然后恰當設置關鍵詞,對應在B3LYP-D3(BJ)/def2-SVP這種稍微像樣的級別下優化。優化完的結果如下所示,之所以這個能量最低是因為形成了分子內氫鍵。實際上從掃描的二維圖上還看到有另一個點的能量也非常低,讀者也可以嘗試對那個點也做一下優化,沒準兒優化之后能量比當前結構還低。
順帶一提,如果你想將這個分子內氫鍵的存在可視化,并考察其本質特征,可以用Multiwfn做RDG、IGM、AIM等分析,見《Multiwfn支持的弱相互作用的分析方法概覽》(http://www.shanxitv.org/252)。
對這個體系做剛性掃描試圖找能量最低結構是沒意義的,因為掃描過程中可能有些原子間出現嚴重不合理的接觸,但這些地方的原子又不會由于限制性優化而自發回避開,故勢能會極高,甚至可能由于原子間距離過近而報錯。
對各種實際分子做構象搜索,往往需要考慮兩個以上可旋轉的鍵,按照上面的方式直接用Gaussian做多維柔性掃描試圖找能量最低構象絕對不是好主意。通常,做分子構象搜索的最佳的做法是使用筆者開發的molclus程序,免費,使用靈活、方便,精度隨意可控,介紹見http://www.keinsci.com/research/molclus.html,特別是要閱讀《gentor:掃描方式做分子構象搜索的便捷工具》(http://bbs.keinsci.com/thread-2388-1-1.html),其中給出了構象搜索實例,并對構象搜索的很多要點做了討論。
5.5 柔性掃描實例3:輔助尋找乙醇脫水的過渡態
乙醇的甲基上的一個氫可以轉移到羥基的氧上,脫掉一個水,剩下的部分成為乙烯。這種氫轉移的過渡態還是比較好找的,初猜不難擺。但有時候碰上比較復雜的情況,不好確定opt=TS找過渡態的初猜結構時,借助柔性掃描幫助判斷合適的初猜結構往往還是有幫助的。這里就拿乙醇脫水作為例子說明一下,乙醇的結構如下。
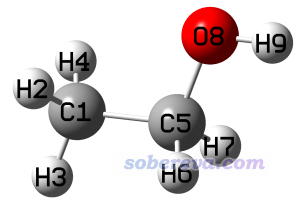
我們假定H2會轉移到O8上,要借助柔性掃描輔助獲得恰當的過渡態初猜,就應該掃描與反應過程對應最直接對應的坐標,對于此例來說應該逐漸縮短H2-O8距離。一開始的結構H2-O8距離2.667埃,而水的O-H鍵長約0.96埃,因此我們可以從初始結構開始掃17步,每步減少0.1埃,最后一步對應2.667-17*0.1=0.967埃。對于粗略確定合適的過渡態初猜的目的,柔性掃描用很便宜的級別就夠了,故這里用很便宜的PM7(對有機體系過渡態研究多數情況定性正確,盡管也有不少反例),輸入文件如下(ethanol_relax.gjf)。
# PM7 opt=modredundant nosymm
...略
2 8 S 17 -0.1
對結果進行繪圖,如下所示
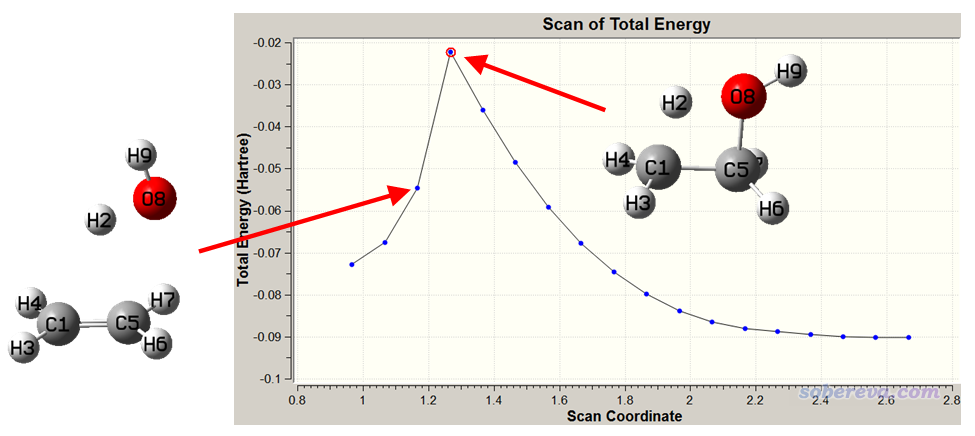
由于被掃描的變量是逐漸減小的,所以圖應該從右往左看。隨著掃描的進行,由于C-H和C-O鍵逐漸被拉長,能量逐漸升高,直到到達圖中的最高峰。再往下走一步,能量突然降低,這是因為O-H距離此時已經比較短了,對其它變量進行優化后產生的結構已很像水+乙烯復合物了。這種斷崖式曲線的形成原因通過本文5.2節的示意圖已經展現得很清楚了。由于憑基本化學直覺就能看出掃描曲線上最高點的樣子差不多就是乙醇脫水的過渡態結構,即處于乙醇結構和水+乙烯復合物結構之間,因此可以用這個點作為找過渡態的初猜結構。
經常有人貼出一個柔性掃描的圖,上來就問最高點能不能作為過渡態初猜結構,這種問法明顯不合適。不看一下最高點對應的具體結構,安知那個點是否有當前研究的反應的過渡態的基本模樣?如果結構看著不像過渡態,顯然不能當做找過渡態的初猜結構。
由于柔性掃描昂貴,不要隨便碰到一個反應就總是試圖通過柔性掃描幫助找適合的過渡態初猜,多數情況花那功夫自己早就試出一個能收斂到過渡態的初猜結構了,應當只有迫不得已時才應該考慮柔性掃描的做法。而且很多反應也不適合或根本沒法通過柔性掃描確定適合的過渡態初猜結構。由于確定過渡態初猜的目的不需要限制性優化得太精確,可以在opt里同時寫上loose用較松的收斂限來降低柔性掃描的耗時。
5.6 柔性掃描實例4:把環丁烷拉成丁烷雙自由基
此例演示怎么實現把環丁烷的一個C-C鍵拉開,最終形成丁烷雙自由基的掃描。最終要拉到差不多是直鏈丁烷的兩端的碳之間的距離。估計了一下距離,比較適合從平衡結構開始掃11步,每步拉長0.2埃。當前任務輸入文件如下(cyclobutane_relax.gjf)
# UM062X/6-31G* guess(mix,always) opt=modredundant pop=always nosymm
...略
1 2 S 11 0.2
由于涉及到共價鍵斷裂,所以關鍵詞必須用guess(mix,always),讓每一步都產生對稱破缺初猜波函數,使得掃描到已經對應雙自由基狀態的結構時,波函數能夠收斂到雙自由基狀態(但是否確實能夠奏效,還需要之后再考察輸出信息)。這里用了M06-2X是因為它很適合研究有機問題,包括雙自由基的情況。用pop=always是為了能在GaussView里直接繪制掃描過程的原子自旋布居。用nosymm一方面使得掃描軌跡看起來連續,另一方面也使得對稱破缺計算更有保障。
在GaussView里顯示結果,如下所示
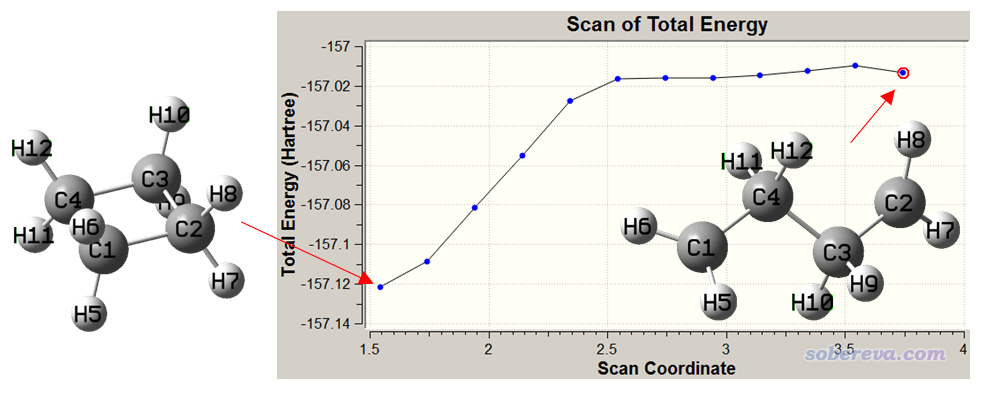
可見一開始,隨著C-C鍵被拉長,能量顯著上升。拉到大約2.5埃的時候,再繼續增加C-C距離時,能量就不怎么顯著上升了。這是因為差不多這個時候C-C化學鍵已經完全被破壞了,繼續拉遠導致的基本上只是構象變化,而構象變化造成的能量變化程度是遠低于破壞化學鍵的。當前圖中最后一個點的能量減去第一個點的能量可以近似作為這個鍵的鍵能,是(-157.01341+157.12159)*2625.5=284.0 kJ/mol。當然這樣算的鍵能并不準,在于兩端結構沒經過優化,而且計算級別比較低(特別是基組)。此外,要和實驗的BDE對比的話需要用焓來求差而非這樣用電子能量求差。
我們再看一下原子的自旋布居在掃描過程中的變化,以了解形成雙自由基的過程。如果不懂什么叫自旋布居、自旋密度,一定要看《談談自旋密度、自旋布居以及在Multiwfn中的繪制和計算》(http://www.shanxitv.org/353)。選擇Plot - Plot Molecular Property - Atomic Charge - Mulliken Spin population,輸入1,可以看到帶顯著單電子的C1的Mulliken自旋布居在掃描過程中的變化,如下所示
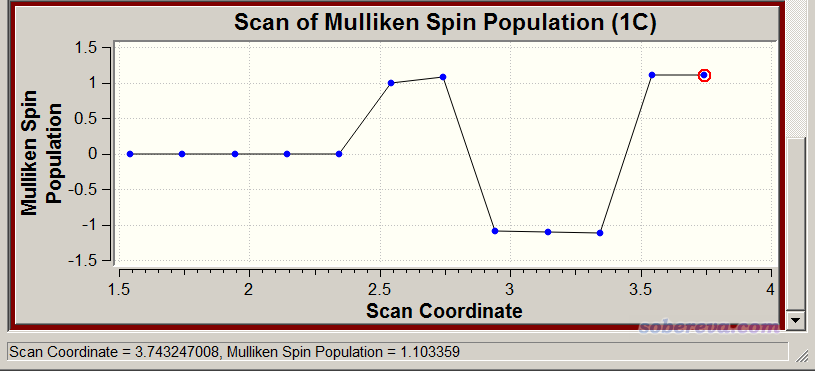
這個圖看起來比較詭異,即中途突然有三個點變成負值了。這絕對不是計算過程出錯了,而是因為我們用了guess(mix,always),每一步都是重新產生初猜波函數而非用上一步收斂的波函數當初猜。因此C1在某個點可能帶的是alpha單電子,但到了下一步,由于產生的初猜的數值巧合性,導致最終帶的是beta單電子。這里假設我們只想考察C1帶的單電子量,因此看這個圖的時候我們不應該考慮自旋布居的正負號,應該只看數值大小。從C1的自旋布居大小上看,前5步都為0,體現由于C-C還沒拉得較遠,尚未呈現雙自由基狀態。從第六步開始自旋布居不再為0,而且數值幾乎為1.0,說明從此開始,雙自由基狀態已經顯著出現了,C1和C2每邊幾乎各分布一個單電子且彼此自旋方向相反。
6 通過廣義化內坐標(GIC)進行柔性掃描
Generalized internal coordinate (GIC)是從Gaussian 16開始加入的特征,利用GIC的設定語句可以比使用冗余內坐標的設定語句更靈活地控制幾何優化和柔性掃描。筆者沒有精力去詳細介紹(北京科音基礎量子化學培訓班里會細說),這里只是給兩個例子體現GIC在柔性掃描中能產生的價值。筆者在《在Gaussian中做限制性優化的方法》(http://www.shanxitv.org/404)文中給出了利用GIC實現限制性優化的例子,讀者有興趣也可以看看。
6.1 GIC掃描實例1:水+氮氣二聚體的幾何中心距離掃描
此例我們利用GIC對水的幾何中心和氮氣二聚體的幾何中心間的距離進行柔性掃描。這種掃描如果不利用GIC設定的話肯定是沒法實現的,因為普通的柔性掃描的掃描設定中不能牽扯虛原子,也因此不可能通過給每個單體中心增加虛原子然后掃二者的距離來實現。
在掃描前,建議先把水+氮氣的二聚體極小點結構優化出來。這里用B3LYP-D3(BJ)/def-TZVP級別,不貴但是可以保證定性正確。將優化完的結構的單體的幾何中心位置都添加虛原子,然后把虛原子與單體中任意一個原子連上鍵,然后調節虛原子間的距離使得兩個片段的幾何中心相距3埃。最終結構如下所示
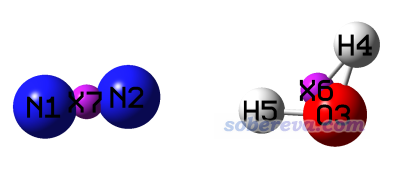
保存gjf文件,然后加上恰當的關鍵詞和GIC設定成為下面這樣(H2O_N2_gic.gjf):
# b3lyp/tzvp em=gd3bj opt=addgic
[空行]
Title Card Required
[空行]
0 1
N -2.09851100 -0.04510800 0.06118400
N -1.02140600 0.07296400 -0.07499500
O 1.65885690 -0.14665701 0.01859668
H 1.94081190 0.77130999 0.08101768
H 0.70784690 -0.10669701 -0.12803832
X 1.43583857 0.17265199 -0.00947465
X -1.55995850 0.01392800 -0.00690550
[空行]
XH2O(inactive)=XCntr(3-5)
YH2O(inactive)=YCntr(3-5)
ZH2O(inactive)=ZCntr(3-5)
XN2(inactive)=XCntr(1,2)
YN2(inactive)=YCntr(1,2)
ZN2(inactive)=ZCntr(1,2)
scan(StepSize=0.1,NSteps=10)=sqrt[(XH2O-XN2)^2+(YH2O-YN2)^2+(ZH2O-ZN2)^2]*0.529177
opt里加上addgic代表讀入額外的GIC設定。上面的設定代表將水分子(3~5號原子)的幾何中心的X,Y,Z坐標分別定義為XH2O、YH2O、ZH2O這三個變量,后面加上(inactive)代表這仨變量本身不是被幾何優化的對象(被優化的對象是原子,而不是幾何中心,幾何中心僅被用來定義約束條件之用)。類似地對氮氣分子也這么定義三個變量。此處scan不是關鍵詞,而是自定義的一個變量名,取其它名字也可以,而括號里的StepSize=...,NSteps=...設定則是GIC里做柔性掃描的選項。我們通過表達式將被掃描的變量定義為了兩個單體幾何中心的距離,最后0.529177是把默認的距離單位由Bohr轉化為埃。此例掃10步,每步增加0.1埃,因此理應最后一個結構下兩個分子幾何中心距離為3.0+10*0.1=4.0埃。我們可以通過GIC支持的運算符和數學函數任意去構造被掃描的變量,所以GIC非常靈活。
用Gaussian跑一下這個任務,發現總共才掃了8個點就報錯停止了。這是因為目前筆者用的Gaussian 16 A.03的GIC代碼不太穩定,也可能存在bug,所以看似合理的GIC設定也往往會莫名其妙出錯中斷,但愿以后會有所改進。對于跑出來的8個GIC掃描點,繪制成動畫是這樣。
由動畫可見,確實每掃一步二者間幾何中心距離都增長一點。而且由于是柔性掃描,所以單體相對朝向都自發做了調整來最小化能量。
下面是scan結果觀看頁面,橫坐標沒有直接顯示幾何中心距離,但根據我們的設定可以知道能量最低的那個點是3.0+(7-1)*0.1=3.6埃處(第一個點是初始結構)。
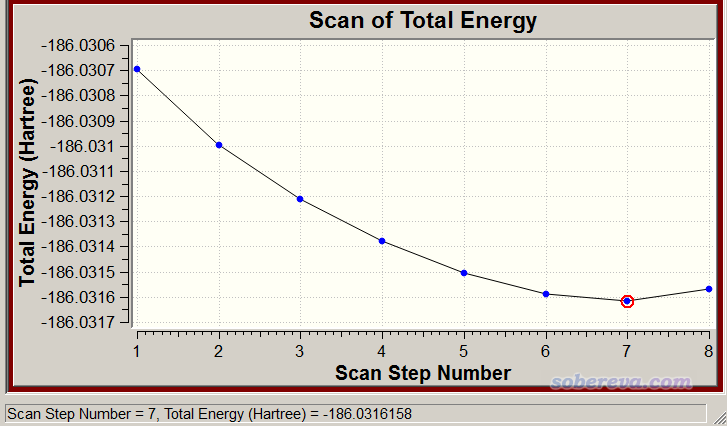
6.2 GIC掃描實例2:令1,3-丁二烯兩個亞甲基同步旋轉的掃描
此例主要目的是演示怎么在GIC里設定約束,使得幾個變量可以被同步地掃描,這在實際中挺有用。比如有時候我們想讓幾個鍵的鍵長,或幾個鍵角/二面角每一步改變相同數值,不利用GIC的話不好實現。
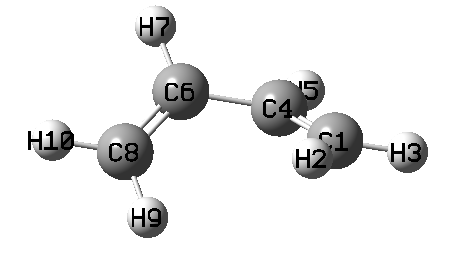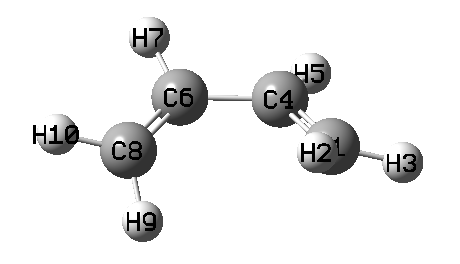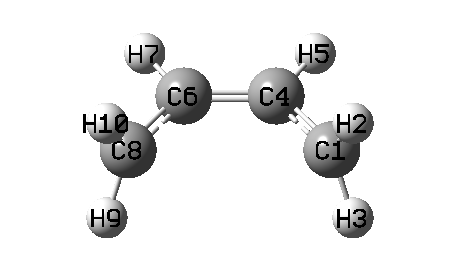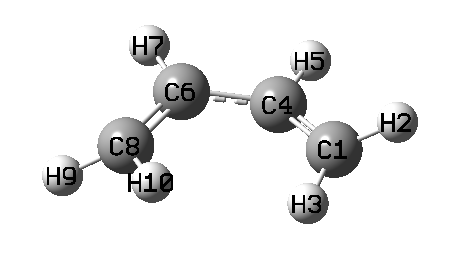
我們畫一個順式1,3-丁二烯,用PM7優化一下,得到如下結構

此例我們想讓左邊和右邊的亞甲基同步順時針旋轉。因此我們可以把被掃描的變量設為6-4-1-2,讓二面角每一步都減小,而且讓4-6-8-9二面角與6-4-1-2二面角初始的差值約束為恒定,這樣右邊亞甲基轉多少度,左邊亞甲基也會同步轉多少度。此例的輸入文件如下(butadiene_gic.gjf)
# opt=addgic UPM7 guess(mix,always)
[空行]
Title Card Required
[空行]
0 1
C 1.52650408 -0.47892841 0.10569634
H 1.19780098 -1.39607531 0.57075384
H 2.58160131 -0.47569529 -0.11790508
C 0.71385757 0.54669829 -0.15773410
H 1.08159899 1.46515075 -0.62106758
C -0.71385135 0.54669658 0.15773254
H -1.08160736 1.46512752 0.62108081
C -1.52650638 -0.47892483 -0.10569716
H -1.19781485 -1.39607552 -0.57075439
H -2.58160268 -0.47568190 0.11790667
[空行]
scan(StepSize=-10.0,NSteps=13)=D(6,4,1,2)
rcons(freeze)=D(4,6,8,9)-D(6,4,1,2)
可見我們要讓亞甲基順時針轉10.0*13=130度。GIC里面R、A、D分別接兩個、三個、四個原子序號,用來選擇距離、角度、二面角。rcons是一個自定義的變量,括號里freeze代表令這個變量的數值在掃描過程中嚴格維持為初始結構中的值。rcons被定義為了能代表左側亞甲基和右側亞甲基的兩個二面角的差值,因此掃描過程中左側亞甲基會與右側亞甲基同步轉動。這里用了U的計算形式和guess(mix,always)關鍵詞,正如我們之前柔性掃描共價鍵斷裂時用的,這是因為當亞甲基旋轉到與C-C-C-C平面接近90度的時候,1,3-丁二烯兩側的C=C雙鍵會很大程度被破壞掉,此時將使體系出現一定雙自由基特征,若當成閉殼層計算的話肯定嚴重不合理。
下面是掃描過程動畫,以及能量變化曲線
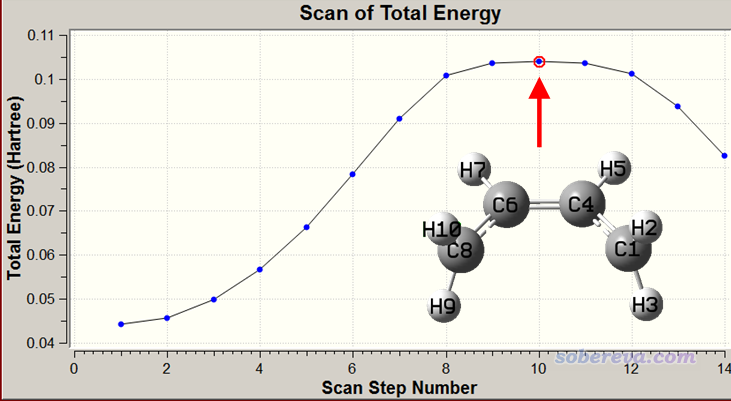
由動畫可見,確實兩個亞甲基同步旋轉了。旋轉到第10個點的時候能量最高,此時對應于兩個亞甲基都垂直于C-C-C-C平面,此時體系兩側的pi鍵被完全破壞掉,在C1、C8上基本上各分布一個單電子而且自旋相反,而原本與C1、C8形成pi鍵的中間兩個碳上的倆p電子此時就用于在中間兩個碳上形成pi鍵了。在第10個點時,C1和C8之間必定有很強的跑到一起形成sigma鍵的趨勢(如果發生了的話就成了環丁烯),但是此現象并沒有出現,這在于如果形成環丁烯的話必須兩個亞甲基的氫往體系兩側顯著偏移來讓C1和C8形成sp3雜化,但當前我們人為控制了亞甲基的二面角數值,因此阻止了C1與C8自發成鍵并形成sp3雜化。
7 總結&其它
本文深入、詳細、系統地對勢能面的剛性掃描和柔性掃描的概念進行了介紹,并結合Gaussian程序以及筆者自行開發的程序舉了大量例子,充分體現出了勢能面掃描的意義和實際用處,其中還介紹了不少特殊技巧、與其它方面知識進行了聯系。望讀者舉一反三,靈活、恰當運用勢能面掃描解決實際問題。
基本上所有主流量子化學程序都支持勢能面掃描,原理都大同小異,只不過大部分程序里的柔性掃描功能沒有Gaussian這么靈活。其它的量化程序往往支持一些自己需要用但是Gaussian又不支持的理論方法,比如CCSD(T)-F12、GFN-xTB、NEVPT2等,此時可以效仿以下兩篇文章的做法讓Gaussian在掃描時調用那些程序去計算能量/梯度,之后能量變化和掃描軌跡可以照常在GaussView里直接觀看:
《將Gaussian與ORCA聯用搜索過渡態、產生IRC、做振動分析》(http://www.shanxitv.org/422)
《將Gaussian與Grimme的xtb程序聯用搜索過渡態、產生IRC、做振動分析》(http://www.shanxitv.org/421)
還有一些特殊情況的勢能面掃描,是使用本文提到的任何方法都沒法直接實現的,比如對于pi-pi堆積二聚體,兩個單體都平行于XY平面,想讓其中一個單體在X,Y方向上進行二維掃描,像這種情況就只能自己寫程序來解決了。具體來說可以產生多幀的xyz軌跡(這是最簡單的記錄多幀結構的格式),每一幀對應把單體在X和Y方向上平移不同的數值,然后再用xyz2QC轉化成Gaussian輸入文件,最后用Gaussian執行、批量提取數據。
本文都是對基態做勢能面掃描,Gaussian里所有支持的理論方法都可以用于做掃描(熱力學組合方法不算,沒有解析梯度的方法也沒法用于柔性掃描),也包括計算激發態的方法。掃描激發態只不過是在常規掃描設定基礎上加上算激發態的關鍵詞或相關設定。比如要在TDDFT下對第2激發態勢能面做柔性掃描,寫比如# PBE1PBE/6-31G* TD(nstates=5,root=2) opt=modredundant即可,詳見《Gaussian中用TDDFT計算激發態和吸收、熒光、磷光光譜的方法》(http://www.shanxitv.org/314(http://bbs.keinsci.com/thread-2413-1-1.html)。另外,對于分子力場,由于在Gaussian里其默認的做優化的模塊、流程和其它方法不同,因此要做柔性掃描的話需要在opt里同時寫nomicro,否則柔性掃描設定都不會生效。