分子動力學初始結構構建程序Packmol的使用
分子動力學初始結構構建程序Packmol的使用
文/Sobereva@北京科音 2019-Mar-23
由于經常有人問Packmol怎么安裝、怎么用,這里就寫一篇文章,做一個完整的介紹,初學者應該都能讀懂,之前用過的人看了可能還能了解一些之前沒注意到的細節和技巧。筆者在北京科音的分子動力學與GROMACS培訓班(http://www.keinsci.com/workshop/KGMX_content.html)里會對Packmol講得更具體、給更多例子。
1 Packmol簡介
Packmol是一個免費的、很有價值的構建分子動力學模擬初始結構的工具,非常流行。簡單來說就是由用戶提供一種或多種分子的結構文件,并且設定一些約束條件,Packmol就會把各種分子按照設定將指定數目的分子堆積到滿足要求的區域中。程序會通過優化算法不斷嘗試各種堆積方式,直到構建出原子間沒有不合理接觸而且能滿足所有約束條件的結構。在堆積過程中分子結構會保持剛性,即構象不會變化。Packmol在堆積過程只考慮空間因素,完全不考慮能量、電荷分布等額外問題。
Packmol的選項設定非常靈活,可以容易地構建出復雜的模型。Packmol產生的結構可以被用于各種主流的分子動力學程序的模擬中,比如GROMACS、AMBER等。M$程序里的Amorphous Cell的用處和Packmol頗有類似之處,但遠遠不及Packmol靈活、強大。
Packmol的網址是https://m3g.github.io/packmol/,在上面有關鍵詞介紹,可以下載到源代碼和預編譯的Windows版,還提供了一些例子。
2 Packmol的安裝與運行
2.1 在Linux下編譯和使用Packmol
在Linux下使用Packmol之前需要先對源代碼進行編譯,如下所示。如果你不方便自己編譯,也可以直接下載我已經編譯好的:http://www.shanxitv.org/soft/packmol_18.169_Linux.zip
Packmol是Fortran寫的,如果你的機子里還沒有Fortran編譯器,需要先安裝Fortran編譯器。比如在CentOS下使用yum install gfortran命令即可安裝gfortran編譯器。
將官網上下載到的packmol.tar.gz解壓,進入解壓后的目錄,在命令行模式下輸入make即可編譯,十秒鐘左右就能編譯完。編譯完成后當前目錄就出現了名為packmol的可執行文件。
修改用戶目錄下的.bashrc文件(比如輸入gedit ~/.bashrc命令),在末尾加上一行export PATH=$PATH:/sob/packmol-18.169,這里假定/sob/packmol-18.169是packmol可執行文件的所在目錄。保存文件后重新進入終端,輸入packmol命令檢查是否能正常啟動,如果能冒出一堆和packmol有關的信息來,就說明已經裝好了。
之后用比如packmol < test.inp命令,就代表以test.inp作為輸入文件運行packmol程序。輸出文件都會產生在當前目錄下。
2.2 在Windows下使用Packmol
Packmol源代碼也可以在Windows下編譯,但我們沒必要這么做,因為在Packmol主頁上已經提供了編譯好的可執行文件,僅適用于64bit Windows系統。你也可以直接在這里下載:http://www.shanxitv.org/soft/packmol_18.169_win64.rar。
解壓此文件包,然后進入Windows的命令行模式下(cmd環境。不會進入的話自行google,屬于Windows使用常識),輸入packmol < test.inp命令即可運行。如果packmol不在當前目錄下,則需要輸入其絕對路徑,或者加入到Windows的Path環境變量里(不會的話參考《GROMACS 2018.4原生Windows版的安裝演示》https://www.bilibili.com/video/av39914815/視頻中的對應部分)。
PS:別問為什么雙擊packmol.exe圖標運行不了,因為packmol根本就不是這么用的!
3 Packmol輸入文件的設置
Packmol的使用需要提供兩類文件
? 輸入文件。文件名隨意,用來定義各種類型分子如何分布、有多少個。
? 各種要加入的分子的結構文件。一般使用pdb格式,用什么程序產生都可以。
輸入文件里的關鍵詞在http://m3g.iqm.unicamp.br/packmol/userguide.shtml#basic有匯總。下面只提及常用的關鍵詞,方括號里的是參數。
3.1 全局設定
tolerance [數值]:要求原子間距離不能小于多少,一般設為2.0。Packmol里所有長度的單位都是埃
output [文件名]:輸出文件的路徑
filetype [類型]:輸出文件的格式。默認為pdb,也可以設比如xyz
seed -1:寫這個的話每次運行產生的結構都會不同
add_box_sides:給輸出的pdb中增加CRYST1字段定義盒子,盒子尺寸對應體系中原子最大、最小坐標。如果寫諸如add_box_sides 1.2,則會給盒子各方向再增加1.2埃,避免做模擬時有某些邊界的原子和鏡像盒子里的原子間存在不合理接觸
3.2 分子設定
每個下面這種字段設置一種分子如何出現在當前體系中。可以寫無數個這種字段。
structure [分子結構文件]
number [分子數]
[分子規則]
end structure
分子規則用來定義分子里的所有原子必須滿足的條件,其常用關鍵詞如下。
inside cube [xmin ymin zmin d]:要求分子出現在長度為d的立方盒子內,盒子x,y,z最小值為xmin ymin zmin
inside box [xmin ymin zmin xmax ymax zmax]:要求分子出現在矩形盒子內,盒子兩個頂角坐標分別為xmin ymin zmin和xmax ymax zmax
inside sphere [x y z r]:要求分子出現在中心為(x,y,z)半徑為r的圓球中。另外,用inside ellipsoid關鍵詞的話還可以要求出現在特定的橢球中
inside cylinder [a1 b1 c1 a2 b2 c2 r l]:要求分子出現在圓柱內。圓柱的定義是從(a1,b1,c1)往(a2,b2,c2)方向伸展l長度,半徑為r
以上inside都可以改為outside,要求不能出現在指定范圍內。上面的空間范圍要求可以同時使用多個,條件會同時滿足。還有一些設置:
over plane [a b c d]:要求分子出現位置滿足平面方程ax+by+cz-d>=0。比如over plane 1 0 0 10.5就相當于要求出現在x>=10.5埃的區域。over可以改成below來反轉條件。
constrain_rotation [x/y/z 平均值 最大偏差]:默認情況下分子可以繞x,y,z軸隨意旋轉任意角度。用這個選項可以設置分子繞x或y或z旋轉的情況,可以同時設多個。如constrain_rotation x 20 5代表允許繞著y和z軸隨意旋轉,而繞著x軸旋轉的范圍必須在15~25度之間,平均值為20度。
fixed [x y z a b c]:用來直接定義分子出現在哪里。此設置代表將分子結構文件里的坐標平移(x,y,z),并繞x,y,z軸分布旋轉a,b,c弧度。如果還同時寫了center關鍵詞,相當于把分子的中心擺到(x,y,z)位置再旋轉
3.3 原子設定
Packmol里還可以要求某種分子的某些特定序號的原子出現的范圍必須滿足的要求,格式為
structure [分子結構文件]
[分子規則]
atoms [第一批原子序號]
[原子規則]
end atoms
atoms [第二批原子序號]
[原子規則]
end atoms
...
end structure
原子規則的設置語法、關鍵詞和分子規則完全一樣。
4 幾個實例
下面舉幾個Packmol使用例子。所有輸入輸出文件都可以在此下載:http://www.shanxitv.org/attach/473/file.rar
4.1 構建5*5*5 nm^3甲醇盒子
首先在GaussView或其它可以對分子建模的程序里畫一個甲醇,然后保存為methanol.pdb。對于這種剛性小分子,用Packmol之前結構不需要非得優化,畢竟GaussView直接畫的甲醇的基本幾何參數是合理的,而且不管對其做不做優化,等動力學跑起來之后就都一樣了。
創建一個名為methanol_box.inp的文本文件,內容如下
tolerance 2.0
add_box_sides 1.2
output methanol_box.pdb
structure methanol.pdb
number 1000
inside cube 0. 0. 0. 50.
end structure
將methanol.pdb和methanol_box.inp都放到當前目錄,運行packmol < methanol_box.inp,經過幾秒鐘,當前目錄下就出現了methanol_box.pdb。將之拖到VMD(http://www.ks.uiuc.edu/Research/vmd/)程序里觀看,并且在VMD的命令行窗口里輸入pbc box顯示出盒子,看到的圖像如下
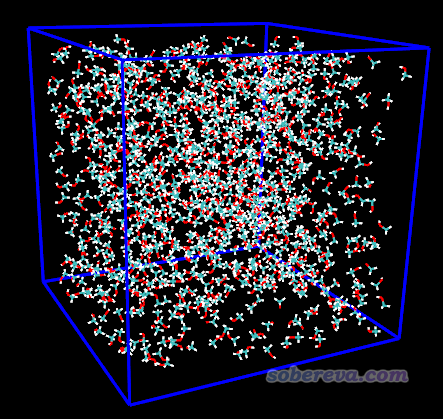
可見構建得很成功。但是1000個甲醇肯定不是Packmol能填充進這個5*5*5 nm^3盒子的上限,因為Packmol很容易就產生成功了,而且從圖上看還有一些空隙。如果大家希望得到更致密的盒子,可以嘗試一點點增加甲醇的number數,看看設到多少的時候Packmol半天也沒法成功產生結構或者提示產生失敗,Packmol最多能插入多少甲醇你心里就有數了。
4.2 構建水+甲醇混合溶液的氣液界面體系
此例我們想構建一個水+甲醇摩爾比1:1溶液的氣液界面體系,界面平行于XY方向,盒子的X、Y長度都為4nm,溶液層厚度是5nm,溶液上下兩側都是真空區,每一側真空區厚度都是3nm。
首先,我們靠Packmol創建一個水+甲醇的4*4*5 nm^3的溶液盒子。甲醇的pdb文件已經有了,我們再用GaussView畫個水,保存為water.pdb。然后編輯一個文件mix.inp,內容如下(經測試,水和甲醇各插入1000個時盒子已經比較致密了,而且已經需要好多輪循環才能成功產生結構,所以筆者就不再嘗試插入更多了)
tolerance 2.0
add_box_sides 1.2
output mix.pdb
structure water.pdb
number 1000
inside box 0. 0. 0. 40. 40. 50.
end structure
structure methanol.pdb
number 1000
inside box 0. 0. 0. 40. 40. 50.
end structure
將得到的mix.pdb弄到VMD里顯示:
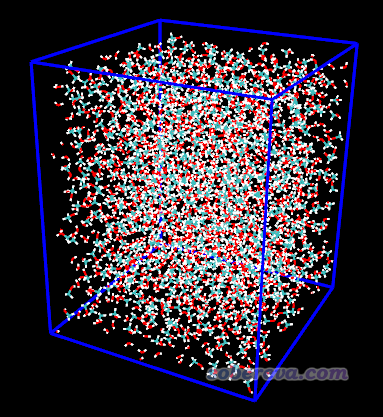
但是目前還沒有真空區。由于溶液層厚度為5nm,每一側真空區厚度為3nm,因此盒子Z方向總長度應為5+3+3=11nm。因此我們在VMD里輸入pbc set {40 40 110}來設置盒子尺寸為4*4*11 nm^3,這里單位是埃。
目前溶劑區域在盒子最底部,我們要將之挪到盒子中間去,也即把所有原子往Z的正方向挪3nm。這可以用VMD的幾何變換命令輕易實現。在VMD里輸入[atomselect top all] moveby {0 0 30},然后將VMD改成正交視角便于觀看(Display - Orthographic),看到下圖。
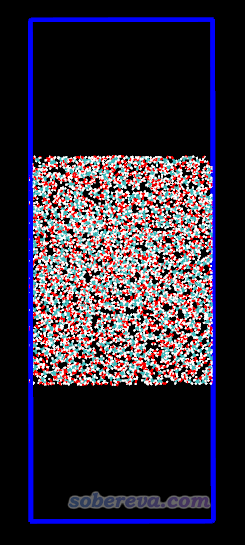
然后在VMD里將當前體系保存成新的pdb文件即可。值得一提的是,動力學模擬開始后,肯定最后達到穩定狀態時溶液層厚度不可能正好是我們最初期望的5nm,但由于目前已經堆積得比較致密了,應該偏離不太大。如果發現偏離的程度超過自己能接受的程度,可以在建模時再適當增加點分子數、在packmol建模時讓盒子在Z方向稍微大一點。
4.3 氯仿+甲醇+富勒烯體系
這是一個稍微復雜的例子,沒有什么實際意義,主要是體現Packmol的靈活性。此例我們要構建一個半徑1.5nm的溶劑球,甲醇和氯仿各一個半球區域,并且富勒烯正好出現在球中央。
此例的輸入文件如下。chloroform.pdb和C60.pdb都是GaussView直接構建的,后者在GaussView的片段庫里直接就有。
tolerance 2.0
output mix.pdb
structure chloroform.pdb
number 200
inside box 0. 0. 0. 40. 40. 20.
inside sphere 20. 20. 20. 15.
end structure
structure methanol.pdb
number 200
inside box 0. 0. 20. 40. 40. 40.
inside sphere 20. 20. 20. 15.
end structure
structure C60.pdb
number 1
center
fixed 20. 20. 20. 0. 0. 0.
end structure
此例我們假定球的中心在(2,2,2) nm的位置,因此直接用center和fixed關鍵詞將C60的中心定位在這個地方。對于氯仿和甲醇,inside sphere要求它們只能出現于距離(2,2,2)半徑1.5 nm的區域內,由于二者的inside box設定范圍正好相接,因此這兩種分子會分布出現在Z=2nm平面的上、下兩側。由于當前模擬的是孤立體系,所以沒有用add_box_sides關鍵詞來產生盒子信息。
用VMD觀看得到的pdb文件,如下所示

4.4 構建水球+十二烷基磺酸鈉
這個例子主要用來體現怎么使用原子設定。十二烷基磺酸鈉(SDS')的頭部基團是親水的,烷烴尾巴是疏水的。此例我們要構建一個半徑為20埃的水球,讓100個SDS'繞著它分布,SDS'的頭部基團都埋在水球里,而且讓烷烴尾巴的方向基本上垂直于水球的界面,最終構建出的結構應該類似于海膽,SDS'就是海膽的刺。
我們先用GaussView畫一個SDS'。由于鈉離子的位置放在哪可能憑直覺不好確定,因此畫完之后可以做一下幾何優化,比如用Gaussian在PM7半經驗方法下粗略優化后得到如下結構,看起來很合理
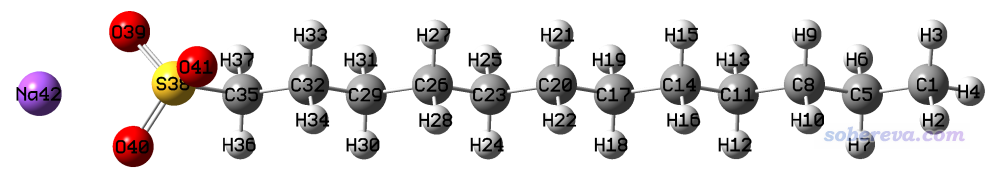
創建packmol輸入文件,內容如下
tolerance 2.0
output mix.pdb
structure water.pdb
number 1200
inside sphere 0. 0. 0. 20.
end structure
structure SDS.pdb
number 100
atoms 42
inside sphere 0. 0. 0. 15.
end atoms
atoms 1
outside sphere 0. 0. 0. 30.
end atoms
end structure
創建水球那部分沒什么好說的,經測試差不多最多就能放1200個水,再多就很難產生成功了。安置SDS'的這一段值得提一下。我們讓42號原子,即鈉離子必須出現在距離水球中心15埃范圍以內,而讓1號原子,即烴鏈最末端的碳原子必須距離水球中心30埃以上。由于在SDS'中這倆原子距離為18埃,僅略大于30-15=15埃,因此這么定義可以起到讓SDS'鏈垂直于水球界面的效果。
在VMD里繪制產生后的pdb,如下所示,結構完全達到了我們的期望。

官網上http://m3g.iqm.unicamp.br/packmol/examples.shtml里也有一些例子,有興趣可以看看。
5 相關問題&注意事項
這里說一些使用Packmol建模應當注意的問題,并且結合最主流的分子動力學程序GROMACS說一些相關事項。
Packmol的執行過程實際上是個循環過程,如果你當前的空間范圍設定比較簡單,Packmol很快就會順利產生最終結構。空間約束設定得越多、越復雜,往往成功產生結構需要的循環次數越多,總耗時越高。如果你把空間范圍約束得過度復雜甚至相互矛盾,或者要加入的分子數較多但允許分子出現的空間范圍相對而言過度狹小,那么Packmol會反復循環,一直也收斂不了,或者最終宣告產生失敗。碰到這種情況,應當將約束條件適當放寬、簡化,或者設的分子數少一點再試。
一般情況下,用Packmol構建的體系肯定是比較松散的,或者說其密度肯定顯著低于真實密度,因為此時還沒有考慮分子間相互作用、構象的變化從而使得分子間能夠接觸得更緊密。但這沒關系,在NPT下跑一下MD就好了,盒子尺寸會自發進行調節。而直接用Packmol產生的結構做NVT模擬一般是無意義的,因為跑起來之后分子會自發緊密聚集,其它地方就成了真空區了,和實際不符。
用Packmol實現構建含有Na+、Cl-離子的溶液體系是可以的。但是肯定沒有用GROMACS里專用的gmx genion命令好,因為gmx genion在插入時會考慮體系電荷分布,會把陽離子和陰離子分別加入到靜電勢較負和較正的地方,這樣比較合理。而Packmol則沒有考慮這點,并且有可能比如產生的結構里恰好Na+出現在顯著帶正電荷的區域,這可能會令模擬初期穩定性較差(此時需要用較謹慎的模擬參數,比如小步長)。
雖然也可以用Packmol構建蛋白質、核酸浸在溶劑環境中的體系,但是這樣做明顯不如用動力學程序自帶的專用做這種事情的程序好,因為Packmol產生的水的密度偏低,水的分布特征和實際體相水相差較大,可能NPT模擬起來之后盒子變形、收縮得厲害,出現溶質與其鏡像最近距離太近之類問題。而如果用比如GROMACS里的gmx solvate命令給蛋白質/核酸加溶劑,由于是直接用事先已經做NPT平衡好的溶劑盒子(比如加水一般用自帶的spc216.gro)通過平移復制來填充真空區,加的溶劑的分布狀態明顯理想得多得多。
GROMACS有一個命令叫gmx insert-molecules,可以由用戶提供分子結構文件、指定插入盒子的分子數,然后試圖將這些分子都插入盒子(但如果設的數目太多,會能插入多少就插入多少)。此命令遠沒有Packmol那么靈活,它能做的事用Packmol都可以實現,而且Packmol會通過優化算法試圖讓分子盡可能好地堆疊,因此對于同樣的盒子范圍,靠Packmol最多能插入的分子數比用gmx insert-molecules明顯要多,故能得到更緊湊的結構。
Packmol原理上也可以產生磷脂雙分子膜,乃至膜蛋白這樣的體系的,但是這需要設置很多約束條件才能產生靠譜的結果,而且耗時很高,結果也不理想。比如磷脂之間或者磷脂與蛋白之間會有好多空隙,這會導致模擬剛開始就有水大量跑進疏水區,后續模擬將毫無意義。構建磷脂膜我最推薦用我寫的genmixmem程序,速度很快,用著很方便,結合GROMACS模擬磷脂體系特別容易,詳見《生成混合組分的磷脂雙層膜結構文件的工具genmixmem》(http://www.shanxitv.org/245)。至于產生膜蛋白,我最推薦使用GROMACS可以實現的membed方法,在北京科音動力學與GROMACS培訓班里會演示操作。
如果你要建模的是分子團簇,用于構型搜索之類目的,用筆者開發的genmer比使用Packmol好得多,一次可以產生一大批,輸入文件寫起來省事得多,參見《genmer:生成團簇初始構型結合molclus做團簇結構搜索的超便捷工具》(http://bbs.keinsci.com/thread-2369-1-1.html)。
如果你用Packmol建模的目的是之后用GROMACS做模擬,別忘了GROMACS的拓撲文件里[molecules]字段里的分子按順序完全展開后,原子的順序必須和結構文件完全相同。為保證這一點,用于Packmol建模的分子結構文件里的原子順序首先就得和這個分子的拓撲信息(具體來說是[atoms]字段)相對應。