使用Multiwfn結合CP2K計算晶體中原子的氧化態
使用Multiwfn結合CP2K計算晶體中原子的氧化態
Using Multiwfn in combination with CP2K to calculate oxidation state for atoms in crystals
文/Sobereva@北京科音 2024-May-23
1 前言
波函數分析程序Multiwfn可以基于電子波函數計算原子和片段的氧化態,這在筆者之前寫的《使用Multiwfn通過LOBA方法計算氧化態》(http://www.shanxitv.org/362)一文中已做了詳細介紹,沒讀過者一定要先閱讀此文,否則無法理解下文的內容。此文介紹的做法已經被廣泛用來準確地考察分子中的氧化態。如今Multiwfn已經支持將這種方法用于CP2K計算的周期性體系的波函數,從而考察晶體中原子的氧化態,這非常有實際價值,具體做法將在本文介紹。注意氧化態和原子電荷完全是兩碼事,周期性體系的原子電荷的計算看《使用Multiwfn對周期性體系計算Hirshfeld(-I)、CM5和MBIS原子電荷》(http://www.shanxitv.org/712)。
Multiwfn可以在官網http://www.shanxitv.org/multiwfn免費下載,本文的讀者必須使用2024年5月23日及以后更新的版本,否則沒有本文介紹的特性。對Multiwfn不了解者看《Multiwfn FAQ》(http://www.shanxitv.org/452)和《Multiwfn入門tips》(http://www.shanxitv.org/167)。
2 用法
在Multiwfn中對周期性體系做LOBA或mLOBA分析氧化態的具體操作流程和對孤立體系做別無二致,都是先用主功能19產生定域化軌道(介紹見《Multiwfn的軌道定域化功能的使用以及與NBO、AdNDP分析的對比》http://www.shanxitv.org/380),然后進入主功能8(軌道成分分析),選擇LOBA/mLOBA分析,之后輸入一個LOBA方法判斷定域化軌道電子歸屬的閾值,或者輸入m讓Multiwfn按照mLOBA的方式判斷歸屬即可。定域化軌道的電子歸屬是什么意思,在前述的《使用Multiwfn通過LOBA方法計算氧化態》里已經專門說過了,這里不再累述。之后各個原子的氧化態就會立馬輸出出來。用戶也可以自定義片段來得到片段的氧化態。
對周期性體系做LOBA/mLOBA分析的輸入文件必須是CP2K產生的molden文件,并且需要自己在里面加入盒子信息。具體方法在《詳談使用CP2K產生給Multiwfn用的molden格式的波函數文件》(http://www.shanxitv.org/651)里有詳細介紹。由于考慮k點時不能產生molden文件,因此原本晶胞較小的話需要擴胞成足夠大的超胞,從而對其計算時只需要考慮gamma點。Multiwfn不支持Quantum ESPRESSO、VASP等其它第一性原理程序產生的波函數,也沒有任何支持的必要,因為對于絕大多數情況,用Multiwfn創建CP2K單點輸入文件并用CP2K計算是非常簡單且快速的事,可參考《使用Multiwfn非常便利地創建CP2K程序的輸入文件》(http://www.shanxitv.org/587)。
對于當前目的,CP2K計算時用贗勢基組也行,用全電子基組也行,沒有什么特別的講究。CP2K計算的時候不要開smearing,否則軌道占據數不再精確為整數、不再是標準的單行列式波函數,此時也就沒法做軌道定域化。如果有特殊原因非開smearing不可,可以在Multiwfn載入molden文件后進入主功能6,選擇選項38,此時Multiwfn會按照軌道能量由低到高重排電子確定占據數,軌道占據數就都為整數了,之后退回到主菜單,再照常按前述步驟做LOBA/mLOBA分析即可。
3 例子
下面給出一系列通過Multiwfn使用LOBA/mLOBA計算固體的氧化態的例子。CP2K的輸入輸出文件在http://www.shanxitv.org/attach/711/file.zip里都給了。只有3.1節例子對應的molden文件我在里面直接給了,如果所有例子的molden文件都給的話壓縮包就太大了。CP2K輸入文件里的各種設置我這里就不細說了,在北京科音CP2K第一性原理計算培訓班(http://www.keinsci.com/workshop/KFP_content.html)里有非常全面系統的講解。
3.1 單層氮化硼
氮化硼(BN)是知名的二維材料,這里完整介紹單層BN的氧化態計算的流程。本文文件包里的BN_bulk.cif是氮化硼晶體的結構文件,用GaussView打開它,可看到晶胞里有兩層BN。刪除其中的一層,然后保存為BN_ml.gjf。之后啟動Multiwfn,載入BN_ml.gjf,輸入
cp2k //創建CP2K輸入文件
[回車] //產生的輸入文件用默認的名字BN_ml.inp
-2 //要求導出molden文件
-11 //進入幾何操作界面
19 //構造超胞
6 //第1個平行于層的方向擴胞成原先的6倍
6 //第2個平行于層的方向擴胞成原先的6倍
1 //垂直于層的方向不擴胞
-10 //返回
-7 //設置周期性
XY
0 //產生輸入文件(計算級別為默認的PBE/DZVP-MOLOPT-SR-GTH)
用CP2K運行Multiwfn產生的BN_ml.inp,我這里用雙路7R32 96核機子4秒鐘就算完了。現在當前目錄下有了BN_ml-MOS-1_0.molden,按照《詳談使用CP2K產生給Multiwfn用的molden格式的波函數文件》(http://www.shanxitv.org/651)所述,把以下信息插入進去,以定義晶胞信息和原子價電子數信息。改好的文件已經在本文的文件包里提供了,是BN_ml-MOS-1_0.molden。
[Cell]
14.98944000 0.00000000 0.00000000
-7.49472000 12.98123580 0.00000000
0.00000000 0.00000000 8.00000000
[Nval]
B 3
N 5
啟動Multiwfn,載入BN_ml-MOS-1_0.molden,然后輸入
19 //軌道定域化
1 //只定域化占據軌道,用默認的Pipek-Mezey方法
8 //軌道成分分析
100 //LOBA/mLOBA分析
輸入一個閾值,比如50,馬上看到各個原子的以LOBA方法計算的氧化態,如下所示。可見B的氧化態為3、N為-3,整個體系所有原子氧化態加和為0,非常符合常識。這里用的閾值50%對于成鍵方式比較典型的情況是適合的,它的意思是如果有某原子對某個定域化軌道貢獻超過50%(Multiwfn以Hirshfeld方法計算貢獻值),則這個軌道的電子就完全歸屬于這個原子。
Oxidation state of atom 1(B ) : 3
Oxidation state of atom 2(N ) : -3
Oxidation state of atom 3(B ) : 3
Oxidation state of atom 4(N ) : -3
Oxidation state of atom 5(B ) : 3
Oxidation state of atom 6(N ) : -3
...略
The sum of oxidation states: 0
我提出的modified LOBA (mLOBA)方法是對LOBA的重要改進,它將每個定域化軌道的電子完全歸屬到對它貢獻最大的那個原子上,可以確保所有原子的氧化態加和總為0,而且也沒有閾值選擇的任意性(有的時候LOBA的結果對閾值敏感),比mLOBA在原理上明顯更好。在當前界面里輸入m即得到mLOBA方法算的氧化態的結果,也是B和N的氧化態分別為3和-3。
3.2 BaTiO3
BaTiO3晶體的cif文件是本文文件包里的BaTiO3.cif。用Multiwfn創建其4*4*4超胞在PBE/DZVP-MOLOPT-SR-GTH下做單點的CP2K計算的輸入文件。當前的超胞在每個方向有16埃,足夠大了。CP2K計算后得到molden文件,在里面加入恰當的[Cell]和[Nval]字段后,啟動Multiwfn并將之載入,然后輸入
19 //軌道定域化
-9 //設置產生定域化軌道后自動計算軌道成份的方法
0 //不輸出軌道成份。這些信息不是當前感興趣的,當前體系又大,不輸出可以節約不少時間
1 //只定域化占據軌道,用默認的Pipek-Mezey方法
由于當前體系大、軌道數很多,因此軌道定域化過程耗時較高。在EPYC 7R32機子上花了約8分鐘。之后輸入
8 //軌道成分分析
100 //LOBA/mLOBA分析
m //顯示mLOBA方法計算的原子氧化態
馬上看到以下結果。可見Ba、Ti、O的氧化態都和常識完全一致
Oxidation state of atom 1(Ba) : 2
Oxidation state of atom 2(Ti) : 4
Oxidation state of atom 3(O ) : -2
Oxidation state of atom 4(O ) : -2
Oxidation state of atom 5(O ) : -2
...略
3.3 MOF-5
MOF-5是知名的金屬有機框架化合物,介紹見https://en.wikipedia.org/wiki/MOF-5。其中含有Zn,這里算算Zn的氧化態是多少。MOF-5晶體的cif文件在本文的文件包里提供了,是立方晶胞,邊長25.86埃。由于邊長已經大到可以只考慮gamma點做計算,因此CP2K計算時不需要擴胞,直接對原胞算單點得到molden文件即可。相應的CP2K輸入文件已提供在了本文的文件包里。氧化態計算過程同前,7R32機子上幾分鐘可以算完整個過程。氧化態結果為Zn=2、O=-2、H=1、C=2/0/2。可見Zn、O、H的氧化態都完全符合預期。當前體系中C的氧化態有三種,和所處的化學環境差異有關。
3.4 單層MoS2
MoS2是知名的二維材料體系,它的體相結構文件是本文文件包里的MoS2_bulk.cif。完全效仿3.1節氮化硼的例子,取單層并基于5*5*1的超胞模型做它的氧化態的計算。如果以LOBA方法計算氧化態并輸入50作為閾值,結果為
Oxidation state of atom 1(Mo) : 6
Oxidation state of atom 2(S ) : -2
Oxidation state of atom 3(Mo) : 6
Oxidation state of atom 4(S ) : -2
Oxidation state of atom 5(S ) : -2
Oxidation state of atom 6(S ) : -2
Oxidation state of atom 7(Mo) : 6
...略
The sum of oxidation states: 50
雖然Mo的價電子有6個,看似Mo的氧化態為6也不是不可能,但實際上明顯是錯的,因為當前所有原子的氧化態加和為100。考慮到S的氧化態不可能比-2更負,明顯當前的結果高估了Mo的氧化態。即便嘗試各種閾值,也始終得不到能令LOBA方法算的氧化態加和為0的情況。因此LOBA對此體系完全失敗。
輸入m使用mLOBA計算氧化態,結果為
Oxidation state of atom 1(Mo) : 4
Oxidation state of atom 2(S ) : -2
Oxidation state of atom 3(S ) : -2
Oxidation state of atom 4(Mo) : 4
Oxidation state of atom 5(S ) : -2
Oxidation state of atom 6(S ) : -2
Oxidation state of atom 7(Mo) : 2
Oxidation state of atom 8(S ) : -2
Oxidation state of atom 9(S ) : -2
...略
The sum of oxidation states: 0
可見mLOBA方法算的所有原子氧化態加和為期望的0,但是Mo有的氧化態是2有的是4,和當前體系中所有Mo等價的事實不符,原因后面會說。解決方法是輸入-1定義片段,然后輸入所有Mo對應的序號。序號的快速查詢方法是在Multiwfn主功能0的菜單里選擇Tools - Get atom indices of a given element,然后輸入Mo,點OK,就會返回下面的內容,將之粘貼到Multiwfn定義片段的窗口里即可。
1,4,7,10,13,16,19,22,25,28,31,34,37,40,43,46,49,52,55,58,61,64,67,70,73
片段定義好后,再次輸入m,可以看到Oxidation state of the fragment: 100,即這個片段總的氧化態為100。由于當前體系里有25個完全等價的Mo,因此Mo的氧化態為100/25=4,是完全正確的值。
為什么mLOBA算出來的明明等價的Mo會有不同的氧化態?這可以通過考察定域化軌道的特征結合mLOBA的原理予以了解。默認情況下,Multiwfn的主功能19做完軌道定域化后會自動輸出各個定域化軌道的成份,可以看到除了單中心、雙中心的定域化軌道外,還有大量三中心的:
More delocalized LMOs: (Three largest contributions are printed)
301: 64(Mo) 24.8% 46(Mo) 24.8% 61(Mo) 24.8%
302: 46(Mo) 24.8% 49(Mo) 24.8% 31(Mo) 24.8%
303: 16(Mo) 24.8% 34(Mo) 24.8% 31(Mo) 24.8%
...略
從軌道成份上明顯可知這些三中心軌道都是等價的。軌道定域化做完后按照《使用Multiwfn觀看分子軌道》(http://www.shanxitv.org/269)說的,在Multiwfn的主功能0里隨便看一個三中心軌道,如下所示
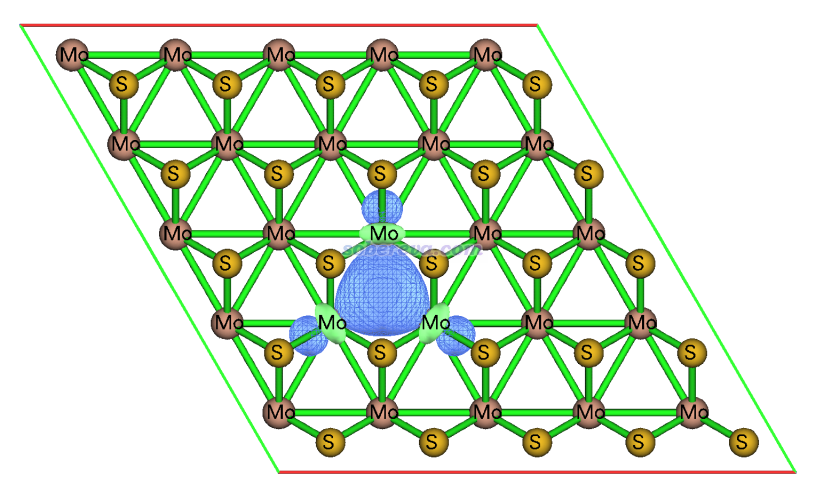
可見這個軌道體現的是三個Mo之間的三中心作用。按照mLOBA的思想,這個軌道上的兩個電子會完全歸屬到對它貢獻最大的原子上,但由于三個Mo的貢獻本質上相同,實際的貢獻量由于數值計算誤差會有極其輕微的差異,所以這個軌道上的電子歸屬到哪個Mo是有隨機性的,這是為什么Mo的氧化態有的為2有的為4。可見,只要把軌道定域化和mLOBA的思想都理解了,遇到可疑的結果自然就能通過具體分析搞明白是怎么回事、知道怎么恰當解決。
4 總結
本文詳細介紹了怎么用Multiwfn基于CP2K對固體做DFT計算得到的波函數通過LOBA和mLOBA方法得到原子的氧化態,給了很多例子。可見本文的方法操作簡單,結果非常合理,這對于討論原子在固體中的狀態、對體系進行分類很有價值。作為練習,讀者可以對NaCl、MgO、AlN、SiO2晶體也都算算氧化態。值得一提的,如本文的例子所體現的,如有了我后來提出的mLOBA方法,原本的LOBA就沒什么使用價值了,我建議總是默認用mLOBA。
使用本文的方法計算在發表結果時務必記得按照Multiwfn程序啟動時的提示對Multiwfn進行正確的引用。