文/Sobereva@北京科音 2024-Feb-29
0 前言
筆者在《衡量芳香性的方法以及在Multiwfn中的計算》(http://www.shanxitv.org/176)中介紹過,磁感生電流是一種重要、直觀的考察分子芳香性的方式。SYSMOIC是一個基于continuous transformation of the origin of the current density (CTOCD)方法考察化學體系的磁感生電流相關問題的程序。本文將結合苯分子為例,介紹這個程序的最關鍵用法。SYSMOIC程序和相關原理的詳細介紹可以看原文J. Chem. Inf. Model., 61, 270 (2021)。
AICD和GIMIC也是兩個重要的考察磁感生電流的程序,筆者在下文有專門的介紹。這些文章里介紹的背景知識對理解本文很有幫助
? 使用AICD 2.0繪制磁感應電流圖(http://www.shanxitv.org/294)
? 使用AICD程序研究電子離域性和磁感應電流密度(http://www.shanxitv.org/147)
? 考察分子磁感生電流的程序GIMIC 2.0的使用(http://www.shanxitv.org/491)
? Utilizing AICD and GIMIC programs to study magnetically induced current density(http://www.shanxitv.org/148)
之所以筆者寫此文介紹SYSMOIC,在于它對以上兩個程序有明顯互補性。AICD程序是將感生電流以箭頭方式繪制在AICD的等值面上,而等值面以外區域的感生電流就無法在圖上體現出來了,而且AICD沒法給出感生電流的鍵截面積分值(鍵電流強度)來定量考察電流大小。SYSMOIC則能夠給出三維空間或二維平面上的感生電流向量場圖,從而能展現出AICD圖表現不出來的信息,而且SYSMOIC還能非常方便地給出鍵電流強度。GIMIC程序雖然能做感生電流積分、結合Paraview程序能給出漂亮的向量場圖,但是GIMIC卻無法將感生電流分解成不同分子軌道的貢獻,而SYSMOIC則能夠方便地指定計算感生電流時只考慮或不考慮哪些分子軌道。可見SYSMOIC有獨特的價值。
本文介紹的情況對應SYSMOIC 2024.1版,對其它版本不一定適用,請結合相應版本程序提示和手冊隨機應變。
1 SYSMOIC的基本特征
SYSMOIC是免費而不開源的程序,Windows、Linux、Mac版都有,可以在http://SYSMOIC.chem.unisa.it/DISTRIB下載。諸如STABIN_LNX-2024.1.tar.gz就是Linux的2024.1版可執行文件包。Linux版需要用較新的系統,筆者用Rocky Linux 9可以正常使用,CentOS 7.4用不了。
SYSMOIC是由一堆子程序組成的,對應壓縮包里不同可執行文件,Windows版有.exe后綴,其它平臺的沒有。例如JBMAP子程序用于計算感生電流格點數據,BOCUST子程序用于計算鍵電流強度,等等。v3d子程序用來對其它子程序計算出來的.3d文件進行可視化,如繪制感生電流矢量圖。各個程序的用法在SYSMOIC的在線手冊http://sysmoic.chem.unisa.it/MANUAL/里都有說明。
SYSMOIC是解壓即用的程序。解壓后只需要把SYSMOIC目錄加入到操作系統的PATH環境變量里(不會的話自行google),然后就可以在任何目錄下輸入子程序名直接使用了。
SYSMOIC有不少官方例子文件,是http://sysmoic.chem.unisa.it/WORKED_EXAMPLES/里的WORKEXA.tar.gz。例子按照體系/理論方法/基組對目錄層級進行命名,里面RUN_TESTS文件是對例子體系做各種分析的shell腳本,可以從中看到都是以什么命令調用程序的,以及傳入的命令文件是什么。REFERENCES目錄下的文件是運行RUN_TESTS腳本后產生的.3d文件和程序輸出信息,對前者可以直接用v3d作圖。
SYSMOIC必須結合最主流的量子化學程序Gaussian使用,目前只支持閉殼層形式的HF和DFT。只需要用Gaussian做NMR計算的同時產生wfx文件,再用SYSMOIC自帶的unpackwfx將之轉化成fort.3、fort.11、fort.23、fort.28這四個二進制文件,之后就可以用JBMAP、BOCUST等子程序做各種計算了,算完了可以用v3d子程序直接作圖。
2 產生苯的SYSMOIC輸入文件
下面以最典型的芳香性分子苯為例,講解SYSMOIC繪制感生電流矢量圖和計算鍵電流強度的方法。其它的用法我覺得在一般研究中用到的機會很少,所以就不在本文介紹了,感興趣者請看程序手冊、原文并結合程序自帶的例子文件摸索。下文涉及的各種輸入輸出文件都可以在http://www.shanxitv.org/attach/702/file.rar中獲得。
首先寫一個在B3LYP/6-31G*級別下做NMR計算的Gaussian輸入文件benzene.gjf,內容如下,在本文的文件包里已經提供了。其中的坐標是在當前級別下已經優化過的。NMR=CSGT要求以CSGT方法做NMR計算,out(wfx,CSGT)要求產生.wfx波函數文件,并且把CSGT計算過程中產生的擾動的軌道展開系數寫入到wfx文件中,輸出位置為末尾指定的D:\benzene.wfx。
# b3lyp/6-31g(d) NMR=CSGT out(wfx,CSGT)
[空行]
benzene
[空行]
0 1
C 0.00000000 1.39621600 0.00000000
C 1.20915900 0.69810800 0.00000000
C 1.20915900 -0.69810800 0.00000000
C 0.00000000 -1.39621600 0.00000000
C -1.20915900 -0.69810800 0.00000000
C -1.20915900 0.69810800 0.00000000
H 0.00000000 2.48297800 0.00000000
H 2.15032200 1.24148900 0.00000000
H 2.15032200 -1.24148900 0.00000000
H 0.00000000 -2.48297800 0.00000000
H -2.15032200 -1.24148900 0.00000000
H -2.15032200 1.24148900 0.00000000
[空行]
D:\benzene.wfx
[空行]
[空行]
算完后就有了benzene.wfx,確保它在當前目錄下,并且已經把SYSMOIC目錄加入到了PATH環境變量中,然后運行unpackwfx benzene命令,當前目錄下立刻就出現了fort.3、fort.11、fort.23、fort.28,它們都已提供在了本文的文件包里。后文說的所有操作都是對于這四個fort文件都處于當前目錄下的情況而言的。
注意當前的苯分子是處在Z=0的XY平面上,用Multiwfn載入Gaussian的out文件然后進主功能0,或者用GaussView載入out文件并顯示笛卡爾軸,都可以確認這一點。后面我們要考察的都是外磁場方向垂直于苯環所導致的感生電流的情況。
3 繪制磁感生電流向量場圖的例子
3.1 默認方式繪制
先來看怎么產生一個最簡單的磁感生電流量場圖,之后再解釋細節。直接在命令行窗口輸入JBMAP運行之,屏幕上會出現好多默認用的參數,比如下面這行,就是告訴你默認的外磁場是(0 0 1)矢量,顯然正垂直于當前苯環,和我們期望的一致
magnetic field...........B 0.0000 0.0000 1.0000
程序問你當前顯示出來的參數是否ok,這里就直接按回車代表用默認的y(yes)。然后再依次問你三個問題:
? external grid points y/n? [n]:即是否讀取外部格點,這里直接按回車代表用默認的n(如果要計算的點不是均勻分布在一個矩形區域,而是比如分布在一個球層表面,就需要輸入y并且提供格點定義文件)
? reference arrow y/n? [n]:含義不明。直接按回車用默認的n
? B direction y/n? [n]:即是否把外磁場箭頭畫在圖上,直接按回車用默認的n。如果你想把外磁場箭頭繪制在圖上就輸入y,并且輸入箭頭的位置和長度
馬上當前目錄下就有了JBM.3d文件。輸入v3d JBM.3d通過v3d程序對這個文件作圖,就蹦出了圖形窗口,如下所示,可以看到在一個矩形區域內均勻分布的每個格點位置都有一個箭頭指示此位置的磁感生電流的方向和大小。

想退出的話在圖形窗口按ESC鍵,或者在文本窗口按ctrl+C。
上面的圖非常丑陋,箭頭特別擁擠,分子結構也看不清楚,完全沒法用。下一節講怎么對JBMAP的計算進行自定義。
3.2 自定義JBMAP的運行參數
JBMAP有兩個方式指定運行參數,比如要把外磁場矢量改為0 1 1(程序自動會做歸一化成為0.0000 0.7071 0.7071),既可以運行JBMAP -B 0 1 1命令,也可以先啟動JBMAP然后直接輸入B 0 1 1后按回車,在屏幕上顯示的參數中都會看到外磁場矢量已經變更成自設的值了。
JBMAP命令后面可以直接接的選項可以輸入JBMAP -h查看,其中幾個關鍵的在這里提一下:
-o:輸出的.3d文件名,默認是-o JBM
-f:原子球大小的倍數,默認為-f 0.2。想讓作出的圖里原子球大一些就把這個改大
-B:外磁場矢量,默認為-B 0 0 1
-j:感生電流類型,默認是-j TOT,即diatropic和paratropic的總和。寫-j PARA代表只考慮paratropic部分,寫-j DIA代表只考慮diatropic部分
-q:考慮的軌道。例如只想考慮17 20 21三個分子軌道對磁感生電流的貢獻,就寫-q +3 17 20 21,只去掉15 16號分子軌道的貢獻就寫-q -2 15 16。默認是考慮所有分子軌道的貢獻(只有占據軌道才有貢獻)
-m:設置SYSMOIC計算感生電流用的CTOCD方法的具體形式,參看http://sysmoic.chem.unisa.it/MANUAL/S4.SS12.html的說明。當基組是完備的時候這些方法結果等價。有些選項是僅對于某些方法才需要設,比如用common gauge-origin的時候需要用-g自定義原點坐標(默認為0 0 0)。默認用的CTOCD-DZ2基本上對于所有情況都是合適的選擇,也不用設額外參數。關于CTOCD不同形式的介紹和差異,可以看前述的SYSMOIC原文以及感生電流綜述文章WIREs Comput Mol Sci, 6, 639 (2016)的Continuous Transformations of the Origin of the Current Density一節。
JBMAP運行后可以修改的參數當中較重要的如下:
? B:外磁場矢量,默認B 0 0 1
? JTERM:感生電流類型,默認是JTERM 0,即diatropic和paratropic的總和。1和2分別代表paratropic和diatropic
? RI和RF:分別指定格點所均勻分布的矩形盒子的X、Y、Z最小笛卡爾坐標和最大笛卡爾坐標。默認是根據體系坐標自動確定以覆蓋整個體系。對上面的苯分子的例子,從屏幕上默認的參數可見,當前對應的是RI -6.0635 -6.6921 -2.0000和RF 6.0635 6.6921 2.0000
? FATT:給感生電流乘的系數。比如大部分箭頭在圖上都顯得很不顯著,就可以設大這個值以更便于觀看
? FMM:磁感生電流過濾范圍。例如FMM 0.01 1就代表忽略感生電流的模小于0.01和大于1的格點(是對乘上FATT之前而言的),免得干擾觀看。默認0 0.1
? STEP:格點間距,數值越小箭頭越密、計算越耗時。默認0.4 Bohr
? LUNA:0和1分別代表箭頭的長度和面積正比于感生電流的模,默認是后者
? METH:計算方法,默認的2對應CTOCD-DZ2
為了方便,我推薦以命令行方式運行JBMAP。交互式程序怎么以命令行方式運行,我在《詳談Multiwfn的命令行方式運行和批量運行的方法》(http://www.shanxitv.org/612)里有很詳細說明。下面以這種方式結合自定義參數用JBMAP再次對苯進行計算。創建一個文本文件叫CDMAP.txt,內容如下,每一行對應一條傳給JBMAP程序的命令。此例RI和RF的Z值都是1,X和Y范圍都是-6到6 Bohr,因此格點會分布在苯分子上方1 Bohr的XY平面上的正方形區域內。此文件后面的y、n、n、n對應交互式界面里程序向你提問時依次輸入的四個指令。
B 0 0 1
RI -6 -6 1
RF 6 6 1
FMM 0.01 1
FATT 3
STEP 0.4
LUNA 1
JTERM 0
y
n
n
n
運行JBMAP -f 0.5 < CDMAP.txt,然后再運行v3d JBM.3d,看到下圖,可見清楚地把苯分子上方的磁感生電流描繪了出來。
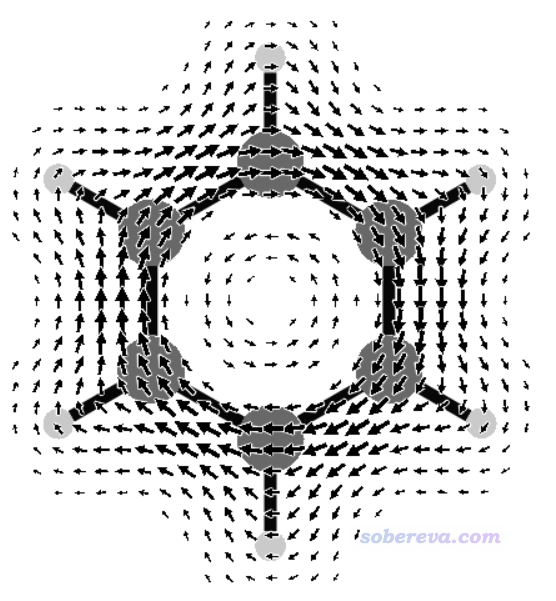
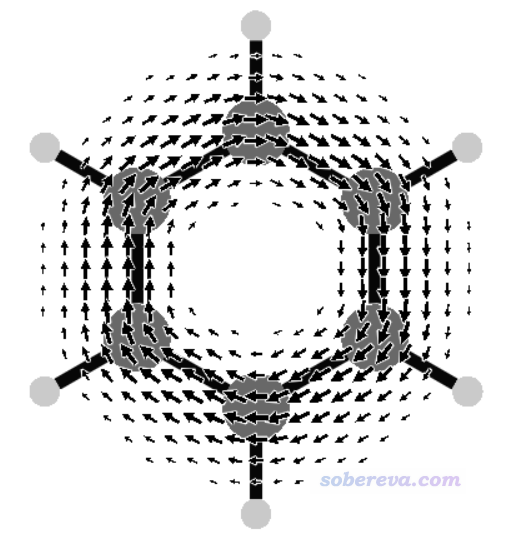
再來看只考慮pi軌道時的磁感生電流圖的繪制。把benzene.wfx文件載入Multiwfn,進入主功能0,按照《使用Multiwfn觀看分子軌道》(http://www.shanxitv.org/269)說的,從HOMO開始往下挨個看軌道,就能找到三個pi軌道,序號為17、20、21。原子數多的時候也可以用《在Multiwfn中單獨考察pi電子結構特征》(http://www.shanxitv.org/432)介紹的做法讓Multiwfn自動找出來所有pi軌道序號。運行JBMAP -q +3 17 20 21 -f 0.5 < CDMAP.txt,然后再用v3d繪圖,得到下圖,這便是苯的pi電子的感生電流圖了。相對于之前的所有電子貢獻的圖,此圖少了sigma電子造成的出現在內環的逆時針(paratropic電流)特征,而且也沒有了氫原子區域的感生電流,因為pi電子不在氫上。
3.3 v3d的使用細節
下面專門細談一下v3d界面的操作。v3d圖形窗口出現后,可以用鼠標拖動體系旋轉視角。還可以在圖形窗口處于激活狀態下按h鍵,此時文本窗口會出現可以用的所有按鍵的功能說明。其中較重要的按鍵在這里說一下。
先說視角調節按鍵:
a、b、g:視角由alpha、beta、gamma值定義,按a、b、g鍵會分別增加它們1.0。如果按住shift鍵再按,也即按A、B、G,會分別增加它們10
? f和F:調節縮放因子。按一下f圖像就會放大一點,按一下F就會放大很多
? d和D:調節視角距離。按一下d就會增加0.1,按一下D就會增加1。視角距離越遠,近大遠小的透視效果越弱、越接近正交視角的效果(據我所知v3d沒法完全用正交視角)
? n和N:設置霧化距離閾值。超過視角一定距離閾值開始就會用霧化效果使得遠處物體被屏蔽。按n增加閾值0.1,按N增加1.0
? C:令圖像居中
每次按以上按鍵調節設置的時候在文本窗口都會看到當前值。如果你想把這些鍵的操作效果反過來,比如按一次f就令圖像縮小一點,那就先按一下c鍵進入反轉操作狀態(文本窗口里會看到c變成了c-1),然后再按f就行了。再次按c則還原到普通操作狀態。
按p鍵可以把當前圖像保存成當前目錄下的.ps文件。這是矢量圖像格式,在Windows下可以用裝了ghostscript插件的irfanview或photoshop等程序打開,也可以用Acrobat distiller轉成pdf再打開。在Rocky Linux、CentOS等系統里也自帶了ps文件觀看程序。
默認情況下產生的ps文件畫出來的感生電流的箭頭會有較厚的白邊,當箭頭多的時候白邊會導致后面的箭頭被遮擋,比較難看。解決辦法是按一下c鍵先進入反轉操作狀態,然后按住k鍵一會兒使得白邊量減小(文本窗口不會提示減小到了多少),然后再導出ps文件,然后就會發現白邊非常細了。
如果想保存當前的作圖設置,按S,當前目錄下就出現了SpecialValues.txt文件,里面記錄了作圖設置,可以看到f、d等數值都記錄在內。當再次啟動v3d時,程序若發現當前目錄下有這個文件,就會自動載入里面的設置。如果你希望每次產生的ps文件里箭頭的白邊都非常細,可以啟動v3d后先直接按S保存一個記錄默認狀態的SpecialValues.txt文件,然后把里面對應白邊粗細的ridfr前面的值改為0.1。這樣每次啟動v3d就會默認用這個設置了。
4 計算鍵電流強度
對鍵截面做感生電流積分的概念我在《考察分子磁感生電流的程序GIMIC 2.0的使用》(http://www.shanxitv.org/491)中已經專門講過了,這可以定量考察穿越特定化學鍵的電流強度從而便于對比,這在SYSMOIC里叫鍵電流強度。SYSMOIC算鍵電流強度遠比用GIMIC更方便,都省得自己定義積分范圍,結果還能直接可視化。下面我們對苯的一個C-C鍵計算鍵電流強度。
直接在命令行窗口中輸入BOCUST命令運行此程序,程序首先會提示
| plotted 12 atoms
| plotted 12 bonds
====> Pair C1 - C2 distance= 1.396 Ang
這說明程序發現有12個原子對兒的距離都處在默認的距離下限(0.9埃)和距離上限(1.6埃)之內。其中C1-C2是第一個原子對,現在問你是否對這個C1-C2鍵計算鍵電流強度。這里輸入y然后按回車,很快就會出現結果
CONTRIBUTION 1 -0.6000526E+00 AU -0.1690912E-07 SI
CONTRIBUTION 3 0.1784109E+00 AU 0.5027513E-08 SI
TOTAL CS ==> -0.4216417E+00 AU -0.1188161E-07 SI
(2) CS/CS0 RELBEN FICS 0.99 3.00 0
上面顯示的兩個CONTRIBUTION分別是diatropic和paratropic對鍵電流強度的貢獻,由于方向相反,顯然符號相反。二者之和取絕對值0.4216417 a.u.就是鍵電流強度大小,換算成常用的nA/T單位就乘以28.179409,即11.88 nA/T。這個值與《考察分子磁感生電流的程序GIMIC 2.0的使用》(http://www.shanxitv.org/491)在B3LYP/6-311G*級別通過GIMIC程序得到的12.086 nA/T非常接近。上面信息中的CS/CS0 RELBEN FICS 0.99代表當前值與參考值的比值是0.99。這里參考值是程序內置的對苯在某個級別計算的C-C鍵電流強度12 nA/T。
接下來程序又問你是否對C1-C6計算鍵電流強度。由于當前所有C-C鍵都是等價的,只需要計算一個就行了,因此輸入N回車,程序就結束運行了。如果你想把所有鍵都進行計算,就接著輸入11次y回車把剩下11個鍵都算完。當前體系一共6個C-C鍵、6個C-H鍵。
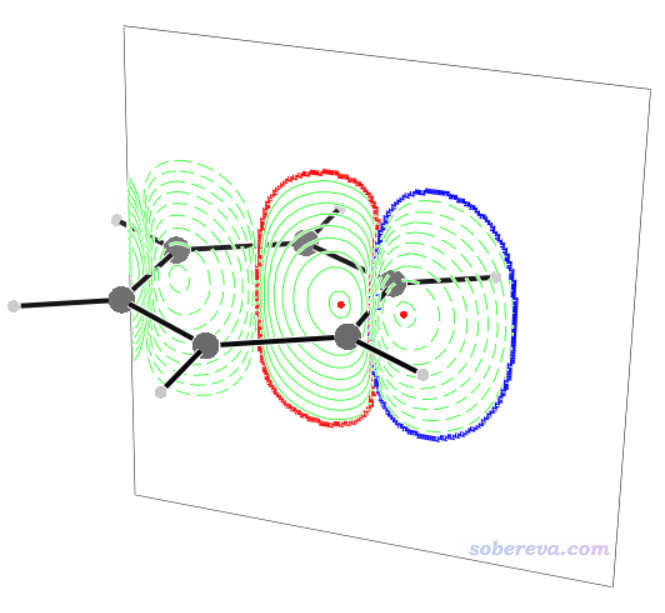
當前目錄下出現了BCS.3d文件。運行v3d BCS.3d,就可以看到下圖

此圖里黑色箭頭體現了C1-C2鍵電流的大小和方向。積分截面處在鍵的正中央并與鍵垂直,圖中的曲線是這個截面上的感生電流0.001 a.u.等值線,曲線內部區域是對感生電流積分的區域,紅色和藍色分別對應paratropic和diatropic部分。可見diatropic分布在環的外側,這與之前從向量場圖中看到的順時針環電流的主要分布是一致的。圖中的紅色小點對應截面上感生電流的極值點。鍵上面標的文字99對應于前述的參考比值0.99乘以100。文字出現的位置在鍵的正中央,不容易分辨,可以在圖形窗口激活狀態下按W鍵關閉文字顯示,然后再自行ps上。
如果你想一次性把所有鍵的電流強度都計算,又不想一次次麻煩地輸入y,那么可以寫一個文本文件比如叫ally.txt,里面有12行,每一行都是y。運行BOCUST < ally.txt,這樣所有鍵就都會被計算了。此時對得到的BCS.3d繪制會看到下圖

下面介紹一下BOCUST運行時可以接的選項中比較重要的:
-o、-f、-B、-m、-q等:和前面介紹的JBMAP程序的情況完全相同。即BOCUST也可以用-B指定外磁場方向、用-q指定考慮哪些軌道,等等。
-d、-D:確定哪些原子對兒被考慮時用的距離下限和上限(埃),默認-d 0.9 -D 1.6
-e:確定鍵截面上做積分的區域用的感生電流等值線的數值(a.u.),默認-e 0.001
-CS0:參考電流強度(nA/T),默認-CS0 12
-nocd:不顯示積分區域和極值點。如果想讓圖上只有箭頭而沒有曲線和小點,應該加上這個
-l [比值]:在截面上繪制感生電流的不同數值的等值線,后面跟的是相鄰等值線數值的比值
-C [粗細]:繪制截面的邊框
例如這里計算C1-C2的sigma電子的鍵電流強度(即扣除三個pi軌道貢獻),并在截面上顯示等值線、顯示邊框,就運行BOCUST -q -3 17 20 21 -l 3 -C 5,然后輸入y回車,再N回車。之后用v3d對BCS.3d作圖看到下圖。可見整個截面,以及里面的感生電流等值線,都顯示得很清楚。由于這個截面上diatropic和paratropic的大小都差不多(畢竟苯分子沒有sigma芳香性特征),所以鍵電流強度僅為0.007 a.u.。
5 總結
此文對分析磁感生電流的程序SYSMOIC做了介紹并給出了具體例子。由例子可見,通過SYSMOIC結合Gaussian繪制感生電流向量場圖很方便,不需要借助第三方程序。SYSMOIC做鍵電流強度計算也很自動化,不需要像GIMIC那樣謹慎地定義積分區域,而且鍵電流大小和方向還能直接通過箭頭直觀顯示出來。SYSMOIC的計算速度也比較快。
本文只涉及了SYSMOIC最常用的功能,還另外有一些其它功能,比如對感生電流密度做拓撲分析,計算各種感生電流密度相關的場(ACID及其各向異性、磁屏蔽密度、vorticity張量的跡及其各向異性等)并繪制成等值面和填色平面圖、計算任意格點上的感生電流張量和導數等等。
]]>文/Sobereva@北京科音
First release: 2024-Feb-16 Last update: 2024-Feb-17
0 前言
Hirshfeld surface分析(以下簡稱HS分析)是展現分子晶體中一個或多個分子與周圍分子間相互作用的方法,它也同樣可以用于展現孤立體系中特定片段與周圍原子間的相互作用。HS分析的原理容易理解,圖像比較直觀,在分子晶體的研究領域已經用得非常普遍。Multiwfn的主功能12中很早以前就已經實現了HS分析,如今已經被不少文章使用。本文將結合許多例子,專門詳細具體講解一下Multiwfn做HS分析的各方面細節、操作和技巧。
本文的讀者請務必使用2024-Feb-16及以后更新的Multiwfn版本,否則與本文所述情況會有很多不同。Multiwfn可以在其主頁http://www.shanxitv.org/multiwfn免費下載。不了解Multiwfn者請參看《Multiwfn FAQ》(http://www.shanxitv.org/452)和《Multiwfn入門tips》(http://www.shanxitv.org/167)。使用Multiwfn做HS分析在寫文章時請按照Multiwfn啟動時的提示對程序進行恰當的引用。
很值得一提的是,筆者提出的IGMH方法也非常適合展現分子晶體中的分子間相互作用,HS分析與IGMH分析的展現形式有明顯區別且有一定程度的互補性,二者亦可以同時使用以提供更多視角。請閱讀《使用Multiwfn做IGMH分析非常清晰直觀地展現化學體系中的相互作用》(http://www.shanxitv.org/621)和《一篇最全面介紹各種弱相互作用可視化分析方法的文章已發表!》(http://www.shanxitv.org/667)提到的綜述了解IGMH的相關知識,里面也專門有IGMH用于分子晶體的實際例子。
1 Hirshfeld surface分析的基本思想
在筆者講授的量子化學波函數分析與Multiwfn程序培訓班(http://www.keinsci.com/workshop/WFN_content.html)和Multiwfn手冊3.15.5節對HS分析的原理有很具體、詳細的講解,下文只是把原理的最關鍵的部分簡要介紹一下,對于正確做HS分析基本夠了。
HS分析最早由Spackman等人于Chem. Phys. Lett., 267, 215 (1997)提出,后來又得到了發展,Acta Cryst., B60, 627 (2004)和CrystEngComm, 11, 19 (2009)是其兩篇綜述文章。HS分析關鍵思想是對分子晶體中的特定分子構造出Hirshfeld surface,它相當于這個分子在分子環境中的表面,然后再將一些有特殊意義的實空間函數映射到這個表面上,由此可以對分子晶體中的分子間相互作用特征進行考察。
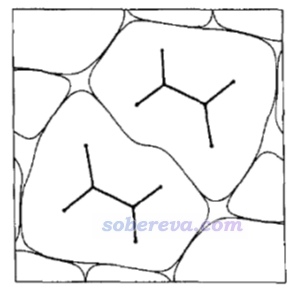
簡要說一下Hirshfeld surface是怎么定義的。Hirshfeld在Theoret. Chim. Acta (Berl.), 44, 129 (1977)最早提出了一種定義化學體系中原子空間的方式,它給每個原子定義了Hirshfeld權重函數來描述這個原子在三維空間中各個位置所占權重,數值從0到1平滑變化,0和1分別對應于此位置完全不屬于和完全屬于這個原子。每個位置所有原子的權重函數加和為1。這是一種典型的模糊式原子空間定義方式。具體定義細節見《原子電荷計算方法的對比》(http://www.whxb.pku.edu.cn/CN/abstract/abstract27818.shtml)的2.5節和Multiwfn手冊3.9.1節,看完了就會知道產生Hirshfeld權重需要有各個原子的坐標,以及體系中的各種元素的原子在孤立狀態下的球對稱化的電子密度。將一個分子中所有原子的Hirshfeld權重函數加和就定義了這個分子的權重,因此分子晶體中各個分子都有各自的權重函數。某個分子的Hirshfeld surface就對應于它的權重函數數值為0.5的等值面。例如下圖的曲線展現了某個平面上各個分子的Hirshfeld surface對應的輪廓。可見,Hirshfeld surface算是分子環境中各個分子與其它分子接觸面的一種定義方式。下文所說的“表面”都是指Hirshfeld surface。
現實當中構造Hirshfeld surface是利用我在J. Mol. Graph. Model., 38, 314 (2012)介紹的Marching Tetrahedron或類似的算法實現的。這個表面被描述為大量小三角形的集合,每個三角形由三個表面頂點構成。由于構造Hirshfeld surface對應的等值面用的算法和《使用Multiwfn的定量分子表面分析功能預測反應位點、分析分子間相互作用》(http://www.shanxitv.org/159)涉及的構造電子密度等值面對應的分子范德華表面的算法在本質上相同,所以Multiwfn中HS分析也是在主功能12(定量分子表面分析)中實現的。
實際中做HS分析的時候是選取一個(也允許是多個)感興趣的分子并對它構造Hirshfeld surface進行分析,這個分子在下文管它叫“中心分子”,它周圍的分子也可以稱為“環境分子”。
HS分析定義了一些映射到Hirshfeld surface上的三維實空間函數,常見的有:
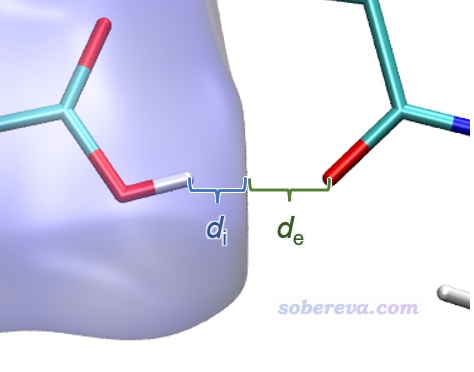
? d_i:表面內部(interior)的原子(也即中心分子的原子)到當前點的最近距離
? d_e:表面外部(exterior)的原子(也即周圍分子的原子)到當前點的最近距離
? d_norm:歸一化的(normalized)分子間接觸距離。由d_i、d_e和原子范德華半徑計算出來。數值越小體現表面上此處附近的內、外原子有越近的接觸,暗示此處的相互作用越強
? shape index(形狀指數):數值在[-1,1]范圍,越負說明此處表面越凹,反之越凸
? curvedness(曲度):在[-4,0.4]范圍,-4對應表面此處完全平坦,越正越凸,0對應單位球面的曲度
d_norm經常用來對Hirshfeld surface進行著色來直觀展現分子環境中的分子間的相互作用,d_norm數值較小的區域對應于較強烈的分子間相互作用。筆者發現用電子密度來著色也很有意義,Hirshfeld surface上電子密度越大的地方體現相互作用越強,效果比用d_norm著色時明顯更好,色彩過渡更為平滑,物理意義也更強,也沒有范德華半徑選取的任意性。由于對大體系做量子化學計算產生比較精確的電子密度比較耗時,因此只需要用準分子近似的電子密度(promolecular density)對Hirshfeld surface著色就夠了,它直接由各個原子孤立狀態的電子密度疊加得到,耗時極低,我實測和使用量子化學計算的電子密度著色的效果差不多。
由于各種元素的孤立狀態的電子密度以及范德華半徑在Multiwfn中是內置的,故產生Hirshfeld surface以及計算以上提及的各種函數只需要原子坐標和元素信息就夠了。由于HS分析依賴的信息非常簡單,不牽扯基于波函數的計算,因此耗時極低,用起來也很方便。
HS分析還經常繪制指紋圖(fingerprint map),是把d_i和d_e作為散點圖的橫軸和縱軸,然后把Hirshfeld surface上的各個頂點根據其d_i和d_e的數值繪制在指紋圖上作為一個個小點。根據指紋圖上散點分布的位置可以對中心和周圍分子間的相互作用特征進行討論,后文會結合具體例子來講。
HS分析中還經常做局部接觸(local contact)分析。完整的HS分析描述了中心分子的所有原子和周圍分子的所有原子間的相互作用,而局部接觸分析可以指定在HS分析中只考慮中心分子的哪些原子和周圍分子的哪些原子的相互作用。比如可以了解在整個Hirshfeld surface中體現中心分子的氧和周圍分子的H之間的相互作用的區域的位置和面積。
Multiwfn還支持Becke surface分析,是我自己提出的概念。它和HS分析的唯一差別是用Becke權重函數而非Hirshfeld權重函數的0.5等值面來定義表面。二者實際效果差別不大,一般沒必要用Becke surface,但它的一個特殊好處是允許表面出現在電子密度為0的區域,此處沒法定義Hirshfeld權重并構造Hirshfeld surface,詳見本文的第5節。Becke權重函數的具體定義方式參見《密度泛函計算中的格點積分方法》(http://www.shanxitv.org/69),它基于原子坐標和原子共價半徑得到。出現在相互作用的原子間的Becke surface會離半徑較小的原子較近、離半徑較大的原子較遠,Hirshfeld surface也有這樣的特點,這是由于定義它的準分子密度分布特征所自然而然帶來的。
2 Multiwfn的HS分析的功能
Multiwfn中做HS分析需要提供含有原子信息的文件作為輸入文件,如.pdb、.xyz、.mwfn、.cif、.fch、.mol2、.gjf等等等等,詳見《詳談Multiwfn支持的輸入文件類型、產生方法以及相互轉換》(http://www.shanxitv.org/379)。
HS分析大多研究的是分子晶體,cif是最常用的記錄晶體結構的格式。通常不能載入cif文件后上來就做HS分析,因為HS分析一般需要提供一個中心分子+環境分子的簇模型,這樣才能靠HS分析考察中心分子與環境分子的相互作用,而cif文件記錄的是晶胞里各個原子的坐標,分子往往是被截斷的,中心分子或環境分子一般都不完整。因此首先需要用《Multiwfn中非常實用的幾何操作和坐標變換功能介紹》(http://www.shanxitv.org/610)中介紹的自動挖團簇的功能構造簇模型。如果你研究的不是分子晶體的情況,就是比如分子二聚體中兩個分子間的相互作用,那么就不牽扯挖簇的過程了,直接提供含有二聚體坐標信息的文件當輸入文件就行了。
HS分析在Multiwfn主功能12實現。進入這個功能后,首先選1把定義表面的方式切換為Hirshfeld surface。此時被映射到表面的函數會自動改為電子密度(對于輸入文件沒有波函數信息的情況具體是指準分子電子密度),你也可以選2把被映射的函數改成其它的。之后選0就開始計算了,Hirshfeld surface會被構造出來,組成它的所有表面頂點上的被映射的函數值會被計算出來,并顯示出Hirshfeld surface的面積和包圍的體積。然后會看到后處理菜單,里面有豐富的選項,提示得都很清楚,Multiwfn手冊3.15.5節都有解釋。利用后處理菜單的選項,可以導出用于在VMD程序中繪制著色的Hirshfeld surface圖要用的.cub文件,還可以繪制指紋圖、做局部接觸分析,這些在后文的例子里都有體現。
后文的例子涉及到的各種文件可以在http://www.shanxitv.org/attach/701/file.zip中獲得。
3 用Multiwfn對NAOB晶體做Hirshfeld surface分析實例
這一節以(Z)-4-((2-nitrophenyl)amino)-4-oxobut-2-enoic acid (NAOB)晶體為例做HS分析。NAOB的分子結構如下,其晶體在DOI: 10.1007/s13738-023-02904-9里進行了研究,文章的補充材料里給了它的cif文件的信息,是本文文件包里的NAOB.cif。
3.0 準備工作:構造簇模型
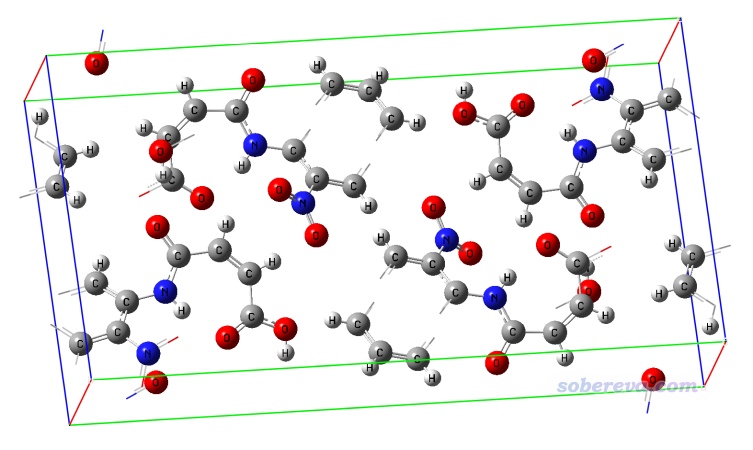
NAOB.cif對應的晶體結構如下,可見連一個完整的分子都沒有,因此在進行HS分析之前,我們顯然得先構造出中心分子+周圍分子的團簇結構才行。
為了構造簇模型,啟動Multiwfn,然后載入NAOB.cif,之后輸入
300 //主功能300
7 //幾何相關操作
25 //構造“中心分子+臨近一圈分子”的團簇
1 //當前晶胞里1號原子所在的分子將被作為中心分子(由于NAOB晶體里所有分子都是等價的,所以當前隨便輸入一個原子序號即可)
[回車] //代表若一個周圍分子與中心分子間最近原子對距離小于這倆原子的范德華半徑和的1.2倍,則這個周圍分子就被整個納入團簇
Multiwfn瞬間就構造出了團簇,從屏幕上的提示可看到這個簇有375個原子,并且屏幕上還巨貼心地把中心分子中的原子序號給了出來,此例為1-10,17-19,22-25,205-210,214,215。把這個序號記下來,之后HS分析時要用到。
現在可以選當前菜單中的選項0看一眼新構造出的團簇是什么樣,如下所示,可見非常理想,確實是中心分子被周圍一層分子所圍繞(為了中心分子看得清楚,在Multiwfn圖形界面的菜單欄里選了Other settings - Set atom highlighting,然后輸入了前述的中心分子里的原子序號,使中心分子用青色高亮了)。

點圖形界面右上角的RETURN按鈕關閉圖形窗口,然后選擇-2 Output system to .pdb file并輸入NAOB_cluster.pdb,以將當前簇結構導出為當前目錄下的這個文件。這個pdb文件在本文的文件包里也提供了。
注:如《實驗測定分子結構的方法以及將實驗結構用于量子化學計算需要注意的問題》(http://www.shanxitv.org/569)所強調的,由于X光衍射實驗一般難以確定氫原子的準確位置,因此原理上做HS分析之前最好先固定重原子而優化一下所有氫原子的位置。用免費高效的CP2K程序對晶體結構優化氫原子位置然后再用Multiwfn摳團簇,或是先摳團簇再用Gaussian等量子化學程序優化氫,都是可以的。
3.1 繪制電子密度著色的Hirshfeld surface圖
這一節演示繪制基于準分子近似的電子密度著色的Hirshfeld surface圖,這是HS分析最重要的一種圖。除了Multiwfn外還會用到非常流行的VMD可視化程序,可以在http://www.ks.uiuc.edu/Research/vmd/下載,使用筆者現在用的VMD 1.9.3版肯定沒問題,用其它版本不保證能按照本文的例子正常作圖。
啟動Multiwfn,載入上一節產生的NAOB_cluster.pdb,然后輸入
12 //定量分子表面分析
1 //選擇定義表面的方式
5 //Hirshfeld surface
1-10,17-19,22-25,205-210,214,215 //中心分子的原子序號
0 //開始分析
從屏幕上可以看到許多信息,以下兩條是值得注意的,第一個是Hirshfeld surface包圍的體積,相當于分子晶體中屬于這個中心分子的體積,第二個是Hirshfeld surface的面積
Volume: 1644.06256 Bohr^3 ( 243.62494 Angstrom^3)
Overall surface area: 859.01840 Bohr^2 ( 240.54965 Angstrom^2)
現在看到了后處理菜單。選擇-2在當前目錄下導出surf.cub,它是中心分子的Hirshfeld權重的格點數據。再選13,Multiwfn會計算被映射的函數的格點數據并導出為當前目錄下的mapfunc.cub。在這個界面里如果選擇-3也可以直接在Multiwfn里預覽Hirshfeld surface,但沒有著色效果。
將剛剛產生的surf.cub和mapfunc.cub以及Multiwfn自帶的examples\scripts\目錄下的hirsh_rho.vmd作圖腳本一起拷到VMD目錄下(即啟動VMD后在其文本窗口里運行pwd命令看到的目錄)。啟動VMD,在文本窗口里輸入source hirsh_rho.vmd來執行作圖腳本,然后就看到了下圖。
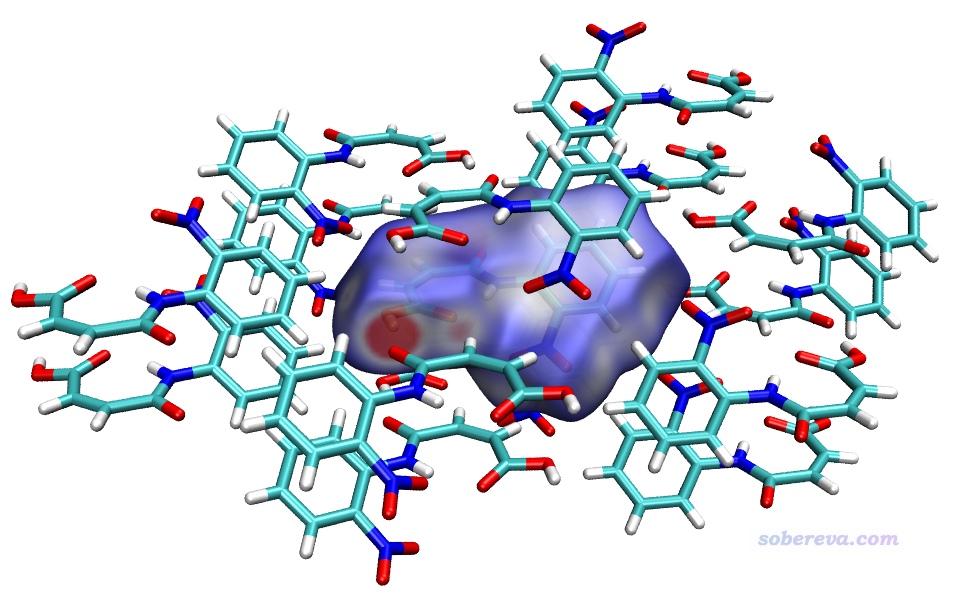
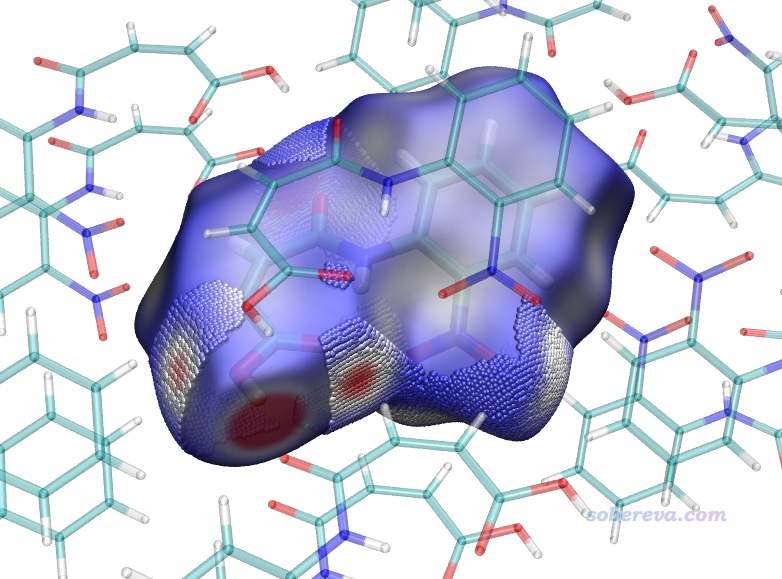
上圖顯示出了中心分子的Hirshfeld surface,并根據電子密度進行了著色。通過hirsh_rho.vmd腳本里的mol scaleminmax top 1 0.0 0.015這條命令可知,默認用的色彩刻度范圍的下限和上限分別為0和0.15。根據腳本里color scale method BWR這行命令可知,當前用的色彩變化是藍-白-紅,色彩變化示意圖在Graphics - Colors - Color Scale里可以看到。因此上圖中越紅的地方就是Hirshfeld surface上電子密度越大的地方,無疑對應于越強的相互作用。上圖中有一塊非常大的紅色,這對應于中心分子羧基的O-H作為氫鍵給體、周圍一個分子的O作為氫鍵受體形成的顯著的氫鍵作用區域。它的旁邊還有一小塊淡紅色區域,對應于中心分子的羧基氧作為氫鍵受體與周圍分子的C-H形成的弱氫鍵。上圖中還有一些發白的區域,在VMD里旋轉圖像仔細觀察的話可以看到對應的是比較遠距離的C-H...O-N很弱氫鍵,以及pi-pi堆積和普通色散吸引作用顯著的區域。這些特征區域都可以自行用powerpoint之類畫個箭頭標注在圖上便于讀者看清楚。上圖還有很多偏藍色的區域,這些地方電子密度接近0,因此不牽扯任何值得一提的分子間相互作用。特別藍的地方也往往對應于分子晶體中的孔洞區域,這種地方電子密度自然特別低,非常建議感興趣的讀者按照《使用Multiwfn計算晶體結構中自由區域的體積、圖形化展現自由區域》(http://www.shanxitv.org/617)介紹的方法作圖考察。
要注意中心分子與各個方向的周圍分子都有相互作用,光是靠一張圖的話很難展現完整,因此文章中可以多給幾張圖展現不同視角的Hirshfeld surface圖。
下面再說一下怎么改進作圖效果。Hirshfeld surface圖的效果受到多方面影響:
(1)光源。可以通過VMD的Display菜單里的Lighting選項設置打開哪些光源。如果選Mouse - Move light,然后在圖形窗口中拖動,還可以移動特點光源的位置。
(2)材質。hirsh_rho.vmd默認對等值面用Translucent材質,可以自行在Graphics - Materials界面里對這個材質的具體定義進行調節。
(3)Graphics - Representation界面里的作圖設置。里面可以創建更多Rep,每個Rep都可以獨立設置顏色和材質,并且通過選擇語句可以定義各個rep顯示的原子,不懂選擇語句怎么寫的話參考《VMD里原子選擇語句的語法和例子》(http://www.shanxitv.org/504)。特別值得一提的是,當前的圖中每個分子都有一個獨立的residue編號并被分子中所有原子所共享,因此可以利用residue選擇特定分子。若想查詢某個分子的residue號,就選Mouse - Query,然后點擊這個分子上任意一個原子的正中央,在VMD的文本窗口中就能看到它的residue號了。
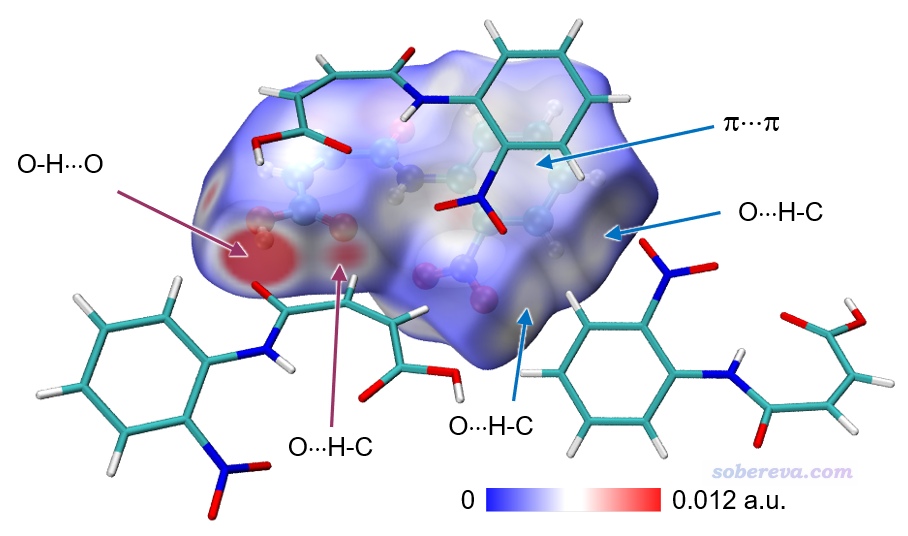
為了讓上面例子的圖像效果更好,我在VMD的Graphics - Representation里做了些修改:把第1個rep設為了residue 0專門用于顯示中心分子,用CPK方式顯示并把Bond Radius設為了0.5。點擊Create Rep按鈕新增一個Rep,選擇語句用residue 7 11 5使得三個與中心分子作用顯著的分子顯示出來,Material設Edgy,Drawing Method用Licorice,把Bond Radius設成0.1。點擊用來顯示Isosurface的那個Rep,再選擇Trajectory標簽頁,在里面把色彩刻度上限設為0.012(注意文本框只顯示兩位小數,輸入0.012后按回車會如實設為0.012),使得著色的色彩顯得更鮮明。在Display菜單里把所有四個光源都打開。最后選File - Render,選擇Tachyon(Internal)進行渲染。之后在圖像上展現特征作用的地方通過powerpoint進行標注,并且把Graphics - Color - Color Scale里顯示的色彩刻度條挪到圖上并適當拉伸、標記上色彩刻度上下限。最終得到下圖,可見對相互作用展現得非常直觀清楚。
如果要繪制d_norm著色的Hirshfeld surface圖,與上面的例子只有兩個差別
(1)進入主功能12并選擇用Hirshfeld surface方式定義分子表面后,選擇2 Select mapped function,再選d_norm。然后再開始分析
(2)作圖使用Multiwfn目錄下examples\scripts\里的hirsh_dnorm.vmd代替hirsh_rho.vmd
由于d_norm著色的圖明顯不如電子密度著色的圖色彩變化平滑,在很多地方有顏色的突越不好看,故這里就不多說了。
Multiwfn還可以給出Hirshfeld surface上被映射的函數(當前為電子密度)的極大點的位置和數值,便于定量對比討論。在Multiwfn后處理菜單中選8 Export all surface vertices and surface extrema as vtx.pqr and extrema.pqr,此時當前目錄下就出現了vtx.pqr和extrema.pqr,分別記錄了所有表面頂點和表面極值點的坐標和被映射的函數數值。如屏幕上的提示所示,extrema.pqr里碳和氧原子分別用于記錄表面極大點和極小點,此文件在本文的文件包里也提供了。按前文用hirsh_rho.vmd作圖后,將extrema.pqr載入VMD,將其顯示方式設為VDW并把Sphere Scale設為0.2,顏色用黃色,此時看到下圖,每個黃色小球都對應Hirshfeld surface上電子密度極大點位置。
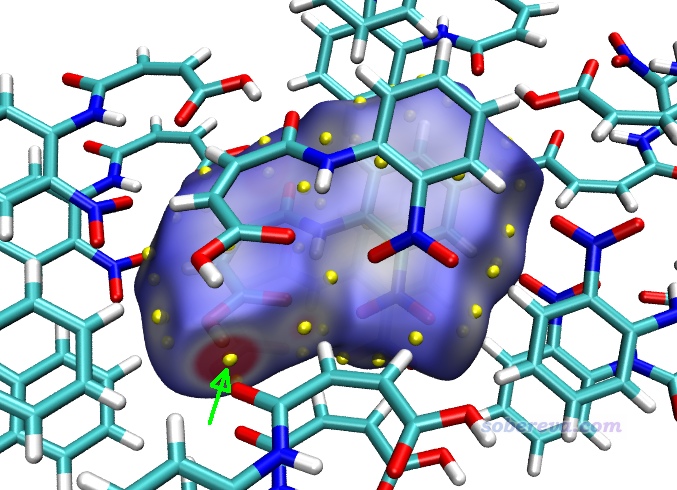
若想查詢表面極值點處的電子密度數值,就在VMD里選Mouse - Query,然后點擊要考察的黃球的正中心,文本窗口就出現了它的index號,從0開始記。上圖箭頭所指的那個對應O-H...O作用的極值點的index為9,相當于從1開始記的編號為10。打開extrema.pqr,找到對應10號碳的下面這一行,倒數第3列的數值0.04228468就是此處的電子密度了,單位為a.u.。
HETATM 10 C MOL A 1 -1.214 3.606 5.014 0.04228468 1.0000 C
以類似的方式查詢旁邊那個C-H...O氫鍵對應的Hirshfeld surface上的電子密度極大點,數值為0.01093053,可見作用顯著弱于O-H...O。
3.2 繪制指紋圖
這一節繪制HS分析中很常見的指紋圖。先按上一節的過程做HS分析并進入到主功能12的后處理菜單,然后輸入
20 //指紋圖與局部接觸分析
0 //開始分析
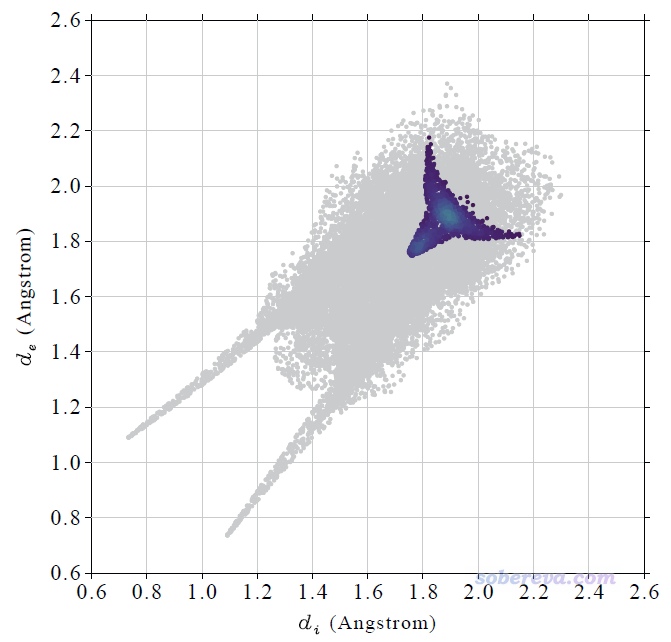
計算很快就完成了,然后看到一個新的后處理菜單,里面的選項不言自明。直接選0就會在屏幕上顯示完整的指紋圖,選1就會把指紋圖保存為當前目錄下的pdf文件(程序用pdf格式是因為它可以無損縮放、線條平滑),此pdf文件已經提供在了本文的文件包里(NAOB_HS.pdf)。當前的圖像如下
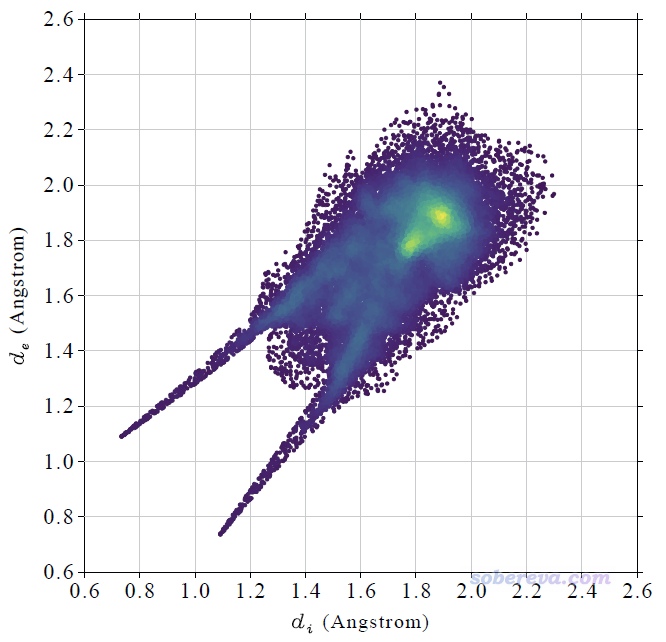
這個指紋圖里每一個小點都是一個Hirshfeld surface上的頂點,并且根據點在指紋圖上的分布密度進行了著色,越密的地方顏色越黃,越稀疏的地方顏色越紫。Multiwfn自動根據指紋圖上的最大分布密度設置色彩變化范圍的上限。Multiwfn畫指紋圖用的這種色彩過度方式稱為viridis,用Google搜圖功能一搜viridis就能找到相應的色條。
上圖在左下角有兩個顯著的尖兒(spike。兒化音明確體現這是個名詞),是圖像的特征區域,相當于“指紋”。靠左的那個尖兒的頂端大約是d_i≈0.7、d_e≈1.1位置,此處d_e顯著大于d_i,這是此體系具有氫鍵給體特征的體現(如上一節所示此體系的羧基氫確實是氫鍵給體)。為什么這個尖兒的存在能體現此分子存在氫鍵給體?因為這說明在Hirshfeld surface的這個區域,中心分子的原子到表面的最近距離(d_i)顯著小于周圍分子的原子到表面的最近距離(d_e),只有當氫鍵跨越這個區域,半徑很小的氫在表面內、半徑較大的重原子在表面外的時候才會出現這種情況,如下圖所示。
顯然不難理解前面的指紋圖中靠下的那個在d_e≈0.7、d_i≈1.1的尖兒體現的是中心分子作為氫鍵受體與周圍分子形成的氫鍵跨越了Hirshfeld surface。所以指紋圖說明當前研究的NAOB分子同時作為氫鍵給體和氫鍵受體。
指紋圖還蘊含了更多信息,但當前的指紋圖的中間區域密密麻麻一片,很難直接討論,進一步考察就需要利用下一節的局部接觸分析了。
順帶一提,如果你想改變指紋圖上的點的密度,可以在主功能12里做分析之前先選擇3 Spacing of grid points for generating molecular surface修改格點間距,格點間距越小則產生的Hirshfeld surface上的點就越多,指紋圖也會越密。
3.3 局部接觸分析與局部指紋圖
這一節演示一下怎么對中心分子特定的原子與周圍原子特定的原子進行局部接觸分析并繪制與之對應的指紋圖。作為例子,這里考察中心分子的氧與周圍分子的氫的局部接觸情況。
按照上一節說的進入到主功能12的后處理菜單中的20 Fingerprint plot and local contact analyses選項后,輸入以下內容
1 //設置內部原子范圍。默認是所有中心分子的原子都包括
[回車] //代表對原子序號不做限制
O //必須是氧元素
現在中心分子的所有氧原子都納入到了要考慮的內部原子范圍了。然后再輸入
2 //設置外部原子范圍。默認是所有周圍分子的原子都包括
[回車] //代表對原子序號不做限制
H //必須是氫元素
現在周圍分子的所有氫原子都納入到了外部原子要考慮的范圍了。
選擇0開始分析,算完后屏幕上顯示以下信息,告訴你當前考察的這種接觸面積是61.5 Angstrom^2,占Hirshfeld surface總面積的25.57%。
The area of the local contact surface is 61.504 Angstrom^2
The area of the total contact surface is 240.550 Angstrom^2
The local surface occupies 25.57% of the total surface
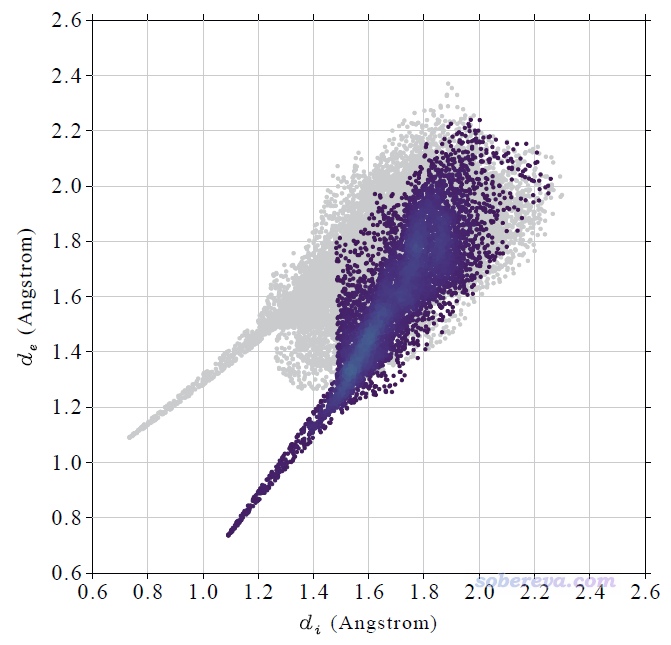
然后在后處理菜單選擇繪制指紋圖,看到下圖(對應本文文件包里的NAOB_HS_O-H.pdf)。此圖中只有對應于當前考察的局部接觸面上的頂點才被繪制為彩色,可見指紋圖中d_e≈0.7、d_i≈1.1的尖兒確實對應于內O與外H的接觸。
這種局部接觸還可以在立體結構圖上體現。在當前界面里選擇4 Export surface points to .pqr file in current folder,然后當前目錄下就產生了finger.pqr和finger_all.pqr,它們都提供在了本文的文件包里。前者記錄的是當前指定的局部接觸表面上的頂點,后者記錄的是完整的Hirshfeld surface上的頂點。它們可以用文本編輯器打開,可以看到每個表面頂點在pqr文件里用一個碳原子表示,倒數第三列記錄的是Charge屬性,當前被用來記錄表面頂點上被映射的函數數值,對當前來說就是準分子電子密度,單位為a.u.。
為了得到同時展現體系結構和局部接觸表面的圖,現在將NAOB_cluster.pdb載入VMD并恰當設置顯示方式,再把finger.pqr載入VMD,在Graphics - Representation里將它的Drawing Method設為Points并恰當設置Size,Coloring Method設為Charge(即根據Charge屬性著色),在Trajectory標簽頁里把色彩刻度設成與之前繪制等值面圖用的相同的0到0.012。確保Display - Rendermode已經設為了GLSL。在Graphics - Colors - Color Scale里把色彩刻度設為與之前相同的BWR,現在看到的圖如下,確實這些局部表面只對應中心分子的氧和周圍分子的氫的接觸。
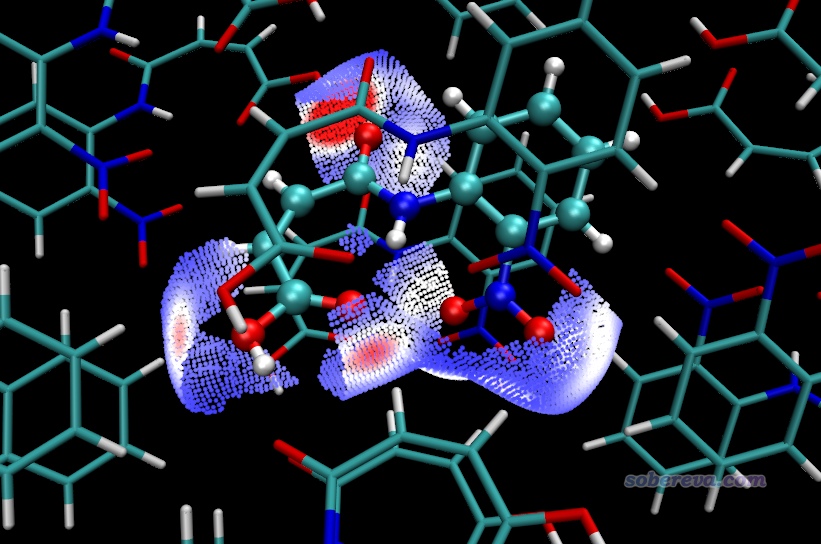
也可以先用hirsh_rho.vmd照常繪制完整的Hirshfeld surface圖,然后再載入finger.pqr,把Drawing Method設為VDW并把Sphere Scale設為0.1,并且對Charge屬性用0到0.012色彩刻度范圍著色,此時看到的圖像如下,局部接觸部分以小球形式著重展現了。
下面再做個演示,對中心分子的芳環和周圍分子的芳環之間考察局部接觸,展現它們之間的pi-pi堆積。在GaussView里,或者在VMD里使用我在《在VMD中顯示原子序號的方法》(http://www.shanxitv.org/197)中提供的腳本,都可以在它們載入NAOB_cluster.pdb后顯示出原子序號,可以看到中心分子的芳環的原子序號是7-9,205,207,209。與中心分子芳環較近因而有pi-pi堆積作用的周圍分子的芳環有兩個,原子序號為11,13,15,112,114,116,201-203,356-358。因此,進入前述的Multiwfn的20 Fingerprint plot and local contact analyses選項后,輸入
1 //設置內部原子范圍
7-9,205,207,209 //原子序號范圍
[回車] //對元素不做限制
2 //設置外部原子范圍
11,13,15,112,114,116,201-203,356-358 //原子序號范圍
[回車] //對元素不做限制
0 //開始分析
4 //導出finger.pqr和finger_all.pqr
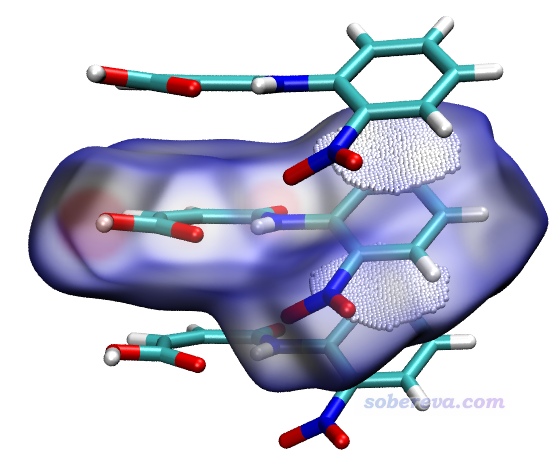
然后按照上一節的方法,用hirsh_rho.vmd繪制出Hirshfeld surface,再將finger.pqr載入VMD并恰當設置顯示方式。顯示體系結構的rep的選擇語句寫residue 0 1 11,其中residue 0是中心分子,residue 1和11對應與它有pi-pi堆積作用的上、下兩個分子。然后在Display - Material里把Translucent材質的Opacity改為0.5以降低透明度,并打開Angle-Modulated Transparency選項使得等值面立體感更強一些。之后看到的圖如下,可見小圓球把中心分子芳環上下兩側的pi-pi堆積對應的接觸區域都清晰展示了出來。
在Multiwfn里顯示出相應的局部接觸表面的指紋圖,如下所示。由于碳的原子半徑不小,因此pi-pi堆積對應的局部表面與表面內和表面外的碳原子都有一定距離,而碳原子間距離若太遠也不會有pi-pi堆積效應,因此這些表面頂點在指紋圖中的位置是d_i和d_e都不大不小的區域。
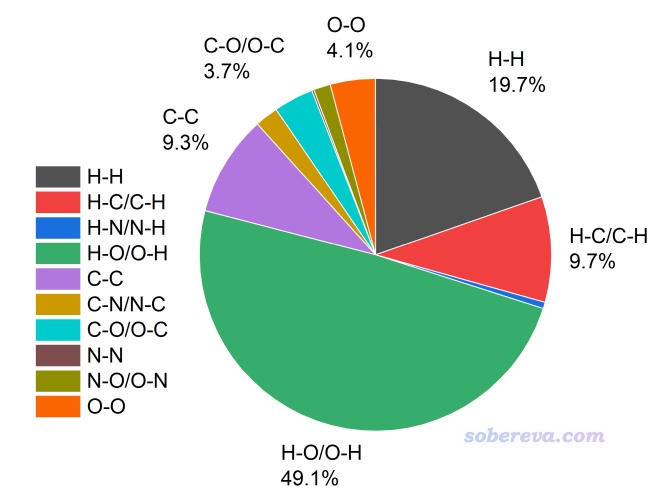
3.4 統計不同元素間的接觸面積
如果想得到中心分子與周圍分子的每一對元素之間的局部接觸面積,雖然通過前面演示的局部接觸分析可以手動實現,但有多少種元素組合就得操作多少次,很麻煩。因此Multiwfn提供了一次性完成的功能。進入前述的Multiwfn的20 Fingerprint plot and local contact analyses選項后,不需要定義內部外部原子,直接選擇選項3 Calculate contact area between different elements,就可以馬上得到以下信息
Inside element, outside element, their contact area (Angstrom^2) and percentage (%)
H-H 47.330 19.676
H-C 10.423 4.333
H-N 0.521 0.216
H-O 56.618 23.537
C-H 12.821 5.330
C-C 22.304 9.272
C-N 2.614 1.087
C-O 4.552 1.892
N-H 0.876 0.364
N-C 2.546 1.059
N-N 0.464 0.193
N-O 1.910 0.794
O-H 61.504 25.568
O-C 4.280 1.779
O-N 1.816 0.755
O-O 9.970 4.145
The same as above, but do not distinguish inside and outside elements
H-H 47.330 19.676
H-C/C-H 23.244 9.663
H-N/N-H 1.396 0.581
H-O/O-H 118.121 49.105
C-C 22.304 9.272
C-N/N-C 5.160 2.145
C-O/O-C 8.831 3.671
N-N 0.464 0.193
N-O/O-N 3.727 1.549
O-O 9.970 4.145
Area of total contact surface is 240.550 Angstrom^2
可見以上信息包含了每一對元素的分析結果。例如O-H 61.504 25.568這一行代表中心分子的氧元素與周圍分子的氫元素之間的局部接觸面積是61.504 Angstrom^2,占總面積的25.568%,這和3.4節我們專門算的結果完全一致。而后面輸出的比如H-O/O-H 118.121 49.105代表不管O和H誰在表面內、誰在表面外,兩種元素之間的接觸面積總共是118.121 Angstrom^2,占總面積的49.105%。
為了看起來更直觀,可以把上面的三列形式的數據拷到txt文件里,然后拖到Origin中導入,再繪制餅形圖,如下所示,一目了然。圖中只有占比相對較大的幾種接觸直接標注了數值。
3.5 Hirshfeld surface等值面的截斷問題
Multiwfn在構造Hirshfeld surface之前,需要先對一個矩形區域(稱為“盒子”)中均勻分布的格點計算你設定的體系片段的Hirshfeld權重。盒子范圍是對你定義的片段往各個方向延展一定距離來確定的,延展距離是位于最邊界的原子的范德華半徑乘以一個系數得到的。默認的系數值是1.7,一般來說夠大了,但如果Hirshfeld surface延伸到距離當前片段較遠的地方,導致超過了盒子范圍,則Hirshfeld surface就會在相應地方被截斷,等值面在那個地方看起來就會有窟窿。此時如果你想要讓等值面完整、完全封閉,顯然就需要增大系數。這一節拿富勒烯晶體舉個例子。
本文文件包里的C60.cif是C60富勒烯晶體結構文件。按照3.0和3.1節的做法摳團簇、繪制電子密度著色的Hirshfeld surface,會得到下圖。這里我在Graphics - Representation里選擇顯示等值面的那個Rep后在界面右下角Show旁邊的下拉框里選擇了Box+Isosurface,這樣除了等值面外,盒子范圍還會用細線同時顯示出來。由下圖可見,等值面上有個難看的窟窿,因為6個富勒烯之間有孔洞區域,Hirshfeld surface實際上會延伸到這里,但當前被尺寸有限的盒子截斷了。
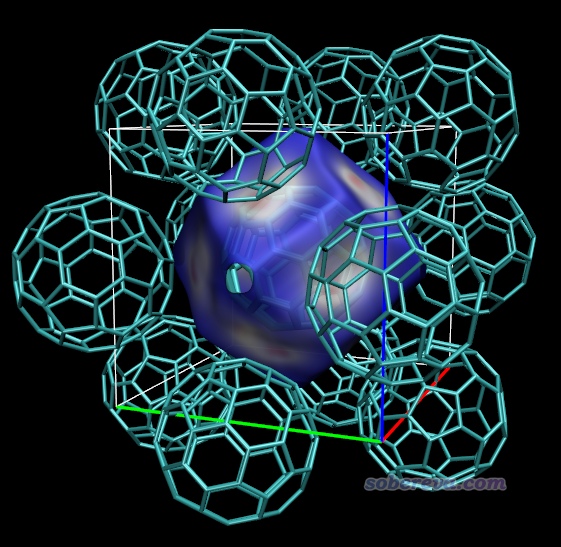
當你發現存在這樣的窟窿,想通過增大延展距離來避免,就應當從主功能12的后處理菜單返回之前的菜單(即剛進入主功能12看到的菜單),然后輸入
4 //高級選項
1 //設置確定延展距離用的范德華半徑的倍數
2.3 //設一個比原本更大的值,數值可以反復嘗試。設得越大,盒子就越大,格點數就越多,計算耗時就越高、產生的cub文件尺寸就越大。由于之前的等值面只有一個小窟窿,所以盒子再大一點就夠了,因此當前嘗試的2.3比之前默認的1.7沒大太多
0 //返回
0 //開始計算
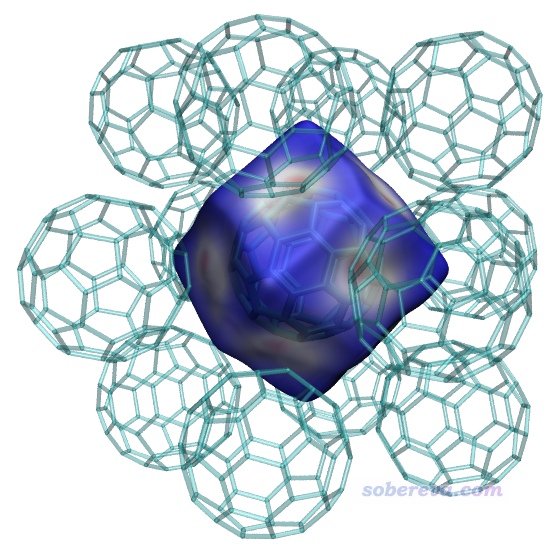
之后按照之前的步驟繪制Hirshfeld surface圖,如下所示,窟窿已經沒了。當前的Hirshfeld surface是個正十二面體,每個面都有塊白色且中間偏紅的區域,清楚地展現出中間的富勒烯和周圍富勒烯在此處有顯著的pi-pi堆積作用,由于這些區域離碳原子不遠因此電子密度不是很小。而表面上藍色部分都是晶體中的縫隙、孔洞區域,電子密度極低。
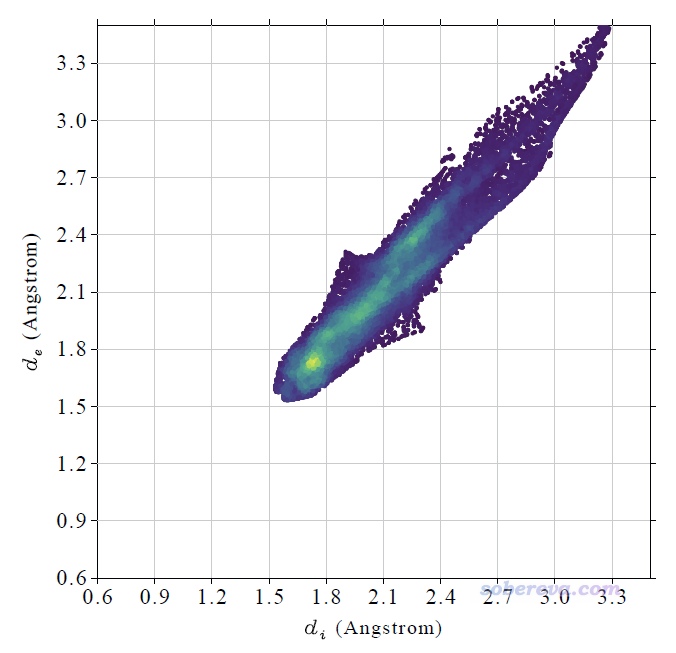
順帶一提,此體系的指紋圖如下所示。繪圖前用Multiwfn菜單里的選項3 Set range of axes適當加大了上限。可見散點的分布區域很窄,各處d_i和d_e數值都差不多大,涵蓋從中等到較大范圍。這體現出表面頂點基本都是位于中間分子和周圍分子之間的正中央區域,并且中間分子和周圍分子有的地方挨得近(d_i和d_e為中等大小),有的地方離得遠(d_i和d_e較大)
4 將Hirshfeld surface分析用于分子復合物
這一節示例HS分析用于展現孤立體系中的片段間相互作用。片段是指一批原子的集合,可以根據需要自由定義,比如既可以是分子復合物中的一個或多個分子,也可以是一個分子中的一個基團,也可以是一個過渡金屬配合物中的一個或多個配體,等等。對于孤立體系,由于感興趣的片段通常不是像晶體環境一樣被其它原子所完整包圍的,所以相應的Hirshfeld surface通常是開放的而不是完全封閉的。
筆者之前做了大量的和18碳環及其衍生物有關的研究工作,匯總見http://www.shanxitv.org/carbon_ring.html。其中研究了具有雙環特征的OPP分子與18碳環的結合問題,成果介紹見《8字形雙環分子對18碳環的獨特吸附行為的量子化學、波函數分析與分子動力學研究》(http://www.shanxitv.org/674)和《理論設計新穎的基于18碳環構成的雙馬達超分子體系》(http://www.shanxitv.org/684)。研究中使用了IGMH方法直觀展現了OPP與18碳環之間的弱相互作用,此例通過繪制電子密度著色的Hirshfeld surface圖也來展現一下。
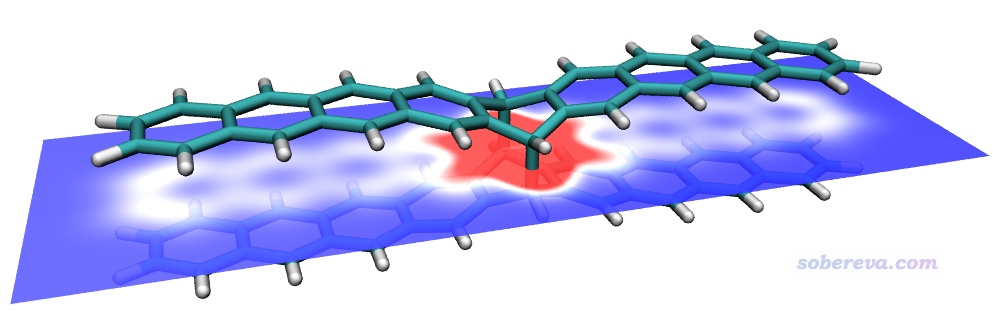
上述研究中使用DFT優化出來的OPP結合一個18碳環的結構文件是本文文件包里的C18-OPP.pdb。將之載入Multiwfn,然后進入主功能0,會發現此結構文件中18碳環是斜著的,而由于HS分析用的盒子的邊框總是平行于笛卡爾軸的,因此直接對它產生Hirshfeld surface的話表面開口的地方也將是斜著的,看著很惡心。因此此例做HS分析之前還需要先用《Multiwfn中非常實用的幾何操作和坐標變換功能介紹》(http://www.shanxitv.org/610)里介紹的功能令18碳環平行于XY平面,如下所示。
啟動Multiwfn,載入C18-OPP.pdb,然后輸入
300 //其它功能(Part 3)
7 //對當前體系做幾何操作
11 //令一批原子擬合的平面平行于某個笛卡爾平面
225-242 //18碳環的原子序號
1 //平行于XY平面
現在可以選0在圖形界面觀看當前結構,會發現確實18碳環已經在XY平面上了。點RETURN關閉圖形窗口,然后接著輸入
-10 //返回
0 //返回到主菜單
12 //定量分子表面分析
1 //設置表面的定義
5 //Hirshfeld surface
225-242 //18碳環的原子序號
4 //高級選項
1 //設置定義盒子延展距離用的范德華半徑的倍數
2.3 //比默認值更大,實測不這樣的話會導致不好看的截斷
0 //返回
0 //開始分析
-2 //導出surf.cub
13 //導出mapfunc.cub
現在基于surf.cub和mapfunc.cub用hirsh_rho.vmd照常繪制Hirshfeld surface圖。這里我把VMD的四個光源都打開了,18碳環用CPK方式顯示,雙環OPP分子用Edgy材質。為了著色效果更鮮明,色彩刻度上限設為了比默認更小的0.01。此時圖像如下所示,可見白色和偏紅的條狀區域把18碳環與OPP的pi-pi作用最強烈的區域展現得挺清楚,此處電子密度比周圍相對更大。由于OPP的大環不是像18碳環一樣理想的圓形,形狀沒有完美匹配,因此它們之間的相互作用明顯不是處處均勻的。通過Hirshfeld surface的顏色可見,在大環末端,特別是兩個大環連接區域,大環與18碳環的pi-pi作用都很弱、電子密度很低。當前的Hirshfeld surface在18碳環上、下方都是開口的,因為在這里被盒子邊界截斷了,顯然這樣的截斷是理應有的,因為上下沒有其它原子了,原理上就不可能是封閉的。
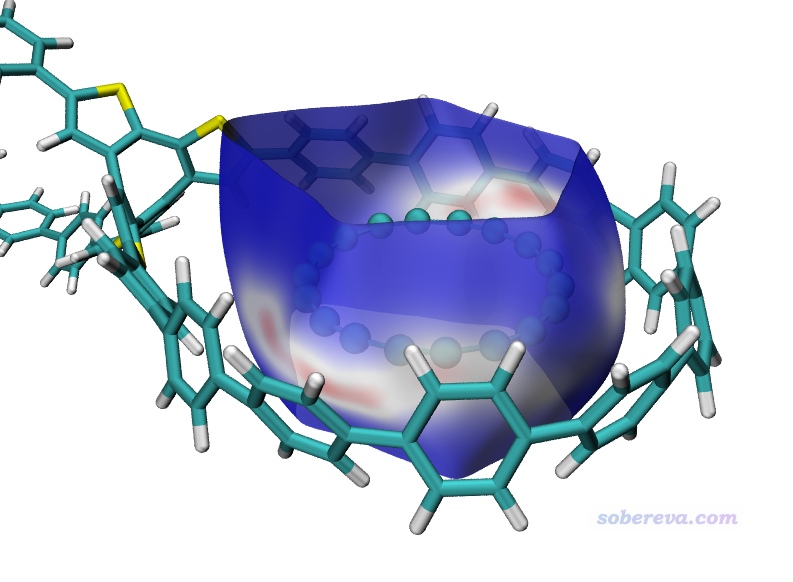
下面這張圖是Comput. Biol. Chem., 101, 107786 (2022)中用Multiwfn+VMD畫的Hirshfeld surface圖,展現了配體和附近的蛋白質的氨基殘基間的相互作用情況,效果不錯,作用位點能看得比較清楚。
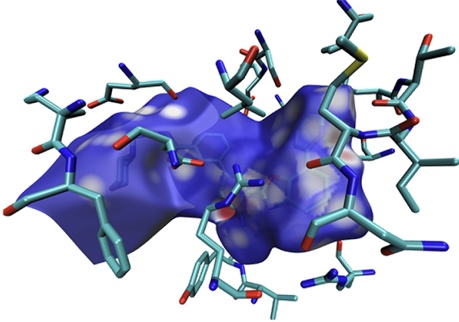
值得一提的是HS分析不僅限于用于考察弱相互作用,讓Hirshfeld surface跨越化學鍵也完全可以。在《一篇最全面介紹各種弱相互作用可視化分析方法的文章已發表!》(http://www.shanxitv.org/667)介紹的我的綜述文章中給出了一個九并苯共價二聚體的例子,這里我也繪制了其電子密度著色的Hirshfeld surface,其中一個九并苯被定義為了片段,色彩刻度用0-0.01,圖像如下所示。可見Hirshfeld surface正好嚴格平行于兩個九并苯、精確處于二者之間。在兩個九并苯之間形成共價鍵的區域及附近電子密度很大,顏色都為紅色,而其它白色區域體現了九并苯之間的明顯的pi-pi堆積作用。
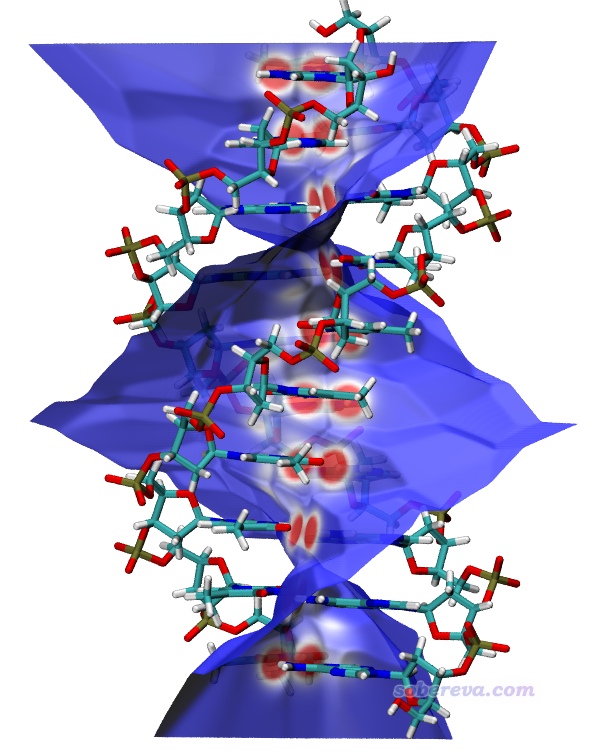
5 Becke surface分析一例:DNA
距離原子非常遠的地方電子密度精確為0,這樣的地方是沒法計算Hirshfeld權重的,Hirshfeld surface也沒法在這個區域出現,這是HS分析的一個缺點。如果極個別情況下被研究的片段與其它部分的接觸面就是會牽扯到距離所有原子都很遠的區域,那么這個時候就必須用我提出的Becke surface代替Hirshfeld surface了,因為在電子密度為0的地方也是可以計算片段的Becke權重的。除了這種極個別情況外沒有用Becke surface的必要,圖像效果不會更好,而且對大體系耗時更高。
這一節就以DNA雙螺旋結構為例繪制做Becke surface分析。Multiwfn自帶的examples目錄下的DNA.pdb是一個DNA片段的結構文件,其中一條鏈的原子序號是1-319,它將被定義為片段。
啟動Multiwfn,載入DNA.pdb,然后輸入
12 //定量分子表面分析
1 //選擇表面的定義
6 //Becke surface
1-319 //第一個片段的原子序號
4 //高級選項
1 //設置定義盒子延展距離用的范德華半徑的倍數
0 //倍數設為0,使得盒子緊貼著邊界原子。因為感興趣的相互作用區域是在兩條鏈間,這樣減小盒子尺寸可以避免Becke surface過大、延伸到不感興趣的區域去
0 //返回
0 //開始計算
在筆者的i9-13980HX筆記本上,用16核并行,花了15分鐘。嫌慢的話可以用核很多的服務器。對于預覽目的,也可以把格點間距設大到比如0.5,耗時只有原本的約八分之一,但此時圖像會明顯更為粗糙。
導出mapfunc.cub和surf.cub并照常用hirsh_rho.vmd繪圖,得到的圖像如下所示,可見Becke surface把兩條DNA鏈間的接觸面非常直觀地展現了出來。在每一層的兩個堿基之間都有兩塊紅色區域,體現出兩個典型氫鍵的存在導致相應地方電子密度相對較大。仔細看的話,會發現有的兩塊紅色區域旁邊還有一小塊白色區域,這對應于C-H...O很弱的氫鍵。Becke surface的其它區域都是藍色,說明兩條鏈在其它地方并沒有值得一提的相互作用(注意不能描述為“沒有相互作用”,用詞必須嚴謹。重原子間的色散作用能即便到了10埃也沒完全衰減到0,兩條鏈的骨架之間的色散作用對相互作用能的貢獻不可完全忽略,只不過強度屬于“不值得專門一提”的程度)
按照HS分析的做法也可以顯示Becke surface的指紋圖,如下所示,可見左下方存在兩個尖兒,體現出每條DNA鏈既作為氫鍵給體也作為氫鍵受體和另一條鏈形成了氫鍵,和HS分析能展現的信息基本一致。但尖兒的具體位置不可能十分一致,畢竟Hirshfeld和Becke權重函數的定義方式差異顯著。
6 總結
本文介紹了HS分析的基本思想,并通過大量例子非常詳細介紹了Multiwfn做HS分析的方法和很多要點。可見HS分析用起來靈活方便,可以較好地直觀展現化學體系的特定片段與周圍其它原子之間的相互作用。同樣的目的用Multiwfn做基于電子波函數的IGMH分析也同樣可以達到,而且IGMH原理更為嚴格,通過sign(lambda2)rho函數對等值面著色還能區分相互作用類型,還能給出各個原子的貢獻量。HS分析可以作為IGMH分析的展現形式的補充,并且由于HS分析只依賴于原子坐標而且計算量很低,因此可以快速地用于很大體系,也不需要事先做量子化學計算產生電子波函數。注:只有原子坐標信息時也可以用Multiwfn做IGM分析,耗時也極低,但圖像效果明顯不及IGMH,參見《使用Multiwfn做IGMH分析非常清晰直觀地展現化學體系中的相互作用》(http://www.shanxitv.org/621)。
最后提醒一下,使用Multiwfn做HS分析或Becke surface分析時請按Multiwfn啟動時的提示恰當引用Multiwfn原文,對于給別人代算的目的也需要明確告知對方這一點。
]]>文/Sobereva@北京科音 2024-Feb-10
在《使用Multiwfn做IGMH分析非常清晰直觀地展現化學體系中的相互作用》(http://www.shanxitv.org/621)中介紹的我在J. Comput. Chem., 43, 539 (2022)中提出的可視化分析弱相互作用的IGMH方法目前已經很流行,在《一篇最全面介紹各種弱相互作用可視化分析方法的文章已發表!》(http://www.shanxitv.org/667)介紹的綜述文章里對此方法也有全面透徹的介紹。
我在去年審了一篇Nature Chemistry的文章,看到作者通過對比對應不同類型相互作用的IGMH等值面來對不對稱催化的原因做了很好的解釋,我覺得很值得作為IGMH分析例子說一下,對一些人可能有啟發。現在這篇文章已經正式發表了:Nature Chemistry, 15, 1672 (2023)。
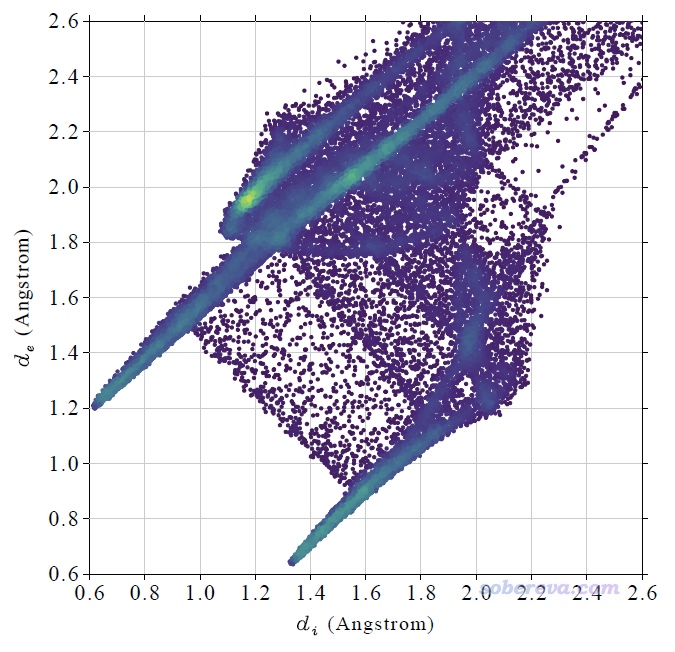
這是一篇實驗為主理論計算為輔的文章,研究對象是下圖的過程。圖的左上角所示的1是個氨基丙二酸類物質,在加入下圖右上角所示的有手性的SPA作為催化劑后,就可以得到脫羧后的結構2,是個有手性的氨基酸類物質。實驗發現,改變底物1中的AG基團會明顯影響產率和對映體過剩率e.e.,如圖所示AG為Bz時是75% e.e.,改為Bz1時80% e.e.,再引入OR取代基后e.e.更高,且R的選擇會進一步影響e.e.,如圖中方框部分所示AG=Bz7時e.e.最高,此時對應于R為CH2-CF2-CF3。下圖右邊示意了這個結構的底物可以與SPA形成顯著的弱相互作用,既有與SPA側鏈的pi-pi堆積作用,底物上的大量的F也與SPA的骨架的C-H有顯著的靜電和色散混合的吸引作用,這些作用可以顯著穩定化SPA對底物的結合。
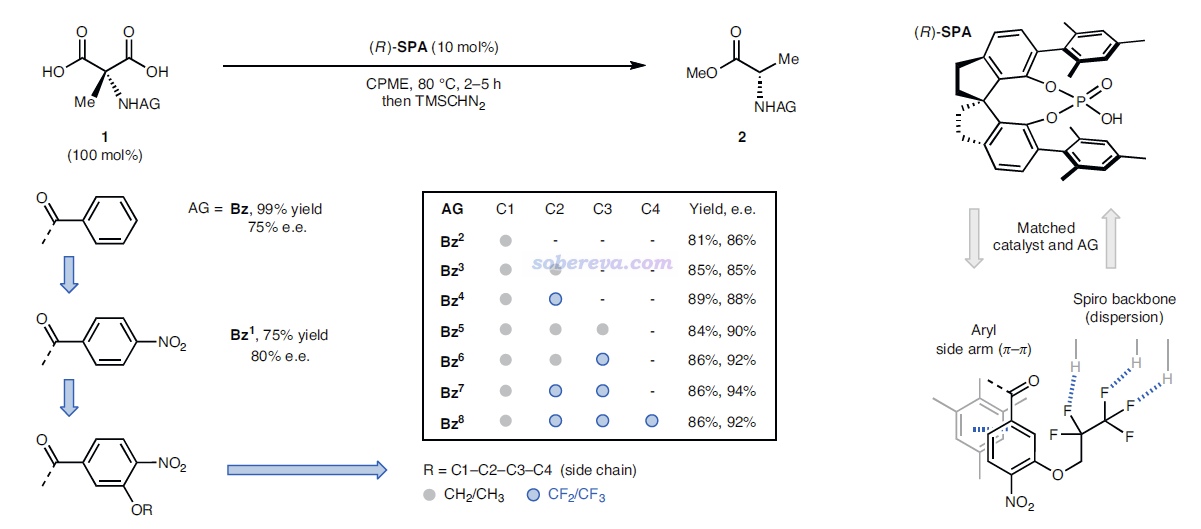
上面那種AG=Bz7的結構在文中稱為S6,能夠自發脫羧變成下圖的Int-1結構。如下圖左側所示,Int-1可以和S6實現自催化反應(經歷TS-5a),也可以自己發生1-4質子轉移(經歷TS-5b),但勢壘都較高,不是研究的重點。重點是圖中所示的Int-1在SPA的作用下,會經歷一系列過渡態和中間體,最終達到氨基酸類似物結構6',產生S和R手性的這個產物經歷的勢壘有很大差別。圖中可見TS-3是決速步,TS-3S的勢壘遠低于TS-3R,這是SPA催化作用下e.e.很大的關鍵原因。
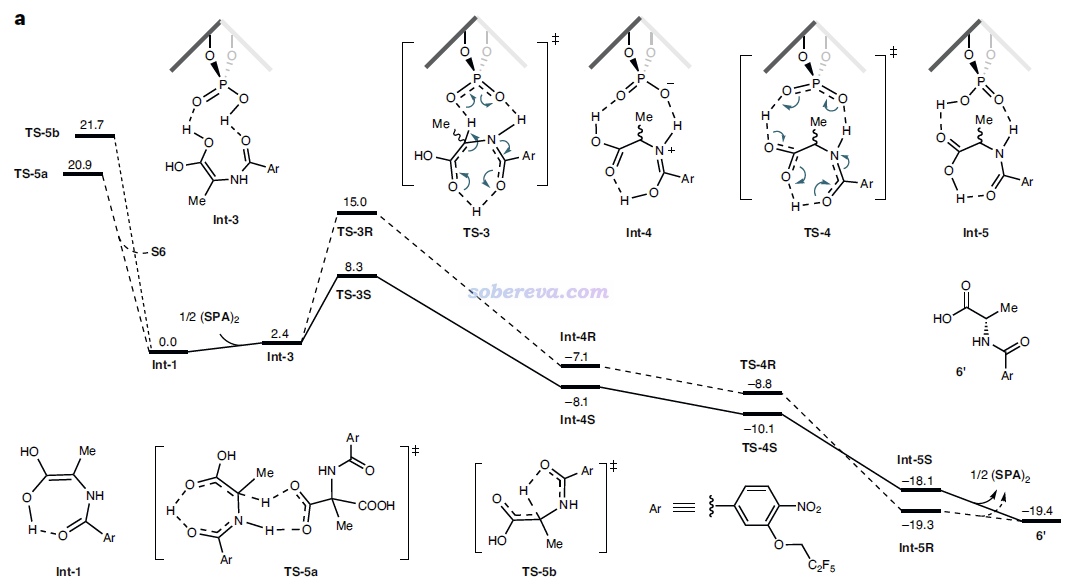
由于反應物相同,因此TS-3S和TS-3R勢壘的差異體現在這兩個結構的能量差別上,差別最主要來自于SPA與Int-1分子的弱相互作用。為了說明這一點,文中用Multiwfn對這兩個過渡態結構做了IGMH分析,給出了下圖,對兩個分子間存在的相互作用類型做了標注。其中N-H...O無疑是挺強的氫鍵,C-H...pi和C-H...O/F都是很弱的氫鍵,苯環間存在pi-pi相互作用,Bz7基團上的一堆F的孤對電子與苯環有色散主導的相互作用。圖中標注Bonding的地方是Int-1與SPA之間發生氫轉移而一定程度成鍵的地方,對TS-3S和TS-3R這個成鍵作用不會有什么明顯差別。
上面兩張IGMH圖的等值面很多,在表面上看起來不太好對比相互作用強度,但如果像本文一樣,把每種相互作用一一羅列出來做細致對比,是完全可以對總作用強度進行區分和解釋的。如上可見,通過仔細觀察等值面可發現TS-3S比TS-3R對應C-H...X作用多出來2個,并且TS-3S的pi-pi堆積作用區域的面積明顯大于TS-3R的,所以前者被標注為strong而后者被標注為weak。
為了能令弱相互作用情況看得更清楚,文中還在分子結構圖上直接用虛線對IGMH呈現的信息做了明確的標注,如下圖所示,字母對應上面的列表里的項,諸如c1、c2...對應不同的C-H...X相互作用。從這個簡化的圖上能更清楚地看到TS3-S中SPA的C-H與底物的Bz7基團上的F之間的相互作用比TS3-R中的更豐富,這一定程度解釋了為什么底物接上F原子較多的Bz7基團時被SPA催化時可以達到較高的e.e.。此外,下面這張圖上還標記了d: C-H...C-H作用的位置,顯然這是普通色散作用,這個作用在上面的IGMH等值面上肉眼不太好辨別(在VMD程序里旋轉等值面時才容易識別),而像下圖這么用箭頭標記一下就容易令讀者看清楚出現在什么位置了。
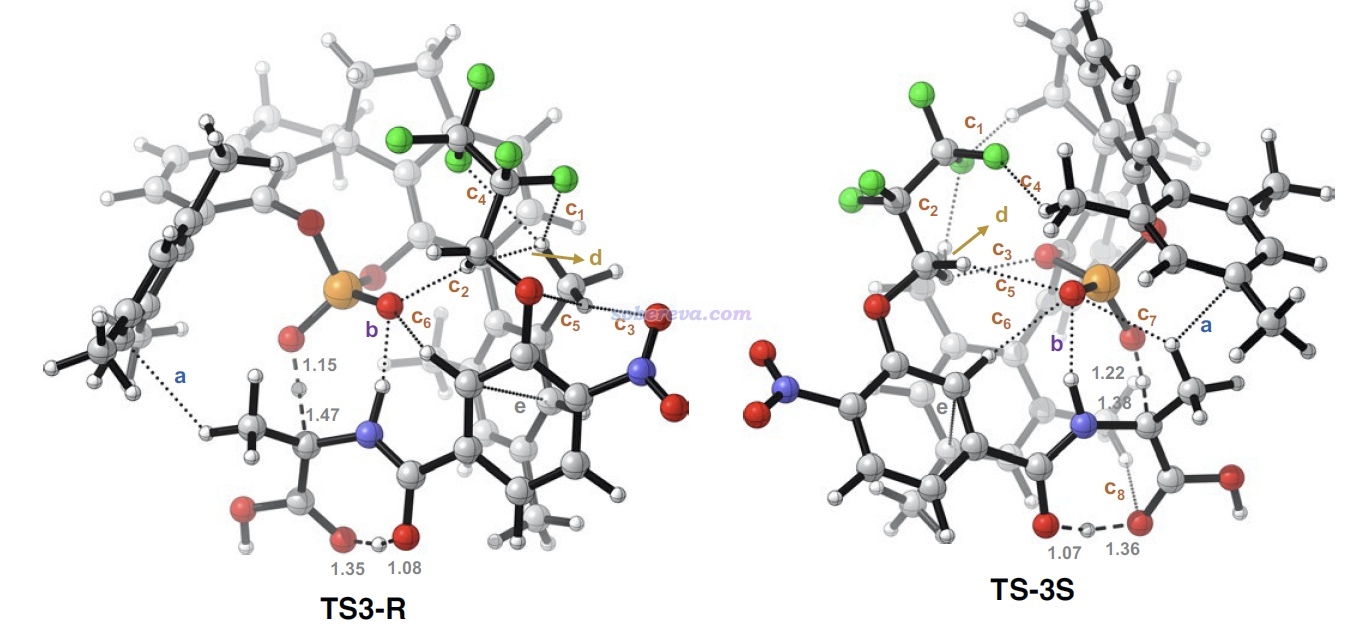
這篇文章體現出基于IGMH分析能清晰直觀地對分子間相互作用導致勢壘存在的差異和由此帶來的實驗現象予以解釋,是很好的IGMH應用范例,值得在其它研究中借鑒。此文的利用弱相互作用實現e.e.的控制也是新穎的催化設計的思路。
]]>文/Sobereva@北京科音 2024-Feb-9
0 前言
量子化學研究中在優化完極小點結構然后做振動分析時遇到虛頻是家常便飯的煩人的問題。之前我專門寫過博文《Gaussian中幾何優化收斂后Freq時出現NO或虛頻的原因和解決方法》(http://www.shanxitv.org/278)講了怎么消虛頻。除非程序存在bug,否則原理上不存在按上文處理完全解決不掉的虛頻。一個現實問題是:雖然靠各種嘗試、折騰,最終確實總能消掉虛頻,但這個過程對于大體系來說過于浪費計算時間,也耗費研究者的精力。那么是否帶有虛頻的結構就一定不能拿來算能量相關的問題呢?是否虛頻總是要不惜一切代價消除呢?此文就談談我個人的看法,給出理論分析,并且通過一個例子對我的觀點予以驗證。
本文牽扯到熱力學數據計算的各種常識知識,沒有這些常識的話先看《使用Shermo結合量子化學程序方便地計算分子的各種熱力學數據》(http://www.shanxitv.org/552),以及Shermo程序手冊的附錄,里面有計算原理的簡潔易懂的概述。在北京科音基礎(中級)量子化學培訓班(http://www.keinsci.com/workshop/KBQC_content.html)里對熱力學量的計算有非常全面深入的介紹,歡迎關注和參加。
本文說的小虛頻和過渡態必然存在的虛頻沒有絲毫聯系,初學者絕對不要把本文內容胡亂套用到過渡態的情況。
1 導致小虛頻的原因
為了避免誤解,首先回顧一下優化極小點后振動分析時出現虛頻的各種常見原因:
(1)振動分析時用的影響勢能面的設置與優化時的不同。這通常是沒有基本常識的外行人才會犯的低級錯誤。注:JCTC, 17, 1701 (2021)中提出的single-point Hessian (SPH)方法通過對勢能面添加偏移勢,從而允許振動分析用的計算級別低于優化用的級別來節約時間,但這個做法非主流、經驗性強,本文不涉及
(2)初始結構的點群高于實際極小點的點群,且如果優化后的結構維持住了初始的對稱性,振動分析后就會發現破壞對稱性的虛頻,即按照虛頻方式移動結構會導致結構的對稱性下降。有時候這種有虛頻的結構恰對應于過渡態
(3)程序bug。比如Gaussian 09 D.01的DFT-D3色散校正對解析Hessian的貢獻的計算有bug,容易導致算勢能面平緩體系出現虛頻。這點在《DFT-D色散校正的使用》(http://www.shanxitv.org/210)我專門做了說明
(4)某些方法對勢能面引入明顯數值噪音。如Gaussian中的SMD溶劑模型的這個問題就比較明顯
(5)幾何優化收斂得不精確。幾何優化收斂限過于寬松或Hessian的質量不太好會導致這種問題。
(6)積分格點質量不夠好。這個問題對于M06-2X等對積分格點敏感的泛函尤為顯著。關于量子化學程序用的原子中心的積分格點參考《密度泛函計算中的格點積分方法》(http://www.shanxitv.org/69)。如果是CP2K等利用平面波的第一性原理程序,用的均勻分布的格點依賴于平面波截斷能的設置。
以上前四種情況導致的虛頻是一定要解決掉的,而且本來解決就非常簡單。只要不犯低級錯誤自然就沒有(1)的情況出現,解決(2)按照虛頻模式調節結構后進一步優化即可,解決(3)就換成沒bug的版本或其它計算程序,解決(4)可以換成其它的能在優化+振動分析中實現基本相同目的而沒嚴重數值噪音的方法,如SMD換成IEFPCM,或者用SMD時結合SAS方式定義溶質孔洞表面。對于(5)和(6)情況帶來的虛頻,只要計算能力和精力允許,能消盡量要消,但如果由于計算資源或精力所限實在太難消掉、被迫要容忍虛頻的存在,那么后續的能量相關問題的計算就是后文討論的內容了。
對于柔性大分子、弱相互作用復合物這樣勢能面的某些維度非常平緩的體系,在做完幾何優化和振動分析后很容易發現存在大小非常小的虛頻,如波數為9.8i、27.3i cm^-1這種,普遍小于50,這一般是前述(5)或(6)情況所導致的。此類虛頻的振動模式通常對應于甲基旋轉運動、柔性骨架的變形運動、分子間相對運動。這類(5)或(6)情況所導致的虛頻以下簡稱為“小虛頻”。但要注意也不是所有波數小于50的虛頻都屬于這種情況,前述的其它四種情況也完全可能導致數值很小的虛頻,所以絕對別光拿虛頻大小來判斷情況。
2 有小虛頻時能量相關問題的計算
當遇到小虛頻時,顯然最完美的做法(具體選擇視情況而定)是用諸如更嚴格的幾何優化收斂限、更好的Hessian、更好的積分格點精度進一步做優化和振動分析,使得虛頻完全消除后再計算能量相關問題。但是對于普通泛函算幾百個原子有機體系,很可能算一次Hessian就要花一整天時間,幾何優化一步也可能花費一個小時左右,最終把虛頻完全消掉可能得花幾天甚至一個禮拜,十分痛苦,甚至不可能實現。那么,在后續算能量相關問題時怎么考慮?下面根據被計算的量專門討論。
2.1 計算電子能量
諸如Gaussian這樣的主流量子化學程序默認的幾何優化收斂限是以受力和位移作為判據的。在默認收斂限下優化正常完成后,即便結構還沒有與實際極小點特別精確一致,但通常相差很小。而且當前的最大和平均受力肯定已經很小了(因為收斂限考慮了它們),由于受力對應于能量梯度的負矢量,所以當前結構稍微再挪一點達到精確的極小點的過程中能量變化肯定非常小。即曰,在有小虛頻的情況下,計算的電子能量一般是可以用的,也即結果和消掉虛頻后再算的能量相差極小。
2.2 計算0 K下的內能U(0)
U(0)是電子能量與零點振動能(ZPE)之和。在諧振近似模型下,每個振動模式對ZPE的貢獻是1/2*h*ν,h是普朗克常數,ν是振動頻率。頻率很低的振動模式對ZPE的貢獻是非常小的,每波數的振動頻率對ZPE的貢獻僅為5.98 J/mol,因此諸如30波數的振動模式只對ZPE有0.18 kJ/mol(0.04 kcal/mol)的貢獻,數量級遠遠低于理論方法、基組、溶劑模型等因素帶來的誤差。順帶一提,Shermo計算熱力學量的時候如果把settings.ini里的prtvib設為1,每個振動模式對熱力學量的貢獻都會明確輸出。
主流量子化學程序,以及Shermo程序在默認情況下,在計算振動對熱力學量的貢獻時都是不考慮虛頻產生的貢獻的,因此小虛頻對熱力學量的貢獻會被直接忽略掉,這不會對ZPE的計算造成實際問題。因為如果把這個小虛頻消了,從而產生一個實頻,其頻率肯定也是非常低的,對ZPE的貢獻可忽略不計。也就是不管消不消這個虛頻,算出的ZPE都沒多大差別。不過,實際情況遠沒這么簡單,因為在虛頻消掉前后,由于二者所在勢能面的位置的些許不同,或改用不同檔次積分格點導致頻率計算精度的差異,其它的實頻模式也會多多少少受到影響,不過頻率值受影響相對明顯的主要是低頻模式(可能影響幾個甚至10個左右波數,而高頻模式受影響一般小于2波數),它們對ZPE的貢獻恰恰又很小,所以虛頻消掉前后算的ZPE雖然未必差異小到諸如前面說的0.2 kJ/mol左右這種程度,但至少也不會大,比如對柔性大體系頂多也就影響個1-2 kJ/mol。
如上所述,鑒于是否小虛頻對電子能量和ZPE的影響都不太明顯,因此算電子能量和U(0)時一般可以容忍一個或多個小虛頻的存在。
2.3 計算從0 K升溫到當前溫度對內能的影響
研究U(0)沒太大實際意義,畢竟實際化學過程也不可能在0 K發生。計算有限溫度,也就是高于0 K時候的熱力學量,就需要考慮從0 K升溫到當前溫度對內能的改變量,其中振動產生的貢獻下面用Uvib(0→T)表示。首先要知道一點,低頻對Uvib(0→T)的貢獻是明顯高于高頻模式的。這點從下面的J. Phys. Chem., 100, 16502 (1996)的圖1可以明顯看出來,圖中ΔHvib(T)和這里說的Uvib(0→T)是一碼事,圖是對T=298.15K的情況繪制的。

雖然低頻模式的貢獻大,但是由圖中放大的0-100波數的區間可見,在小幾十波數范圍以內,頻率值對Uvib(0→T)的影響雖然不可忽視,但不算多大,都在2.0-2.4 kJ/mol范圍內。這也就是說,由于存在一個小虛頻導致少考慮一個實低頻模式,會給Uvib(0→298.15K)帶來-2.2 kJ/mol左右的誤差,這雖然不算小,但還算可以湊合接受。如前所述消虛頻后還可能會對其它低頻模式產生不可忽視的影響,這對Uvib(0→T)的改變一般也不至于太大。
綜上所述,若帶著小虛頻計算U(T),會造成一定低估。對于有一個小虛頻的情況,低估程度在0.5 kcal/mol左右。如果有多個小虛頻,那還是別計算U(T)了,除非T遠低于常溫,或者結合后文說的把小虛頻當成實頻的做法。
2.4 計算熵、自由能
低頻對熵的貢獻遠大于高頻,而且在很低頻區間內,頻率越低貢獻明顯越大。在包括Gaussian在內,大多數量子化學程序(ORCA除外)計算熱力學量用的是傳統的rigid-rotor harmonic oscillator (RRHO)模型,振動熵的計算完全基于諧振近似模型。此時在低頻區間內,隨著頻率趨近于0,振動模式對熵的貢獻猛增,如下面的Chem. Eur. J., 18, 9955 (2012)文中圖2的虛線所示。Grimme在這篇文章中還提出了一種quasi-RRHO模型(QRRHO,后來也叫modified RRHO、mRRHO),以下稱為QRRHO(Grimme),它將自由轉子和RRHO用的諧振近似模型算的振動熵做插值,頻率越接近0自由轉子的權重越大、頻率越高諧振近似的權重越大,如下圖實線所示。QRRHO(Grimme)對高頻模式算的熵貢獻和RRHO完全一樣,而對于低頻模式(特別是波數在100以下的)考慮得比RRHO明顯更合理。由下圖還可見QRRHO(Grimme)算的低頻模式的熵比RRHO整體小得多,沒有RRHO的嚴重高估的問題。此外,QRRHO(Grimme)下低頻模式的振動熵對頻率值的敏感性低于RRHO很多。

直接計算熵的用處不大,算熵的主要目的是用來算自由能,因為自由能里有-T*S項。顯然低頻對熵的貢獻計算的準確度顯著影響到自由能的計算準確度。假設體系有一個小虛頻,并且在消掉之后會多一個20 cm^-1的實頻,并且用的是QRRHO(Grimme)算298.15 K的熵,那么在有虛頻的結構下由于缺失了這個實頻,根據上圖可知會造成熵的誤差約為-18 J/mol/K,對自由能造成的誤差約為-298.15*(-18)/1000=5.4 kJ/mol,略高于1 kcal/mol,這就不小了,都能趕上高質量ωB97M-V泛函結合大基組算普通有機反應的誤差了。不過實際中小虛頻帶來的誤差未必那么大,因為有和沒有小虛頻的結構下實低頻模式的頻率也往往有不可忽視的不同,例如下面第3節的例子在有虛頻的結構下實低頻模式的頻率比精確極小點結構下的整體偏小(勢能面形狀原因所致),導致高估了不少實低頻模式的熵,藉由-T*S項進而對自由能造成低估,于是和少考慮一個低實頻對自由能的高估產生了很大程度的相互抵消,下面管這稱為“熵的誤差抵消”。
根據以上討論,有小虛頻的情況下計算熵和自由能都有一定風險。如果就有一個小虛頻,算常溫的情況往往還說得過去,而如果有多個小虛頻,或計算高溫的情況(注意T越大-T*S越大,熵的誤差造成自由能的誤差越大),我就不建議冒險帶著虛頻了。而且就算非要帶著小虛頻算,也至少應當用QRRHO模型,而切勿用RRHO模型,因為RRHO算的振動熵對低頻頻率值的敏感程度顯著高于QRRHO,因而用有虛頻的結構算的熵的誤差上限比QRRHO的明顯要大。
要注意QRRHO模型不止Grimme提出的這一種。如Shermo手冊附錄部分所介紹的,Truhlar的QRRHO是把所有小于一定閾值(通常為100 cm^-1,可以由Shermo的ravib參數控制)的低頻統一提升為這個閾值再算它們對Uvib(0→T)和熵的貢獻,這個做法的物理意義不甚明確,沒有QRRHO(Grimme)的插值做法優雅。目前還有一種QRRHO是Minenkov等人在J. Comput. Chem., 44, 1807 (2023)中提出的,它對振動熵部分的考慮和QRRHO(Grimme)一樣,但還同時將這種自由轉子與諧振近似做插值的思想同時應用到了計算振動對內能的熱校正量Ucorr上(此時沒法單獨計算ZPE),原理更理想,實際結果還略好一點點。這些QRRHO模型在最新版Shermo中都是支持的,通過ilowfreq參數控制。
2.5 將小虛頻視為實頻的做法
在Angew. Chem. Int. Ed. 2022, e202205735中的3.2節Grimme等人認為存在小虛頻的情況下,只要使用QRRHO模型并且將小虛頻視為實頻處理,比如18i cm^-1直接當做18 cm^-1,則得到的熵就是可以用的、小虛頻的存在就不再是個問題。這種做法表面看起來似乎有點莫名其妙、太任性了,但確實有一定道理,可以這么理解:在消除小虛頻后,必定會得到相同數目的波數也很小的實頻模式,它們都對熵有明顯貢獻。相對于完全忽略掉這些小虛頻的貢獻而導致明顯低估熵,干脆把他們當做實頻處理,從而“湊”出來相應數目的實低頻是更好的做法,而且本身在QRRHO模型下,數值在十幾、幾十波數區間的頻率對熵的貢獻也都相差不懸殊,因此也用不著對這么人為湊的實頻頻率太較真。由于如前所述,Uvib(0→T)也對低頻數值不敏感而低頻的貢獻又沒小到可以隨便忽略的程度,所以計算Uvib(0→298.15K)時也使用這種處理是有道理的。從Shermo 2.5版開始,可以通過settings.ini里的imagfreq選項設置視為實頻的虛頻閾值,比如設為50,就代表大小在50 cm^-1以內的虛頻都當做實頻處理來計算振動對各種熱力學量的貢獻,此時從Shermo程序輸出的頻率信息中也會看到已經沒有虛頻了。
然而,這種將小虛頻視為實頻的做法目前很非主流,其思想在我來看過于主觀和樂觀、把問題想得太簡單了,目前也缺乏系統性的檢驗,我個人不建議輕易使用。這種做法有一個明顯問題:如前所述,有小虛頻和消掉虛頻的兩種情況相差的不僅僅是一個小虛頻轉變成一個小實頻,許多其它低頻模式的頻率也會有不小變化,而且有時是整體明顯增加的。不做這個虛頻到實頻的轉換的情況下能夠有上一節說的熵的誤差抵消效應,而這么做了之后,忽略虛頻模式導致對熵的低估問題確實很大程度解決了,但在有小虛頻結構下其它頻率往往偏低而導致熵的高估帶來的誤差就沒處抵消了。此外,如果當前體系的低頻還有很多都是10 cm^-1 左右這種特別低的,這個范圍附近即便是QRRHO(Grimme)或QRRHO(Minenkov)模型算的熵也對頻率很敏感,問題會更大。
所以,將小虛頻視為實頻的做法雖然確實對一些情況更好地計算熵和自由能是有幫助的,但也很可能起到負面效果。所以這絕不是普適、穩健、理想的做法,更千萬不能一看到存在小虛頻就總是想靠這個做法來完全無視虛頻。
3 有虛頻時算熱力學量的實例
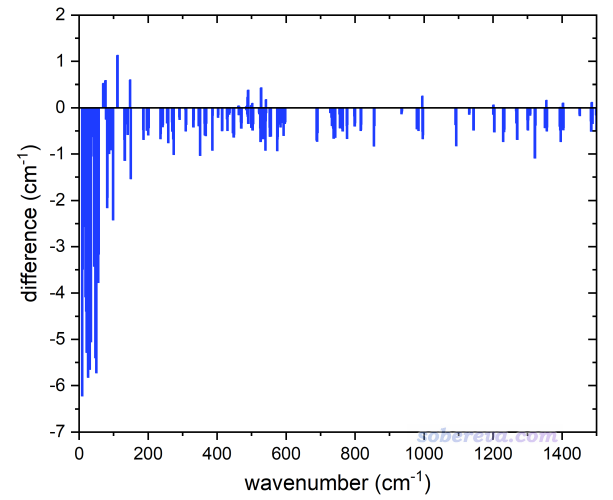
筆者最近通過Gaussian 16在ωB97XD/6-311G*級別研究的一個以pi-pi堆積方式形成的共128個原子的三分子復合物,在優化和振動分析后發現存在6.44i cm^-1的小虛頻,之后進一步優化時用opt=calcfc關鍵詞提供精確的初始Hessian,優化完再做振動分析就發現沒虛頻了,此時最低的頻率成了10.35 cm^-1。正好這個例子可以拿來考察消虛頻前后計算的熱力學量的差異,由此檢驗帶著那個小虛頻時以不同方式算的熱力學量有多大誤差。要強調的是,僅僅這么一個測試,顯然不足以充分證明某種做法理想,因為這必須通過大量體系做統計分析,但至少誤差較大的情況可以用來體現相應做法不夠穩健和普適。
先看一下有虛頻相對于無虛頻時各個實頻的頻率變化情況。先把實頻頻率按照由低到高排列,用有虛頻時候它們的頻率減去無虛頻時候的頻率作為縱坐標值,而兩種情況頻率的平均值作為橫坐標,作的圖如下所示。可見,至少對于此例,有虛頻時數值較低的實頻是普遍明顯偏低不少的,而>200 cm^-1的相對而言的中、高頻則受到的影響很小。這種情形雖然不能說是普遍情況,但至少也是挺常見的一種。
此例有虛頻相對于無虛頻的情況計算的電子能量僅僅相差0.04 kcal/mol,小到完全可以忽略不計。用Shermo程序基于Gaussian振動分析輸出文件計算的有虛頻相對于無虛頻時的ZPE和氣態標況下的熵、內能、自由能熱校正量的差異如以下表格所示。RRHO以及Truhlar、Grimme、Minenkov三種QRRHO分別對應Shermo的ilowfreq=0/1/2/3的情況,imagreal=0和50相當于忽略那個虛頻以及把那個虛頻當大小相同的實頻考慮的情況。

首先看常規的忽略虛頻貢獻的情況。由表格可見:
? ZPE:有虛頻時和無虛頻時ZPE相差僅-0.3 kcal/mol,屬于通常可以接受的差異
? 熵:由于誤差抵消的巧合,恰好RRHO下有小虛頻時的熵和無虛頻時的差異很小。QRRHO(Truhlar)的情況差異挺大,在于它把實低頻都統一當成了100 cm^-1,這部分在有和沒有小虛頻情況之間幾乎都抵消了,而有小虛頻時相當于少考慮了一個100 cm^-1振動模式對熵的貢獻,因此造成熵的嚴重低估。QRRHO(Grimme)和QRRHO(Minenkov)對熵的部分處理相同,由于前述的熵的誤差抵消效應導致有和沒有小虛頻時熵的差異不算太大
? 內能的熱校正量Ucorr:等于ZPE+Uvib(0→T)。由于Uvib(0→T)對低頻的具體數值不太敏感,有無小虛頻情況下實低頻的貢獻變化很小,而有小虛頻時由于缺失了一個低實頻對Uvib(0→T)的貢獻,導致有小虛頻時Uvib(0→T)偏低不少,Ucorr因此比ZPE偏低得明顯更多。QRRHO(Minenkov)用的計算Ucorr的方式和其它模型不同,對于此例使得有小虛頻時Ucorr偏低程度比其它模型更小,僅為-0.5 kcal/mol。因此在計算有小虛頻情況下的U(T)時,用QRRHO(Minenkov)可能會比其它模型更有優勢。
? 自由能的熱校正量Gcorr:相對于其它模型,有小虛頻相對于無虛頻時Gcorr的差異在RRHO下是最大的,這在于有無小虛頻時RRHO算的S的差異對此例恰好頗小,導致-T*S項的差異才0.03 kcal/mol,因此沒能充分對Ucorr的差異產生抵消效應。而各種QRRHO模型下Gcorr的差異明顯較小,這在于這種情況下S的差異不小,而且-T*S項的差異的符號和Ucorr的差異相反,因此相互抵消了很多。其中Gcorr差異最小的是QRRHO(Minenkov),才-0.11 kcal/mol,體現出它可能適合作為有小虛頻情況下算自由能的優先選擇。
再來看將小虛頻當實頻處理的情況。根據以上表格可見,這種處理使得Ucorr在有和沒有小虛頻情況下計算的結果差異減小很多,因此對于算內能的目的,這種做法有一定意義。但是,除了QRRHO(Truhlar)外,這個做法卻造成了有小虛頻時算的S和Gcorr與無虛頻時的差異變得顯著更大,對RRHO最為嚴重,對QRRHO(Grimme)和QRRHO(Minenkov)問題也挺嚴重,這來自于有小虛頻時實頻模式偏低造成的對振動熵的高估無法與因為少考慮一個實低頻導致的低估一定程度相抵消。把小虛頻當實頻處理時結合QRRHO(Truhlar)對此例倒是很不錯,有小虛頻相對于無虛頻時的熵只相差0.27 cal/mol/K,這在于此種QRRHO將低頻全都上拉到100 cm^-1,此時又把小虛頻轉化成了小低頻,因此無虛頻和有虛頻時相當于有同等數目的100 cm^-1的實頻,自然兩種情況下算的熵很接近。
4 總結
根據以上討論和實際體系的測試可知,對于因(5)和(6)情況造成有小虛頻的情況,算電子能量和U(0)沒大問題,不是非得消虛頻。
對于計算Ucorr或與之相差RT的焓的熱校正量Hcorr,用QRRHO(Minenkov)或QRRHO(Grimme)并同時把小虛頻當實頻處理是較好選擇,不會因為存在小虛頻的問題而帶來太大誤差。
有小虛頻時最難搞的是熵,以及溫度不很低時候Gcorr的計算,難點在于小虛頻消掉前后實低頻模式頻率也可能受到不小影響且影響情況不易估計,然而即便用QRRHO(Grimme或Minenkov)時低頻的具體數值對熵的影響也不算太小,所以明顯不是把小虛頻當成實頻處理以避免少考慮一個實低頻模式就能較好解決的,而且如第3節的測試可見,這種做法還可能造成熵和Gcorr在有小虛頻時計算結果更差。所以當你對熵、Gcorr要求精度高時,小虛頻要盡量消。實在搞不定的話,那就姑且用原理上最好的QRRHO(Minenkov),并且建議不用小虛頻當實頻的處理,畢竟此做法非主流而且起到正面效果的可能性很有限。雖然把小虛頻當實頻并結合QRRHO(Truhlar)的情況下小虛頻和無虛頻時熵的差異很小,但在J, Comput. Chem., 44, 1807 (2023)的測試中QRRHO(Truhlar)算團簇反應自由能表現得遠不如QRRHO(Minenkov),甚至比RRHO沒明顯改進,所以我也不推薦。
]]>文/Sobereva@北京科音 2024-Jan-29
0 前言
環狀化學體系具有不同程度的環張力,環張力和環狀體系的諸多問題緊密相關,諸如生成焓、穩定性、合成難易程度等。環張力能(strain energy, SE)是衡量環張力大小的最直接的指標,它對應當前環狀結構轉變為假想的沒有環張力的結構過程中能量變化的負值。
北京科音自然科學研究中心(www.keinsci.com)的盧天等人之前受邀發表了一篇專門精確考察碳環(cyclocarbon)體系環張力能的文章,文中對環張力的計算思想有詳細介紹并給出了諸多討論:
Accurate theoretical evaluation of strain energy of all-carboatomic ring (cyclo[2n]carbon), boron nitride ring, and cyclic polyacetylene, Chin. Phys. B, 31, 126101 (2022) DOI: 10.1088/1674-1056/ac873a(不方便獲取者也可以看預印版https://doi.org/10.26434/chemrxiv-2022-v8w9h,內容基本一樣)
下面,將基于此論文中的內容簡要說明環張力能的計算原理并給出具體例子。感興趣的讀者請務必仔細閱讀論文原文以了解更多信息,也推薦以類似方式計算其它體系的環張力能時引用此論文。另外,讀者如果對碳環體系感興趣,很建議查看同作者之前發表的此類體系的各種研究文章和相關博文:http://www.shanxitv.org/carbon_ring.html。
1 同聯反應法
設計同聯(homodesmotic)反應是計算環張力能的很常用方法。這個方法設計一個化學反應,既使得環被打開使得環張力被完全釋放,同時又盡可能不影響原本的成鍵情況,顯然這個反應能的負值就對應了環張力能。
例如,要計算氧雜環丁烷的環張力能,就可以設計以下同聯反應。紅色部分對應具有顯著環張力的氧雜環丁烷,將它插入到甲乙醚所示位置就變成了右邊的產物,顯然此時環張力就被完全釋放掉了。與此同時,反應物和產物的成鍵特征并未發生變化,例如下圖紅字的第一個碳在反應前后始終是sp3雜化狀態、始終連著一個氧原子和一個CH2,因此化學環境變化導致的成鍵特征的極輕微改變對這個反應能的影響可以忽略不計,也即這個反應能幾乎只體現出環張力的釋放。
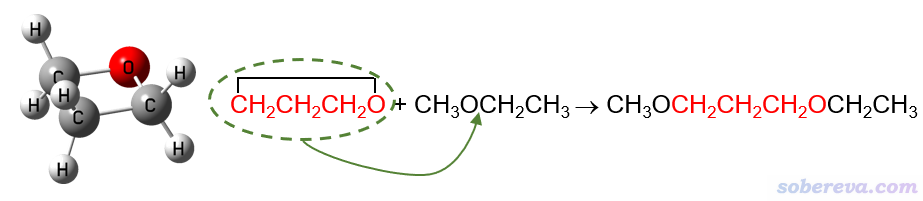
同聯反應法計算環張力能的缺點是反應的設計并不唯一,比如以上反應的甲乙醚也可以是甲醚、乙醚等等,結果或多或少會有點差異。顯然反應應當設計得盡可能令各個鍵的特征在反應過程中不發生改變,但這并非總能很好實現,后文提到的碳環體系就是一例。
2 同聯反應法計算實例:[6]CPP
這一節示例使用同聯反應法計算[6]CPP的環張力能。此體系的幾何結構,以及設計的同聯反應如下所示。可見是把[6]CPP插入到了聯苯中間變成了直線的八聯苯,從而完全釋放了環張力。
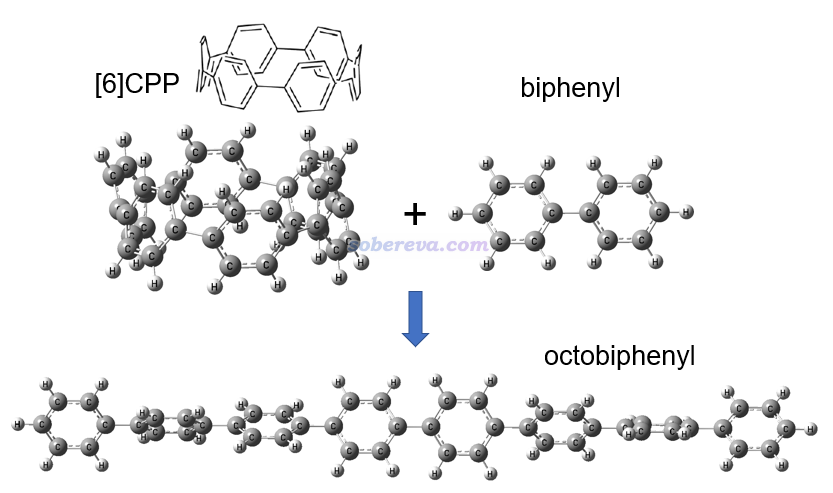
上圖涉及的三個分子都在ωB97XD/6-31G*級別下用Gaussian 16程序進行了幾何優化,并做振動分析確認了無虛頻,輸入輸出文件都在http://www.shanxitv.org/attach/698/file.rar里。基于輸出文件里最后一次輸出的電子能量,用反應物能量之和減去產物能量,就得到了環張力能:627.51*(-463.1444911-1385.7232492+1849.0260518) = 99.3 kcal/mol。這個值比C-C單鍵的鍵能還要大一些,可見其環張力是相當強的。
如果想得到更精確結果,可以基于優化完的結構在更好的級別下算高精度單點再求差,比如基組可以改用明顯更好的def2-TZVP。這對結果可能造成幾kcal/mol程度的影響。
3 外推法
如果被考察的環狀體系是由重復單元構成的,比如前面的[6]CPP是苯環作為重復單元,就也可以用這一節介紹的外推法計算其環張力能。這個方法計算過程如下
(1)計算從小到大不同重復單元數(n)的環的每單元的能量E/n,這里E是整體能量。顯然E/n是依賴于n的,n較小時環尺寸較小,自然環張力大,平均到每個單元的環張力能也大
(2)用Origin之類程序通過恰當的函數(一般是二次函數)擬合E/n與n之間的解析關系,并外推得到n無窮大時的E/n,這對應于沒有環張力時候的每單元能量
(3)將當前考察的環狀體系的E/n減去n無窮大時的E/n,就得到了當前體系的每單元環張力能(SE/n)
(4)將當前體系的SE/n乘以當前的體系的n即是當前體系的環張力能
相較于同聯反應法,外推法的優點:
?原理非常嚴格
?沒有同聯反應法設計反應的任意性,并可以用于同聯反應法不適合的體系
?可以順帶得到環張力能與n的解析關系,由此可以直接預測出任意重復單元數的環張力能
缺點:
?需要計算從小到大不同尺寸的環狀體系,總耗時明顯高于同聯反應法
?計算步驟略繁瑣,得自己做擬合
如果你只是要考察當前一個環狀體系的環張力能,而且可以設計出很合適的同聯反應,那么就沒必要用昂貴且略麻煩的外推法。
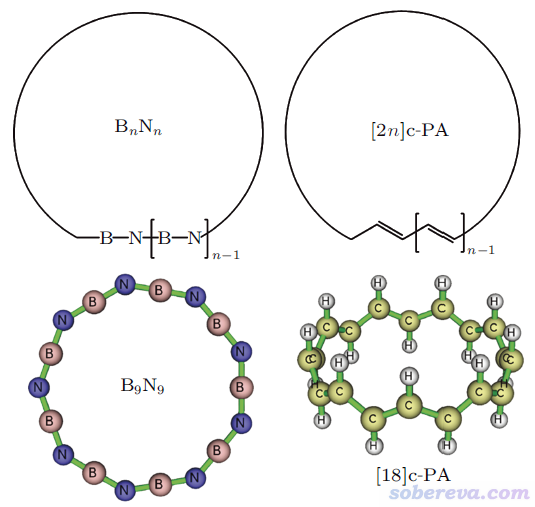
4 外推法計算實例:18碳環
2019年首次在凝聚相實驗中觀測到的18碳環分子由18個sp雜化的碳依次相連構成環狀。由于sp雜化的碳明顯傾向于形成180度鍵角,因此18碳環必然有顯著的環張力,具體環張力能是多少非常值得研究,這正是前述Chin. Phys. B, 31, 126101 (2022)文章研究的主要內容。
此文中指出碳環類體系不適合同聯反應法計算環張力能。碳環類體系是長、短C-C鍵交替構成,兩個碳是一個重復單元,若用同聯反應法計算化學式為C_2n的碳環的環張力能,可以設計如下反應式,相當于把碳環的每個重復單元插入到了一定長度的碳炔里。碳炔是sp雜化的碳形成的鏈狀分子,兩端由氫封端。PS:對碳炔感興趣者建議閱讀《氫封端碳鏈H-(C≡C)n-H (n = 3-9, 15)的電子光譜的尺寸依賴性:性質分析及對碳炔的預測》(http://www.shanxitv.org/679)
![]()
然而,如下圖可見,將18碳環的重復單元插入到不同長度(通過m體現)的碳炔里,按照同聯反應算出來的環張力能有明顯區別,因此靠同聯反應法難以得到嚴格的18碳環的環張力能。
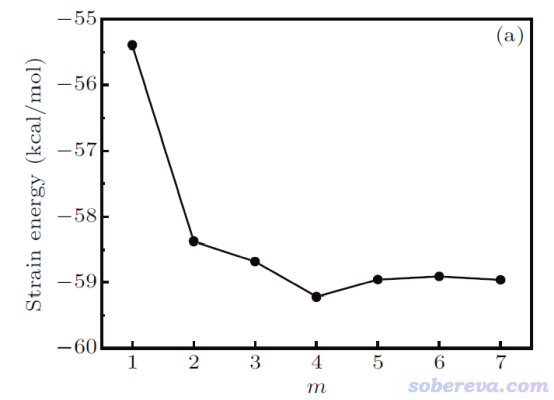
碳炔和18碳環都具有C-C鍵長、短交替的特征。文中還發現,不管碳炔的m取多少,處于它中央的較長的C-C鍵和18碳環中的較長的C-C鍵之間的鍵長差異都不可忽視,對較短的C-C鍵也是如此。因此原理上沒法設計一個同聯反應使得18碳環的C-C鍵特征在轉移到產物時幾乎完全不發生改變,這也必然導致靠同聯反應法不可能對此體系得到完全精確的環張力能,也即成鍵特征差異造成的能量改變會不可避免地摻入到計算出的環張力能中。
由于發現了以上問題,文中使用外推法計算碳環的環張力能。為了追求盡可能好的精度,文中用ORCA程序在精度很好的DLPNO-CCSD(T)/cc-pVTZ級別下對n=7到n=24的碳環都計算了單點能(基于ωB97XD/def2-TZVP優化的結構),ORCA輸出文件在http://www.shanxitv.org/attach/699/file.rar里都提供了。每重復單元能量(E/n)與n的關系如下所示。由于n較小的碳環的電子結構特征有特殊性、和較大的碳環存在一定差異,因此文中只對n>=13的數據點做了二次函數的擬合,由下圖的紅色曲線可見擬合質量超極好,R^2近乎精確為1。
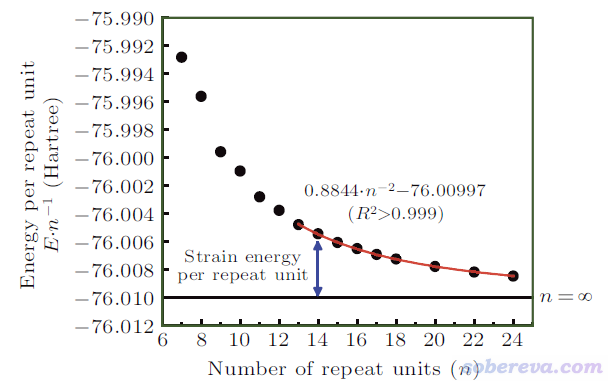
令擬合公式E/n = 0.8844/n^2 - 76.00997中的n為無窮大,就得到了無環張力對應的無窮大的碳環的每單元能量-76.00997 Hartree。每單元環張力能SE/n = E/n - 76.00997,于是有SE/n = 0.8844/n^2,也即碳環類體系的環張力能SE = 0.8844/n Hartree。18碳環的n=9,代進去就得到了其環張力能0.8844/9 Hartree = 61.7 kcal/mol。
值得一提的是,如果令前述同聯反應法中碳炔的m設得很大,從而使得環張力能隨m收斂,最終算出來的結果是59 kcal/mol,可見和嚴格的外推法的結果相比誤差并不可忽略。
前述論文中也測試了用ωB97XD/def2-TZVP的能量結合外推法算碳環的環張力能,結果和上面用的昂貴的DLPNO-CCSD(T)/cc-pVTZ能量的結果沒多大差別。這在一定程度上體現出ωB97XD/def2-TZVP級別算碳環類體系很不錯。
文中還基于不同尺寸碳環的環張力能和鍵角之間的關系,推導出了適合碳環體系用的C-C-C鍵角力常數56.23 kcal/mol/rad^2。
此外,文中還對以下所示的18碳環的等電子體硼-氮環,以及具有單套pi共軛特征的環聚乙炔的環張力做了計算,發現硼-氮環類體系的環張力明顯小于碳環類,而環聚乙炔類體系的環張力則略大于碳環類。文中利用Multiwfn程序(http://www.shanxitv.org/multiwfn)做波函數分析從電子結構角度清晰解釋了它們的環張力與碳環之間存在差異的原因。文中還發現用常用的B3LYP泛函算全局pi共軛的環狀聚合物的合理性較差,E/n隨n的變化明顯不合理。這些方面這里就不多提了,請讀者務必閱讀論文原文。
5 總結
環張力是環狀分子的重要特性。本文簡明扼要地介紹了兩種最常用的計算環狀分子的環張力能的方法,并以[6]CPP和18碳環作為具體例子進行了演示。同聯反應法比較方便省事,但對于一些全局pi共軛的環狀體系,諸如18碳環,則更適合外推法,明顯嚴格得多,同時外推法還很有助于探究環尺寸與環張力之間的關系。還應當注意并不是所有環狀體系都能用這兩種方法之一計算環張力能,比如苯分子,它存在顯著的和其環狀結構緊密相關的六中心鍵,因而環張力本來就是難以被定義的。
]]>文/Sobereva@北京科音 2024-Jan-26
在《18碳環等電子體B6N6C6獨特的芳香性:揭示碳原子橋聯硼-氮對電子離域的關鍵影響》(http://www.shanxitv.org/696)中提到,筆者在Inorg. Chem., 62, 19986 (2023)一文考察B6C6N6分子的電荷分布時,專門計算了此分子的平面內π軌道、平面外π軌道、σ軌道和內核軌道上的電子是怎么分布在各個原子上的。原子空間定義的方法不唯一,此文用的是流行的Hirshfeld-I原子空間劃分,這種方式劃分的原子空間物理意義較強,可以較合理體現外環境導致的原子空間收縮和膨脹。這種分析方法對于讀者研究很多其它體系也很有益處。本文就演示一下怎么用Multiwfn實現這種計算,以計算B6C6N6的平面內π電子(π-in)的分布為例。由于這個計算需要充分利用Multiwfn的靈活性,牽扯一些細節,這是為什么我專門寫個文章來具體說明。
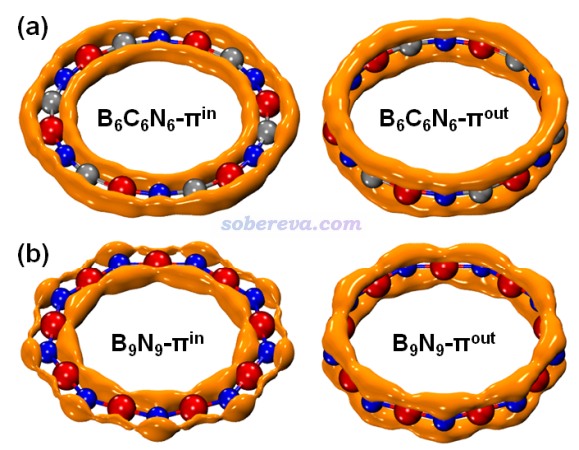
上述文章考察的B6N6C6的波函數文件是http://www.shanxitv.org/attach/697/B6C6N6_OS.rar,解壓后是B6C6N6_OS.fchk,是由Gaussian 16在wB97XD/def2-TZVP級別下以對稱破缺方式計算得到的。本文例子用的Multiwfn是2024-Jan-21更新的版本。Multiwfn可以在http://www.shanxitv.org/multiwfn免費下載,不了解者看《Multiwfn FAQ》(http://www.shanxitv.org/452)。
啟動Multiwfn,載入B6C6N6_OS.fchk。首先要做的是構造Hirshfeld-I原子空間,把Multiwfn的examples目錄下的atmrad子目錄挪到當前目錄下,這是因為此目錄下有各種元素不同價態的徑向電子密度信息,在構造Hirshfeld-I原子空間時要用到(若缺乏計算機常識不了解什么叫“當前目錄”,看http://www.shanxitv.org/237)。然后在Multiwfn里依次輸入
15 //模糊空間分析
-1 //選擇原子空間
1 //開始構造Hirshfeld-I原子空間
很快就構造完了。Hirshfeld-I原子空間權重數據現已被儲存在了內存里,輸入0返回主菜單。
因為我們要考察π-in分子軌道上的電子分布,故接下來需要把這類軌道以外的分子軌道的占據數都清零。可以按照《使用Multiwfn觀看分子軌道》(http://www.shanxitv.org/269)說的,在Multiwfn主功能0里一個一個觀看占據軌道的圖形判斷,把π-in軌道的序號都記錄下來,最終找到的序號如下。注意當前是對稱破缺計算,因此兩種自旋要分別考察。
Alpha自旋的π-in占據軌道:38,41,42,45,46,48,50,53,54
Beta自旋的π-in占據軌道:596,599,600,603,604,606,608,611,612(注意beta軌道序號在Multiwfn中的記錄規則,在http://www.shanxitv.org/269專門說了)
在Multiwfn里接著輸入
6 //修改波函數
26 //修改軌道占據數
0 //選擇所有軌道
0 //把所有軌道占據數清零
38,41,42,45,46,48,50,53,54,596,599,600,603,604,606,608,611,612 //所有π-in軌道序號
1 //占據數還原為原本的1.0
q //返回
-1 //返回主菜單
現在就可以開始正式計算了。在Multiwfn里接著輸入
15 //模糊空間分析。進入后從選項-1的文字上可以看到當前的原子空間仍是Hirshfeld-I
1 //對特定實空間函數在各個原子空間中積分
1 //被積函數是電子密度。顯然當前對應的是π-in電子密度
馬上看到如下結果
Atomic space Value % of sum % of sum abs
1(B ) 0.64940465 3.607804 3.607804
2(C ) 1.00713759 5.595209 5.595209
3(N ) 1.34345778 7.463655 7.463655
4(B ) 0.64926116 3.607007 3.607007
5(C ) 1.00733834 5.596325 5.596325
6(N ) 1.34338216 7.463235 7.463235
7(B ) 0.64944181 3.608010 3.608010
8(C ) 1.00710182 5.595011 5.595011
9(N ) 1.34348431 7.463802 7.463802
10(B ) 0.64924542 3.606919 3.606919
11(C ) 1.00734233 5.596347 5.596347
12(N ) 1.34339152 7.463287 7.463287
13(B ) 0.64942036 3.607891 3.607891
14(C ) 1.00713369 5.595188 5.595188
15(N ) 1.34344824 7.463602 7.463602
16(B ) 0.64928283 3.607127 3.607127
17(C ) 1.00730639 5.596147 5.596147
18(N ) 1.34341808 7.463434 7.463434
Summing up above values: 17.99999847
Summing up absolute value of above values: 17.99999847
如上可見,B、C、N原子的π-in電子數分別是0.649、1.007、1.343,和如下所示的Inorg. Chem., 62, 19986 (2023)中的表2中的數據完全一致。

最后再提醒一下,必須按以上說明先產生Hirshfeld-I原子空間、修改軌道占據數,最后再在Hirshfeld-I原子空間里積分,而不能先修改軌道占據數然后再進入子功能15產生Hirshfeld-I原子空間并做積分。因為修改軌道占據數之后,此時的電子密度函數就不再是總電子密度了,而Hirshfeld-I原子空間在構造時用到的電子密度函數必須對應總電子密度。
]]>文/Sobereva@北京科音 2024-Jan-26
0 前言
由18個碳原子連接形成的環狀體系18碳環(cyclo[18]carbon)具有十分特殊的幾何和電子結構,自從它于2019年首次在凝聚相中被觀測到后,18碳環及其衍生物就得到了化學界的廣泛關注,并陸續有大量理論研究工作發表。特別是在Carbon, 165, 468-475 (2020)中,北京科音自然科學研究中心(http://www.keinsci.com)的盧天和江蘇科技大學的劉澤玉等人證實18碳環具有明顯的雙pi芳香性特征,這是18碳環非常與眾不同的一點。
18碳環的一種等電子體是B9N9,由B和N原子交替相連構成環狀。此體系早已被研究過,被證實幾乎不具備像18碳環那樣的明顯的芳香性。最近,盧天、劉澤玉和西班牙多諾斯蒂亞國際物理中心的Mesías Orozco-Ic發現,由6個(硼-碳-氮)重復單元依次相連構成的18碳環的另一種等電子體B6C6N6具有和18碳環很接近的芳香性。為什么在無芳香性的B9N9中摻雜進去碳原子后,或者說讓碳原子橋聯硼和氮后,B9N9的芳香性就能得到巨大提升?這是非常有意思的問題。于是以上研究者對B6C6N6的特征做了全面深入的理論研究,詳細分析討論了其芳香性特征和本質,成果近期在美國化學會的知名期刊Inorganic Chemistry上發表,非常推薦閱讀:
Exploring the Aromaticity Differences of Isoelectronic Species of Cyclo[18]carbon (C18), B6C6N6, and B9N9: The Role of Carbon Atoms as Connecting Bridges, Inorg. Chem., 62, 19986 (2023) https://doi.org/10.1021/acs.inorgchem.3c02675
此研究的研究意義不僅在于討論了B6C6N6本身,還在于揭示了碳原子橋聯硼和氮原子對電子離域特征的關鍵性影響,這對于通過碳摻雜的方式對純硼、氮構成的體系(如硼-氮納米管、二維層狀h-BN體系等)進行改性提供了重要啟示。此研究通過強大的Multiwfn程序(http://www.shanxitv.org/multiwfn)利用了各種波函數分析手段對B6C6N6進行了充分的考察并和18碳環、B9N9進行了對比,這也是充分運用波函數分析探究新穎體系的很好的范例。
下面本文就對Inorg. Chem., 62, 19986 (2023)這篇文章的主要研究思想、結論進行深入淺出的介紹,并對一些研究細節進行附加說明,以便于讀者更好地理解此文,以及將此文的研究手段用于自己的研究。同作者之前還對18碳環及其衍生物的各方面特征做過充分的理論研究并得到了同行的廣泛關注,成果匯總見http://www.shanxitv.org/carbon_ring.html。
1 B6N6C6的幾何結構
做B6N6C6的各方面研究之前先得得到其可靠的幾何結構。18碳環及衍生物用ωB97XD/def2-TZVP級別做幾何優化是很穩妥的,在Carbon, 165, 468-475 (2020)以及http://www.shanxitv.org/carbon_ring.html里列舉的此類體系的大量研究工作中都已經充分證明了這點。因此此文用Gaussian 16在此級別下優化了B6N6C6的結構。特別需要注意的一點是,B6N6C6基態是單重態,但是其閉殼層單重態波函數存在不穩定性,用DFT計算時需要作為對稱破缺單重態來處理方可得到真正的基態,相關討論見《談談片段組合波函數與自旋極化單重態》(http://www.shanxitv.org/82)。因此,此文對B6N6C6基態的各種研究都是基于對稱破缺單重態的結構和波函數做的。如果誤當成了閉殼層單重態來算,作者發現得到的幾何結構將明顯不合理,而且電子結構也明顯錯誤,比如芳香性嚴重偏高。實際上之前有一篇論文也算過B6N6C6,但由于那些作者沒注意這一點而誤當成了閉殼層計算,導致結果是沒有任何意義的。此例體現出DFT計算B6N6C6這種電子結構不同尋常的體系一定要注意做波函數穩定性測試。而ωB97XD/def2-TZVP級別下的B9N9、18碳環的穩定波函數都是單重態閉殼層的。
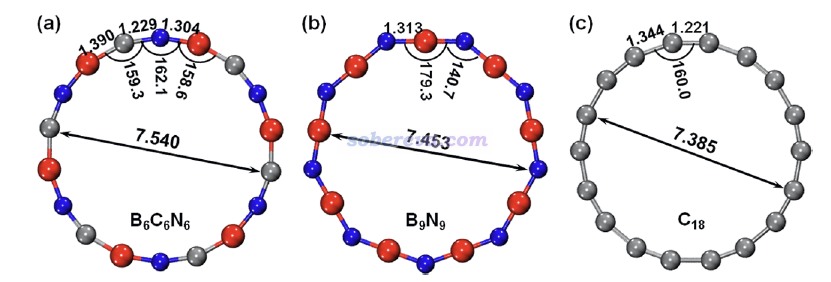
ωB97XD/def2-TZVP級別下優化得到的無虛頻的B6N6C6、B9N9和18碳環的結構如下所示,紅/灰/藍分別對應硼/碳/氮,笛卡爾坐標在文章的SI里提供了。可見B6N6C6屬于C6h點群,而B9N9屬于D9h點群。
畢竟B6N6C6是一個新穎的結構,不排除靜態相關很顯著導致ωB97XD描述不理想的可能,故為了絕對穩妥,此文還用ORCA在CASSCF(12,12)/def2-TZVP級別下也做了幾何優化。此活性空間設定下,最終會有12個占據數明顯偏離整數的pi型自然軌道,而若活性空間進一步擴大到CAS(14,14),則會再多出兩個占據數接近整數的pi型自然軌道,這體現CAS(12,12)恰好足夠充分描述B6N6C6的pi電子的靜態相關效應。CASSCF(12,12)優化出的結構和ωB97XD的非常相近,體現出ωB97XD得到的此體系的幾何結構靠譜。為了進一步體現ωB97XD計算出的波函數也合理,文中也順帶對比了一下這兩個級別得到的單電子分布情況。文章按照《談談自旋密度、自旋布居以及在Multiwfn中的繪制和計算》(http://www.shanxitv.org/353)計算了ωB97XD波函數的自旋密度,而由于CASSCF沒法得到自旋密度,故文章使用《使用Multiwfn計算odd electron density考察激發態單電子分布》(http://www.shanxitv.org/583)介紹的方法對CASSCF(12,12)波函數產生了單電子密度(ODD),等值面圖對比如下所示。在不考慮自旋密度符號(體現在等值面顏色上,不同顏色對應單電子不同自旋方向)的情況下,可見二者展現出的單電子分布是基本吻合的,即單電子主要都分布在碳上。這在很大程度上體現出ωB97XD對稱破缺計算產生的波函數也是合理的,適合用于之后的波函數分析。
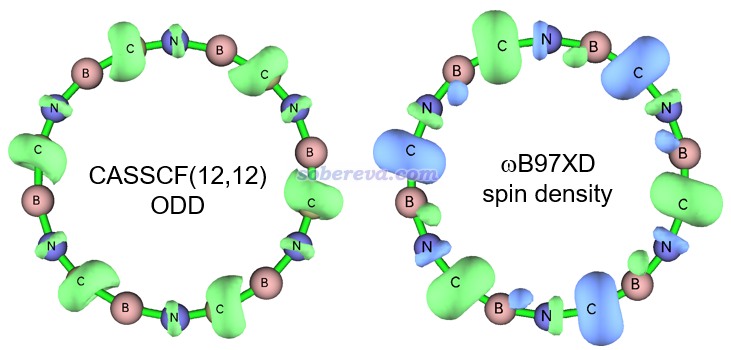
2 B6N6C6的電荷分布
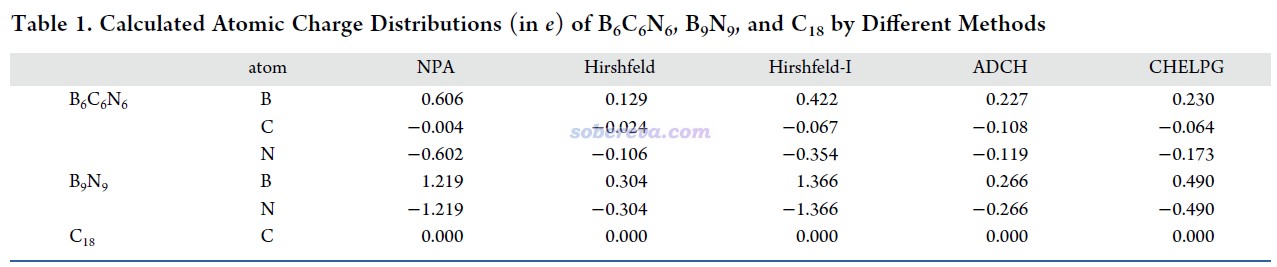
原子電荷是定量考察化學體系中電荷分布最常用的指標之一。18碳環由于所有的碳都是等價的,顯然原子電荷都為0,而對B6N6C6和B9N9計算原子電荷,可以直接考察這倆體系中不同原子帶的凈電量、了解二者電荷分布的差異。原子電荷計算方法并不唯一,不同方法基于不同的思想、有不同的特點,在《原子電荷計算方法的對比》(DOI: 10.3866/PKU.WHXB2012281)中有大量介紹和對比。考慮到B6N6C6電子結構的特殊性,為了得到穩妥的結論,文中用Multiwfn計算了很常用的ADCH、Hirshfeld、Hirshfeld-I、CHELPG原子電荷(它們的計算例子看Multiwfn手冊4.7節)、用NBO程序計算了常見的NPA原子電荷,結果如下所示。可見雖然不同原子電荷給出的具體數值不同,但可以得到共同的結論,就是氮明顯帶負電,硼明顯帶正電,這體現出了它們顯著的電負性差異。而在B6N6C6中,硼和氮的凈電荷差異明顯小于它們在B9N9中,這體現出碳元素在硼、氮之間的插入明顯促進了體系整體電荷分布的均衡性。而B6N6C6中碳自己的原子電荷則接近于0,即沒怎么受周圍化學環境的影響。
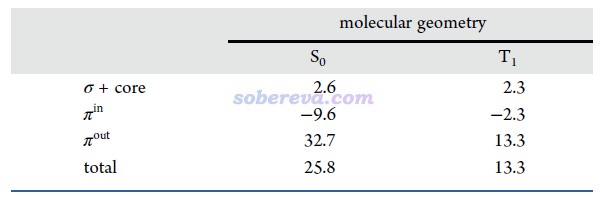
18碳環具有四類分子軌道:平面內和平面外兩套π軌道,分別稱為π-in和π-out,此外還有σ軌道,以及碳的內核原子軌道組成的內核分子軌道。B6C6N6和B9N9也有這四類軌道。為了深入考察各個原子上的電子是怎么分布在這些軌道上的,文中用Multiwfn基于Hirshfeld-I原子空間劃分對三個體系計算了電子在這些軌道上的布居數,如下所示。具體計算方法我專門寫了篇文章說明:《使用Multiwfn基于Hirshfeld-I劃分計算特定類型電子在各個原子上的分布量》(http://www.shanxitv.org/697)。
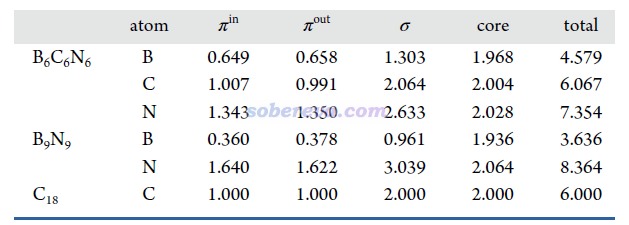
從以上數據可以看到,在每個原子上,π-in和π-out電子分布量都是基本一致的,沒有什么偏向性,體現出這些體系里π-in和π-out電子特征的近似等價性。B6C6N6和18碳環中的碳的電子結構特征非常相近,π-in和π-out電子數在兩種體系里都基本為1.0,明顯都處于sp雜化的狀態。
3 B6N6C6的成鍵情況
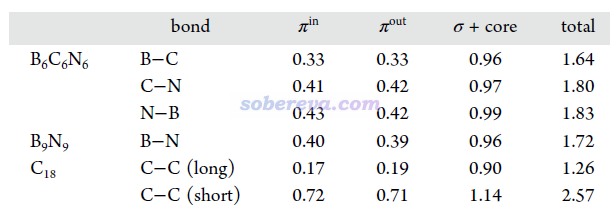
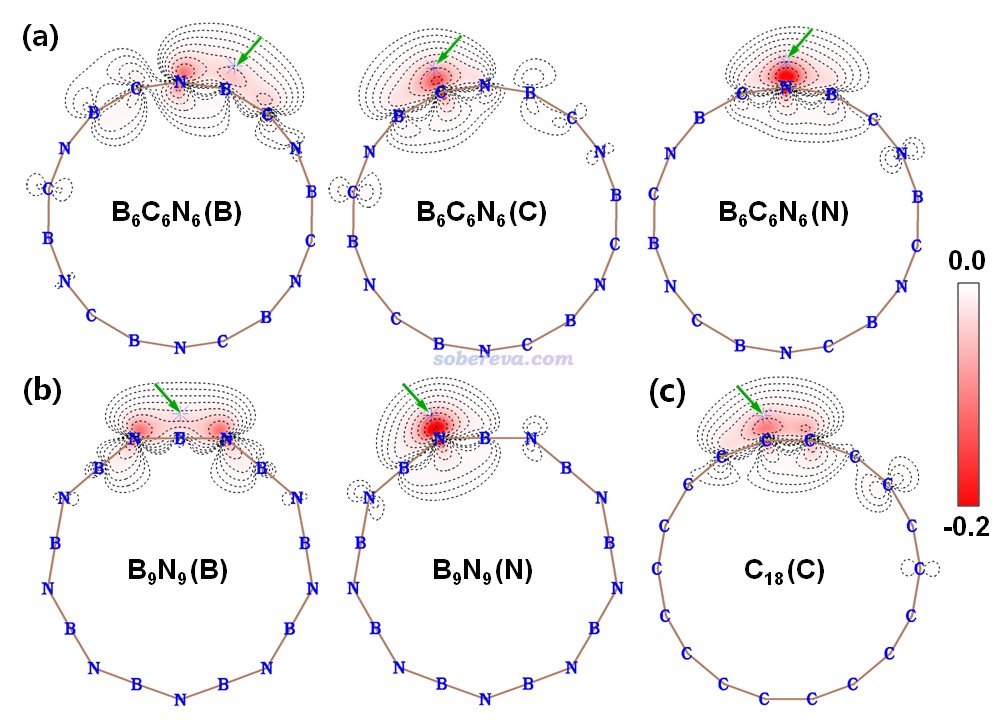
鍵級是考察化學鍵特征非常常見的方式,參見《Multiwfn支持的分析化學鍵的方法一覽》(http://www.shanxitv.org/471)。文中用Multiwfn對B6C6N6、B9N9和18碳環計算了流行的Mayer鍵級,并且分解為了不同類型軌道的貢獻,如下所示。Mayer鍵級體現了原子間等效的共享電子對數,由數據可見,三個體系里所有鍵都包含典型的單個σ鍵特征(即σ+core電子貢獻的Mayer鍵級接近非常接近于1),同時π-in和π-out電子都構成了一定程度的π鍵特征,且二者貢獻相近。B6C6N6中的π作用程度是N-B ≈ C-N > B-C。B9N9中所有B-N等價。18碳環由長、短兩種C-C鍵構成,后者的π作用顯著強于前者。文中還用Multiwfn計算了模糊鍵級以進一步確認了這些結論,數據見補充材料。
《使用IRI方法圖形化考察化學體系中的化學鍵和弱相互作用》(http://www.shanxitv.org/598)中介紹了盧天提出的IRI-π函數,可以直觀地展現化學體系中π電子相互作用情況。此文對B6C6N6和B9N9繪制了IRI-π=1.2的等值面圖并用sign(λ2)ρ函數著色,如下所示。可見每個鍵上都出現了環狀的等值面,非常直觀地體現出這些鍵都存在明顯的π作用,這和Mayer鍵級展現出的信息相吻合。
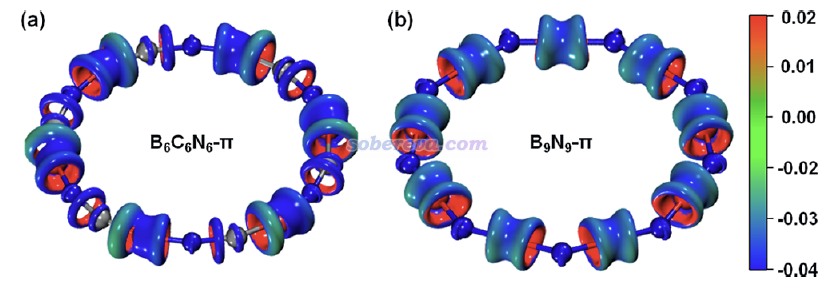
4 B6N6C6的芳香性
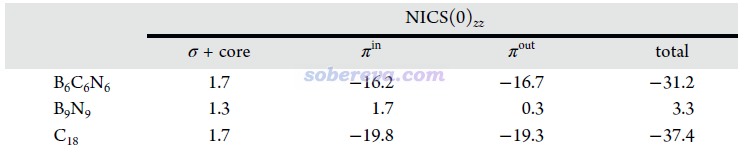
B6N6C6的芳香性是當前研究的重點。和18碳環一樣,B6N6C6和B9N9的π-in和π-out電子皆為18個,都滿足Hückel的4n+2芳香性規則,因此都有可能存在雙π芳香性。衡量芳香性的方法非常多,在《衡量芳香性的方法以及在Multiwfn中的計算》(http://www.shanxitv.org/176)里有充分的介紹。其中基于電子結構和基于磁性質的芳香性衡量方法的可靠性和普適性普遍較好,可以用于考察B6N6C6的芳香性并與18碳環和B9N9的進行對比。
AV1245*1000在《使用Multiwfn計算AV1245指數研究大環的芳香性》(http://www.shanxitv.org/519)里專門介紹過,是基于波函數定義的適合用于定量考察共軛大環體系芳香性的指數。文中首先使用Multiwfn計算了AV1245*1000,結果如下。除了總值外,還分別給出了π-out電子和其它電子的貢獻(注:π-in、σ、core電子對AV1245的貢獻無法精確分離故沒有單獨給出。但顯然σ和core的離域性極低,不會對AV1245有什么貢獻,故“其它電子貢獻”實質上等于π-in電子的貢獻)。由數據可見,B6C6N6的芳香性顯著強于B9N9,并且π-out的芳香性比π-in還略強一點,這可能在于體系的π-in電子的共軛程度相對略低,源自于環的曲率使得平行于環的p原子軌道彼此交疊程度低于垂直于環平面的p原子軌道。單從AV1245*1000的數值來看,B6C6N6的芳香性甚至比18碳環還要更強一點。

NICS是非常流行和穩健的基于磁屬性衡量芳香性的指標,它的不同形式在http://www.shanxitv.org/176里有專門介紹。其中NICS(0)zz對本文研究的這些環狀共軛體系最為適合,結果如下所示,數值越負說明芳香性越強。為了更充分了解芳香性的來源,文中還按照《基于Gaussian的NMR=CSGT任務得到各個軌道對NICS貢獻的方法》(http://www.shanxitv.org/670)介紹的方法分別計算了π-in、π-out和σ+core電子的貢獻。NICS(0)zz體現出B6C6N6的芳香性接近18碳環,并且π-in和π-out的貢獻相仿佛。而B9N9雖然形式上也滿足4n+2規則,但由于其NICS(0)zz基本為0,因此可斷定是非芳香性的,這必然是由于其π電子缺乏整體離域能力所致。雖然B9N9的各個B-N鍵如前述Mayer鍵級和IRI-π所示有明顯π作用,但顯然不意味著其π電子能夠容易地離域。對這些體系,σ和core電子都由于其高度定域性而對芳香性基本沒有影響。
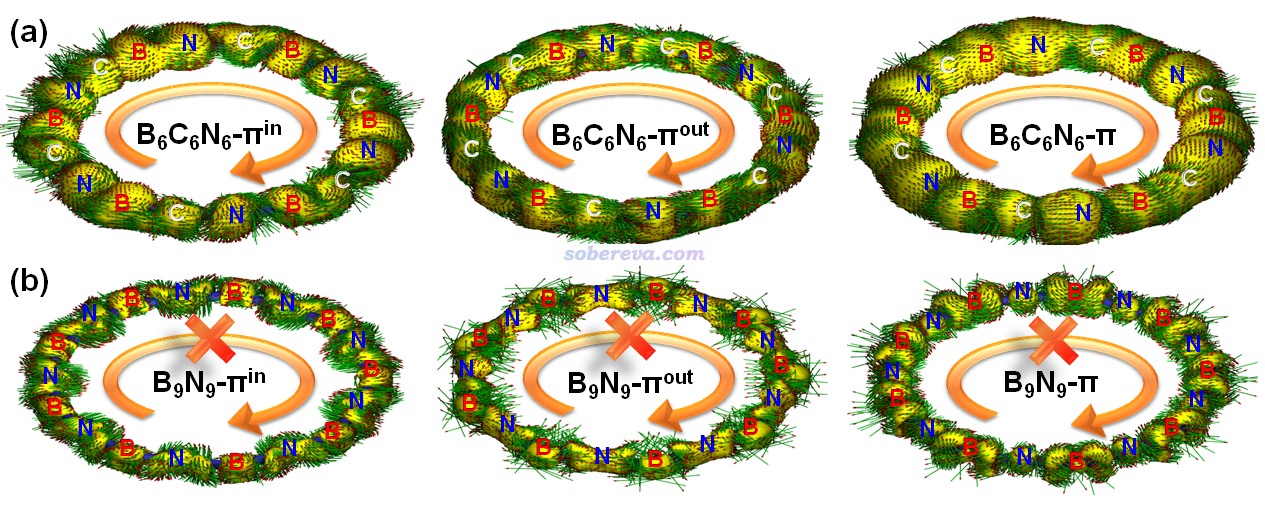
《使用AICD程序研究電子離域性和磁感應電流密度》(http://www.shanxitv.org/147)和《使用AICD 2.0繪制磁感應電流圖》(http://www.shanxitv.org/294)介紹的AICD圖是使用得較普遍的圖形化直觀展現化學體系磁感生電流的方法。為了進一步從磁感生電流的角度展現B6C6N6的芳香性以及和B9N9的差異,文中對這些體系的π-in、π-out以及全部π電子分別繪制了AICD圖,如下所示。外磁場方向垂直于環由下朝上施加。由于等值面上的箭頭太小,為了看得清楚,圖中還用大的橙色箭頭做了清晰的標注。此圖直觀體現出,B6C6N6確實存在顯著的π芳香性,因為感生電流方向符合左手定則并且連貫地環繞整個體系,這正是典型的π芳香性體系具備的特征。而缺乏芳香性的B9N9則明顯不具備這樣的特征,感生電流幾乎只是繞著原子核或原子間區域轉。
此文還按照《考察分子磁感生電流的程序GIMIC 2.0的使用(含24分鐘演示視頻)》(http://www.shanxitv.org/491)介紹的方法繪制了GIMIC磁感生電流的動畫,如下所示,更進一步地體現出芳香性明顯的B6C6N6能夠形成整體感生環電流而非芳香性的B9N9不具備這樣的特征。
B6C6N6:http://www.shanxitv.org/attach/696/B6C6N6_GIMIC.mp4
B9N9:http://www.shanxitv.org/attach/696/B9N9_GIMIC.mp4
文中還對穿越B6C6N6、B9N9和18碳環的化學鍵的感生電流進行了積分,數值分別為21.64、0.70、22.50 nA/T,這定量體現出B6C6N6和18碳環的芳香性相仿佛,而B9N9完全不具備芳香性。
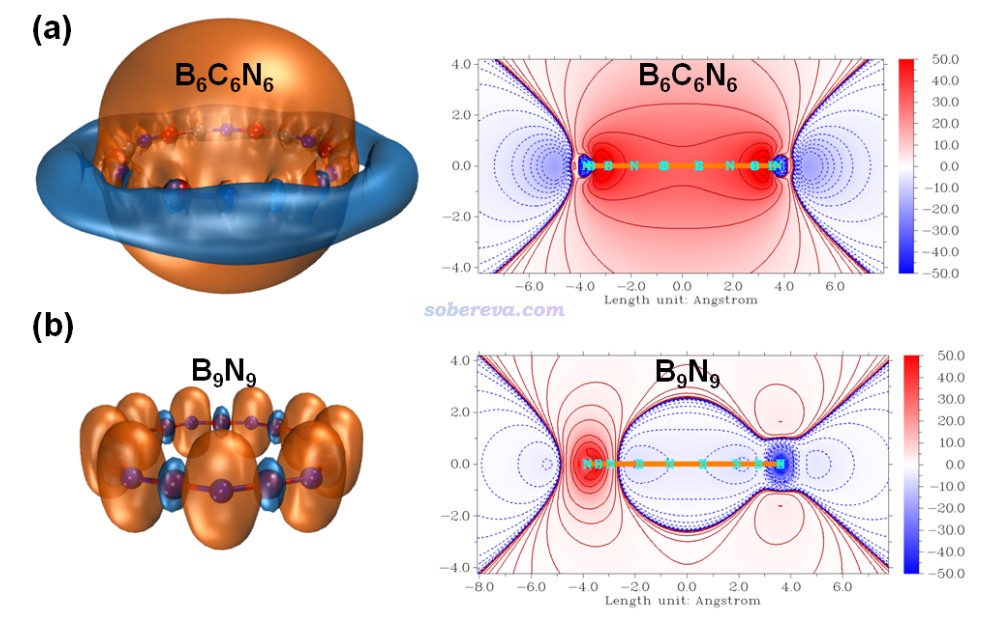
在《通過Multiwfn繪制等化學屏蔽表面(ICSS)研究芳香性》(http://www.shanxitv.org/216)和《使用Multiwfn巨方便地繪制二維NICS平面圖考察芳香性》(http://www.shanxitv.org/682)中介紹了ICSS函數,它直觀地體現了化學體系的磁屏蔽和去屏蔽區域的位置和強度,是NICS的重要擴展,已被非常廣泛應用于討論芳香性問題。此研究使用Multiwfn對B6C6N6和B9N9都繪制了ICSS_ZZ = 10 ppm的等值面圖以及垂直于環的截面的填色等值線圖,如下所示。等值面圖中橙色和藍色分別對應屏蔽和去屏蔽區域。由圖可見,B6C6N6的環內部完全是屏蔽區域,環外面完全是去屏蔽區域,這種特征和苯這樣的典型芳香性體系完全相同,更進一步嚴格地證明了B6C6N6有顯著的芳香性。而且B6C6N6的ICSS的這種特征和Carbon, 165, 468-475 (2020)報道的18碳環的高度一致,體現出二者芳香性的高度共性。而B9N9的ICSS等值面圖的屏蔽和去屏蔽區域彼此交錯,沒有形成遍及整體的連貫的等值面,體現出此體系既沒有芳香性也沒有反芳香性,而是非芳香性。
5 B6N6C6的電子離域性
芳香性的本質來自于電子的離域,因此文中還特意對B6N6C6的電子離域性從多方面進行了考察。在《在Multiwfn中單獨考察pi電子結構特征》(http://www.shanxitv.org/432)中介紹了Multiwfn可以計算和繪制的LOL-π函數,這對于考察π電子的離域情況極其有用,如今已經非常流行。下圖對比了B6N6C6和B9N9的LOL-π=0.5等值面,明顯可見B6N6C6的等值面比B9N9顯得連貫得多,明顯體現出B6N6C6的π電子有強得多的在18個原子上全局離域的能力,也一定程度解釋了前者的芳香性比后者強得多的原因。
LOL-π函數比較適合直觀定性考察,而《在Multiwfn中單獨考察pi電子結構特征》里介紹的ELF-π函數的二分點數值則適合定量分析對比。下圖是B6N6C6和B9N9的ELF-π函數等值面圖,等值數值設為了令等值面剛好分裂時的數值,這被稱為二分值,數值越大說明電子越容易跨越二分點區域發生離域。在圖上也標注了二分點位置和二分值。由圖可見,B6N6C6的π-in和π-out的二分值都明顯大于B9N9的,這給B6N6C6的整體電子離域性更強又進一步提供了定量證據。

電子的費米穴是考察電子離域特征的一個重要的函數,它包含兩個三維位置坐標,作圖考察時通常將一個位置作為參考點,然后對另一個坐標繪圖。Atoms-in-molecules理論的提出者Bader指出an electron can go where its hole goes and, if the Fermi hole is localized, then so is the electron,也就是假設一個電子出現在某個參考點位置,那么其費米穴函數明顯分布在哪,這個電子就容易離域到哪去。因此,通過繪制費米穴圖,可以更進一步了解體系處于不同位置的電子的離域能力。文中通過Multiwfn對B6N6C6、B9N9和18碳環分別繪制了費米穴的二維圖,如下所示。參考點位置以綠色箭頭標注,設在了不同原子的價層區域距離原子核特定距離的位置,此時圖中的費米穴的分布就體現了這個位置的價電子容易離域到哪去。用Multiwfn繪制這種圖的操作在《制作動畫分析電子結構特征》(http://www.shanxitv.org/86)里有相關說明。
由上圖等值線分布可見,B6N6C6的硼和碳的價電子的費米穴分布范圍較廣,體現出電子有往較遠區域離域的能力,特別是其碳的費米穴分布特征和已知具有較顯著全局離域性的18碳環的很接近。相比之下,B9N9的硼和氮的費米穴分布廣度明顯不及B6N6C6的相應元素的,等值線只分布在距離參考點較近的原子上,體現出B9N9的價電子的離域能力相對較差。
上面的等值線圖適合定性討論,為了能把以上信息轉化成定量形式來更好地對比分析,文中定義了一個新的量叫做原子遠程離域指數(atomic remote delocalization index, ARDI)。離域性指數(DI)在《Multiwfn支持的分析化學鍵的方法一覽》(http://www.shanxitv.org/471)里專門介紹過,是衡量特定兩個原子間等效共享電子對數用的,而ARDI則是對單個原子定義的,定義為距離它兩個化學鍵以上的原子與當前原子的所有DI之和,顯然基于Multiwfn計算的所有原子間的DI可以簡單地手算出來各個原子的ARDI。ARDI體現了特定原子上的電子的遠程離域能力,數值越大說明遠程離域能力越強。環上各個原子上的電子有顯著的遠程離域能力顯然是這個環具備明顯芳香性的前提。各原子ARDI如下所示,可見B6N6C6上的原子的ARDI都較大,平均值與18碳環的碳的相仿佛,而B9N9的硼和氮的ARDI都遠小于B6N6C6相應元素的原子,這更充分體現了B9N9的電子的全局電子離域能力遠不如B6N6C6,進一步展現了B9N9芳香性遠低于B6N6C6的原因。

6 碳摻雜進B9N9使得芳香性巨幅提升的本質
根據前面的討論,文章已經充分證明了芳香性的關系是B6C6N6 ≈ C18 ? B9N9。為什么將碳元素摻雜進入無芳香性的B9N9,就能使其芳香性巨幅提升、電子全局離域能力大幅增加?實際上原因并不難理解。盡管前面的Mayer鍵級體現出B9N9中的硼-氮之間有一定π作用,但由于硼和氮的電負性差異非常大,這無疑會導致電子傾向于在氮上定域而難以往相鄰的硼上離域,至于往更遠的原子上離域就更是難上加難了。而碳的電負性則正好介于硼和氮之間,顯然硼-碳和氮-碳鍵的共價性顯著強于硼-氮鍵,因此電子更容易離域過去,相當于碳給硼和氮之間架設了能夠令電子容易離域過去的橋梁,充分調和了硼和氮之間電負性差異過大導致的電子跨鍵離域太難的矛盾。
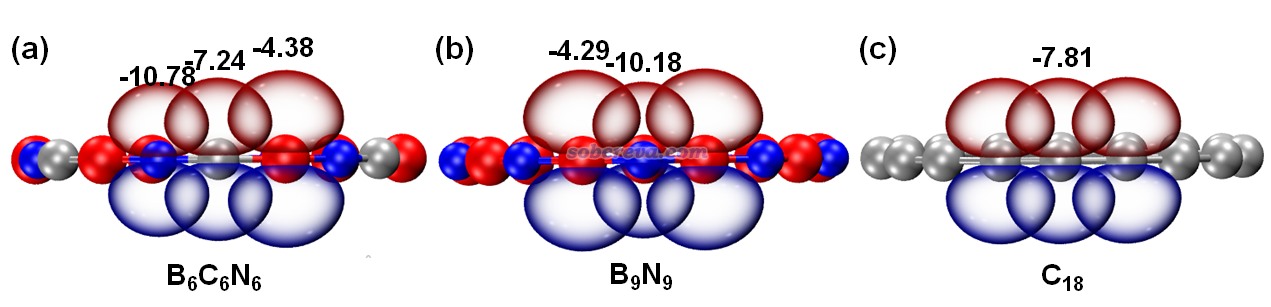
為了更好地展現碳原子起到的價值,文中將垂直于環的各原子的2p原子軌道繪制了出來,π-out電子正是分布在這些p軌道上,π-out分子軌道也正是它們線性組合產生的。具體來說,由于NBO程序產生的預自然原子軌道(PNAO)可以較合理地描述分子環境中的原子軌道,因此文中按照《使用Multiwfn繪制NBO及相關軌道》(http://www.shanxitv.org/134)中的做法,用Multiwfn結合VMD將相應的PNAO軌道用等值面圖形式做了展示,如下所示。等值面數值選為了適合對比的0.1。圖中也把PNAO的能量(eV)標注在了上面。由圖可清楚地看出,B9N9中的氮和硼的p軌道的空間延展程度差異很大,而且軌道能量差異也很大,勢必電子難以在它們之間離域。而從B6C6N6的圖中可見,碳的p軌道延展范圍和軌道能量都恰好介于硼和氮之間,它的引入無疑能夠顯著幫助電子離域過去。
7 B6N6C6異構體的芳香性
以上研究的那種以B-C-N作為重復單元排列的B6N6C6結構是否真的是B6N6C6的能量最低結構?是否B6N6C6化學組成的體系還有芳香性更強的異構體?這是個值得考察的有趣的問題。為此,文中還另外考慮了兩種B6N6C6構型,優化后得到的無虛頻結構如下所示,可見B6N6C6'構型讓所有碳盡可能連在一起,其它部分部分保持B9N9那樣硼-氮交錯的結構,而B6N6C6''構型則是由三個C-C-B-N-B-N重復單元構成的結構。這兩個結構的基態是閉殼層的,能量分別比前面研究的B-C-N為重復單元構成的B6N6C6結構能量低280.0和250.2 kcal/mol。這體現出B6N6C6中B-C和C-N的平均鍵能比B-N和C-C是要更小的。雖然前面研究的B6N6C6結構能量不是這種化學組成的能量最低結構,但并不意味著它是不穩定的。根據《使用ORCA做從頭算動力學(AIMD)的簡單例子》(http://www.shanxitv.org/576)中介紹的方法,文中對此體系做了較高溫度(500 K)下20 ps長度的基于wB97X-D3/6-311G*級別的從頭算分子動力學模擬,用VMD每1 ps繪制一次的結構疊加圖如下所示(顏色按照藍-白-紅變化),可見環狀結構始終很好維持著,并沒有發生異構化、解離等現象,體現出B6N6C6具有一定熱力學穩定性。

文中還對B6N6C6'和B6N6C6''構型計算了NICS(0)zz,發現分別僅有1.8和1.4 ppm,可見這倆構型沒有芳香性。這說明只有讓碳插入到硼和氮中間,從而以B-C-N作為重復單元時,碳才能真正起到顯著提升純硼-氮環體系的電子離域性的作用。
8 B6N6C6最低三重態激發態的芳香性
前面考察的都是B6N6C6的單重態基態(S0)。最后,文章還研究了B6N6C6最低三重態激發態(T1態)的芳香性。計算發現S0-T1垂直和絕熱激發能分別為1.62和1.17 eV。下圖(a)是優化后的T1態B6N6C6的結構,可見對稱性相對于S0態發生了下降。下圖(b)是按照《使用Multiwfn作電子密度差圖》(http://www.shanxitv.org/113)繪制的在B6N6C6的S0結構下T1態與S0態的密度差,藍色和綠色分別為電子密度降低和增加部分。由圖可見S0到T1態的電子激發是π-in → π-out的激發。由于垂直激發導致的電子密度變化破壞了B6N6C6基態的C6h對稱性,這也是為什么在T1態勢能面上從Franck-Condon點往極小點弛豫過程中幾何結構對稱性發生了下降。
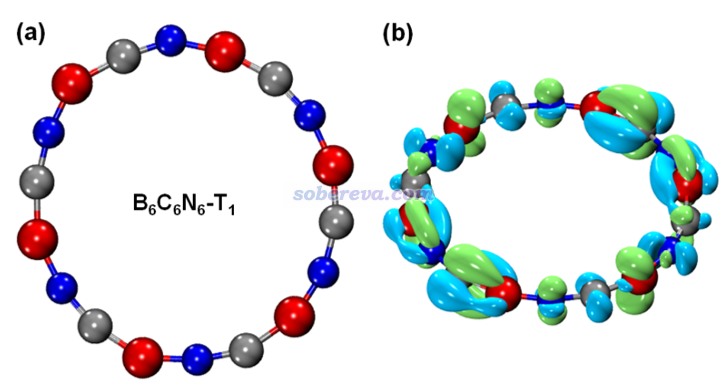
文中對B6N6C6的T1態計算了NICS(0)zz,并分解為了不同類型電子的貢獻。而且為了考察幾何結構弛豫的影響,在S0極小點和T1極小點分別做了這種計算,結果如下所示。可見無論在哪種結構下,T1態由于NICS(0)zz遠大于0,因此是顯著反芳香性的,這和Baird規則所述情況一致。Baird規則指出單重態基態滿足Hückel 4n+2規則的體系的最低三重態是反芳香性的。從NICS(0)zz還看到,T1態的反芳香性特征在T1極小點結構下比S0極小點結構下更弱,這體現出B6N6C6自發減小T1態的反芳香性可能是其S0→T1垂直激發后結構弛豫的重要驅動力。
9 總結
此文基于量子化學計算和Multiwfn等程序做波函數分析,非常全面系統研究了18碳環的等電子體B6N6C6的幾何結構和電子結構,從不同角度全面考察了其芳香性和電子離域性特征,并且和18碳環及其另外一個重要的等電子體B9N9進行了細致的對比。本文體現出以碳原子橋聯硼和氮原子,可以顯著提升純硼-氮體系的電子離域性。這不僅對電子結構產生重要影響,必然也同時會影響其它性質,諸如光學性質、電子輸運性質等。本文雖然研究的只是一維體系,可以預想電負性介于硼和氮之間的碳元素的摻雜必定也會對二維、三維純硼-氮體系產生顯著影響,而且摻雜方式的選取可能會對這些體系的性質起到一定調控作用。
如果讀者對于18碳環及其相關體系感興趣,非常推薦查看http://www.shanxitv.org/carbon_ring.html里匯總的本文作者的巨量其它研究工作,包含大量深入淺出的論文內容和研究思想介紹文章,以及對計算技術細節的附加說明。
]]>
此文對筆者完整看過的2023年度完結的79部全長版TV動畫和7部泡面番進行總評,約20萬字。劇場版、OVA之類的都沒有納入其中,因為另有文章。此文只有“2024年1月完結”部分是新寫的,2023年4月、7月、10月完結的動畫的評論在作品剛完結之時就已經寫好并發在了筆者的blog上,在此文只是匯總而已(所以有些文字現在看起來已經out了),不過很多作品的總評分為了保持評價的一致性,在此文又做了微調。以前寫的總評見(不同年份的評分標準有一些系統性不同,不適合橫向對比):
2022年度完結動畫總評:http://www.shanxitv.org/669
2021年度完結動畫總評:http://www.shanxitv.org/633
2020年度完結動畫總評:待發布
2019年度完結動畫總評:http://www.shanxitv.org/544
2018年度完結動畫總評:http://www.shanxitv.org/470
2017年度完結動畫總評:http://www.shanxitv.org/401
2016年度完結動畫總評:http://www.shanxitv.org/358
2015年度完結動畫總評:http://www.shanxitv.org/316
2014年度完結動畫總評:http://www.shanxitv.org/273
2013年度完結動畫總評:http://www.shanxitv.org/219
2012年度完結動畫總評:http://www.shanxitv.org/174
2011年度完結動畫總評:http://www.shanxitv.org/118
2023年筆者觀看的非TV版動畫另有文章進行評論:
2022下半年和2023上半年觀看的非TV版動畫簡評(http://www.shanxitv.org/673)
2023下半年觀看的非TV版動畫簡評(http://www.shanxitv.org/694)
筆者正在追的2023年10月開播的《葬送的芙莉蓮》和《藥屋少女的呢喃》尚未完結,所以不納入本文。
筆者是堅定的掃番黨,所有新番無論喜好至少看第一話,不少都觀望到第3話才決定是否棄。沒有納入此文的動畫,按筆者的觀點來說大部分就屬于無聊、跟風、庸俗、粗制濫造之作(可能讀者會發現在視頻網站上播放數高的作品,反倒出現在本文的幾率低,因為主流的品味實在是不敢恭維,而真正的好作品卻總是受到冷落)。也有極少數作品雖然可能做得不錯卻沒出現在此文,是因為那些題材著實不是灑家的菜,諸如恐怖惡心之類,所以也就沒看下去。
評分主體區間是90~95分(低于90分根本不追,超過95的則是超神作水準,極度罕見),不了解此系列文章評分傳統的勿試圖吐槽這點。評分依據是作品畫面、音樂、表現力、創意、劇情、人設等因素,以及很少部分個人主觀觀感和興趣因素。
注意,本文的評論有的包含一些劇透成份,會影響觀賞樂趣。如果目的是想找一些好看的,只要看頒獎部分就夠了,等看完了再看評論為宜。
每個獎項里面的順序是隨意的,不分先后。
題外話:我只看日本動畫,因此此文里看不到其它國家的動畫。我總看到有一些人無視每年層出不窮的大量杰出的日本動畫作品,卻往往拿撐死了也就占70%比例的那些很俗套的商業化日本動畫作品(這些我也基本都不追)為由貶低和唱衰日本近年來的動畫,對于他們我表示極度遺憾,錯過了太多佳作,而且他們的言論也嚴重偏激和誤導他人,令我十分反感。若做不到像我一樣完整掃番(就算哪怕只掃1/3也行)、認真觀賞,就根本沒有做評價的資格。上述這樣的人也有些是因為自己年齡和心態發生變化導致觀賞體驗發生變化,卻歸咎于是當下日本動畫本身的問題,這顯然也是很不負責任的。還有些人對日本動畫主觀地懷有惡意,甚至會因為一些動畫里有一些其不喜歡的元素(如賣萌賣肉)就無腦地批判和否定整部作品乃至業界。比如有人說“現在日本動畫千篇一律,都是賣萌賣肉、穿越異世界,沒意思”,我就想反問這些人,你完整看過《石紀元》?完整看過《16bit的感動》?看過《藥屋少女的呢喃》?這算賣萌賣肉?這是穿越異世界?它們的出色,明明靠的是杰出的創意、劇情、人設以及畫面/音樂/配音等各方面高水準的制作質量。倘若你看到了自己不喜歡類型的作品,別張口就隨便否定業界,近年來的日本動畫里有的是其它類型的非常優秀的作品可推薦的。我從2008年開始認真掃番至今,每年完整追100部左右TV版動畫(另加許多劇場版、OVA),可以非常準確客觀地在此表示,日本動畫整體品質的平均值從未有任何下降,而且出色作品的數量也根本沒有減少過,依然是百花齊放,繁榮昌盛的狀態。我想強調的是,做出評論一定要負責,諸如我雖然不看其它國家的動畫,并且對某些國家的動畫有一些長久以來的負面印象和看法,但由于我沒認真全面觀賞過其近年的作品,無法確定是否有一些優秀作品是我不知道的、乃至我看了后說不定會追的,所以我就不會輕易做出評價。我一直致力于寫動畫評論,主要目的之一是為了讓真正優秀的日本動畫得到更多的關注,給觀賞時間有限的(而且觀賞品味不離譜的)人提供指引,讓他們更容易接觸到出色且適合他們觀賞口味的作品,我認為這是很有意義的。
超神作獎:幻日的夜羽 -SUNSHINE in the MIRROR-
神作獎:浪客劍心 -明治劍客浪漫譚-,Dr.STONE 石紀元 第三季,16bit的感動 ANOTHER LAYER
杰作獎:間諜過家家 第二季,超超超超喜歡你的100個女朋友,女友成堆 第二季,我的幸福婚約,租借女友 第三季,尼爾:機械紀元 Ver1.1a,海盜戰記 第二季,我推的孩子,我家的英雄,小鳥之翼 BIRDIE WING -Golf Girls’Story- 第二季,鬼滅之刃 刀匠村篇,異世界舅舅,小智是女孩啦!,藍色監獄,不要欺負我,長瀞同學 2nd Attack
一等獎:我們的雨色協議,我的推是壞人大小姐。,東京復仇者 天竺篇,盾之勇者成名錄 第三季,銀砂糖師與黑妖精 Part 2,間諜教室 第二季,無職轉生Ⅱ ~到了異世界就拿出真本事~,Lv1魔王與獨居廢勇者,輝夜姬想讓人告白-永不結束的初吻,百合是我的工作!,黃金神威 第四季,總之就是非常可愛 第二季,賽馬娘 Pretty Derby ROAD TO THE TOP,女神的露天咖啡廳,和山田談場Lv999的戀愛,弦音 -連結的一射-,轉生王女與天才千金的魔法革命,銀砂糖師與黑妖精~sugar apple fairy tale~,在地下城尋求邂逅是否搞錯了什么 第四季(迷宮篇) ,再得一勝!,傲嬌反派千金莉潔洛特與實況主遠藤同學及解說員小林同學,間諜教室
二等獎:鴨乃橋論的禁忌推理,賽馬娘 Pretty Derby 第三季,大小姐與看門狗,家里蹲吸血姬的苦悶,哥布林殺手 第二季,Secret Mission~潛入捜査官絕對不會輸!~,公司的小小前輩,我喜歡的女孩忘記戴眼鏡,能干貓貓今天也憂郁,堀與宮村 -piece-,妖幻三重奏,熊熊勇闖異世界 PUNCH!,放學后失眠的你,江戶前精靈,偶像大師 灰姑娘女孩 U149,擁有超常技能的異世界流浪美食家,東京復仇者 圣夜決戰篇,飆速宅男 LIMIT BREAK,TechnoRoid: 超越意志(overmind),極主夫道 第二季,冰屬性男子與無表情女子,關于我在無意間被隔壁的天使變成廢柴這件事,不當哥哥了!
三等獎:破滅之國,轉生成自動販賣機的我今天也在迷宮徘徊,廟不可言,夫妻交* ~一旦做過就回不去了,幼女社長R,我心里危險的東西,我與機器子,怕痛的我,把防御力點滿就對了。 第二季,無意間變成狗,被喜歡的女生撿回家。(生而為狗 我很幸福),D4DJ All Mix,被解雇的暗黑士兵(30多歲)開始了慢生活的第二人生,虛構推理 第二季,Love Live!虹咲學園學園偶像同好會 虹四格動畫
鼓勵獎:特搜組大吾 救國的橘色部隊,藍色管弦樂,艦隊Collection 總有一天,在那片海,呆萌酷男孩,Showtime!2~唱歌的大姐姐也想做,某天早上變成人頭麥克風的我君的人生
超神級女主角:夜羽(幻日的夜羽)
神級女主角:秋里木葉(16bit的感動 ANOTHER LAYER)
最佳女主角:神谷熏(浪客劍心 -明治劍客浪漫譚-),星崎理香&桐生紫乃(女友成堆 第二季),零緹拉&克蕾雅(我的推是壞人大小姐。),好本靜&榮逢凪乃(超超超超喜歡你的100個女朋友),約爾&阿尼婭(間諜過家家 第二季),三枝悠宇(我們的雨色協議),琥珀(Dr.STONE 石紀元 第三季),2B(尼爾:機械紀元 Ver1.1a),三重愛(我喜歡的女孩忘記戴眼鏡),水原千鶴(租借女友 第三季),安(銀砂糖師與黑妖精 Part 2),齋森美世(我的幸福婚約),由崎司(總之就是非常可愛 第二季),白鷺陽芽(百合是我的工作!),鳥棲歌仙(我家的英雄),鳳凰寺紅葉(女神的露天咖啡廳),伊芙(小鳥之翼 BIRDIE WING -Golf Girls’Story- 第二季),安·哈魯佛德(銀砂糖師與黑妖精~sugar apple fairy tale~),琉小姐(在地下城尋求邂逅是否搞錯了什么 第四季),長瀞(不要欺負我,長瀞同學 2nd Attack),相澤智(小智是女孩啦!)
優秀女主角:雷土吏子(Secret Mission~潛入捜査官絕對不會輸!~),瀨名垣一咲(大小姐與看門狗),花園羽香里&院田唐音&藥膳楠莉&羽羽里(超超超超喜歡你的100個女朋友),拉芙塔莉雅(盾之勇者成名錄 第三季),精靈弓手(哥布林殺手 第二季),佐木咲(女友成堆 第二季),中村雪(特搜組大吾 救國的橘色部隊),北部玄駒(賽馬娘 Pretty Derby 第三季),可瑪麗(家里蹲吸血姬的苦悶),緹婭(間諜教室 第二季),風卷祭里(妖幻三重奏),希露菲(無職轉生Ⅱ ~到了異世界就拿出真本事~),片瀨詩織里(公司的小小前輩),魔王(Lv1魔王與獨居廢勇者),福澤幸來(能干貓貓今天也憂郁),堀京子(堀與宮村 -piece-),機器子(我與機器子),優奈(熊熊勇闖異世界 PUNCH!),灶門禰豆子(鬼滅之刃 刀匠村篇),山田杏奈(我心里危險的東西),木之下茜(和山田談場Lv999的戀愛),阿西麗帕(黃金神威 第四季),艾爾達(江戶前精靈),曲伊咲(放學后失眠的你),莉潔洛特&小林詩帆乃(傲嬌反派千金莉潔洛特與實況主遠藤同學及解說員小林同學),犬飼加戀(無意間變成狗,被喜歡的女生撿回家。),愛本鄰久(D4DJ All Mix),巖永琴子(虛構推理 第二季),艾妮絲菲亞&尤菲莉亞(轉生王女與天才千金的魔法革命),冰浦永遠(再得一勝!),冬月(冰屬性男子與無表情女子),椎名真晝(關于我在無意間被隔壁的天使變成廢柴這件事),艾爾加&藤宮澄夏(異世界舅舅)
最佳女配角:時野谷美櫻(我們的雨色協議),瑪娜麗婭(我的推是壞人大小姐。),黑澤戴雅&小原鞠莉&櫻內梨子(幻日的夜羽),橘純加(百合是我的工作!),愛&有馬佳奈&MEM 啾(我推的孩子),天鷲葵(小鳥之翼 BIRDIE WING -Golf Girls’Story- 第二季),雪女(虛構推理 第二季),阿伊莎(在地下城尋求邂逅是否搞錯了什么 第四季),群堂美鈴(小智是女孩啦!),妹尾梨可(弦音 -連結的一射-)
優秀女配角:香織&冬夜(16bit的感動 ANOTHER LAYER),薩迪娜&菲洛&亞朵拉(盾之勇者成名錄 第三季),劍之圣女(哥布林殺手 第二季),星崎理沙(女友成堆 第二季),艾爾達(浪客劍心 -明治劍客浪漫譚-),薇兒赫澤(家里蹲吸血姬的苦悶),稻月望(我們的雨色協議),渡邊曜&松浦果南&黑澤露比&國木田花丸&萊拉普斯(幻日的夜羽),一之瀨小百合&八重森彌妮&更科瑠夏(租借女友 第三季),潔尼亞(Lv1魔王與獨居廢勇者),由里江(我的幸福婚約),阿妮特(間諜教室 第二季),美月(百合是我的工作!),甘露寺蜜璃(鬼滅之刃 刀匠村篇),月島流星&鶴河秋水(女神的露天咖啡廳),早乙女一奈&新莊雨音(小鳥之翼 BIRDIE WING -Golf Girls’Story- 第二季),命小姐(在地下城尋求邂逅是否搞錯了什么 第四季),卡洛爾(小智是女孩啦!),社長(不要欺負我,長瀞同學 2nd Attack),伊莉雅&緹爾蒂(轉生王女與天才千金的魔法革命),柚葉(東京復仇者 圣夜決戰篇),緹婭(間諜教室),狐森(冰屬性男子與無表情女子),梅貝爾(異世界舅舅)
神級男主角:緋村劍心(浪客劍心 -明治劍客浪漫譚-),石神千空(Dr.STONE 石紀元 第三季)
最佳男主角:六田守(16bit的感動 ANOTHER LAYER),花垣武道(東京復仇者 天竺篇),勞埃德(間諜過家家 第二季),愛城戀太郎(超超超超喜歡你的100個女朋友),久堂清霞(我的幸福婚約),阿庫亞(我推的孩子),鳥棲哲雄(我家的英雄),舅舅(異世界舅舅),貝爾(在地下城尋求邂逅是否搞錯了什么 第四季),潔世一(藍色監獄),夏爾·斐恩·夏爾(銀砂糖師與黑妖精~sugar apple fairy tale~)
優秀男主角:宇藤啟彌(大小姐與看門狗),巖谷尚文(盾之勇者成名錄 第三季),哥布林殺手(哥布林殺手 第二季),鴨乃橋論(鴨乃橋論的禁忌推理),向井直也(女友成堆 第二季),9S(尼爾:機械紀元 Ver1.1a),木之下和也(租借女友 第三季),夏爾(銀砂糖師與黑妖精 Part 2),阿箱(轉生成自動販賣機的我今天也在迷宮徘徊),克勞斯(間諜教室 第二季),麥克斯(Lv1魔王與獨居廢勇者),諭吉(能干貓貓今天也憂郁),托爾芬(海盜戰記 第二季),灶門炭治郎(鬼滅之刃 刀匠村篇),山田秋斗(和山田談場Lv999的戀愛),由崎星空(總之就是非常可愛 第二季),粕壁隼(女神的露天咖啡廳),遠藤碧人&齊格瓦托(傲嬌反派千金莉潔洛特與實況主遠藤同學及解說員小林同學),芝浦繪空&氖恩(TechnoRoid: 超越意志),緒山真尋(不當哥哥了!),小野田(飆速宅男 LIMIT BREAK),克勞斯(間諜教室),龍(極主夫道 第二季),冰室(冰屬性男子與無表情女子),藤宮周(關于我在無意間被隔壁的天使變成廢柴這件事),達利艾爾(被解雇的暗黑士兵(30多歲)開始了慢生活的第二人生),穆寇達(擁有超常技能的異世界流浪美食家)
最佳男配角:相樂左之助&明神彌彥(浪客劍心 -明治劍客浪漫譚-),馬狼照英(藍色監獄)
優秀男配角:Mikey(東京復仇者 天竺篇),四乃森蒼紫(浪客劍心 -明治劍客浪漫譚-),秋那千尋(公司的小小前輩),蛇&凱迪爾&大老爺(海盜戰記 第二季),龍水&卡瑟吉(Dr. STONE 石紀元 第三季),白石由竹(黃金神威 第四季),千切豹馬&蜂樂回&凪誠士郎(藍色監獄),室井昌幸(虛構推理 第二季),瀧川雅貴(弦音 -連結的一射-),御堂筋(飆速宅男 LIMIT BREAK),芬里爾(擁有超常技能的異世界流浪美食家),敬文(異世界舅舅)
神級女CV:古賀葵(秋里木葉-16bit的感動 ANOTHER LAYER),小林愛香(津島善子)
最佳女CV:竹達彩奈(星崎理香-女友成堆 第二季),水樹奈奈(瑪娜麗婭-我的推是壞人大小姐。),早見紗織(約爾-間諜過家家 第二季),種崎敦美(阿尼婭-間諜過家家 第二季),鈴木愛奈(小原鞠莉),小宮有紗(黑澤戴雅-幻日的夜羽),逢田梨香子(櫻內梨子-幻日的夜羽),若山詩音(三重愛-我喜歡的女孩忘記戴眼鏡),小倉唯(白鷺陽芽-百合是我的工作!),鬼頭明里(伊芙-小鳥之翼 BIRDIE WING -Golf Girls’Story- 第二季),日高里菜(群堂美鈴-小智是女孩啦!),花澤香菜(小林詩帆乃-傲嬌反派千金莉潔洛特與實況主遠藤同學及解說員小林同學),高橋李依(相澤智-小智是女孩啦!)
優秀女CV:小市真琴(明神彌彥-浪客劍心 -明治劍客浪漫譚-),御苑生メイ(雷土吏子-Secret Mission~潛入捜査官絕對不會輸!~),芹澤優(零緹拉-我的推是壞人大小姐。),奈波果林(克蕾雅-我的推是壞人大小姐。),鬼頭明里(瀨名垣一咲-大小姐與看門狗),小清水亞美(薩迪娜-盾之勇者成名錄 第三季),麻倉桃(時野谷美櫻-我們的雨色協議),朝井彩加(藥膳楠莉-超超超超喜歡你的100個女朋友),瀨戶麻沙美(榮逢凪乃-超超超超喜歡你的100個女朋友),高橋李依(桐生紫乃-女友成堆 第二季),佐倉綾音(佐木咲-女友成堆 第二季),齊藤朱夏(渡邊曜-幻日的夜羽),降幡愛(黑澤露比-幻日的夜羽),諏訪奈奈香(松浦果南-幻日的夜羽),高槻加奈子(國木田花丸-幻日的夜羽),伊波杏樹(高海千歌-幻日的夜羽),日笠陽子(萊拉普斯-幻日的夜羽),石川由依(2B-尼爾:機械紀元 Ver1.1a),貫井柚佳(安-銀砂糖師與黑妖精 Part 2),雨宮天(水原千鶴-租借女友 第三季),大空直美(魔王-Lv1魔王與獨居廢勇者),戶松遙(堀京子-堀與宮村 -piece-),上田麗奈(齋森美世-我的幸福婚約),桑島法子(由里江-我的幸福婚約),石川由依(福澤幸來-能干貓貓今天也憂郁),德井青空(好歌劇-賽馬娘 Pretty Derby ROAD TO THE TOP),鬼頭明里(由崎司-總之就是非常可愛 第二季),大久保瑠美(MEM 啾-我推的孩子),花澤香菜(甘露寺蜜璃-鬼滅之刃 刀匠村篇),水瀨祈(木之下茜-和山田談場Lv999的戀愛),小市真琴(橘純加-百合是我的工作!),小清水亞美(艾爾達-江戶前精靈),瀨戶麻沙美(天鷲葵-小鳥之翼 BIRDIE WING -Golf Girls’Story- 第二季),藤田咲(早乙女一奈-小鳥之翼 BIRDIE WING -Golf Girls’Story- 第二季),渡邊明乃(阿伊莎-在地下城尋求邂逅是否搞錯了什么 第四季),早見紗織(琉小姐-在地下城尋求邂逅是否搞錯了什么 第四季),悠木碧(雪女-虛構推理 第二季),貫井柚佳(安·哈魯佛德-銀砂糖師與黑妖精~sugar apple fairy tale~),西尾夕香(愛本鄰久-D4DJ All Mix),上坂堇(長瀞-不要欺負我,長瀞同學 2nd Attack),水樹奈奈(社長-不要欺負我,長瀞同學 2nd Attack),田村睦心(芝浦繪空-TechnoRoid: 超越意志),高野麻里佳(緒山真尋-不當哥哥了!),內山夕實(狐森-冰屬性男子與無表情女子),木野日菜(史伊-擁有超常技能的異世界流浪美食家),天城莎莉(卡洛爾-小智是女孩啦!)
最佳男CV:齊藤壯馬(緋村劍心-浪客劍心 -明治劍客浪漫譚-),福山潤(阿箱-轉生成自動販賣機的我今天也在迷宮徘徊),松岡禎丞(貝爾-在地下城尋求邂逅是否搞錯了什么 第四季),津田健次郎(龍-極主夫道 第二季),子安武人(舅舅-異世界舅舅)
優秀男CV:新祐樹(花垣武道-東京復仇者 天竺篇),小林裕介(石神千空-Dr.STONE 石紀元 第三季),榎木淳彌(向井直也-女友成堆 第二季),阿部敦(六田守-16bit的感動 ANOTHER LAYER),花江夏樹(9S-尼爾:機械紀元 Ver1.1a),玄田哲章(白金-妖幻三重奏),堀江瞬(木之下和也-租借女友 第三季),安元洋貴(諭吉-能干貓貓今天也憂郁),松尾駿(機器子-我與機器子),諏訪部順一(鳥棲哲雄-我家的英雄),花江夏樹(灶門炭治郎-鬼滅之刃 刀匠村篇),Kayto(氖恩-TechnoRoid: 超越意志),杉田智和(達利艾爾-被解雇的暗黑士兵(30多歲)開始了慢生活的第二人生),日野聰(芬里爾-擁有超常技能的異世界流浪美食家),福山潤(敬文-異世界舅舅)
最佳BG CP:勞埃德X約爾(間諜過家家 第二季),星崎理香&桐生紫乃X向井直也(女友成堆 第二季),緋村劍心X神谷熏(浪客劍心 -明治劍客浪漫譚-),愛城戀太郎X花園羽香里&榮逢凪乃&好本靜&藥膳楠莉&羽羽里(超超超超喜歡你的100個女朋友),六田守X秋里木葉(16bit的感動 ANOTHER LAYER),木之下和也X水原千鶴(租借女友 第三季),夏爾X安(銀砂糖師與黑妖精 Part 2),久堂清霞X齋森美世(我的幸福婚約),諭吉X福澤幸來(能干貓貓今天也憂郁),由崎星空X由崎司(總之就是非常可愛 第二季),白銀御行X四宮輝夜(輝夜姬想讓人告白-永不結束的初吻),安·哈魯佛德X夏爾·斐恩·夏爾(銀砂糖師與黑妖精~sugar apple fairy tale~),前輩X長瀞(不要欺負我,長瀞同學 2nd Attack),齊格瓦托X莉潔洛特(傲嬌反派千金莉潔洛特與實況主遠藤同學及解說員小林同學),久保田淳一郎X相澤智(小智是女孩啦!)
優秀BG CP:宇藤啟彌X瀨名垣一咲(大小姐與看門狗),時野谷瞬X三枝悠宇(我們的雨色協議),向井直也X佐木咲&水瀨渚(女友成堆 第二季),愛城戀太郎X院田唐音(超超超超喜歡你的100個女朋友),拉密絲X阿箱(轉生成自動販賣機的我今天也在迷宮徘徊),篠崎拓馬X片瀨詩織里&秋那千尋X早川千夏(公司的小小前輩),小村楓X三重愛(我喜歡的女孩忘記戴眼鏡),堀京子X宮村伊澄(堀與宮村 -piece-),山田秋斗X木之下茜(和山田談場Lv999的戀愛),粕壁隼&紅葉(女神的露天咖啡廳),中見丸太X曲伊咲(放學后失眠的你),室井昌幸X雪女(虛構推理 第二季),貝爾X琉小姐(在地下城尋求邂逅是否搞錯了什么 第四季),格雷特X克勞斯(間諜教室),冰室X冬月(冰屬性男子與無表情女子),藤宮周X椎名真晝(關于我在無意間被隔壁的天使變成廢柴這件事),遠藤碧人X小林詩帆乃(傲嬌反派千金莉潔洛特與實況主遠藤同學及解說員小林同學),舅舅X艾爾加X&高丘敬文X藤宮澄夏(異世界舅舅),達利艾爾X瑪麗卡(被解雇的暗黑士兵(30多歲)開始了慢生活的第二人生)
優秀bl CP:鴨乃橋論X一色都都丸(鴨乃橋論的禁忌推理),手島X葦木場(飆速宅男 LIMIT BREAK)
最佳百合CP:零緹拉X克蕾雅(我的推是壞人大小姐。),夜羽X小原鞠莉(幻日的夜羽),白鷺陽芽X美月(百合是我的工作!),純加X果乃子(百合是我的工作!),伊芙X天鷲葵(小鳥之翼 BIRDIE WING -Golf Girls’Story- 第二季),艾妮絲菲亞X尤菲莉亞(轉生王女與天才千金的魔法革命)
優秀百合CP:瑪娜麗婭X克蕾雅(我的推是壞人大小姐。),薇兒赫澤X可瑪麗(家里蹲吸血姬的苦悶),風卷祭里X花奏鈴(妖幻三重奏),福澤幸來X柴咲由里&福澤幸來X仁科理央(能干貓貓今天也憂郁),小金井小糸X艾爾達(江戶前精靈),新莊雨音X天鷲葵(小鳥之翼 BIRDIE WING -Golf Girls’Story- 第二季),早乙女一奈X伊芙(小鳥之翼 BIRDIE WING -Golf Girls’Story- 第二季),伊莉雅X蕾妮(轉生王女與天才千金的魔法革命),梅普露X莎莉(怕痛的我,把防御力點滿就對了。 第二季)
最佳OP:16bit的感動 ANOTHER LAYER,幻日的夜羽,TechnoRoid: 超越意志,艦隊Collection 總有一天,在那片海
優秀OP:幻日的夜羽,尼爾:機械紀元 Ver1.1a,黃金神威 第四季,總之就是非常可愛 第二季,海盜戰記 第二季 OP1(指畫面),我推的孩子
最佳ED:東京復仇者 天竺篇,放學后失眠的你,關于我在無意間被隔壁的天使變成廢柴這件事
優秀ED:地下忍者,浪客劍心 -明治劍客浪漫譚- ED2,16bit的感動 ANOTHER LAYER(指的是畫面),BanG Dream! It’s MyGO!!!!!(沒追完),江戶前精靈,弦音 -連結的一射-
最佳百合番:我的推是壞人大小姐。,幻日的夜羽,百合是我的工作!,小鳥之翼 BIRDIE WING -Golf Girls’Story- 第二季,轉生王女與天才千金的魔法革命
優秀百合番:家里蹲吸血姬的苦悶,江戶前精靈,熊熊勇闖異世界 PUNCH!
神級音樂:幻日的夜羽
最佳音樂:艦隊Collection 總有一天 在那片海
優秀音樂:Dr.STONE 石紀元 第三季,海盜戰記 第二季,熊熊勇闖異世界 PUNCH!,小鳥之翼 BIRDIE WING -Golf Girls’Story- 第二季,弦音 -連結的一射-,TechnoRoid: 超越意志
Dr. STONE 石紀元 第三季 后半
總評:96
這一季后半繼續講千空一行人在島上和滿肚子壞水的宰相荊棘斗智斗勇的故事,內容依然非常精彩,最終終于千空他們拿到了石化裝置,并且解決掉了荊棘。然后發現不明的電波居然來自于月球,于是他們即將踏上登陸月球的旅程。
千空繼續表現得超機智,繼續用原始的工具造出了各種現代文明產物,連無人機、氧氣瓶都出來了,令人嘆為觀止,還表現得非常有勇氣和膽識,再次令人感嘆他真是超級了不起的角色。
荊棘真是從里到外無處不惡心,極度邪惡,石化了島主后自己當了皇帝,此作倒是沒有明確交代他是怎么享用那些美女的。他雖然不懂科學但非常有計謀、格外敏銳,還有不俗的戰斗力,想不到他曾經竟然還是戰士。他實在太難對付了,絲毫不亞于之前對付獅子王司。他居然還能想出把整個島范圍石化的主意,還挺有想法。
最后千空跟荊棘的決戰真是太拼了,好在還是千空的隨機應變能力、頭腦思考速度更勝一籌,最后把這家伙石化的一幕真心感覺巨爽,總算把這個超老奸巨猾、窮兇極惡,還謹慎得無與倫比的大BOSS滅掉了,這真是不可思議的壯舉。
荊棘那邊的戰士伯勞的戰斗力竟然超乎想象的強,沒想到比琥珀還厲害,甚至把冰月復活了都打不過他。我以為只有把獅子王復活才可能贏他,想不到后來冰月獲得了潤滑的竹筒,使得他能夠使出利用杠桿原理的祖傳的槍術把伯勞打得沒有招架之力。我挺想看有朝一日獅子王和他打一架的。
伯勞還不是頭腦簡單的家伙,很是棘手,居然摸到了千空他們藏身的洞穴,差點就把他們團滅。欺詐師淺霧幻純靠嘴皮子把伯勞弄走真是大功一件。
硝酸復活液也太厲害了,銀狼都被荊棘戳穿了,居然復活液用了之后能滿狀態復活。最后還把冷凍狀態的獅子王也給順利滿狀態復活了,實在是神器。石化裝置和硝酸復活液的設定,真是從頭到尾充滿科學元素的此作最最最不科學的地方。既然石化裝置都拿到了,我好奇千空怎么不趕緊造個放大鏡深入研究一下那到底是什么原理?
琥珀從石化狀態中復活,居然一上來就抱上千空了,怎么看琥珀都應該對他有喜歡的感覺才對,畢竟千空那么聰明和威風,不斷創造奇跡。不過此作的官設卻似乎完全沒有這一層感情。
霧雨之前一直被荊棘蒙騙,幫他用石化裝置排除異己。后來被千空等人解除了她的石化,認清真相后加入了科學王國,這結局挺不錯,她的武力值可以和琥珀媲美,給科學王國增添了新的有價值的戰斗力。
后來居然還從海底下撈出來一個很厲害的日本武士style的原始人,將他復活后居然能把琥珀和霧雨瞬間打飛,真是撿到寶了。我還以為他的戰斗力深不可測,原來還是比復活后的獅子王弱一些,也許能和冰月或伯勞打得不分勝負,早知如此應該先把他復活用來牽制伯勞。
銀狼雖然在被荊棘戳穿時形象曾經高大了一回,但復活后果然又跟以前一樣形象十分矮小,卑微得不得了,還特別好大喜功。其實不復活他也罷,估計他以后也不會派上什么用場。
真難以想象之后千空他們怎么上月球,這難度和之前制造那些玩意根本不是一個次元的。我感覺去世界各地收集材料、制造宇宙飛船的劇情制作2、3季都完全可以,沒想到下一季就是最終一季了。如果真是從頭制造宇宙飛船,那應當是有生之年程度的工程,牽扯到的技術、材料、學科、人材實在太多了,某國到現在都還沒登月。相對于制造宇宙飛船,我感覺先制造一個大型望遠鏡,弄清楚月球上現在到底有什么東西,是明顯更可行的。我還覺得如果登月,最可行的辦法是先找到JAXA或NASA,把這里的專業人士復活,很可能在幾年時間內能翻新3000多年前的火箭和宇宙飛船然后送千空等人上天。我感覺此作的劇情總是把復雜物件的制造高度簡單化,也就是諸如制造一個成品所需要的可能也就1/3的東西湊齊了(idea、最主要原料),在此作的世界里這東西就算造出來了,而所有技術細節、工程問題直接忽略掉。但即便如此,我還是無法預料制造宇宙飛船這事在此作里將會被簡化到什么程度,即便簡化到1/10的復雜度依然對目前的科學王國來說是挺遙不可及的難度。如果下一季千空他們真是靠土法制造的火箭登上月球,要是《宇宙兄弟》的男主看到的話估計要吐血了。
浪客劍心 -明治劍客浪漫譚-
總評:95.7
我第一次知道《浪客劍心》還是上小學的時候,那時看了小伙伴的《浪客劍心》的漫畫書,當時我就挺喜歡的,感覺劍心很帥氣,臉上的十字刀疤很有型。雖然我以前也看過《浪客劍心》的番外動畫,但沒有完整看過《浪客劍心》的老版本TV動畫。當前這部作品是《浪客劍心》劇情重頭開始做的動畫,真想不到這年頭居然會出新動畫,而且還不是什么外傳之類的。
此作總共24話,講述了曾經明治維新時的傳說中的劊子手緋村拔刀齋放下屠刀后自稱緋村劍心,作為浪人游蕩,巧遇了開劍術道場的神谷熏,將她從危機中解救后就住在了道場里。之后劍心不斷攤上各種事情,和一個又一個強敵交手。在此期間他展現出的強大和卓越的個人魅力,也使得彌彥、惠、左之助成為了他堅實的伙伴。
這個新版動畫觀感極佳,是我強烈推薦認真一看的極品!雖然一開始看會感覺其并沒什么驚艷之處,會覺得畫面和表現力一般,但越往后看越會覺得此作真是極其出色,人物塑造滿分,劇情相當引人入勝,絕對稱得上是神作!我真心感覺此作真是非常有重制的意義,要不然年輕一代大多可能都不知道曾經有一部如此不朽的經典。
此作的故事背景是日本明治維新時期,有非常濃郁的時代感,從中能體會到那個時代的文化、外交、商業、建筑等元素,很有種實感,設定上非常考究。這些元素使得此作有種底蘊感。
此作的畫面制作、演出效果雖然不像《鬼滅之刃》之類的在戰斗場面上能給人巨大的沖擊感,但也已經算得上優秀,完全配得上此作的劇情。但如果畫面表現力做得更驚艷,此作妥妥能上96分。
此作中有有各種出乎意料的戰斗方式、招式,非常有想象力,但又不顯得完全脫離實際(雖然還是超越了人類極限),這方面的構思相當優秀,又非常有看頭但又不覺得太玄幻。
緋村劍心的人設絕對是經典中的經典,是各個方面都格外出類拔萃的完美男主,我特別喜歡。他的面龐精致,甚至都有一點點女性的柔美感,那個十字形的傷疤不僅不損傷顏值,反倒還是很個性的點綴。他無比強大卻又無比溫柔,對身邊的人特別關愛和重視,性格還很風趣,非常隨和和友善,說話還總是很禮貌客氣,總用“在下”來自稱。緋村劍心真是很有兩面性,在極個別情況下他真正發怒的時候展露的氣勢逼人、令人不寒而栗的眼神和氣場,和平時笑呵呵的他形成了截然的反差,顯示出曾經最強殺手的本色。雖然如今溫柔的他已幾乎覆蓋掉了曾經冷血無情的他,但壓倒性的強大真是一點也沒變,在面臨危機,特別是他重視的人即將受到傷害的時候,那個傳說中的劊子手的設定會再次登場。
如今的劍心對劍道主張的是用劍拯救他人,而非常憎惡用劍殺人的行為,這正是最了不起的劍客應當持有的理念。這和作為劊子手時的他有截然的反差,不知道是因為什么令他產生了這么大的改變,在這一季的劇情中從未有過明確交代。臉上的十字傷疤是怎么來的在這一季也沒有交代。
劍心的CV齊藤壯馬表現得很可圈可點。剛開始看此作的時候還感覺他的聲音有點太柔了,覺得不是很配劍心的形象,但把此作看下去后,真心覺得齊藤壯馬配得很好,把劍心既強大又充滿柔情的設定展現得很理想。而且當劍心突然認真甚至憤怒起來時,那種語氣和氣勢的轉換也很棒,很好地表現出了他人格的雙重性。
神谷熏真是出色的女子,外表美麗,性格可愛,有時還顯得楚楚動人,而同時她的內在又非常堅強,同時又挺有脾氣,表現出鮮明的個性,他真是個很容易令人喜歡上的角色。她深深愛慕著劍心,不想失去他變得孤獨一人,還對劍心很體貼。此作里也把她和劍心之間的羈絆描寫得很好。劍心雖然是云游四方的浪人,但如今的熏已經算是他最重要且最想守護的人。熏和劍心非常般配,但可惜劍心始終對熏沒有產生過男女之情。
一開始我不怎么喜歡彌彥這角色一般,感覺咋咋呼呼的,不知天高地厚,發型也亂七八糟,是個煩人的小鬼,但看到后來覺得他的人設也相當不錯。他的正義感很強,敢說敢做,勇氣過人,從不退縮,看到惡行不會袖手旁觀,很有可圈可點的地方。
第14話的彌彥真是了不起,居然敢憑自己一個人對抗盜賊團伙,最后居然還把對方BOSS干掉了,令我對他相當刮目相看。特別是BOSS要砍他腳時他一腳踩在BOSS的刀上化解其招式這一刻特別帥,這反應可真夠快的。他小小年紀劍術竟然已經相當不俗了,前途無量,感覺再過一、兩年劍術都得超過熏了。
明神彌彥的CV小市真琴配得真不錯,把堅韌而且非常有骨氣的少年的感覺表現得很好。這CV之前一直都不怎么出名,能碰上這個角色展現出她的潛在的實力真是很難得。
相樂左之助也是個很不錯的角色。一開始的他是個黑道打手,十分自負、桀驁不馴,被劍心實力碾壓后,折服于劍心的強大,也認同了劍心的人格魅力,然后成了他靠得住的伙伴,很講義氣,并且還很懂劍心的想法和心理,成了劍心的很好的助力。雖然左之助表面上看起來是個粗人,但頭腦一點也不糊涂,對于社會、時局還認識得挺清楚的,難得。左之助用的斬馬刀的設定很有魄力,很適合用來秒殺一群小嘍啰,可惜被劍心廢掉后就再也沒用上。
高荷惠挺漂亮的,不是熏的那種清純可愛型,而是大和美女那種氣質。熏對她還挺提防的,就怕她對劍心產生了愛意。高荷惠也挺討我喜歡的,對劍心溫柔且表現得主動,而且頭腦清醒,內心正值,有很好的品行和品德。
率領御庭番眾的蒼紫刻畫得不錯,不甘心曾經幕末時經歷的失敗,而且不肯為了獲得自己的利益拋棄曾經跟隨他的弟兄們,是個很有信念和尊嚴的人。可惜如今他被雇傭他的黑心的拜金男觀柳所出賣,這么了不簡單人卻跟隨了那么個黑心商人,真是太看走眼了。
御庭番眾四個人剛洗白不多久,結果就都被觀柳的加特林機關槍給突突了,挺慘的。感覺他們有點笨,如果同時撲上去,而且蛇形跑位的話,觀柳肯定招架不住,不至于全都斃命。最后蒼紫不知道去哪了,不知道還會不會要挑戰劍心。按理說劍心對他那么寬容,應該很大程度放下了敵意和競爭心理才對。
第20話出場的艾爾達人設很優秀,一個西洋妹子用自己高超的醫術對日本人救死扶傷,還不要錢,醫德十分高尚。她為了隱藏自己是女性、使得患者不因她是女性而質疑她的醫術,于是平時戴著面具、用男性聲線,真是深藏功與名。摘下面具后的她的相貌真漂亮,像精致的洋娃娃一樣。艾爾達積德行善使得她在離開日本的時候引來了大量百姓自發為她送行,這一幕配合著溫馨的BGM我覺得還挺感人的,她真是個很了不起的女孩。
艾爾達的善行觸動了唯利是圖的黑心醫生的利益,居然那廝還找來殺手殺她。那個殺手艾斯皮拉爾一開始看起來是個惡人,但很快就洗白了,此作對這個小配角的刻畫很不錯,體現出了他雖然在劍術上挺走火入魔的,但本性挺好,并且有著武人的尊嚴,最后他在經歷了被劍心碾壓后重新審視了自己,和劍心化敵為友,這劇情令我看得挺舒服的。感覺艾斯皮拉爾可能有點喜歡上了艾爾達,居然主動要求陪伴她踏上美國的旅途,說不定之后這倆人會在一起。艾斯皮拉爾除了長得丑,其它方面都還基本配得上艾爾達。雖然故事里沒有明示,但我感覺艾爾達應該對劍心也產生了好感才對,畢竟劍心又溫柔又帥氣又保護了她,還知道了她秘而不宣的秘密。
齋藤一真惡心。曾經是新選組的最強,如今卻茍且偷生,放棄劍士的尊嚴。他雖然嘴上說嫉惡如仇,但其實也在做著惡行,并沒有立場說別人。我很不喜歡這個家伙。我納悶的是,齋藤一那么厲害,又是給zf效命的人,怎么大久保不讓齋藤一去京都對付志志雄?
大久保看起來是個很淡定穩重的人,沒想到被那么容易就被志志雄手下的小鬼刺殺了,死時一臉慘樣。我對這角色本來沒什么好感,但看到后來他說要在國力充盈后把日本變成nation state,也就是變成民選,有這樣的決心、遠見,這么開明,算是挺難得的領袖。
那個惡棍志志雄之前被潑上油燒了,居然沒死,現在開始搞起事來,真是令我匪夷所思,當時干嘛不把他的腦袋砍下來以絕后患?而且志志雄現在這么囂張,直接威脅zf,怎么不派軍隊收拾他?
ED2的人物水墨畫畫得真不錯,人物顯得英氣十足。這個ED2歌曲也不賴,叫《存在證明》,挺有氣勢,挺振奮的。最后一話的最后一幕,即將遠去京都對付志志雄的劍心抱著熏的那個畫面正好是這首歌的高潮部分,感覺真是教科書級別的超神插入,營造出的情景特別特別特別感人,看得我淚目(我都懷疑選這首歌曲作為ED2的原因之一是不是就是為了匹配這一幕)。當歌唱到“存在證明”這句歌詞的時候畫面上正好是劍心遠去的背影,感覺劍心真是在熏這里留下了他短暫的作為一介普通浪人的存在證明,令人唏噓不已。最后一個人痛哭的熏真是挺令人心碎的,她最重視、最愛的人就這么遠去了,而且未來是生是死都不可預料,太扎心了~
這一季完整講述了一個精彩的故事,期待此作出第二季,但估計劇情會很虐。
16bit的感動 ANOTHER LAYER
總評:95.4
這是一部講述女主秋里木葉和六田守等其他人一起制作美少女游戲為核心的動畫。此作絕對是超超超級黑馬,設定太新穎了,劇情發展太精彩了,充滿各種意想不到,高能不斷,跌宕起伏。女主木葉的人設太棒了、太可愛了!我吐血推薦一看,說此作是神作在我來看都不過分!最后一話更是極其有愛,把我感動得不得了(真的是應了標題里的“感動”兩個字),我都是流著淚看完的!此作真是絕了,看完此作后我都想鼓掌。
此作女主木葉是酷愛美少女游戲的宅女,對美少女游戲有著非常深刻的見解,也深愛著游戲里的美少女們。她的愿望是能讓自己創作的美少女角色出現在正經的游戲中,被大家認識和喜愛。然而現實是殘酷的,此作一開始的她只是一個小型游戲公司的插畫師,每天都在干著重復的給路人上色的這種無聊的工作。盡管其工作卑微,所在公司毫無大志只想做一些廉價游戲撈錢,但木葉始終心懷理想、不忘初心,從未失去成為游戲創作者的熱忱。突然有一天,她的命運發生了改變,在她打開傾聽她訴說對美少女游戲感想的老婆婆留給她的《同級生》PC98版的游戲盒子時,她突然穿越到了1992年,那正是美少女游戲逐漸邁入黃金時期的始端,這無疑是她實現理想最完美的沃土。此作主要內容講述木葉從現實(2023年)穿越到過去不同年代,和包括男主小守在內的alcohol soft公司的眾人一起做游戲的事,他們之間誕生了深厚情誼,共享了成功的喜悅,也一起經歷過困境,同時還改變了歷史。雖然故事有許多大膽和超現實的元素,但給我感覺一點也不顯得突兀,情節發展、人物心理的變化都顯得非常自然,這很難能可貴,劇本質量太高了。
此作的背景真是很有時代氣息,在展示現階段秋葉原以及木葉房間的時候,可以看到各種如今當紅動畫游戲角色,大紅大紫的Lycoris Recoil、小圓、FGO都有,真是很真實。此作對如今美少女游戲產業的現狀描寫得也很真實,玩家的注意力越來越多地被虛擬主播之類的奪走,大投資高收益的鴻篇巨制如今也難以像曾經那樣誕生。在這樣的年代,有木葉這樣堅定不移摯愛美少女游戲而且還那么有理想的玩家,真是太難得了,這樣的玩家本身就是galgame產業的寶藏!第1話看著她在老婆婆開的游戲店里興奮地抒發對美少女游戲的見解、理念并且談及現狀,都把我看得感動了,令我深感這妹子真棒!
此作里看到了《同級生》的游戲,里面還有3.5寸軟盤,真是勾起了我的回憶。想當年,第一次得知《同級生》的時候我才上小學,還不很懂戀愛,在商場里免費領到一根同級生的宣傳筆時我還挺高興。在場景進入1992年后,還看到了5寸大軟,那都是我在幼兒園的時候家里的中華學習機上用的。感覺此作的作者是很有閱歷的人,對30年前的事物了解頗多,也見證了galgame領域的發展歷史。此作還有很多80年代及更早出生的人才有的共鳴,也常激起我對過去軟硬件的懷念。木葉在現代世界里熟練掌握的繪圖技法在穿越到的90年代初那會兒全都用不上,卻還得用那個時代的抖色之類古法辛苦地繪圖、甚至有時還得一個像素一個像素地著色,我深感要是那個時候有photoshop、painter就好了。
木葉的外形給我的第一印象并不是那種特別可愛和閃耀的感覺,但剛看此作不久,她就開始特別吸引我,那簡單、天真、開朗的性格,以及她對美少女游戲的赤誠和心動,在我眼里有非常獨特的魅力,這個人設真是絕了,而且是越看越覺得可愛,令我喜歡極了。值得一提的是,其它作品里許多女角色的虎牙顯得是故意裝上去的,有點假和俗套,而木葉的虎牙我感覺是真的非常自然,那就是她天生的虎牙,和她的角色感覺太搭了。這個小虎牙充分增添了她的萌感。
特別要大夸特夸的是古賀葵給木葉配音實在是超贊!聲線有一種很積極明亮又有輕微稚氣的感覺,對木葉性格的表現超級到位,我完全無法想象還怎么能配得更完美,所以忍不住為她在此作的表現授予了神級女CV賞。古賀葵對木葉的演繹絕對值得納入CV屆的教科書。
木葉在此作里說過“我看到可愛的女孩子就會精神振奮”、“游戲中的女孩子是最為耀眼的”,看到這種場面我會很高興,我還曾想如果她是百合妹子就好了。不知道她如果穿越到了游戲世界里,會不會真的有和她們交往的想法。
小守的人設也特別出色。木葉第一次的時空穿越是到了90年代初,那時候的小守還只是個少年,很看不起galgame,覺得那都不是游戲,是做不出其它游戲的人才做的只能在短時間內博人眼球的作品,他萬萬想不到之后正要迎來galgame的黃金年代。小守對可愛的女孩子們似乎沒任何感覺似的,腦子里只有編程,難以置信。面對木葉真誠的制作galgame的邀請,他雖然表面上還在抗拒,但態度還是軟化了下來,表現得很傲嬌。到了第4話,他仔細看了木葉帶的平板電腦,才終于信了她是未來世界穿越而來的,對她的態度有所改觀,之后還親眼見證了她突然消失的一幕。第6話,試圖為負債10億的alcohol soft力挽狂瀾的木葉終于第一次得到了小守的完全認同,小守表示讓她放手去做,會盡自己全部所能幫她實現,這是她和公司轉折點,看到這一刻,木葉興奮的神采令我著實很高興,二人的關系也進入了新的stage。
此作里木葉和小守有很多近距離接觸,但一直沒有戀愛描寫,小守也一直表現得很傲嬌,不禁令人懷疑此作是否真的會有BG。第11話終于令我感到他倆有了發展戀情的苗頭。木葉從1999年回到2023年時,小守已經又帥又十分富有,很有成功、穩重男人魅力。此時的他都已經看上去像木葉的父親的樣子,居然仍然都還沒結婚,令我都懷疑他會不會是在一直等著木葉的重新出現(亦或是他根本就沒有戀愛的概念?)。這一話里有一幕,木葉對臨走的小守說“路上小心”,小守微微一驚。這一幕的設計真是很棒,這一句簡單的話令我感覺真甜!出門前有自己重視的人對自己說這么一句溫柔的話語,不可能令孤獨的小守的內心完全沒有觸動。這是木葉第一次沖著小守臉紅,也是他倆從友情轉變到愛情的開端。
第9話腦洞真夠大的,居然在木葉參與制作了爆紅的《最后的華爾茲》回到原先世界后,發現二次元的審美已經走上了邪路,全都是歐美風格的粗獷粗野的形象,居然還出現了看著像邪神般的Saber(不知道川澄綾子配音的時候作何感想!)。這哪叫美少女?要是二次元都變成了這種風格,我就不可能在二次元的坑里了!木葉發現自己的穿越導致毀掉了整個日本galgame產業,感到特別自責。
為了修正歷史,木葉和小守以及從美國趕來alcohol soft的大家制作了另一款可以與之抗恒的游戲,準備回到1999年投放。在最后一話,木葉即將穿越回1999年時,她一瞬間沖著小守羞澀地臉紅,目光蕩漾,這一刻我再次覺得她真是可愛得不得了!好在這不是他倆的最后一面、不是緣分的盡頭。在木葉重新回到2023年、發現秋葉原已回歸正軌后,卻看到alcohol soft已人去樓空,內心正在失落之時,竟看到了在那里一直等候她回來的小守的背影。驚喜的木葉立刻尖叫著撲上去,趴在小守懷里感動得痛哭,這是整部作品最高潮的一幕,太催淚了。小守溫柔地笑著對木葉說“愿意和我一起做游戲嗎?”,令我極為吃驚,這完全就是求婚啊!這也太甜了!!!然后木葉吃驚地臉紅了,并立刻回答“嗯!”,同時此作令人印象深刻的OP的前奏響起,這一幕我真的是看一遍哭一遍,哭了又哭,看后心里久久不能平靜,這絕對是能在歷史上留名的名場面!(我甚至都納悶,怎么這一幕對我的催淚效果那么強的?一定是木葉太令我喜歡,而且此作劇情對他倆關系的鋪墊做得太好了,所以他們對彼此的感情在最后引爆的時候殺傷力才那么強)我也真是感嘆,最初是木葉軟磨硬泡求著小守做游戲,而到最后,竟然是小守主動邀請木葉一起做游戲,轉變太大了。雖然此作13話算不上長,卻令我感覺他倆共同經歷了太多,這倆人也是超般配,是命中注定要在一起的!
小守雖然是個年輕人,居然極度保守和守舊,起初只做PC98平臺的游戲,不肯接受開發windows程序,執著程度驚人。而且他對PC98有著極度的狂熱,把這機器當情人似的,也因此不肯換平臺。總算到后來意識到PC98注定要沒落,要消失在未來人們的視野中,于是他在cosplay成為PC98樣子參加CM50后,開始轉向windows了。居然在10話,看到小守收藏了一倉庫的PC98,畫面感真是夠壯觀的。他居然還把PC98啟動聲音錄下來當做舒緩壓力的音樂來用,不可思議。他還把這些PC98組成了集群一起運行,想法和行動力真是不得了。真不明白他為啥非要用這一堆PC98的運算能力去尋找木葉的下落,難道是為了儀式感?然后他還讓PC98連接到了壞人的實驗室控制機器人幫助自己和木葉逃跑,有種PC98拯救世界的感覺。我感覺galgame對木葉的重要程度,就是PC98對小守的重要程度,小守似乎都把PC98當成戀人了。
小守真是技術大拿,編程和動手能力極強。第7話居然還攢了個古典集群出來,解決了99年時候機能不足、photoshop無法創建多達200個圖層的制約。2023年的他居然還弄了AI模型出來,真是相當與時俱進。甚至他還有黑進安防系統的本領,太厲害了。
第3話很有趣的一段劇情是,自稱工口游戲博士的木葉在96年遇到不好意思一個人進店買galgame的清純可愛的眼鏡妹冬夜,帶著她進店買到了她心儀的游戲,令木葉覺得遇到了福利事件,和她分別后還在心里一直夸她可愛。這段劇情都有點橘氣。像這樣對工口游戲有興趣又很害羞的楚楚動人的妹子誰能不喜歡呢?第4話,冬夜直言說“我喜歡可愛的女孩子”,居然她后來還主動管木葉叫姐姐大人,令木葉非常開心,她深深覺得木葉是非常難得的galgame的知己。當時我還想要是她倆能有百合發展就好了。
第6話,居然冬夜以cosplay形象出場,從土宅妹變成了那種宅男心儀的清純美女形象,也不戴眼鏡了,性格上也顛覆了之前內向害羞的設定,真是有了180度轉變。可惜她在作為插畫家獲得成功后變得不再那么單純了,知道怎么利用男性的心理。
想不到在第9話,冬夜想要創立游戲制作公司,意外地相當有想法。她極力拉攏木葉和她一起做游戲的那一幕有點像求交往似的。可惜,第12話,看到成年時的冬夜,她已經不是因為熱忱而制作游戲,做游戲反倒成了她的負擔,是為了養活員工和他人的期待而制作游戲,真是可悲。令我感嘆很多行業也是,很多人都因為熱情和興趣而進入那個行業,但久而久之心態和目的就都完全變化了。她雖然依然特別重視和尊敬木葉,但想不到她會做出背叛木葉的決定。好在是冬夜最終重新找回了自我,幫助木葉脫險,木葉再次敞開胸懷接納她,還邀請她一起制作游戲。木葉始終沒有忘記冬夜想和她一起做游戲的約定,可見木葉對她的重視,這也令冬夜很感動。有點可惜的是直到最后也沒看到她倆共同制作游戲的情景,要是在重新回到原本的2023年后冬夜能有和木葉共事的機會就好了。
alcohol soft的那個瞇瞇眼角色香織的身材真好,特別是第5話到了99年的時候,她穿露腰裝,身材真是火辣,令我眼前一亮。好像香織和另一個負責原畫的芽衣子都不喜歡工口畫面,只喜歡可愛的美少女畫面,06話里香織負責游戲向主機平臺移植的時候她居然試圖把禁忌部位都和諧掉。第7話,看到木葉的畫之后,香織居然開眼了!瞇瞇眼設定果然就是留待有朝一日開眼。開眼后真漂亮!
alcohol soft里那個戴面罩的男子挺神秘的,此作對他沒多少描寫。似乎游戲里的床戲劇情都是他負責寫的,他對于游戲移植過程中刪除工口畫面這事挺不滿。他是alcohol soft的元老之一,對這公司蠻忠誠的,看到他在最后也熱心和木葉等人一起參與木葉主導的作品,感覺這個人還挺不錯。
小守他爸真是人渣,就想著賺錢,對于美少女作品沒有真愛。而相反,alcohol soft里除了對galgame一直有偏見的小守外,都是真心喜歡著galgame,將制作galgame作為傾心投入的事業。結果小守爸愈發糊涂和張揚,誤入歧途,第6話被騙導致公司負債10億,令之前員工們打拼的積累付之東流,真是可惡。對著犯下滔天罪行的小守爸時,第一次看到了香織發怒的樣子,陰著臉瞇著眼再加上巨大的歐派,展現出巨大的威壓感。
我挺喜歡alcohol soft后來招的似乎是負責財務、運營的那個黑短發黑瞳孔的女角色橋本的,顯得帥氣穩重,這種形象估計尤為討女觀眾喜歡。她之前因為所在的公司破產才偶然到了alcohol soft,但逐漸地認識到了這家公司是多么有愛。我挺好奇她對galgame是什么看法,怎么看待可愛的女孩子。我覺得無論男女都不可能不喜歡可愛的美少女才對,美少女是無上的瑰寶。
alcohol soft的大家很團結一心這一點我挺喜歡。最后一話為了制作木葉的游戲,小守一個電話就把alcohol soft的大家都叫回來了。雖然時隔20多年,大家重新見到木葉這個老朋友都挺開心,芽衣子還夸木葉是天使,看到她們如今依然能其樂融融地在一起感覺真好。
第7話小守穿越到了85年見到的echo 1和echo 2是什么來頭挺迷的,似乎是外星人?此作始終沒有交代,我感覺穿越時空這事一定和他們有所關聯。他們對于創造力的觀點我是很認可的。人類(非那些NPC般的人類)最了不起之處在于有創造力。人所在的時間、空間,放眼宇宙以及時間長河來看都是微不足道的一個點,但是人的創造力則是沒有邊界的。創造力真是人類擁有的無比寶貴的特質。
此作故事的時間跨度很大,差不多30年,從16bit的年代進展到了最后AI制作游戲的時代,業界的變化真是相當大,令人感慨。小守說AI制作游戲可以廉價地創作出高質量的作品,但是沒有熱量。這令我感覺就如同通過機器雕刻可以快速地產生精準完美的物件,但卻少了手工匠人的靈魂,因此機器制作得再怎么精妙,其價值的認可度也遠不及人類大師的作品。此作對未來世界游戲制作的方式的構想蠻科幻的,把許多活人關在玻璃缸里,大腦神經連著機器,用人類大腦提供AI制作游戲所需要的創作力。這情景挺可悲的,人類被物化、腦力被機械性地采集,真的跟human礦一樣。這也稍微有點像CPU跟GPU的關系,AI是GPU,負責高通量處理,人腦就像CPU,有處理復雜邏輯的能力,原本GPU作為CPU的輔助,而到了未來,反倒很大程度是GPU作為計算主力而CPU成為輔助了。
OP的開場旋律很有16bit的風味,很不錯,很配此作,特別是前奏令我印象太深刻了。OP里我特別喜歡的一個畫面是剛開始時木葉在漆黑的屋子里睜大了眼睛緊盯著屏幕,就像看著閃閃發亮的珍稀財寶一樣,那種夜深人靜時一個人暗自入迷的感覺真的是超棒!那是一種特別美好的興奮體驗(我當年也有過)。而且這個畫面里的木葉的樣子令我很著迷,可愛的少女沉迷于一個技術性的非主流事物,那副樣子在我眼中實在是特別有魅力。OP的歌曲演唱稍微有點搞笑意味,這種唱法令我馬上想起中川翔子,結果一看還真是她!其實有很多人比她唱這首歌能唱得更好聽,但我倒覺得這種非常樸實的、不帶修飾甚至粗糙的歌唱反倒somewhat恰好映襯此作獨特的題材,所以說不定是官方有意這么安排。
ED的福利真好,真大。有像素畫風格效果的加成感覺更有味道了,居然還有把3.5存軟盤塞到歐派中間夾著的畫面,過于工口,誘惑力太強了。這種像素風格可真好。
要是官方出一個alcohol soft制作的游戲試玩版就好了,我對那些游戲還蠻好奇的。
此作里的跟二次元有關的梗很多,很有樂子,尤其是第7話木葉的手機里居然播放了《小圓》的OP,突如起來的熟悉的歌曲傳出來令我大驚,而歌詞完全應景,可謂是神插入。
間諜過家家 第二季
總評:94.8
這一季沒什么可特別評價的,就是上一季內容的延續,還是由各種相互獨立的任務組成。整體制作質量依舊非常出色,劇情質量依然很高。期待下一季。
第30話約爾的西裝造型挺不錯,有點小帥。戴太陽鏡和遮陽帽的約爾顯得真是漂亮又可愛,令我眼前一亮。
第33、34話約爾在船上的戰斗做得真是很出彩。沒想到此作的世界里也有能令約爾陷入苦戰的對手,我還以為只要約爾認真起來所有敵人都是可以被輕易秒殺的。約爾被那個刀客在胸口砍了一刀令我心疼,但愿不會留下傷疤。
約爾的弟弟依然痛并快樂著,真是心理畸形的抖M。
這一季的OP畫面做得極差,很丑陋,尤其是三個人走在一起時由下朝上視角的畸變過頭的畫面相當難看,令我絕對不想再看第二遍。
超超超超喜歡你的100個女朋友
總評:94.8
此作男主愛城戀太郎已經失戀過100次了,非常郁悶。有一天神明告訴他他有100個真命之人,結果剛上高中頭一天他的人生就出現了逆轉,在走廊撞上了兩個漂亮妹子羽里香和唐音。她們迅速向戀太郎狂熱倒貼、瘋狂發動攻勢,戀太郎從戀愛的最悲催狀態一下子變成了人生贏家。這倆妹子的同時告白令戀太郎束手無策,他得知如果只選擇一個交往的話另一個就得死,于是像《女友成堆》的男主直也一樣經過思想斗爭后決定腳踏兩條船。再然后,通過各種各樣的邂逅,戀太郎的后宮不斷增員,好本靜、榮逢凪乃、藥膳楠莉依次加入,在故事最后連羽里香她媽都成了戀太郎的戀人。最終達成了6后宮的完美happy end。此作和《女友成堆》略有一定共性,都是男主光明正大地同時和多個妹子交往的重度搞笑番。
如果把此作簡單看成庸俗老套后宮番于是根本看都不看,那絕對是重大損失!此作真是部質量極高的超級有趣的戀愛喜劇番,我強烈推薦。此作的作畫和演出極佳,劇情極富創意,故事中驚喜不斷,超超超超超歡樂!此作還里有大量水準極高的賣萌賣肉賣可愛的元素。此作也絕對不膚淺,在戀愛感情描寫方面也非常出色,很多劇情特別有愛,甚至令人感動。戀太郎的六個后宮每個角色都塑造得非常好。總的來說此作真是不可多得的佳作。
戀太郎乍一看在外型上很普通,沒啥亮點,但把此作認真看下去后,我真心覺得他是很優秀的少年。戀太郎的性格和心靈真是很不錯,非常溫柔,特別有責任心,對每個和他有緣分的少女都最大程度付出真心,盡最大努力帶給她們幸福,有很強的人格魅力。在11話他救羽香里的時候甚至豁出自己性命。雖然他的女朋友是神明給他的安排,但那我想即便沒有神明的助攻,只要他們能有偶然的邂逅,憑借戀太郎出色的人格,他們最終也同樣能成為深愛彼此的戀人。我想也是因為戀太郎的極致溫柔,他的后宮妹子們才能緊密圍繞在戀太郎的身邊并相處得和諧融洽,彼此像朋友一樣相互關照,并且其中一個妹子遇到困難時其他后宮妹子也會和戀太郎一樣全力幫助,這樣的相處方式真是相當理想。
一開始出現在戀太郎身邊的羽里香和唐音的性格和身材兩極分化嚴重。羽里香體態十分豐腴,表現得對戀太郎相當熱忱,是個溫度特別高的妹子。她總是說戀太郎的各種好話,各種包容和褒獎他,居然戀太郎新招來了其她妹子入后宮時她還會感激戀太郎沒有忘掉她。而唐音則是與羽里香相反,外形有點不良少女的她的性格剛烈而且還暴力(挺有點像《女友成堆》的男主后宮之一母猩猩咲咲),特別傲嬌,脾氣很倔。雖然她心里也對戀太郎喜歡得不得了,但總是試圖隱瞞這種感情,跟她戀愛有些辛苦。
羽里香雖然表面看起來很溫和純真,但卻有個心機婊的設定,她自己心里都說自己的可愛是極盡努力恰到好處地表現出來的。羽里香的這個性格搞得唐音老和她斗氣和爭風吃醋。不過二人的針鋒相對的關系隨著故事的進行逐漸變得緩和多了,還潛移默化地誕生了友情。最后羽里香被母親囚禁、不許她再見到戀太郎時,唐音都不肯袖手旁觀自己的情敵就這樣離開戀太郎了,這也表現出唐音內在挺美好的一面。此作里生化危機那一話以及最后一話,居然這倆人還彼此纏繞著嘴對嘴緊密吸到一起熱吻,是令我很喜聞樂見的畫面。這倆人什么時候也正式發展出百合關系就有意思了。
唐音的歐派和羽香里真是兩個相距無窮遠的極端,感覺就像是呈倒數關系似的。第11話曝光唐音的歐派只有B偏A,真是殘念。羽里香一個得頂她至少十個。
loli體型的圖書館管理員好本靜雖然不是我一貫中意的類型,我也很難將這樣的loli當戀愛對象看待,但越往后越覺得這個角色非常出色,越來越令我喜歡,后宮里能有這樣一個妹子很好。此作把她的性格和神情展現得相當可愛,面對她閃亮亮的大眼睛沒人能抗拒得了,她還經常有非常萌的表情出現,非常討人喜歡。她原本是個超級無口屬性,不是像《租借女友》里的小墨那樣極個別時候才會說話,而是完全不說話,要表達的話語直接在手里的厚書中找對應的詞句并指出,她甚至連笑都不帶出聲的,這個設定真是新奇。第3話里,戀太郎和她的關系發展的構思和描寫真心非常優秀,令我很贊嘆。她由于不會像一般人那樣說話,被同學嘲笑、被母親呵斥,也沒有朋友,唯有戀太郎對她展現出了極致的溫柔,充分體諒她的困難,甚至還為了讓她和別人說話能夠更方便,把她天天抱著的整部書都錄入了可以朗讀文字的app里,真是GJ!想不到好本靜用朗讀app對戀太郎說的第一句話就是“我喜歡你”,這一幕太甜了!好本靜之前因為看到戀太郎和另外兩個妹子甜蜜地膩在一起而一度非常難過和受傷,明明才剛剛萌生戀情卻馬上失戀,她覺得自己注定無法和她傾慕的戀太郎相愛,是悲劇中的女主。她萬萬想不到后來戀太郎竟然會把她也納入到其后宮里,這徹底打開了她的心扉、解凍了她的心靈。我真心感覺戀太郎和好本靜之間的緣分是命中注定的,她能遇見戀太郎真是太幸運、太幸福了,誰都不可能取代戀太郎在她心里的位置。
好本靜真是無口到離譜,居然第4話戀太郎撓她癢癢,她發笑的聲音都是手機打字發出來的,真是可愛。
第5話出場的榮逢凪乃相當優秀,是個極有個性的冷美人,是此作里我最喜歡的角色。她的顏值是此作里最高的,再加上本作作畫本來就非常好,令她的面龐經常顯得精致美麗到驚人,第5話這一話我就給她截了40多張圖。榮逢的身材也和顏值一樣是無與倫比的完美。她的性格非常獨特,情感顯得非常淡漠,思維極度理性,對做的任何事情都追求價值,總是以最有效率的方式思考和解決問題,不肯做低效率、無意義的事,她也完全無法理解普通人的很多想法、做法和感受,同學甚至懷疑她的真身是AI(她的姓的發音聽起來很像AI,這個設定有趣)。戀太郎對她的攻略過程構思得很不錯,令她認識到體會當下的快樂和幸福并不是一件無意義的事。這點確實不假,這些幸福快樂的記憶本身也是很有價值的,甚至是無價的。最后戀太郎終于成功打開了她的心扉,令她這個“AI”主動說出了喜歡。并且她還按照其一貫的效率最大化的作風,立馬就吻了戀太郎,并順利加入后宮。戀太郎收了她可真是幸福,光是每天能看到閉月羞花的她那都是至上的眼福。這一話榮逢還用柔軟濕滑的舌頭吮吸了戀太郎的手指,畫面真是瑟琴,令他享受到了絕倫的觸感,令人羨慕。瀨戶麻沙美給榮逢配音配得很可圈可點,把那種高冷且缺乏豐富感情的她演繹得很到位。
值得一提的是榮逢對學校讓學生做實驗的態度。她認為這是浪費時間,對此我深表贊同!我一直以來就這么認為!讓學生現場做實驗有何意義?明明結果都是明確已知的,直接看實驗視頻、教學片不就完了?學習效率能提高100倍。讓學生體會那么短暫且膚淺的實驗過程根本一點意義都沒有,完全就是為了走形式。對于真需要這部分知識的學生,以后需要的時候再針對性地學就完了。也別說什么做這樣短暫時間的實驗就能提升科學素養因而是有益的話,同樣的時間若讓學生看緊湊、內容豐富的教學片(youtube上大把),學到的知識、對眼界的開闊程度,遠高于浪費時間做這種微不足道的實驗!
第6話泳著回,榮逢的泳著裝顯得歐派緊實且很有美感,魅惑力無限。而羽香里的泳著裝顯得r量頗為下流,尺寸太離譜了,呼之欲出,感覺就像胸前支了個小桌子,放N個手機都滑不下來。我感覺榮逢的尺寸是最符合理性的完美(或者說,歐派size最大化時考慮了理性的約束條件),而羽香里的我感覺過于夸張和下作了。這話居然榮逢直接讓戀太郎抓她最柔軟的地方,過于直球,真有她的個性,羽香里也立刻照搬。戀太郎一手一個極致的柔軟,幸福到鼻血染紅泳池,這樣的死法真是死而無憾了。羽香里這一話泳著跑步的r搖真是觀賞性極強。相比之下唐音就太慘了,r量遠小于路人的平均值(后來知道是B-),這令她非常自卑,都不愿意以泳著出場。當戀太郎以一番溫柔且誠懇的話語解開她的心結后,她露出的羞紅的樣子真可愛,和平時剛烈強勢的她展現出了極大反差,之后跟戀太郎接吻吻得那叫個酣暢淋漓,太過激了。
第7話出場的楠莉人設也是充滿亮點。她極度癡迷于研究藥物,能夠隨心所欲制造各種藥物的能力真是有趣,給此作帶來了大量樂子,特別是變得性感的藥水用在羽香里的身上時真是給觀眾帶來了巨大福利(可惜的是她發明的enlarge歐派的藥水并不能挽救唐音的貧瘠)。她還說能做男女性別互換的藥水,可惜沒在此作里實際展示一下效果。她平時是像好本靜一樣的幼兒體,但實際上真身是個非常豐饒、胸懷不輸于羽香里的性感學姐,第7話那個原本姿態剛在畫面上出現時令我瞬間眼前一亮,這種性感理科生的人設很對我胃口(令我想起《心跳回憶》里的紐緒結奈,是我的二次元初戀)。楠莉居然平時天天穿著尿褲,這習慣挺奇葩。第8話還有點小感人,雖然楠莉制造的藥水導致了生化危機情景的出現,惹了大麻煩,但戀太郎對她表現得非常溫柔和包容,并且還說特別喜歡忠于自己熱愛事物的她,這使得由于以前制造藥水總闖禍、被同學嫌棄的楠莉感受到了她特別需要的來自他人的認可,由此更是對戀太郎喜歡得不得了。楠莉平時個子小小的,和好本靜是尺寸相同的幼兒體,活潑可愛的感覺非常充盈,她說話還總帶著“諾達”的口癖更給其增添了萌點,朝井彩加給她配音配得很不錯。
最后一個加入后宮的是羽香里她媽羽羽里,現年29歲。她是個超級富婆,size居然有I,比羽香里還大,不愧是母女。她居然13歲時生下了羽香里,真是離譜!沒想到一開始特別敵視戀太郎的她,竟然也是戀太郎命中注定的人,第一次正式對視時她瞬間感受到觸電并向戀太郎求交往,真是令人意想不到的展開,戀太郎的后宮也立馬多了個超級白富美。母女同時在后宮里,這在我看過的眾多后宮番里真是前所未見的。她居然還把第11話ED買下來成了個人秀,在里面熱吻戀太郎,羽香里還拿個牌子擋住。居然親媽成了自己的情敵,不知道羽香里作何感想。
戀太郎是非常認真地和羽羽里成為情侶,而完全不是隨便迎合一下、玩玩的心態。他對羽羽里的跨輩分的愛情毫無顧忌,并且真心愿意成為她心靈的慰藉,完全像對待其它后宮妹子一樣付出真心愛她、決心給予她一生的幸福,這也彌補了羽羽里13歲時就失去丈夫后一直留下的心靈的空虛。這略帶禁忌的CP很不錯,戀太郎又一次表現得值得佩服。戀愛郎的真誠和執著甚至把羽羽里那個當年初中生丈夫的一直沒能升天的靈魂都感動了,我甚至都心想會不會戀太郎的個人魅力最后把羽羽里的丈夫都給吸引了,再給此作添個bl元素。羽羽里的前夫在成佛時居然特別驚慌和不情愿,我還是首次見到如此不想升天的角色。
看到最后,戀太郎等一行人從羽羽里那里離開了,回到學校,我就心想會不會羽羽里把學校買下來(此刻我想到當年aqrous的學校廢校后,鞠莉仗著他爸的鈔能力,買下了并入的那個學校并成了理事長),結果還真把學校買下來了并成了理事長,這樣天天都能和戀太郎在一起。妻子成了學校理事長,我感覺戀太郎就像成了學校皇帝似的。
目前看到此作漫畫還是連載狀態,令我納悶后宮現在都這么多了,接下來還能怎么編。如果真有下一季,估計后宮得增員到10人左右了。
女友成堆 第二季
總評:94.8
此作的第一季我相當喜歡,太有笑點了,腦回路相當清奇,想象力非常豐富,是極有樂趣的絕對不可錯過的戀愛喜劇番。這一季也完全沒有令我失望,情節過于歡樂,高能和槽點滿滿,同時對于人物性格、魅力以及愛情的表現都非常優秀。
這一季里直也繼續天天一驚一乍跟得了甲亢似的,榎木淳彌給他配得真好。
星崎理香(米莉卡)是此作里我最喜歡的,上一季我就特別喜歡她,這一季更是如此。她不僅漂亮極了,性格還超超超可愛,特別是看她為了能賴在直也家而在門口撒潑耍賴的樣子我真心覺得可愛爆了。看這一季時我還反復感嘆彩喵配她太合適了,性格被表現得超級鮮活,稱得上絕配。
米莉卡對直也真是超執著。面對直也的言辭拒絕,很有金的她居然把隔壁的房子買了下來,真是行動力超強。第6話米莉卡表現出的對直也的愛、為此付出的努力以及展現出的覺悟真是令人欽佩。她明知道追求直也很可能最終毫無結果,并且使自己受傷,但依然不肯放棄直也,而是全力以赴去攻略他,即便最后失敗也要盡可能多地留下和他在一起的回憶。看到這里我更加贊嘆米莉卡真是很有想法的非常了不起的妹子,真叫我喜歡。
米莉卡的真心終于令她獲得了和直也相處5個月并住在一起的機會,這真是良機。以米莉卡的魅力,直也再怎么忠心于咲和渚,對米莉卡完全不產生好感、不被她吸引都是不可能的。畢竟之前渚能打動直也令其加入后宮,沒理由更執著、更有魅力的米莉卡做不到。
第7話,米莉卡為了誘惑直也,把布偶裝一把扯開,歐派彈出來的畫面真叫人哦呼!那真是無上的超勁爆誘惑,直也居然把持得住,難以置信!米莉卡的發言尺度真的是超大,極度open,毫不掩飾地將自己的歐派作為大規模殺傷性武器來使用,這就對了!米莉卡開出的條件非常簡單直白:“那只要我問你你是喜歡還是討厭,你說了我就給你摸啦!”,可直也就是不爭氣,唉!米莉卡這種直率的性格我就是喜歡。
第10話,米莉卡成功把直也綁在了床上,可以對他為所欲為了。可惜米莉卡表現得意外地有點放不開,只是親了直也的臉頰和胸口,這居然就令她已經臉紅害羞不已了,明明她有機會干更過激的事,比如把直也的胖次拽下來。能被米莉卡這么對待,直也可真幸福。咲咲趕來捉奸,看到直也的褲子還穿著,突然放心了,她對直也的貞操真是很重視。
這一季米莉卡的妹妹星崎理沙新出場了,顏值和她姐姐一樣高,但性格比較安穩,完全沒有米莉卡那么聒噪,反倒比姐姐顯得成熟得多。她雖然完全不性感,但可愛度極高,賣起萌來令直也都無法抗拒。理沙也是youtuber,不賣肉而光靠可愛居然訂閱量都上萬了,是個不輸于姐姐的人才,前途無量。米莉卡使喚妹妹攻略直也從而令自己能上位的想法挺絕的,但果然還是失敗了,米莉卡光靠鬼點子試圖攻略正直到極點的直也還是行不通。理沙真是三觀特別正,腦子是所有人里最正常的,吐槽精準犀利,她在第5話對姐姐喜歡腳踏兩條船的行為表現出憤怒和難以理喻,還同時把接受腳踏兩條船的咲咲給罵了。
咲咲在這一季里繼續表現得很像母猩猩。她真是挺不爭氣的,第7話其他妹子都在和直也共寢時向他以各種方式獻出了自己,唯獨咲咲去橋下撿了貧r工口書給直也供他“使用”,讓里面的模特代替自己令直也泄火。其實最令直也表現得主動的是咲咲,明明自己有著第一后宮的得天獨厚的優勢,可惜她又過于害羞又過于暴力,還老用暴力掩飾自己的害羞,導致她和直也老也沒有實質性的愛情上的進展,連一次像樣的接吻都沒有。雖然最后在海灘上確實吻了一次,但那也是直也意識恍惚的狀態。
這一季對紫乃的描寫真的太出色了。看完這一季,紫乃成了此作中我第二喜歡的少女。
看上一季的時候我總覺得紫乃的發型不好看,對她也沒多少興趣。而看了這一季,我對紫乃的好感逐漸增加,真心感覺她真是內在很可愛的少女,很有魅力,她對直也真的是純純的真愛,太有少女心了。此作雖然是很搞笑的戀愛喜劇番,但對于紫乃對直也的感情的表現實在是超級棒,充分展現了她喜歡直也卻又不能表達的非常矛盾復雜的心情,也不禁令人逐漸會喜歡上她。
紫乃起初顯得非常反感直也腳踏兩條船,并且表面上顯得討厭直也。這一季她為了令直也放棄腳踏兩條船和監視他們,和直也、咲咲、渚住在了一起。這成了令紫乃近距離接觸直也的契機,也是增加她對渚了解的契機,無疑會逐漸接受了他們的相處方式,并且最后自己也身陷其中,成了后宮的一員,果不其然確實劇情是這種展開。本來紫乃是想和直也一直嚴格保持距離的,但是卻總由于機緣巧合被直也摸到敏感部位或被看光光之類,直也真是幸運色wolf。紫乃總是莫名地被直也牽著鼻子走,想保持距離都無法如愿,很樂。
看了第2話,才明白為啥紫乃也喜歡直也。直也總是毫不掩飾地誠實地夸贊對方的優點,而且很能發現對方的優點。他非常認真直率地夸獎紫乃的魅力,令紫乃對他好感度大增。紫乃為什么不允許直也腳踏兩條船的原因在這一話我才終于明白,并不光是因為他這樣是對自己重要的好友咲的不執著、不一心一意因而感到生氣,還在于紫乃其實也喜歡上了直也,因而如果直也確實一心一意愛著咲、沒有二心,紫乃就可以對直也徹底死心,從而不必再因為喜歡他又不能在一起的感情所折磨。而紫乃如今看到直也并非僅限于和咲交往,而在認真地腳踏兩條船,這導致紫乃意識到自己也有和他交往的可能性,這令她感到糾結。紫乃想避免自己誕生這種煩惱,想克制自己對直也產生戀愛感情,因此更加無法接受他對咲有二心。此作的這個人物關系真是構思得非常特別,甚是有趣。
紫乃總對咲咲怒其不爭,怎么慫恿她、怎么助攻她和直也的關系,就是沒效果,始終連接吻都做不到,結果好處凈被渚占去了,又加上現在又有米莉卡這個強大外敵,更加深了紫乃的危機意識。紫乃也真是難,總是自己干著急,心里各種想法只能一個人煩悶。逐漸地,她覺得自己其實比咲咲還更喜歡直也,而且和直也相處時間越長,越按捺不住這種感情,于是她也開始旁敲側擊接近直也了。
想不到直也和紫乃從初中就認識,頭上的緞帶正是直也曾經送她的。原來紫乃是為了不離開直也因而才選擇上和直也相同的高中,居然能暗戀到這份兒上,真不容易。令我深感這么執著的愛意如果最終不能表達出來而永遠被埋沒在心里,那實在是太可惜了。
第10話無人島的劇情中把紫乃的心意表現得真好。居然最后紫乃撲倒直也,嘴對嘴吻了他,這進展得太突然了,明明之前她的進度是排在最后面的,這一下子反倒把直也的正宮咲咲遠遠甩在了后面。
直也X紫乃這一對真是令人著急,就像咲咲總是親不上直也似的,紫乃老也沒法向直也表達真實的感情。第11話紫乃都果著壓在直也身上了,再多兩秒鐘告白估計就成功了,結果又縮回去了,唉。直也也真是不會看氣氛。很奇怪的是第11話紫乃是果體暈過去的,等天亮了她的泳著又穿回去了,難道是直也給她穿的?那豈不都看遍了?看紫乃那么虛弱,應該沒有力氣自己穿才對。
雖然直也對紫乃沒有戀愛感情,但是他發自真心想全力以赴保護和關照紫乃。第11話他背著紫乃從無人島游回去的一幕中,勇往直前的直也的樣子很有魅力,這段還令我有點小感動。無疑這下紫乃更是要下決心以身相許了。可惜紫乃向直也再一次試圖表白時被跑來的咲咲打斷了,紫乃看到摯友咲咲那么深愛和關心著直也,于是又縮了,唉!
好在是最后一話,紫乃在眾人面前,做了好長思想斗爭、做了好多好多鋪墊后,總算訴說了心中隱藏了太久的感情,這段表現得實在太精彩了!是這一季一個重要的加分點。咲咲不僅一點都沒有因為紫乃喜歡直也而生氣,反倒因為紫乃一直不肯說出這份心意而生氣,可見她對紫乃是多么的珍重。
咲咲居然還自作多情以為紫乃一直隱藏、死活不肯說的心意是她喜歡自己,突然冒出來的這個腦回路真是超搞笑,也蠻令我吃驚。其實我倒是原本也懷疑紫乃是對咲咲有百合情感,而如今看來二人之間確實只有純粹的摯友的關系。
紫乃當場被直也秒拒后,后來還是在海邊說出了希望自己也能進入直也的后宮的心意,這挺不錯的,好在她沒在被拒之后就灰溜溜消失了。看到紫乃現在變得坦誠起來,不再像以前那樣拼命隱藏心意,而是肯努力直率表達出自己對直也的真心和想法,我著實挺為她高興的。紫乃以前是連直也腳踏兩條船都死活不肯接受的人,現在倒變成了希望他能腳踏三條船,這轉變看起來真是太大了。以前是渚希望直也在喜歡咲咲的同時能分一些感情給自己,為此努力幫助直也X咲咲,而現在則是紫乃希望直也在喜歡咲咲和渚的同時能分一些感情給自己,為此努力協助直也X咲咲和渚,這發展真是有趣。
看上一季的時候我都沒怎么注意紫乃的CV,這一季到最后幾話,突然感覺高橋李依配紫乃配得真好,把那種微妙的心情以及復雜矛盾的心理展現得非常到位,給這個角色增色很多。
第5話居然咲咲饒有興致地挑弄紫乃敏感部位,不知道佐倉大法在配的時候作何感想。居然紫乃還露出了一副羞澀弱氣的樣子,和平時有很大反差。這倆人真是沒頭腦和不高興組合。這一季里還有好多次咲咲揉捏紫乃的柔軟的行為,咲咲在最后一話還挑釁說紫乃貧瘠,我都納悶咲咲哪來的這個立場?我感覺咲咲還沒紫乃的大!
真想不到米莉卡和紫乃竟然會產生交集,敏銳的米莉卡接觸紫乃不久很快就發現了紫乃喜歡直也的真相,并利用她懼怕這份感情被曝光的弱點脅迫她做為自己與直也的感情發展的助力,米莉卡可真是小機靈鬼兒。我挺喜歡看到米莉卡這么使計的。此作最后,紫乃為了逃避對直也的感情說要轉學,然后一個人跑到海灘邊傷心的時候,米莉卡主動過去試圖安慰她,還用激將法來讓她留下來,還溫柔地摸她頭,之后還給她做助攻,并覺得她和自己是相同戰線上的伙伴,有點惺惺相惜。米莉卡的這些行為給她又明顯加分了,本來紫乃只是個被她利用的工具人而已,而內心善良的米莉卡現在卻這么關心起她,令我深感米莉卡的心腸真好。我還想要是米莉卡和紫乃能發展百合關系也一定很好,顯然自信張揚的米莉卡是攻而弱氣內斂的紫乃是受。
按理來說應該還有下一季,到時候大抵內容是紫乃在后宮里的位置進一步穩固,然后米莉卡就要正式被納入后宮了。此作的原作已經完結了,如果有下一季的話應該就是最后一季了。
我們的雨色協議
總評:93.95
這番的題材挺新穎,講述年輕FPS電競選手們的故事。此作上高中的男主時野谷瞬在一個貧窮、瀕臨倒閉的電競咖啡館店打工。可惜他家庭條件差,是在單親家庭里,還有個腿有毛病的妹妹需要照顧,因而沒法自由追求自己的理想。他里在電競咖啡館老板邀請下,有了成為職業選手的契機。此作內容主要就是講他和其他四個人組成了FOXONE電競隊伍參賽的曲折經歷,以及他和身邊的三個女人,妹妹、悠宇、望姐之間的感情關系發展。
此作又是一個我特別慶幸沒因為第一觀感比較粗糙而棄掉的作品!別看此作作畫、演出等方面顯得特別樸素,劇情可真是絕了!這是為什么我能給這么一部看似廉價的動畫打這么高分的原因(如果經費更加充裕,分數肯定還能更高)。此作的劇情構思真是極其出彩,內容相當有看頭,引人入勝,情節中槽點甚多,還有很多令人特別喜聞樂見的狗血劇情和十分令人胃疼的劇情,這是我每周都相當期待的作品。到后期,很多劇情更是稱得上令我拍案叫絕!此作的人設也很好,尤其是妹妹和悠宇的人設極佳,瞬和她倆關系的發展是此作的關鍵看點。
制作方為了此作似乎還專門做了個FPS游戲,蠻有意思。雖然這游戲畫面很粗糙(也就是CS初代的水準),但作為電競用途完全夠了。我都挺希望官方把這個游戲在steam上廉價發行一下,令此作愛好者有機會體驗體驗。
此作的電競過程很有看頭,蠻有觀賞性,但這部分的演出和腳本還有很大的提升空間,可以令此作更加出色。比賽過程節奏挺快的,一會兒功夫好幾局就打完了,一點都不拖戲,我覺得其實還可以更好地控制一下節奏和心理描寫、增加一些特寫,使得觀感更血脈噴張一些,但需要把此作變成兩個季度的長度才行。PS:但也不能太學《飆速宅男》,演出效果過多,導致拖戲拖得太厲害,100米的沖刺都能講兩話。不過,此作最后一話的演出效果真是絕了,果不其然和我想象的一樣變成了瞬單挑睦生作為生死決勝,瞬控制的角色居然拔出了刀子,刺殺睦生的角色時突然渲染方式大變,那一幕上演得真是超絕贊,創意和構思真是神了!令我感嘆這不是好鋼用在刀刃上,此作沒那么多好剛(經費),而是把僅有的一點好剛完全用在了刀尖上。
悠宇這個角色真是特別有個性和魅力,很閃亮。她的性格跟一般妹子截然不同,而且是與此前我看過的所有動畫里的妹子都非常不同。她雖然不是那種令我會很主動喜歡上的類型,但是她真的很吸引人。雖然悠宇是個妹子,卻很喜歡FPS游戲,實力居然還很強。像這種美女電競高手的設定,哪怕不是悠宇,一般也是會令人喜歡上的。
悠宇相當喜歡瞬,第3話看到她以爆裂君的網友身份旁敲側擊問他對女隊友望姐的感覺,來試探他對自己有沒有交往意愿的時候令我感到蠻有趣。第5話,她居然把瞬的頭ps到她自己的臉旁邊,這可真行,令我感嘆居然悠宇喜歡瞬都到了這份兒上。在黑暗的屋子里,對著屏幕含情脈脈地以爆裂君的身份和瞬打字對話的她真是顯得挺少女心的,這幅樣子很不錯,我很喜歡。第8話,悠宇在屏幕前看到瞬說“喜歡哦”兩個字的時候,興奮得像觸電一樣,然后又露出一臉爽態,那反應真是超可愛!誰能想得到,平時顯得非常自信和獨立的大美女悠宇,會對一個普通男孩瞬如此癡情?如果我也能像瞬一樣被悠宇如此喜歡上,那肯定是超爽的事。
悠宇是個當紅大明星,作為演員時表現得又溫柔又天然,竟然在現實中卻是個內在有些暴力,不爽時說話還很粗魯的妹子,電競對戰時她甚至還總是噴出粗鄙之語,特別是第10話還給這個設定進行了充分的特寫。如此清純的妹子的嘴里總噴糞真令人瞠目結舌。她的生活作風還特別糟蹋,內衣隨地亂丟,顯得蠻粗野的,過于隨性,和普通同齡日本櫻花妹有天壤之別,不過她在戀愛感情上卻和普通妹子沒有太大差別。值得一提的是,悠宇在家穿露單側肩服裝的骨感+邋遢的造型風格挺性感的。
悠宇在作為FOXONE的成員參加比賽時為了不暴露自己是明星的身份,一直戴著面具。在最后一話時,FOXONE面臨絕境,我原以為悠宇會說如果輸了比賽就摘下面具,從而不給自己留退路,想不到比賽過程中她就主動摘了面具。我以為這意味著其人設崩塌,演藝道路就此終結,沒想到結果意外地好,雖然掉粉不少,但也收獲了很多新粉絲。不過,她帶著面具時曾說的那些粗鄙之語,這都已經成了既定事實,無論如何也是極度有損形象的事,沒法洗,以后粉絲們若看到她拍的裝作清純可愛妹子演的影視作品時,心里回想到比賽時她滿嘴噴糞的樣子,心里得感到多違和啊!
電競咖啡廳店長的女兒稻月望(望姐)人設不錯,現在是高三,比上高一的悠宇和高二的瞬都大不少,平時作為姐姐的身份和瞬相處。我挺喜歡望姐的,她沒有大多數女生的活潑、柔美、靚麗,作為擔負店面營業的人,她的性格顯得比較沉穩理性,責任心很強。望姐雖然不怎么打扮,一直素面朝天,但底子很好,完全不輸于悠宇,而且歐派明顯比悠宇大幾號。望姐有這么好的自身條件,不去演藝圈發展一下我都覺得有點可惜。望姐作為玩家的實力不俗,玩的時候那副認真的樣子有點小帥氣,這樣踏實穩重的FPS妹子的設定誰能不喜歡呢?
悠宇和望姐與瞬之間的愛情發展,尤其是悠宇和望姐變得爭風吃醋的情節,是此作里我特別喜歡看的劇情,太有意思了!瞬從小和望姐一起長大,令望姐一直過于安逸了,把瞬在她身邊視為理所當然。前幾話的時候,望姐對他沒有表現出戀愛感情,但悠宇每次看到她和瞬有親近行為的時候都會吃醋,挺樂的,令我感嘆悠宇真是醋壇子。到了第5話,想不到一直挺淡定穩重的望姐居然開始慌了。她看到悠宇離瞬越來越近,越來越顯得沒距離感,開始意識到問題的嚴重性、產生了嚴重的危機感,她擔心搞不好從小就在自己身邊的瞬真的就會被悠宇搶走了,這令她在洗澡的時候一個人煩悶了半天。望姐雖然之前沒有表現出明確的對瞬的心儀,但是她容不得瞬被別的女人奪走。此刻這副心急、焦躁樣子的望姐真是和之前有很大反差,很有意思。看來表面上成熟的望姐其實也是一個有正常少女心思的人。在第5話過后,悠宇在望姐眼里算是正式確立了競爭關系了,二人看望完瞬走在回去的路上都相互笑得很尷尬。第8話,這倆人都因對方而吃醋,樂死了。悠宇和望姐目前在表面上還是友好的隊友關系,尚沒有形成對立和沖突,但時間長了不可能不會因為爭奪瞬的感情而起矛盾,畢竟瞬的妻子早晚得從這倆人之中選一個。
瞬的妹妹美櫻真是超級軟萌的粉色系美少女,可愛極了,瞬有這樣一個超兄控漂亮可愛的妹妹真是太幸福了,真羨慕瞬老能抱她。第4話她還說“我要胖到哥哥抱不動才好”,她可真會撒嬌!第5話,居然看到妹妹和瞬睡在同一個被窩里還抱著他,然后她還偷拍哥哥的睡顏,還聞著哥哥的衣服暗爽。看到瞬抱著妹妹那么和諧幸福的畫面,我心想他倆結婚得了。美櫻還是個能干的妹妹,第5話看到她在瞬起床之前就起來給他洗衣服熨衣服,簡直像是他的賢妻。瞬有這么可愛+貼心+能干+會撒嬌的妹妹,真是太爽。不僅如此,瞬還有女明星悠宇暗戀他,還有唯獨對他溫柔的望姐照顧他,令我感覺瞬簡直是夢幻級的人生贏家。
然而,好景不長,隨著劇情發展,妹妹開始逐漸黑化,劇情變得開始胃疼起來。第7話真是顛覆了我對妹妹的認知。她開始覺得最愛的哥哥想讓自己離開他,覺得哥哥不想再和自己在一起,于是心情變得陰暗沉重,脾氣也開始變差,和哥哥發生了口角。居然她還在廁所里把那個療養院的宣傳單用打火機燒了,當時真令我想不出來這是天使般軟萌的妹妹會做出的事情,令我覺得她有點粉切黑,擔心她之后變得病嬌,乃至做出過激的無法挽回的事出來。妹妹真是不惜一切代價想要和哥哥在一起,對他的獨占欲還特別強,甚至都不容忍哥哥的衣服上有不熟悉的味道。
第8話,兄妹的矛盾徹底激化,變得無法調和,真是老刺激了,又太扎心了。我感覺雖然妹妹性格任性、執拗有一定自身問題,但哥哥和母親都應該考慮妹妹自己的感受,靠針鋒相對解決不了問題,只會令彼此更受傷。妹妹原本一直以為自己是獨占哥哥的,而在這一話看到哥哥和望姐、悠宇在廚房關系親密,更顯得可憐。這一話里愚蠢的瞬還犯了一個致命的錯誤,說了一句“沒有美櫻在,我也是可以的”,這是妹妹最害怕聽到的話。妹妹離不開哥哥,也特別希望哥哥成為離不開自己的人,能永遠粘在一起對妹妹來說是再好不過的。只要能夠如此,妹妹覺得是否能重新走路都是無所謂的事。可是偏偏瞬非要讓妹妹治腿、讓她能自立,還說了一句經典的普遍特別令對方感到討厭的話“我這可都是為了你好”,結果糟至妹妹憤怒的回擊。唉,瞬過于木訥,完全不懂妹妹的心思。瞬被妹妹說“你這不都是為了自己嗎”之后陷入恐慌和混亂而跑走。妹妹為了追哥哥,驚慌地從屋里爬出去,然后從公寓的樓梯跌落,真是太可憐了,當時都把我看哭了。好在最后妹妹沒摔骨折之類,要不然瞬真是錯上加錯。若是妹妹摔死了,或者摔成更嚴重的殘疾,瞬恐怕一輩子都無法原諒自己了,而且也沒有辦法贖罪了。
第10話,看到妹妹呆坐在門口,無神的眼睛直愣愣地看著門,表情一臉淡漠,心中期盼著心愛的哥哥早日回來,然而瞬卻住在網咖始終不回家,美櫻的那副失神的樣子太令人心疼了!之前我看過的動畫里雖然也有很多關于兄控的,但能把兄控的感情描寫到此作的深度,真是非常罕見。
隨著劇情發展,有一層關系開始顯現:瞬總是不惜代價讓美櫻能站起來走路,為此試圖靠職業電競賺錢,這是為了他自己贖罪(曾經因為他參加電競間接導致了父親和妹妹遇到事故,從此妹妹走不了路),而本身不是為了美櫻的幸福。第11話,意識到這一點的美櫻徹底跟瞬鬧掰了,還把美櫻氣得夠嗆。居然瞬還說贏了比賽就要把美櫻送到療養院住院,輸了比賽就再也不回家、不見到美櫻,明顯無論哪種情況對美櫻都是無法接受的,瞬做出這種決斷真是超級腦殘,一點都沒真心考慮妹妹的主觀感受和想法。望姐知曉后,馬上給瞬一個大逼兜,這做得太對了,真是只有望姐能懂得妹妹現在的感受。
妹妹雖然也有點扭曲和病態,但內在還是挺好的,最后一話令我進一步增加了對她的好感度。妹妹看到比賽中哥哥面臨困境,于是在視頻電話里拼盡全力試圖站起來,以此鼓舞哥哥。雖然哥哥深深傷害了她,也肯定已經明白了哥哥并沒有把自己當做唯一、并不會一直陪伴在自己身邊,但依然還是全力支持哥哥的比賽,表現得很值得贊賞。令一方面,妹妹可能也是真怕哥哥履行他自作主張立下的腦殘決定,即輸了比賽就永遠離家出走再也不見自己。
此作里這對兄妹注定發展不起來雙向的BG,瞬對妹妹完全沒有家人以外的想法,永遠不會把她當做戀人,這很可惜。只能希望瞬在和悠宇或望姐交往、結婚后,還是能分一些愛給妹妹。瞬的隊友曉斗對美櫻喜歡得不得了,還老是揚言要娶了她,但無疑妹妹不可能看得上這么輕浮的家伙,喜歡的應該是個踏實且對她很溫柔的人,像此前一直照顧她的那個版本的瞬那樣。
美櫻的腿似乎不用治了,沒有什么生理上的毛病。雖然美櫻自己說并不是心理問題,努力試過就是站不起來,但感覺她經過長期康復訓練應該還是能走路的,希望妹妹有朝一日不用再坐輪椅。妹妹其實可以試圖和瞬立個約定,什么時候能站起來走路了,瞬就必須和她交往。
松崗配的那個小崽子洛克斯真是個心機boy,是個令人討厭的家伙,還很自負。他的戰術真是夠雞賊的,讓隊友當誘餌,他在后面偷襲對方。他居然直接喝拌飯用的咖喱汁,這行為還把瞬給傳染了。
睦生對瞬惺惺相惜,他覺得瞬有和自己成為一生的宿敵的潛質,想讓瞬脫離家庭和一般的人生,和自己一樣把一切都獻給電子競技。睦生可真是惡心,把瞬的家人和過往情況調查了個底兒掉,第6話末尾bb半天,居然勸誘瞬成為他的伙伴。第9話更顯示出睦生過于自私,為了獲得瞬不擇手段、不惜挑撥離間瞬與妹妹的關系,各種蠱惑瞬,想讓瞬和他一起走上無法回頭的路,表現得跟惡魔一樣。我真想看到瞬給陰險的他一記友情頗顏拳,結果第9話末尾這一拳還真的來了,我很高興!
睦生對瞬的重視除了游戲方面,明顯還有額外的感情,這在第8話的對白中展露得極度露骨,基情四射,跟表白似的:“我倒是看好你,作為我在這世上陪著一直活下去的對象...我要的是能殺得有來有回的伙伴(PS:在哪里“殺”得有來有回?呵呵...),能夠互相度過能沉浸其中時光的對象...你就是能夠滿足我的,唯一的宿敵”。不僅如此,居然他還來了個能令人驚掉下巴、令腐女看了能噴鼻血的描述:“搞不好,那可是比夫婦和戀人更要親近的關系”。睦生本身未必是gay,但至少在面對瞬時,他絕對是gay!
睦生真的是很機靈,洞察力非常強,他對瞬和妹妹的關系確實看得很透,說了真話,道破了美櫻走不了路本質上是心理問題。正是因為瞬在妹妹發生事故后才開始溫柔地照顧起她,生理上已經沒問題的妹妹在潛意識里才拒絕康復,由此可以一直像這樣被哥哥照顧和關懷。
起初一直當觀眾的星詩流我原以為只是個喜歡電競帥哥的膚淺+壞脾氣的辣妹,在第8話她真令我刮目相看,居然相當懂電競和戰術,是個很有頭腦的狠角色。她在噴了一通FOXONE發泄后,被店長請去當了FOXONE的教練,立馬令隊伍水平大漲,星詩流真是很不簡單。她跟瞬并沒什么交集,她要是也踏入瞬的爭奪戰中那就更火爆了。星詩流對以前的FOXONE真是狂熱,那時候的5個隊員確實顏值和氣質不錯,作為男偶像團體都能出道。第8話她得知她推的選手不僅有老婆,連娃都有了,備受打擊,這一幕真逗,曾經她打賞的錢都打了水漂了。不過即便星詩流在,FOXONE的大家也遲遲沒有看透睦生的戰術,令我對她的能力稍微有點失望。
和星詩流始終在一起的那個一部分藍頭發的妹子不知道是否也有過人之處,打扮得蠻靚的。這齊劉海彩頭發的風格令我想起了Bang Dream的RAS中的PAREO。她在此作里始終都是打醬油,希望能看到她也參與進主線里。
從最后一話的ED上看,廢貓居然染了粉頭發,似乎星詩流還和他關系親密,給他投食,不知道是不是交往上了。可惜此作太短,沒有劇情描寫這倆人的發展。
這電競咖啡廳真是貧窮,顯卡壞了還試圖自己修。瞬居然把顯卡放到烤箱里烤,真樂。店里干脆再弄臺BGA機吧!
我始終沒明白此作的標題的含義何在,此作也沒體現什么“雨色”、“協議”的成份,感覺這標題有些詭異。
此作是完全原創作品,劇情構思水準如此之高真是很難得了。我真是非常希望此作能出第二季,但感覺希望渺茫,而且這一季的末尾已經是大happy end了,從最后一話ED畫面里看到每個人都很開心,妹妹和哥哥的矛盾似乎也解決了,悠宇的演藝和電競事業的沖突也解決了。倘若真有下一季,可能就要充分描寫悠宇和望姐爭奪瞬的事了,而且到時候悠宇在瞬面前隱藏自己用的爆裂君的這個身份估計就要揭開了。
此作ED雖然沒有驚艷之處,但聽感和觀感還比較舒服。
我的推是壞人大小姐。
總評:93.7
之前早就在百合會上聽聞此作的漫畫,看了這動畫,發現是一部超有趣的百合番。我起初還有點擔心此作會不會只是賣設定,只是前幾話會給人新鮮感,這擔心真是多余的,此作劇情從始至終一直都非常有趣,甚至越往后越好看,真是完全沒有令我失望。此作對兩位女主的性格塑造和她們的感情關系描寫相當出色。雖然此作畫面比較拉胯,有不少崩壞,戰斗部分顯得頗為貧窮,但其實不明顯影響觀感。此作真是看得我很開心,是歡樂百合番里的精品,百合眾絕對不能錯過。如果此作做得更精致一些,我的評分還會明顯更高。此作可以說得上是在非常有限的預算下將觀感做得盡可能好了。有人說此作是慘遭動畫化,而我覺得這動畫明顯是做得挺成功的。整部作品看完,我可以說我非常喜歡此作。
此作女主零緹拉超級喜歡某部galgame游戲里的一個反派女角色克蕾雅(就像我喜歡夜羽的程度),甚至都為她做過同人作品參加過comiket。緹拉喜歡克蕾雅到這份兒上,緣自于每日被工作折磨得很辛苦的她真心感到被游戲中的克蕾雅的音容笑貌所救贖。有一天緹拉莫名其妙穿越到了這部游戲里,成為了克蕾雅的同學,得到這樣天賜良緣的她立刻展開了向克蕾雅各式各樣的特別簡單粗暴的示愛,天天用令人害羞的臺詞表白,倒處沒完沒了纏著她,而且表現得死皮賴臉乃至毫無尊嚴。被克蕾雅欺負對她來說是饋贈、是享受,都有點像抖M,甚至顯得有點惡心。被緹拉天天粘著,在克蕾雅眼里是自己被她調戲,令脾氣本來就不好的克蕾雅又氣又惱,但是又擺脫不掉她,這相處方式真是很有趣。此作主要內容就是講述在學園里這倆人關系的逐步發展。
克蕾雅一開始對緹拉很抗拒,對她各種訓斥,也充滿戒心,但一點點認識到了她的真心。她總是說緹拉對她的示愛是在戲弄她,其實她已經逐漸一點點感受到緹拉是付出真心的,只不過她不想承認這一點而一直欺騙自己,擔心傲嬌的自己可能會真的對她動情。
緹拉對克蕾雅的死纏爛打的攻略方式還是挺奏效的,在潛移默化中關系不斷得到進展,令克蕾雅逐漸放下戒心,然后還逐步對她開始在意起來。到后來,有時候克蕾雅還有點被緹拉牽著鼻子走。比如第9話,向陛下求情使得蕾妮免死后,二人離開皇宮時,緹拉拉著她歡快地跳舞慶祝,終于此時的克蕾雅完全放下架子了,露出了開心的笑容,好感度明顯進一步增加。到故事最后,克蕾雅都已經非常不想讓緹拉離開自己身邊了,甚至在埃莫之祭上都等著緹拉奪走自己了。
最后一話真是超級happy end,比我想象得還要happy,超級幸福圓滿,看得我很高興。終于克蕾雅不再全力掩飾自己對緹拉的喜歡了,直接表現得臉紅羞澀,在瑪娜麗婭要親緹拉的時候居然還馬上把緹拉拽走宣誓主權,慌忙說“零是我的東西”。克蕾雅甚至在別人打趣說到自己要嫁給緹拉時,她馬上回口說應該是緹拉嫁給自己才對,這場面真是太甜了!不過克蕾雅的傲嬌本性十分難改,最后在教室里緹拉再次對她在耳邊示愛,居然克蕾雅還紅著臉假裝沒聽到,這一幕真是太有愛了!
克蕾雅真是挺漂亮,有一些鏡頭里克蕾雅畫得很賞心悅目,面容相當精致。而且越往后看我越覺得她漂亮。雖然克蕾雅在游戲設定里是反派,但骨子里真是蠻溫柔的,欺負零的時候都怕她受傷,向她潑水都只潑常溫白水而不潑熱茶怕傷著她。無疑也正是因為她這樣良好的內心,緹拉才會那么深愛和癡迷于她。克蕾雅的戰斗力意外地還挺強的,而且有時戰斗的身姿還顯得挺瀟灑帥氣的,她在火焰魔法方面算是top了,這些方面她一點都不像普通的養尊處優的大小姐。雖然克蕾雅作為貴族,表面上就算為了人設也得表現得高傲一些、在平民面前擺出一些架子,但其實內在挺平易近人的,挺討人喜歡。
有趣的是,雖然克蕾雅原本是BG,心儀的是王子,但她倒似乎對于女性之間發生戀愛這樣的不同尋常的感情完全沒有覺得有什么禁忌的地方,也沒因為緹拉是女孩而覺得和她交往有什么不妥、需要在意別人什么眼光。不知道該說是她觀念開放,還是她本來就沒有“同性交往屬于不同尋常的事”的這種意識。反倒是緹拉對于自己作為女性和克蕾雅交往有點顧忌,擔心這會是個阻礙。此作里的同學們都顯得很開明,不僅沒有因為克蕾雅和緹拉同性交往而說閑話,反倒在最后一話里大家在教室里看到緹拉對克蕾雅大聲示愛時表現得還挺支持的。
緹拉對克蕾雅的日常百般示愛總是令她感到羞恥,表現得很狡猾和機靈,無與倫比的皮。此作故事中總見緹拉各種犯賤似的向克蕾雅表白、各種逗她,故意招她跟自己拌嘴,怒刷自己的存在感,反復磨好感度,這真是緹拉的獨特的攻略手法。剛看此作時我都覺得克蕾雅有點可憐,突然被緹拉這樣的怪人黏上,怎么甩都甩不開,結果她還通過和自己父親的交易成了自己的貼身女仆,更是徹底擺脫不掉了。
緹拉不僅靠日常死皮賴臉的方式攻略,在一些關鍵時候還顯得很有一手,表現得非常有智慧,有點像情圣的感覺,能迅速拉高克蕾雅對自己的好感度。比如第7話緹拉溫柔地給克蕾雅梳頭,充分利用這個難得的機會進行推心置腹的交流,然后還單膝跪地邀請她逛創立祭,表現得很紳士。再比如第9話,失去了前女仆蕾妮的克蕾雅正在傷心時,緹拉從后面突然抱住心理防線最低、最脆弱時候的她,令她放下偽裝的堅強,給予她在自己看不到的那一面哭泣、釋放壓抑的感情的機會。我深感緹拉真是非常能抓住機會拉進自己和克蕾雅的距離、消除她心中的屏障,提升自己在她心中的信任以及默契。
雖然緹拉的表面樣子看上去顯得簡單膚淺,但其實肚子里挺老謀深算的,只不過這方面從她發自真心的臺詞和真誠無邪的眼神上一點都看不出來。比如第6話緹拉計劃通,巧妙地利用鬧鬼的事,成功實現和怕鬼的克蕾雅同床。在這個劇情后,居然克蕾雅因為看到緹拉和蕾妮親切地交流而開始吃醋了。克蕾雅殊不知自己已經中計了。
我真心感覺,如果不是緹拉,面對心理防線高聳、特別傲嬌而且性取向很直的克蕾雅,真是沒有任何攻略的可能。這倆真是命中注定的CP,克蕾雅內心的鎖只有緹拉能打開。
緹拉曾表示出很支持克蕾雅和王子交往,而非表現出對克蕾雅的獨占欲,這看起來非常矛盾。緹拉自己表示理由是她最希望看到克蕾雅幸福,始終把克蕾雅的幸福放在第一位。雖然這看似說得通,但我總懷疑是不是這種說辭其實是聰明的緹拉攻略克蕾雅的一個精妙的手段,因為好處有三:(1)這會令克蕾雅多多少少有些覺得虧欠緹拉,因此會引得她想分一些感情給緹拉 (2)會引起傲嬌的克蕾雅的逆反心理,使之淡化和王子交往的意愿 (3)會令克蕾雅微妙地覺得自己并不是緹拉絕對放不下的人,因而使得克蕾雅會主動想和緹拉拉近關系,有點欲擒故縱的意味。但后來我發現我想多了,緹拉的心機還沒有到這個地步。
故事最后,瑪娜麗婭對緹拉X克蕾雅的助攻真是關鍵性的。她告訴緹拉,不求回報的愛情最后一定會扭曲,這點醒了緹拉。之前緹拉雖然總是奔放地向克蕾雅示愛,但其實她內心有所保留、隱藏了自己真實的心意,一直給自己留出退路,所以才覺得只要克蕾雅幸福的話,和她交往的即便不是自己也沒關系。可是真心愛一個人,怎么會心甘情愿、坦然地看到戀愛對象的位置被他人占據?明顯應該竭盡全力去爭取才對。緹拉聽了瑪娜麗婭的一番話后,再加上她的激將法,總算重新有了勇氣和動力了,變回了赤誠熱愛克蕾雅的她本該有的樣子。最后在贏了瑪娜麗婭后,緹拉對克蕾雅說“我不希望只是女仆,可以讓我成為您的伴侶嗎”,聽到這話,克蕾雅都幸福得流出了激動的淚水,無疑這就是此時此刻克蕾雅最想聽到的話語!要是緹拉沒抱著成為克蕾雅交往對象的決心最后一搏,那絕對是一輩子的悔恨。
第9話轉學來的瑪娜麗婭的人設真是驚艷,外形顯得特別瀟灑帥氣,魔法實力無與倫比,同時擁有四種屬性,運動萬能,頭腦還極好,而且性格層面(表面上)還顯得非常完美,行為落落大方,待人溫柔,顯得端莊高貴又平易近人,簡直是各方面能力值都滿格的終極完美的存在。瑪娜麗婭的CV居然還是水樹奈奈,其溫厚又自信的聲線進一步把她的氣質拔高到無與倫比的程度。原本克蕾雅就特別傾慕于瑪娜麗婭,從小第一次見到她的時候就愛上她了,而如今瑪娜麗婭還主動表示喜歡克蕾雅,這一對簡直是閃瞎眼的天作之合。緹拉面對其他競爭對手都顯得非常自信和游刃有余,唯獨碰上瑪娜麗婭這個狠角色時覺得非常棘手、完全沒有了曾經的自信。她看到自己最愛的克蕾雅和瑪娜麗婭經常放閃、克蕾雅各種夸贊瑪娜麗婭、瑪娜麗婭各種撩撥克蕾雅,感到憂心忡忡,被醋意搞得渾身難受。瑪娜麗婭還挺壞心眼、很腹黑,在得知緹拉喜歡克蕾雅后,各種針對她挑釁,在第10話最后還故意在她面前宣誓對克蕾雅占有主權,看得緹拉都有點急哭了,卻又手足無措,這場面真是很樂,想不到超級皮的緹拉也有這么一天啊!瑪娜麗婭真可謂是唯一能令緹拉吃癟的BOSS般的存在。緹拉雖然是游戲的骨灰玩家,卻沒想到自己進入游戲世界后會遇到這樣的超狠角色。
我在看此作的過程中一直懷疑是不是瑪娜麗婭其實并沒打算和克蕾雅作為情侶一生在一起,只是希望克蕾雅未來能夠安全和幸福,所以想試探緹拉,看看她是否有能力和決心一直守護克蕾雅,是的話就可以將克蕾雅放心地托付給她了,相當于助攻。第11話瑪娜麗婭把緹拉打得半死,可能也是檢驗克蕾雅究竟如何看待緹拉,也可能希望讓克蕾雅恍然意識到緹拉對她的重要,促進她們關系的發展。
第11話真是太吊人胃口了,沒想到緹拉在瑪娜麗婭面前完全沒有招架之力,被徹底碾壓。居然瑪娜麗婭的戰斗力如此離譜,給我感覺她就像是GM般的存在。戰斗失敗后的緹拉居然自暴自棄,徹底失去了平日的光彩和自信,還主動離克蕾雅遠遠的,逃避和她在一起。之前一直對緹拉避猶不及的克蕾雅開始特別擔心和關注起緹拉來,這二人的關系都有點反轉的感覺了。最后居然緹拉說了無法挽回的致命的氣話,辭掉了克蕾雅女仆的工作、放棄了她最愛的克蕾雅,想不到一向表現得游刃有余的緹拉居然也有控制不住自己情緒的一天。這一幕真是太扎心了,緹拉苦心經營的和克蕾雅的感情徹底破裂,也令克蕾雅對緹拉失望至極,憤恨地說她是個騙子,并深刻感受到內心孤獨帶來的痛楚。本來克蕾雅兒時失去家人時就很難過,如今蕾妮走了,緹拉也拋棄了她,想想都知道克蕾雅得有多難受。這一幕真心挺催淚的。
第11話爆料居然瑪娜麗婭在別的國家逛窯子玩弄女性。以她瀟灑的外形、高超的撩撥技能,以及性格上壞壞的一面,真覺得這確實是她干得出來的事。想不到第12話她還自己主動說她因為曾經對她喜歡的女仆出手,導致女仆主動辭職離開,然后失意的她云游各國花錢玩弄女性以試圖得到滿足。看到她如此風流,令我還曾懷疑她那么撩克蕾雅、那么快就主動湊得那么近,是不是是真的對她的身子感興趣。第12話瑪娜麗婭居然還說如果贏了緹拉,以后就要將克蕾雅作為泄yu對象(怎么個瀉yu法?),這話可真是夠刺激的!不過其實我覺得能被瑪娜麗婭玩弄是蠻幸福的事,是常人求之不得的獎勵。
最后一話終于揭開謎底了,果不其然瑪娜麗婭的真正想法就是自己扮黑臉給緹拉X克蕾雅做助攻,可喜可賀她的目的圓滿達成了。我挺好奇的是,最后瑪娜麗婭并沒有放水,如果最后緹拉沒能贏過她,會不會她就真的要占有克蕾雅、真的玩弄她了(也可能用心守護她)?或許這真的就是瑪娜麗婭的備選方案。雖然緹拉X克蕾雅是No.1的CP,但我也真想看看倘若緹拉失敗,瑪娜麗婭X克蕾雅最后能進展到何種地步,會不會上壘。
瑪娜麗婭一開始就對緹拉表現出明顯興趣,但想不到她在最后一話竟然還對緹拉直白地示愛,就像是緹拉對克蕾雅做出的行為一樣(挺有趣,某種程度構成了三角戀了)。雖然瑪娜麗婭顯得有點像是開玩笑,但說不定她真的對緹拉有好感。畢竟緹拉實在太特殊了,這肯定是瑪娜麗婭目前見過的最奇葩、最神奇的女孩,瑪娜麗婭在心底愛上緹拉真不是沒有可能。不過瑪娜麗婭的心思實在過于狡猾,很難猜得透她對緹拉究竟有幾分是真心的。我挺想看看瑪娜麗婭X緹拉能如果真能結成的話能發展成什么樣,可以作為出本子的素材。瑪娜麗婭還說如果埃莫之祭贏了緹拉的話她就要成為自己的東西,出本子的話劇情可以是緹拉最后輸了,然后就成瑪娜麗婭的奴隸,成了被她玩弄乃至泄yu的對象。
克蕾雅的女仆蕾妮居然和哥哥是戀人關系,想不到同一個作品里竟會同時有百合和兄妹兩種禁忌的戀情。蕾妮真是蠢,自己的行為導致整個家族的人都最終被流放。最后蕾妮看到緹拉完美地代替了自己的專屬女仆的位置,算是放心了,將克蕾雅托付給了緹拉。蕾妮雖然對克蕾雅忠心耿耿,但對她倒是沒表現出戀愛感情。
第6話出現的緹拉的丸子頭造型真是挺帥氣的,有點man,令我眼前一亮,不知道克蕾雅對她的這副形象有何看法。第7話克蕾雅、緹拉、米夏的男執事造型很不錯,又美又帥氣,特別是男裝克蕾雅的特寫結合帶有冰晶圖案的藍色背景的那個畫面真是魅力驚人!幾個妹子的男裝都很出色,但王子們的女裝實在糟糕,除了那個黃毛小子還說得過去。
緹拉真是挺適合男裝的。學園祭,以及第9話的男裝造型很不錯,連傲嬌的克蕾雅都不禁紅著臉說“真是的,如果不說話,倒也不是不能看”,明顯已經被這副姿態的緹拉吸引了。
OP里有兩幅緹拉和克蕾雅在一起的插圖我很喜歡,一副是克蕾雅穿盔甲、緹拉穿軍裝,二人顯得非常英姿颯爽。另一幅圖則顯示出她倆的非常甜美可愛的一面,克蕾雅的紅蝴蝶結+雙馬尾的形象真是巨棒!這圖令我覺得她倆結成偶像組合出道很不錯。
此作的幾個王子還挺不錯,雖然沒啥亮點,但至少完全沒干擾到緹拉X克蕾雅的發展,這就已經難得了(反觀諸如《京吹》里的秀一的存在討厭死了)。在后來他們都知道了緹拉對克蕾雅的心意后,還當吃瓜群眾,偶爾還稍微助攻一下,在最后一話他們還高高興興地看著她倆進入交往模式。百合作品中的男角色都像他們這樣就挺好,一點都不搗亂、不招人煩。
說來以反派大小姐為題材的動畫太多了,看此作之前我還擔心此作會有些套路,但看完后感覺此作真是挺標新立異的,和之前的此類作品截然不同。此外,此作和《傲嬌反派千金莉潔洛特與實況主遠藤同學及解說員小林同學》有那么點相似,都是一個日本的顏值普通的黑發妹子喜歡游戲世界里反派的美貌的金發傲嬌貴族大小姐。只不過此作是百合,并且穿越到游戲世界,而那個是BG,是隔著現實和虛擬世界靠傳話推動劇情。
東京復仇者 天竺篇
總評:93.6
前幾季一直都追了,此作的劇情真是相當漫長。這一季主要講述東卍和天竺的爭斗,以及男主武小道繼續破壞稀咲陰謀的故事。最終在兩個幫派的死斗結束后,稀咲徹底完蛋。罪魁禍首死掉,感覺此作按理說應該完結了,但感覺這么結束了又有點太突然,不知道還有沒有下一季。這一季警察依然完全下線,是一個沒有警察的世界。
在第一話看到在未來的世界中大壽居然洗白了,現在的他一直想念著死去的弟弟,還幫了武道,給他提供信息,居然還搞起了經營,走上了正路,令我感嘆歲月真是能改變人。此時居然東卍的小頭目還帶著手下去打當警察的橘直人,堂堂警察被一群混混追殺,還只能逃跑,此作里的警察真是毫無威信可言,太丟人了。第2話武道真是智商下線,非要自己挨一槍才跟直人握手穿越回去,搞不懂干嘛非得要白挨這一槍。要是被爆頭,一下子掛掉,豈不就再也回不去了?
黑川伊佐那是這一季關鍵的新角色,稀咲和他沆瀣一氣。我挺討厭他的,非常自負,性格差勁,長相尤其是眼睫毛還特別難看。后來知道原來伊佐那的生母是菲律賓人,難怪那么黑。
伊佐那對曾經關照他的Mikey的哥哥真一郎始終念念不忘,那程度簡直就像愛情似的,他要讓真一郎關愛的包括Mikey在內的其他人受折磨,自己獨享真一郎的愛,真是變態,太扭曲了!稀咲正好找到了這么個家伙成就他的目的,這組合太惡劣了。伊佐那的內心完全被仇恨和孤獨感所浸染,完全喪失了是非觀和人性,雖然仗著自己非凡的武斗的實力獲得了成果成了天竺的老大,但實際上只是個很丑陋和可悲的人。最后看到他被Mikey打到沒有還手之力、被實力碾壓的時候感覺很爽,想不到他也有今天的下場,在眾目睽睽之下被虐得盡顯丑態、人設崩塌,平時高傲和自負徹底喪失。他變成這副模樣了竟然還想使喚手下,真是丟死人了。這一幕也令我感嘆果然Mikey確實是此作不容置疑的戰斗力的天花板,他揍伊佐那的時候真是顯得很有型。伊佐那看到Mikey哥哥和妹妹都靈魂附體的那個樣子尤為帥氣。
伊佐那倒是在中了稀咲三槍后,在臨死前努力洗白了,也令我覺得他是個挺悲情的人物。原來他從小就被很不負責任的養母拋棄,連自己的父母都不知道是誰,難怪成長成這樣。伊佐那的內心非常孤獨,沒有親人的他特別希望能與他人形成緊密的紐帶。一直陪在他身邊的阿鶴在真一郎死后也成了他心中最重要的人,難怪伊佐那會為他擋子彈。
阿鶴人品還不錯,一直跟隨從小就和他在一起的伊佐那,對他忠心耿耿,但頭腦還是清醒、有理智的,比伊佐那強得多,最后還阻止了伊佐那試圖槍殺Mikey。可惜阿鶴中了一槍,按理說沒打到要害應該不會死,但居然磨磨唧唧也不去趕緊醫院,血流了很多,懷疑是打中什么大血管了,然后就好像死在那了。我心想按理來說還可以搶救一下。好在確實最后阿鶴沒掛,不知道怎么撿了條命,伊佐那沒白給他擋槍。
我感覺伊佐那他媽和艾瑪真像,簡直跟艾瑪她媽似的。不過最后并沒有出現伊佐那的養母是艾瑪她媽的這種設定,要不然人物關系就更復雜了。
花垣武道在這一季雖然還是戰斗力非常弱,但氣概和決心很值得稱贊,令我覺得他不再窩囊,所以這次給了他一個最佳男主角賞。
第4話,武道眼睛炯炯有神,對著曾經在天竺和黑龍作為干部的阿乾說“黑龍就由我來繼承...擊垮天竺!你能跟上我的腳步嗎”,我感覺這一刻是武道迄今為止的形象最閃耀的一次,很有勇氣和覺悟。阿乾說“我的命就交給你了”,這令我看到了武道改變未來的希望。之前武道自己又弱,又沒有手下,啥都干不好,我覺得他就得靠自己的德行多收一些阿乾這樣骨子里不邪惡、對他忠心,又有戰斗力和行動力的幫手才行,否則什么也干不成。不過最后黑龍倒也沒復活,并沒有出現武道自立門戶的劇情。
這一季的武道真算是有一定英雄的感覺了。他面對強大的天竺,敢于自己帶一幫人去挑戰,也不再那么畏懼戰斗。第10話他的表現得真是令人佩服,特別不屈不撓,只要身體不被徹底破壞就始終拼盡全力站起來。他堅信只要他還站著,東卍就還沒有結束。他在這一幕表現出的豁出性命也要保護東卍和Mikey的決心令我贊嘆。我深感武道啥都不靈,但唯獨決心和意志力是此作里最強大的,劇情發展到這里他終于發光了。好在他的努力沒有白費,終于等到了Mikey的到來,這重新相會的一幕真可謂是名場面,我很欣賞。
稀咲太壞了,第5話居然掄金屬棒打死了Mikey的妹妹艾瑪,為了利用Mikey完全不擇手段,這下無論如何都不可能洗白了。艾瑪都死了,居然警察還不趕緊抓人,真是邪乎。
最后武道把稀咲逼到走投無路了,想不到第12話時二人居然開始1v1肉搏了,我感嘆稀咲終于也有淪落到這種地步的一天啊!我之前感覺他倆戰斗力差不多,但由于武道的意志力驚人地頑強,理應不會輸給稀咲,果然沒有落入下風。
每次武道回到原世界時日向總是死掉的這個此作的最大謎題終于徹底揭開了。萬萬想不到,居然稀咲喜歡日向!稀咲自我感覺良好,以為靠錢和權力就能得到日向,然而扭曲、缺德、陰暗的他怎么可能獲得日向的芳心?果然告白被拒。一般來說,內心美好的人,就算獲得不了心愛的人的心,也應當祝愿對方幸福,然而稀咲卻邪惡且極端得無可救藥,居然得不到日向就要殺死她。最后無比猖獗的他的報應真是太棒了,被大貨車撞飛,血流一地,身體變得畸形,還在那里念叨不想死。看到終于稀咲也有這么一天,我感到極度愉悅!本來這一季一開始武道還意識到稀咲可能也是時間穿越者,看來稀咲沒有這個能耐,也并沒有任何后路,我還一度以為稀咲在這個世界死后會觸發什么機制進行時空穿越。但愿別出下一季然后稀咲又活過來!
其實我有點疑惑,稀咲眼里只有錢和權力,還想成為日本第一的不良少年,他難道真的會喜歡日向?那真的是戀愛、心動的感覺?我覺得稀咲和戀愛這個詞都不沾邊。我懷疑稀咲只是想不惜一切代價得到日向這個令他很在意的女人,令她成為自己的所有物而已,而并不是希望和她成為對等的戀人、夫妻。
半間這家伙居然跟稀咲關系那么鐵,稀咲當眾做了不可原諒的惡行,被逼到走投無路、只能在地上狼狽驚慌地爬時,居然半間開摩托帶他逃跑。也不知道稀咲給了他什么好處。看來他的戰斗力還是比不上Draken,最后Draken只受了輕傷就把他打到蹲地上了,這一幕看得我挺高興。此時的半間的頭發都垂下來了,感覺這個形象比平時豎著頭發的樣子好看很多。
乾青宗的表現不錯,自己的密友可可迷失了自我,后來投靠了天竺,而青宗還有著自己的骨氣和執著。
第8話看到,九井一小時候喜歡比他大5歲的青宗他姐赤音,赤音因火災而死后,上了中學的阿一居然趁青宗睡覺的時候嘴對嘴吻他,此作居然有bl,真是難以置信!阿一對赤音的一心一意這一點很不錯,但為了籌錢救她而走上邪道,變得無法自拔和回頭,真是很糊涂。赤音還真是挺漂亮的,氣質頗佳,難怪阿一被她所吸引,被燒了真是可惜。也不知道為啥著火時她沒逃出去,又不是傻子。赤音的眼睛風格有點像艾瑪。
沒想到藍頭發的那個angry居然在哭后突然戰斗力爆表,秒殺天竺的四大天王,太科幻了,我感覺此刻的戰斗力和Mikey都不相上下了。但是他之后卻被阿鶴一擊打飛,真是很迷(不知道是不是angry只在剛變身的時候超強,然后很快就沒電了)。然而顯得強到bug的阿鶴居然打最弱的武小道打半天也沒把他擊倒,真是迷啊!難道武小道的真實防御力和血量都高得離譜?我都完全搞不清楚此作的戰斗力關系了。
這一季ED不錯,挺有激情,特別是一開頭的那一句say my name特別有氣勢。
盾之勇者成名錄 第三季
總評:93.5
盾勇的上一季做得實在拉胯,令我大失所望,看得挺不爽,都想棄了。沒想到這一季的制作質量很不錯,用心程度至少是第二季的兩倍以上,光是看OP就覺得挺上檔次的,不知道這一季的質量大幅提升會不會是因為上一季的失敗而令官方覺得必須在這一季好好認真做一做,要不然這個企劃就毀了。這一季的劇情和看頭比上一季強多了,許多戰斗做得著實很不錯,特別是第9話跟惡龍的戰斗過程做得相當出彩,視覺沖擊力很好,甩上一季N條街。
麥茵那個婊子居然禍害完盾勇禍害槍勇,禍害完槍勇禍害劍勇,禍害完劍勇禍害箭勇,全都禍害一遍。居然槍勇劍勇箭勇都跟腦殘一樣,竟然會那么輕易被她的花言巧語所蠱惑、喪失心智。感覺她的計謀也并不多厲害,我看更可能是由于她的歐派的魅惑力而失去了基本的判斷能力和智商了。這一季一多半劇情就是講盾勇一行人把被禍害的另外三個勇者找回來的事。此外還有參加競技場、買亞朵拉、和黑龍戰斗的事。
槍勇現在太落魄了,眾叛親離,現在變得毫無自尊,那個婊子都唾棄他。居然他在失意的時候被唱歌跳舞的天使般菲洛的可愛所吸引,向這幼女求婚,還管尚文叫岳父,太違和了,這轉折也太大了!這展開真是過于離譜,真是想不到他有這么一天。總之他算是改邪歸正了,還算有救,沒有令我那么討厭了。
之前真想不到那個婊子居然在迷惑槍勇后又纏上了劍之勇者,而劍勇居然意外地巨蠢,輕輕松松就被她的謊言所蒙騙,令我納悶怎么傳送到這個世界的勇者除了男主外都是腦殘。第6話里他的裝備被婊子偷走后,落魄不堪,最后還黑化了,顯得性格好爛。他被漂亮威風劍術高超的紅發女領主好好教訓了一通。意外的是他倆居然組了CP,之后開始總在一起出現,感覺劍勇完全配不上那個女領主。
第6話很奇怪的是,在劍勇被教訓后,突然有兩個雜兵跑出來,然后被槍勇利落地秒殺了。這倆雜兵的行為實在太迷惑了,完全是自己來送死。這劇情真是令人摸不到頭腦,很沒邏輯,完全不知道他倆為啥這時候要出現、想干啥。
令我想不到箭勇川澄樹會如此之中二,超級固執地堅持自己的所謂的正義,干的事跟瞎了眼似的。婊子不僅利用很中二的箭勇的天真的正義感賺了大錢,居然在拋棄他時還以他的名義借了很多債,真是忒惡毒了。
這一季新出現的角色薩迪娜這大姐真棒!非常有風韻,又漂亮又特性感,武藝高強,還挺有善心,對男主還挺心儀。她的著裝特別好,我非常喜歡這設計,低腰,凸顯出特別緊致的腹部線條,并且還秀出她極度誘人的南半球,真是超養眼的角色!這個角色cosplay的話肯定很炫。薩迪娜的性格也很不錯,又成熟又風趣的,而且頭腦正值、有理性。這角色真是里里外外都令我挺喜歡,設定得很棒。沒想到她的真身是只海豚,第2話第一次看到那形象真是感到很出戲。
那個白虎女亞朵拉居然喜歡尚文,偷偷跑到他被窩里同睡,這令她哥大為吃醋,也令拉芙塔莉雅看到后很著急,挺有趣的。亞朵拉一開始因病臥床不起,看起來是個廢人,想不到被尚文買走、把身體養好后居然戰斗力那么強,是武學奇才,會點穴,又聰明,比她哥都厲害得多,并且在和黑龍一戰的時候使出了不知哪習來的大招將黑龍擊破,成了致勝的關鍵,令我更是刮目相看。亞朵拉的顏值也挺高的,如果什么時候開眼的話可能會令人覺得漂亮到驚艷。亞朵拉真是很不錯的角色,設定相當可圈可點,我也感嘆尚文買了她可真是意外撿到寶了。而相比之下亞朵拉她哥就用處就不大了,沒看出有啥長處,戰斗力也很平庸。亞朵拉目前還是瞇瞇眼角色,如果什么時候睜開眼睛,可能戰斗力還要提升不少、獲得什么新技能,也許這是個伏筆。
這一季尚文收獲了薩迪娜和亞朵拉,令隊伍的戰斗力大增,而且還是兩個出色的角色,真心感覺很不錯,此作令我喜歡的角色增多了。
尚文和拉芙塔莉雅之前以及這一季都從未有過戀愛展開。最后一話居然薩迪娜大姐專門把話挑明了,試探尚文是否想讓拉芙塔莉雅當妻子,居然只是換來了尚文“我只把她當女兒”這句話,樂~ 看來在可預見的未來這一對沒什么希望了,感覺拉芙塔莉雅在戀愛感情方面沒怎么發展。大多數異世界動畫里男主都開后宮,但此作的男主尚文真是很正經,好好旅行勇者職責,一點都不花心。他身邊出現的女人雖然不少,但倒貼的不多。我覺得以尚文的魅力,按理說應該能吸引莉希雅喜歡上她才對,居然莉希雅在箭勇還是跟著很蠢的箭勇。
最后一話拉芙塔莉雅的巫女裝很驚艷,如果下一季也一直以這副裝扮出場就好了,真心很適合她。這副刀使巫女的樣子很帥氣。想不到曾經落魄成為奴隸的她居然是九天樓的王族,這設定的出現真是太突然了。九天樓的刺客居然因為她隨便穿上了寓意稱王的巫女服就突然冒出來暗殺她,這劇情有點離譜,都不知道這么多刺客以前藏在哪,完全沒有任何伏筆。收拾做虐的九天樓的人的這個支線劇情,看來就是下一季的主要內容了。
我挺納悶,難道婊子也不想想,得罪了四個勇者,還不早晚得被他們聯合給搞了?也或許這婊子現在有了什么后臺,因此開始肆意妄為了,這可能要在未來的劇情中揭露。總之我特希望看到這廝終有一日被盾勇等人虐到永無翻身之日。
鴨乃橋論的禁忌推理
總評:93.35
此作是個劇情和整體制作質量都不錯的破案推理番。此作的不少案件還蠻有看頭的,犯人們的作案手法總是設計得頗為精妙。
此作的男主鴨乃橋論是個推理天才,還對破案有癮,但每次在抓住真兇時都會被動地發動超能力導致犯人自盡,因而無法從事偵探職業,只能在偵探學校畢業后一直荒廢技能,一個人頹廢地獨居。直到有一天,警察遇到了始終無法破獲的連環殺人案,愣頭青警察一色都都丸被老警察建議去找鴨乃幫忙,這促成了鴨乃重新出山的契機。成功合作后鴨乃還對天真的都都丸產生了興趣,覺得他正是自己所需要的搭檔,此作之后就是講他倆一起不斷破各種疑難案子的故事。由于鴨乃橋論不能作為名義上的偵探,因此每次都是他進行分析推理并告訴都都丸,以都都丸為名義破案。
都都丸的人設普普通通,性格還不錯,但沒什么閃光點,只是個稱職的警察而已。而鴨乃橋論的人設相當突出,個性鮮明,特立獨行,平時樣子邋遢但頭發偶爾掀開時顯得相貌格外帥氣,頭腦極度靈活,推理和預見能力極強,有非常敏銳的觀察力,真是個頗為神奇的人,充分展現出推理奇才的感覺。橋論和都都丸總在一起,二人都沒有女性伙伴,而且二人都把彼此看得極其重要,因而盡管此作沒有什么露骨的bl描寫,但無疑會有bl本子出現。
此作一個挺bug的地方是每次鴨乃橋論識破真兇,然后發動被動技能令其自殺居然都都丸就一直在那觀望,總是到最后最危機時才出手阻止。既然他都知道鴨乃橋論有這個毛病,咋就不能早點做準備,當真兇剛被接觸按時就準備攔住他?
警視廳的女長官雨宮的人設不錯,形象干練,性格直率,看起來是女精英、很獨立的她卻喜歡鴨乃橋論,令人意外,不過這感情不可能開花結果。鴨乃橋論意外地看到她果體的那一幕里她的身材顯得很不錯,相當白皙圓潤。
那個叫翡翠臣疾的精英刑警的設定很有特點,待人特別苛刻,總是拿著彎頭鑷子挑別人的毛病,這道具的構思很有趣。
第12話橋論到了一個村子尋找修比茲失蹤的哥哥,那里的村民都在抗議zf在當地建立水壩的項目,然后派來的公務員不知怎么回事死掉了。這劇情實在太太太像《寒蟬鳴泣之時》了!不知道此作的作者是否有所借鑒。
雖然此作的案件是獨立的,但是主線在后來一點點浮現,牽扯到橋論的血緣、世界最強犯罪組織,以及他在偵探學校被陷害的真相,下一季可能會更精彩。居然最后一話連福爾摩斯和莫里亞蒂的名字都出現了,令我立馬想到《憂國的莫里亞蒂》。
此作OP歌曲難聽,ED歌曲也不怎么樣,不過ED剛開頭的那幾句清唱結合著人物特寫帶給我的感覺很好。
賽馬娘 Pretty Derby 第三季
總評:93.3
這一季主角又變了。看前作時我挺喜歡的可愛的小馬駒(當時感覺還有點像nico)北部玄駒長大了,成了這一季的女主。此作講述她作為spica隊伍一員的成長歷程,從起初有一定實力但和top還有顯著差距的狀態,逐漸靠努力成為了這個時代最強的賽馬娘,取得了極其輝煌的成就,甚至都達到了略微超過東海帝皇的高度。
雖然小北現在這個形象倒也還行,但還是感覺外形遠沒有小時候那么可愛了,尤其是發型顯得比較亂,令我是稍微有點失望的。這一季制作質量依舊很不錯,有很多可圈可點之處,但是也有很多劇情令我感覺略微普通,沒之前的作品劇情那么扣人心弦、吊人胃口、喜聞樂見或者有話題性,總的來說看點不如幾季多。另外,除了spica隊伍里的角色外,這一季其它的賽馬娘大多令我沒啥興趣。
小北最大的優點就是超級拼命,總是靠不屈不撓的毅力強行克服困境,在比賽時也異常的拼,這個閃光點使得她從觀眾那里獲得了極高的人氣,長期霸占人氣No.1。她的內在并不算多有個性、性格在我眼里不算特別有魅力,不過優點是很爽朗,很open,很明亮,也很熱心,這使得她和父老鄉親們的關系搞得很好,有好多大爺大媽成了她的堅定支持者,這些方面在此作中做了著重描寫。觀眾和父老鄉親們對她的熱忱和堅定的支持成了她不斷拼搏的動力。
小北居然發育得意外的好,沖刺時候看她的脂肪搖擺得挺兇猛的,絕對在F以上。
小北起初在自信滿滿地出道后很快就遇到了嚴重挫折,實力離大鳴大放差得太遠,我本以為大抵這一季要講她怎么一點點努力去追趕大鳴大放并重新樹立信心,從而最終能有實力一決高下。沒想到后來大鳴大放就萎了,淡出了劇情。雖然二馬最終并沒有再次同臺對決,但無疑起初氣勢洶洶像BOSS一樣的大鳴大放的實力已經明顯不敵小北了。大鳴大放的性格顯得很冷淡,充滿野性和霸氣,完全沒有可愛的女孩子的感覺,不知道她比賽勝利后的live表演是什么樣,她若穿著漂漂亮亮的idol服裝在臺上蹦蹦跳跳一定很違和,估計表演是cool風格。
第5話的比賽結果真是離譜,居然小北和大鳴大放都沒得第一,竟然殺出個名不見經傳的光明河流,這結果真離譜。雖然她得了第一,卻沒什么存在感。不知道光明河流什么來頭、以后的動畫里會不會專門描寫一下。經過這一賽,之前完全不把小北放在眼里的大鳴大放終于對小北刮目相看了,還建立了友誼。
之前基本每一季的主角都有個CP,這一季小北的CP是里間光鉆,二人感情很深厚,十分要好和看重彼此,不過二人的百合元素不明顯,我對光鉆這角色也完全沒興趣,因此我對這對CP也不怎么推。
從外表來看我本覺得光鉆應該也沒有多強,居然第6話她以壓倒性的優勢獲得G1冠軍,挺出乎我的意料的。第9話不錯,總算小北拿了天皇賞第一名,令光鉆知道小北有多強。我也沒想到小北的實力能成長得如此之快,而且上限非常高,竟然還打破了記錄。當年的可愛的小馬駒能達到這樣的高度,想來挺不可思議的。
光鉆參加凱旋門賞居然只得了第15名,太丟人了,不知道算是倒數第幾。我心想好在小北沒參加凱旋門賞,她的實力比光鉆也強不了太多,到那里如果因為實力原因或水土不服原因最后排在十名開外,還可能給她留下心里陰影,弄得她羞于面對家鄉父老。似乎光鉆因為這個比賽失利,導致一蹶不振了挺長時間。
我感覺小北明明還很年輕,居然在凱旋門賞之前就已經過了巔峰期了。相比之下,和小北同期的其它馬娘似乎都還沒過巔峰期,不知道怎么就小北的巔峰期過得那么快。
最后一話做得很優秀,講述將要隱退的小北最后的比賽。這場比賽很驚心動魄,這大抵是小北此生最拼的一次,沖刺時面容都完全猙獰了,想不到這是可愛的小北能露出來的面孔,在表情管理上已經完全不管不顧了。最后小北總算拿了第一,成為了她最完美的隱退之戰,這令我很是欣慰和欣喜,也令觀眾和父老鄉親們欣喜至極。這勝利可謂是眾望所歸,有點像阿根廷拿下世界杯一樣。獲勝后小北作為center的live演出的畫面做得相當優秀,近距離的舞蹈全手繪,非常流暢,這可真是燒錢,太良心了,可能是把剩下來的所有經費都搭進這里了。
這一季里能看到前作出場的spica隊伍里所有角色,這點不錯,她們都很支持小北。再次看到特別周,感覺她真可愛,也再次看到東海帝皇,感覺她很閃耀。可惜她們都只是打醬油,估計都是處于養老狀態,一次都沒參加比賽。
這一季有一話第一次認真描寫了黃金船這個角色。雖然之前她都只是打醬油,但早就令我產生了興趣。想不到她如今的實力平平但積累的人氣卻頗高,我原以為她只是人氣很一般的邊緣角色。這一話出現的過場圖不錯,黃金船顯得英姿颯爽。這一話的好幾副圖里里還出現了黃金船過去的一些嚴重出戲行為和不可描述的顏藝,第一次令我了解到黃金船是這么離奇的馬娘。
看此作的過程中我有一回突然覺得服裝不應該花里胡哨,應當盡可能減小空氣阻力,按理說該穿比基尼才對。
大小姐與看門狗
總評:92.9
此作的女主瀨名垣一咲從小父母出事故雙亡,于是被黑道的祖父養大,組織中年輕的黑道男子宇藤啟彌從那時起負責照顧她(這種黑道男子養女兒的設定之前就有過其它動畫用過,但此作和那些明顯不同)。如今15歲的一咲剛上了高中,現在已經26歲的啟彌靠關系也混入這所學校,擅自作為她的男同學以守護她,以免有男同學向她伸出魔爪。此作內容講述這兩個人的感情發展,在經歷各種故事后,他倆從起初的有點父女或者哥哥妹妹的關系,逐漸轉變成了戀人的關系。
此作劇情挺有趣的,一咲和啟彌的人設也挺不錯,是個值得一看的題材很特殊的戀愛作品。此作比較偏女性向,是典型少女漫畫的感覺,啟彌顯得像黑道的白馬王子,對一咲保護得無微不至,能充分滿足女生的被保護欲。同時啟彌也顯得有點像狗,有時候還向一咲撒嬌求寵,這也迎合了不少女性想寵愛自己的對象的想法。
此作的最大缺點是作畫,且不說很多地方人物神情表現不到位,很多畫面還崩壞嚴重,看起來非常詭異。啟彌畫得有點丑,而一咲本身設定得很美,但許多畫面里卻看起來怪怪的。畫面是此作的明顯減分點,沒這樣缺點的話此作能不低于93.3分。此作作畫監督是國人,而且原畫人數很少,看得出預算很有限。
一咲設定得很不錯,眼睛很大,皮膚白皙,清秀漂亮,性格溫柔,靦腆內向,顯得楚楚動人,令人有保護欲,里里外外都挺討我喜歡的。鬼頭明里的聲線把一咲的性格表現得很好,還有種很好的微妙的朦朧和慵懶感(稍微有一點點近江彼方的style)。第7話一咲在額頭前面編兩個辮子分開的發型不錯,挺有新鮮感,比平時她的中分顯得更漂亮。她的嘴的畫風總是有貓嘴的感覺,有時候顯得略怪。
一咲雖然是個弱女子,但是在一些關鍵時刻倒也表現得不錯,不簡單。她被壞人綁架、要被田貫侵犯時,并沒有驚慌、尖叫,反倒比較淡定,在最佳機會給對方要害暴擊,使得啟彌有機會救她。
啟彌看起來相當成熟穩重,假扮高一學生實在太違和了,扮演成老師我看還差不多,沒準他比班主任歲數都大。他還戴耳釘,學校居然都不管。他居然還渾身是紋身(黑道標配),為了隱藏紋身不被發現也挺不容易,游泳課都得穿連體的泳衣。啟彌的身體素質和能力近乎超人,體格強壯,肉搏和射擊都精通,能輕輕松松捏爆一咲身邊的害蟲,只要他在旁邊一咲就絕對安全。
啟彌經常顯得有點愣頭愣腦的,甚至腦子少根筋,第12話居然在愛慕他的女同學的要求下親了那個人,把偷窺的一咲氣壞了。而且在一咲發自真心向他表達感情時,啟彌馬上就要撩她的衣服,感覺就像是被下半身控制的生物一樣,也令一咲不高興。這人真是太奇葩了,一點都不懂女孩子的心思。
啟彌似乎平時經常玩弄異性,對此毫不顧忌,居然對班主任女老師都施展男色,差點解了她的泳著。第11話風俗店老板娘還揭露了啟彌曾經和自己的rou體關系,真是令人吃驚。啟彌表面上顯得一本正經的,但其實也夠風流的。但盡管如此,啟彌倒是只對一咲一個人動過真情。
感覺啟彌挺會濫用自己的看門狗的設定的,對一咲在肢體接觸上肆無忌憚,比如第6話直接在她床上抱住她,第8話他坐著把一咲抱住,身體彼此交叉,重要部位都接觸上了,然后還把頭埋在一咲柔軟的部位上吸。第11話他居然還把一咲抱上床要強行接吻,他伸出舌頭被一咲推開的樣子巨鬼畜和滑稽,啟彌表現得真是像發q的大狗一樣,真是頗有獸性的一面。明明名義上還不是戀人,啟彌卻干出這種事,簡直跟明目張膽耍流氓一樣,他的膽子還真大。似乎他也是由于自己的特殊的監護人的位置顯得有點有恃無恐,不擔心會被一咲揍。
劇情中啟彌和一咲的關系變化是重要看點。啟彌是看著一咲從小女孩一點點長大的,因而在故事一開始,啟彌仍只把她當小孩子看待,表現得像是父親關愛和守護女兒。一咲雖然喜歡啟彌,戀愛感情已經萌發,但她認為啟彌對她不可能有戀人般的喜歡的感覺,同時啟彌對她表現得也沒有對異性的距離感,因而一咲起初認為啟彌始終只是以監護人的立場看待她,就沒有考慮過和啟彌交往的可能。不過,如今一咲已經從小孩漸漸長大成了亭亭玉立的少女,從來沒真心喜歡過女孩的啟彌天天和她在一起,顯然不可能對她一直沒有想法,果不其然二人的關系逐漸發生了轉變,感情和身體帖得越來越近。
雖然一咲一開始并沒有主動向啟彌表示出愛意,但啟彌卻經常變相甚至直接地撩她,老弄得她羞澀臉紅,還說不許任何男人把她搶走之類的話。這種戀人才說出來的話語,到底啟彌是懷著什么樣的心情和想法說的,到底他是怎么看待一咲的,令一咲捉摸不透,導致一咲對啟彌感情的流露也是半遮半掩,關系顯得非常朦朧和曖昧,這模糊的關系正是此作最有戲劇性的地方。而啟彌也一度弄不明白自己對一咲是怎么想的,但明白她對自己來說是極為與眾不同的,不能容忍有其他男人靠近她。到了故事中期,二人都明確知道自己喜歡對方了,都間接親對方了,但始終雙方都不點破關系,挺令人著急的。好在是后來此作沒有像很多作品一樣在這個階段停留很久,主角沒有表現得特窩囊,而是最終確認了感情。而且還是一咲主動先表達的感情,有點令我沒想到這是羞澀內斂的她會做的事。不過話說回來,其實啟彌倒是早就表達感情了,只不過是用身體接觸來表達的。
第7話出現的跟一咲同齡的黑社會少爺田貫干男很討厭,輕浮得不得了,居然一開始就對一咲毛手毛腳的,心機挺重,試圖占有漂亮的一咲但對她又不是付出真心實意的戀情,還破壞她和啟彌的關系。這家伙在此作里被形容成貍貓,十分貼切。要是看到他被啟彌好好教訓一頓重新做人就好了。他要是玩玩也就罷了,第8話,居然他還把小刀頂在一咲的脖子上,太過分了!我覺得啟彌就應該把田貫強吻了,表現得自己像gay佬,把他惡心到,又顯得偽同性戀的啟彌對一咲不是戀人般的喜歡,田貫肯定自己就主動跑掉了。一咲對田貫的態度過于軟弱,這點令我覺得窩囊,田貫毛手毛腳過分時一咲就該給他一巴掌,要不然他覺得一咲好欺負,騷擾顯然會變本加厲。
一咲太圣母了。第9話田貫當著啟彌的面要侵犯她(劇情被我精準猜中了,我就知道田貫肯定要干這事,一方面自己爽了一方面還令啟彌極度受辱),還拿刀對著她的脖子,被一咲踢了要害后脫險。居然之后一咲那么容易就原諒了田貫,理由是沒了他舞臺劇就沒法演了,這邏輯很迷,難道舞臺劇演出成功比自己的人身安全還重要?田貫真是惡心的要死,一咲應該多踢他要害幾腳,最好踢廢了,把那里踢炸了,以此作為其懲罰。之后田貫顯得完全改邪歸正了,主動把自己演羅密歐的位置讓給了啟彌(PS:啟彌演羅密歐的樣子真是很帥),不知道田貫到底是怎么想的,最后也沒徹底交代清楚,令我納悶他挾持一咲本意是為了試探他們的關系,還是真的就是因為愉悅目的要做出犯罪行為。
家里蹲吸血姬的苦悶
總評:92.7
此作的女主可瑪麗是吸血鬼將軍家的女兒,但她不肯喝血,導致能力不行、發育受阻,個子小小的。她之前一直處于家里蹲廢柴狀態。有一天突然來了個女仆薇兒赫澤闖入了她的生活,并且可瑪麗突然得知自己已成為了七紅天大將軍之一,從而不得不帶兵打仗。從此薇兒赫澤就開始監督可瑪麗的生活作風,禁止她繼續懶散,并幫助廢柴的她偽裝成了不起的統領的模樣,各種給她救場和圓場,還確實取得了威信和戰果。此作的劇情就是講她倆共同經歷的各種各樣的事情,一起跨越了很多困難、度過了許多危機,在此期間感情不斷加深。之后又有兩個妹子佐久奈和娜莉婭出現在了可瑪麗身邊并且愛著她,最終可瑪麗算是有了三個后宮。
此作前期制作質量不錯,內容有趣,百合度極高,妹子們的作畫很精致,特別是可瑪麗的面容真心令我覺得很養眼。但是到了后期,感覺制作變得漸漸不太走心,崩壞愈發明顯,并且后期劇情有點混亂,且出現了一些我不怎么喜歡的角色,令我有點失望。此作前期我能打93.4分。
可瑪麗的人設很不錯,這是此作關鍵的優點,令我一看就喜歡。她的外形顯得嬌小可愛,擁有超一流偶像般的顏值,金發很漂亮,腦袋兩側兩個錐形的小角進一步增添了萌點。可瑪麗起初特別頹廢不肯上進,這個樣子非常有雙葉杏的感覺。她還挺會賣萌,做出羞答答的樣子時相當撩人。她的CV是楠木燈,感覺在此作里的發揮一般,氣勢弱了些(但后來聽習慣了倒也還行),如果讓釘宮來配可能會明顯更好。
可瑪麗內在挺善良的,這點很不錯。雖然她自己很弱小,但看到陷于痛苦的人不會袖手旁觀,曾經在學校時看到受欺凌的陌生人都不顧自己可能面臨的危險而出手相助。雖然可瑪麗平時的性格非常懦弱,但到了面對生死存亡,最需要鼓起勇氣的時候,卻比誰都勇敢。第4話她救薇兒赫澤和惡女布魯奈特殊死對決的那一幕很不錯。可瑪麗扔寶石和惡女戰斗的樣子令我想起了遠坂凜,想不到家里蹲的她居然還能如此利落地戰斗,刷新了我對她的認知。
第4話可瑪麗喝了薇兒的血后烈核解放,相當于小宇宙爆發,想不到懦弱的家里蹲可瑪麗居然也能有這么帥氣的一面,和平時的形象真是兩個極端。她把布魯奈特的胳膊斬下來的場面看得我很有快感。最后她把惡女的脖子捏爆,甚爽,我還以為可瑪麗會因為圣母心留她一命。布魯奈特真是太惡劣了,上學時候欺負弱小,長大后拿著匕首倒處捅人,必須遭報應,感覺她遭的報應還不夠多。
此作里的人似乎都能靠魔核無限復活,這設定挺離譜。我搞不明白魔核到底長怎么樣、在哪,對復活有什么限制。如果不管什么情況掛了之后靠魔核就能復活,還認真打什么啊,戰爭豈不就跟鬧著玩似的?這個設定令我感覺挺迷。幸好有這個設定,可瑪麗三歲的時候因為不慎烈核解放把在場的父親都殺了,要是不能復活就糟糕了。也可惜由于這個復活設定,導致布魯奈特在最后一話又出現了,感覺她還是挺囂張的,要是徹底完蛋就好了。
此作的另一個亮點是薇兒赫澤的人設,非常出眾,相貌非常精致養眼,歐派很大,玉腿修長,真是美如畫,她是此作里和可瑪麗我同等喜歡的角色。薇兒對可瑪麗忠心耿耿,雖然起初可瑪麗對薇兒完全沒印象,但薇兒曾經在學校時就因為可瑪麗幫助了受布魯奈特欺凌的她而喜歡上可瑪麗了,并由此下定決心成為可瑪麗的女仆。此作里薇兒作為女仆對懶散的可瑪麗的生活起居總是顯得很苛刻和嚴厲,甚至有時冷酷無情,但實則內心喜歡可瑪麗不得了。她內心里有著和表面形成截然反差的重度癡漢的一面,第1話里她看到可瑪麗紅著臉對她說“你說什么我都聽啦”的時候興奮得猛噴鼻血,然后又摟又抱,完全沒了平日的矜持,很有趣。薇兒赫澤還沒什么羞恥心,居然第1話還在可瑪麗床上一絲不掛地果睡,第2話果穿圍裙。薇兒平時的情緒總顯得很冰冷,有點機器人的感覺,但由于她總展現出對可瑪麗的癡心和癡態,這冰冷的形象在此作中沒多久就完全破滅了。薇兒赫澤X可瑪麗真是非常好的CP。
經歷第4話和布魯奈特慘烈的一戰后,可瑪麗和薇兒更增加了一層生死之交的關系,哪怕就算薇兒想走,可瑪麗都不肯了,還明確對她說“要一直陪在我身邊”,被紅著臉的薇兒吐槽“這是求婚嗎”,這可不就相當于求婚!真是很甜!
薇兒穿女仆服的時候我還沒特別注意到她的歐派,只覺得她的腿很漂亮。第9話薇兒的泳著真是把我驚到了,沒想到竟然又大又白到如此夸張!滿分!要是薇兒多穿著一些露出度較高的服裝就好了,女仆裝不夠顯身材。
我對佐久奈的感覺一般,不怎么喜歡她。可瑪麗倒是很喜歡她,感覺她很可愛,佐久奈稱可瑪麗姐姐的時候更令她被射中紅心。可瑪麗對她的態度令薇兒很警覺和不爽,要是可瑪麗也這么欣賞薇兒的美貌就好了,在我來看明明薇兒明顯更美。佐久奈和可瑪麗正好相反,可瑪麗是外強中干(&內超強),而佐久奈是外干中強。可瑪麗始終不敢展示自己孱弱的戰斗力居然反倒令佐久奈覺得是真正強大的表現,很巧。
第6話出現了佐久奈的房間,想不到她竟然喜歡可瑪麗到顯得像hentai一樣,屋子貼滿了可瑪麗的照片,有等身抱枕,還有十幾個等身人偶,實在太癡狂了。可惜她的家人被惡勢力逆月黨挾持,居然要殺死可瑪麗,其實明明能找到更好的方法的。最后可瑪麗把控制佐久奈的逆月黨的大叔抹殺的那一段不錯,可瑪麗吸了佐久奈的血之后居然頭發變成白色,結合微微發光的赤瞳,顯得無限強大又有些神圣感(令我想起了《十字架與吸血鬼》的白萌香,同為吸血鬼),最后她把大叔脖子瞬間干凈利落地捏斷的一刻又爽快又令我感到有趣。這一戰幾天后,佐久奈說“可瑪麗小姐 能懲罰我嗎?”,居然可瑪麗說“那就和我交往吧”,這可真是夠離譜的!居然還當著薇兒的面對佐久奈說,想不到可瑪麗居然如此調皮和不懂事,太傷薇兒的心了!薇兒看著她倆親密地握手時都哭了,真可憐!
第10話里看到,居然娜莉婭從小時候第一次見到可瑪麗就喜歡上她了,如今又對她親上去,把旁邊的薇兒驚了,挺有趣。居然娜莉婭說那是卡爾卡一貫的問候方式,薇兒立刻機智地試圖以同樣的方式問候可瑪麗,可惜她完全不接受。第11話,居然可瑪麗讓娜莉婭吸自己的血以獲得力量的一幕表現得那么姬情,簡直像是告白+接吻的場面,可瑪麗竟然如此信賴她。不知道薇兒在旁邊在干啥,可能還處于昏厥狀態,她要是知道這二人有這一幕肯定垂死病中驚坐起。第12話,娜莉婭自豪地說“我跟可瑪麗是共享血液的關系了”,還引誘可瑪麗吸她的血,顯得太姬了。娜莉婭在可瑪麗面前總是一副愛她的姐姐的姿態,似乎有意以這種身份哄和誘騙可瑪麗和她毫無距離感地在一起。
女王這個角色設定蠻不錯,挺有意思的。她十分隨性,挺老謀深算的,她似乎知道可瑪麗烈核解放是多么的無敵,因而很放心地利用她。她的造型挺贊的,整體顯得挺華麗的,歐派也太大了,太夸張了。她雖然身居女王要位,卻表現得很不正經,居然似乎還是個姬佬,光明正大地覬覦可瑪麗的身子,真是女王失格,太不穩重了。
可瑪麗手下有個紅頭發的將領,對她很是赤誠,居然第2話還說希望得到她獎勵的玉足狂舔券,真變態!
可瑪麗的母親只在回憶中出現過,不知道現在哪去了,也可能是我漏看了。她母親的形象很出色,又漂亮又高挑,有些御姐氣質,要是作為此作的主要配角能夠多出場就好了。
此作里有可瑪麗的周邊,是印著她一臉傻笑的白T恤,每次我看到這圖案都覺得很出戲。淘寶上一搜,果然有賣這個的。
此作ED的歌曲雖然說不上很動聽,但是那種溫柔月夜的韻味感表現得不錯。
哥布林殺手 第二季
總評:92.7
之前看哥布林殺手第一季和劇場版感覺都很好。這一季男主和他的小隊繼續在執著地殺哥布林,期間又經歷了各種各樣的事情。這一季評分沒之前高,主要是覺得這一季劇情吸引力整體來說感覺沒上一季高,從中期開始制作質量有點粗糙、畫面拉胯,甚至第8話連劇情都有點沒邏輯,也不知道女神官對著哥布林薩滿使出來法術令其歇菜后她在那里哭什么。不過下一季我肯定還是會繼續追。
上一季我就對圣女這個角色的美麗豐饒的外形設定印象挺深的,這一季她還成了個挺重要的配角,很難得,更令我喜歡她了。她的身材可真棒,個子高挑,大長腿,巨r,細腰,而且性格溫柔,還會倒貼。一直以為她完全沒有戰斗力,沒想到最后一話使大招壓制了一堆哥布林。
這一季里圣女對哥布林殺手的戀愛感情有充分描寫。第5話看到她對男主臉紅,對他依依不舍,當時令我覺得真可惜男主不解風情,對殺哥布林太執著了、腦子里只有哥布林。第9、10話,圣女對男主更是倒貼得極其露骨,一臉緋紅,感覺都恨不得把自己的美好的歐派貼上去直接誘惑他了。要不是男主一直帶著頭盔,估計圣女都要對他親上去。面對如此魅惑的圣女,男主依然對她完全沒有任何想法,還是只想著殺哥布林,唉。似乎不把世界上的哥布林除盡,他就沒有和誰談戀愛的可能,但哥布林就是無窮無盡。
精靈弓手這個角色一直令我感覺不錯,長相清秀漂亮,身材纖細,性格也不錯,挺開朗和挺利落的,從來不磨磨唧唧,對戰斗還有重要作用。她對哥布林殺手的感情沒有描寫得很明確,感覺目前對他并尚沒有暗戀的感覺,只停留在把他當做可靠也挺喜歡的隊友的階段。畢竟她的壽命遠比人類要長,對人類可能很難產生出愛情。
第8話居然精靈弓手在中了毒霧后被哥布林用刀戳,按理說應該立馬就掛了才對,但居然沒事,也沒有永久性傷害,真是走運但也蹊蹺,令我納悶難道哥布林的攻擊力那么弱?還是說故意不把她殺死以供之后那啥?不過她倒是被打到爆衣了,關鍵部位差點就要暴露了。
精靈弓手她姐在這一季第一次出場,還蠻漂亮的,有種神圣的美感,不過性格不太理想,由于偏見還不想讓自己的妹妹和優秀的男主在一起。
女神官在這一季利用地母神的力量,在戰斗中派了不少用場,甚至成為了團隊脫險的關鍵,作用已不僅限于治療了,表現得整體還不錯。感覺她還是應該練練肉搏,要不然什么時候若趁隊友沒注意,有哥布林偷襲她的時候,她完全招架不了也不行。從最后一話可以看得出,她對哥布林殺手已經有不可忽略的喜歡的感覺了,都已經想一直陪著他了。
第5話里,一直心儀男主的牧場妹子總算換了件衣服,顯得蠻漂亮的,和平時判若兩人。第6話出現了她的泳著,真是給力,巨猛。相比之下精靈弓手完全是殘念,size還沒她的1/10大,倒是和女神官半斤八兩。
這一季里柜臺女已經明顯對哥布林殺手顯得有意思了,我挺希望看到她什么時候也像圣女一樣主動倒貼。我以為她始終都是不會進入主要劇情的次要小配角,沒想到她在這一季也進入了主線,和哥布林殺手他們一起去參加精靈弓手姐姐的婚禮,還看到了她的泳著,感覺很難得。她的胸懷也不容小覷。
雖然哥布林殺手完全不解風情,也不會說任何漂亮話、討女孩子歡心,但還蠻令長期接觸他的妹子們中意的。現在柜臺女、牧場妹、圣女全都明確喜歡著他,女神官和精靈弓手雖然還沒正式對他沒表現出顯著戀情但也無疑是潛在后宮。
第10話,國王的傻妹妹居然偷了女神官的衣服,跑到城外試圖當冒險者,結果中途被哥布林給埋伏了,一猜之后就要被哥布林糟蹋,為自己的愚蠢付出代價。要是身份高貴的公主被哥布林給糟蹋了的事傳出去,那皇族真是徹底丟死人了,完全失去威嚴。第11話,看到被綁到祭臺上的她的衣服還在,也不知道到底被糟蹋了沒有。第12話看到她的眼神空洞,令我懷疑可能確實已經被糟蹋了。
最后一話突然出場的鉆石騎士不知道是啥來頭,感覺像是個伏筆。這一季還出現了三個妹子組成的隊伍,看似戰斗力高強,但也沒交代身份,也沒和哥布林殺手的隊伍產生交集,令我挺好奇的。
第1話新出現的那個不知天高地厚的紅毛小子的性格真招人討厭,明明是個什么都不懂的新人冒險者,對老前輩還不謙虛,真希望看到他自己一個人去殺哥布林時被圍攻并被干掉,為自己的狂妄付出代價。我挺不想看到此作加入這么個沒魅力又制杖的角色。看這一季一開始還擔心這小子會成為這一季的主要角色,好在是過了幾話后就見不到了。
不知道殺哥布林何時是個頭,哥布林的數目一點都沒少,也沒找到源頭。我要是國王,就讓軍隊沒事就倒處掃蕩哥布林,將哥布林完全清零。
此作完全沒出現母哥布林,不知道哪去了,亦或是從外形上無法判斷公母?不知道哥布林是怎么出生的,不由得聯想到哥布林抓人類的女性,而且抓得那么積極,就是為了用于繁衍,但這樣的話為什么生出來的都是哥布林就不知道了。我有時會擔心哥布林殺手小隊里的女神官和弓箭手什么時候失手,被哥布林捉去,然后就出現糟糕的事了...
總感覺哥布林殺手的隊伍里缺乏一個會大規模殺傷性魔法的法師,就像魔炮少女那種,或者會使用流星雨那種魔法的。沒這樣的角色的話,每次面對一群哥布林時,一個個殺太費勁了,效率也太低了。
破滅之國
總評:92.45
此作的世界里上帝創造了人類和魔女。人類在古代一直順從于擁有魔力的魔女,但自從人類科技革命,獲得了超越魔女的能力后,就開始倒處獵殺魔女。魔女克羅耶曾經撿到了還是個男孩子的阿德尼斯,并成為他的師傅兼家人將他養大。雖然阿德尼斯只是個未熟的孩子,但在他眼里克羅耶就是自己最喜歡,而且還是帶有戀愛感情的最喜歡的人。但不幸的是克羅耶作為魔女終于也沒能逃過被人類消滅的命運,國王凱特在眾目睽睽之下將她的衣服撕爆,然后將她崩死了并且還讓小兵虐遺體,這沖擊性的一幕給阿德尼斯心中留下了極度仇恨的種子。此作之后主要就是講具有極強書寫魔法天賦的阿德尼斯對這個國家復仇同時也被追殺的故事,期間魔女多羅卡結識了他并一直喜歡和追隨他。
此作總的來說比較虎頭蛇尾。一開始幾話做得很精良,劇情還各種出現驚人的場面和超展開,令我明顯感到是個高水準動畫,但到了中、后期開始變得粗糙,第10話的戰斗甚至顯得沙雕、很迷。這導致最終我對此作的評價不太高。如果整部作品質量都能像前幾話一樣,估計打分能有94左右。
此作在死人方面夠快夠利落的,也沒有什么老套的救場,此作尺度也蠻大的,有不少血腥畫面。比如第1話克羅耶就那么直白地被活生生地亂槍打死,第2話剛具體描寫不久的多羅卡居然那么快就被亂槍打沒了,第5話保安局局長一個大招把多羅卡以外的魔女全都斬成兩半了(可惜喜歡她的那個藍毛妹子也因此掛了,百合CP沒了)。
克羅耶又溫柔又美麗又強大,是個相當出色的御姐,令我極其遺憾的是這么好的角色在第一話就被寫死了。我早就有預感她要掛,果不其然,死得真是太可惜了!如果她是此作的女主多好!雖然克羅耶是為了保全阿德尼斯而給國王下跪,但這個舉動在我心中還是多多少少令她的堅毅的形象有所減分。畢竟她當時只是失去了魔法而已,靠肉搏也能給國王帶來一些不可逆的損傷,還不如在生命的最后拼一把。
一個堂堂國王,居然撕毫無抵抗力的女性的衣服,真不要臉,克羅耶怎么不當眾給他一個大逼兜呢?或者插爆他的雙眼。這個國家的yi shi xing tai真是極度扭曲,上梁不正下梁歪,惡心的國王的統治也導致此國的國民內心變得和他一樣丑惡,希望在此作里看到這些民眾因自己的邪惡愚昧而遭到狠狠的報應。后來這個國王中了魔女多羅緹拉的愛情魔法,表現得像下賤的狗一樣,最后自己跳樓掛了,大快人心。
阿德尼斯心中充滿了仇恨,殺敵十分殘酷且不擇手段,冷酷無情,毫無憐憫之心,此作前半甚至把他描寫得很像反派。他把復仇的對象肆意擴大化,殃及那個國家的所有普通民眾,這就過分了。實際上只要把軍隊、高官和國王干掉就夠了,如若能找到當年那些在底下湊熱鬧的好事者可以也都消滅。不過看到后面,感覺人類和魔女在常年的戰爭過程中,心態都變得十分扭曲,都有點無可救藥,多羅卡想要他們和平共處實在是很難,全都滅絕倒是簡單痛快。
雖然阿德尼斯對于害他、想利用他的一方是極度殘忍無情,但是此作里他對于受害者倒也展現出一定溫柔,畢竟他也是受害者。
阿德尼斯對克羅耶真是一心一意,對她既是特別的尊敬,對她也有深厚的戀情。克羅耶就是他的世界,為了克羅耶,他既可以與人類為敵,也可以與魔女為敵。他在第7話甚至說“克羅耶的性命比全人類的命都還要高貴,奪走她生命的這個世界不如毀了算了”。本以為克羅耶真的能復活,可惜生命樹被阿德尼斯燒了,以后就真是復活不了了,太可惜了。此作的下一季我都不太想追,如果克羅耶能復活,可能我才會繼續追。
阿德尼斯把物體巨大化的能力很不錯,能把子彈變成戰艦的大小。還有反轉的能力也很厲害,把敵人的子彈都反向打回去。他施法都靠筆,那根筆的耐久度還真夠高的,如果哪一天筆尖折了就完了。
多羅卡和阿德尼斯真是兩個極端,多羅卡是完全的博愛,堅信人類和魔女可以互相溝通消除戰爭,相當圣母心,在我來看圣母得過頭了,太傻白甜了,現實和人心遠比她想象得殘酷。她被阿德尼斯的殘忍行為氣到、失望多次,阿德尼斯則認為她天真幼稚,這倆人都太過激了。不過在二人旅途過程中,想法水火不容的他們逐漸了解了對方,逐漸相互調和,都改變了最初對對方的看法,到這一季末尾時彼此都成了自己相當重視的人,阿德尼斯變得也比之前溫順多了,感覺這倆到最后還算比較有愛。多羅卡對阿德尼斯真是極度倒貼,哪怕阿德尼斯展只現出一點點善意和溫度都能令她心花怒放,也不知道是為啥。
想不到多羅緹拉居然成了偶像女王,靠著美貌&超頂的身材以及控制人心的能力令國民對其如癡如醉,就像宅男fans心中的女神,這做法可真行。她本身是魔女,卻利用魔女的能力消滅了其它魔女,成為唯一的魔女以及人類的主宰,還挺有想法的。
那個白兔先生真是惡心得要死,樣子丑陋,性格卑劣,很納悶他身體里怎么能放出來那么多火箭手,會令有密集恐懼癥的人十分不適。居然第10話多羅卡的眼睛就那么容易被火箭手給戳瞎了,太扯了,她當時身子又不是不能動,居然也不反抗一下(比如用腳踢)。更想不到居然她的倆眼珠子被完整地掏了出來,竟然還被白兔當溜溜球玩,那一幕太鬼畜了。當時我心想還好,眼珠子是完整的而不是被戳破的狀態,說不定真有辦法塞回去,不料緊接著就被白兔捏爆了,這是完全沒復原希望了(只能期盼諸如用什么魔法令多羅卡的靈魂替換到多羅緹拉身上,由此重見光明)。這段劇情真是令我看得很郁悶。
那個大和局長令我很討厭,耳垂巨大,難看死了,性格也討厭。被阿德尼斯一刀扎穿了腦袋看得我挺爽。
特搜組大吾 救國的橘色部隊(12話之后沒繼續追)
總評:92
此作主要講述東京消防廳的三名隊員,十朱大吾、斧田駿和中村雪經歷的故事,劇情中有訓練也有現場救援,還有人物關系和情感的描寫。此作制作質量還不錯,內容挺有新意,不少劇情挺有看頭,不少救援場面被展現得驚心動魄。但我從12話之后沒繼續追(沒想到此作居然有兩季長度),主要是感覺此作不少劇情太拖沓,故事節奏不好,到后來都快進看。而且居然每一話開頭的前情提要回顧時間都將近兩分鐘,太浪費時間了。到后來就沒什么繼續看的動力和興致了。此外,此作對人物情感和人物關系的描寫略顯生硬,而且大吾、中村的內心想法和過往,都顯得有點神秘兮兮的,看得我感覺有點迷。
此作在救援方面表現得特別硬核,非常考究。從此作中能感受到救援真的是相當專業的事,講究真夠多的,甚至在救援時還得管理自己的表情,訓練過程也真是極為艱苦。救援隊員的整體素質要求真是高,不僅必須身體素質極佳,還必須懂得各種知識才能真正出色完成救援,諸如大吾在救手臂被鋼筋刺穿的廚師時,在切割鋼筋的時候還得考慮不傷到他的手臂神經,免得他以后沒法再掌勺。看了此作我也深感這些救援隊員真是難,凈是得冒著生命危險救人,很可能人沒救出來,自己就被嗆死或者落石砸死之類。
大吾的人設不錯,是個挺完美化的男主,少言寡語,嚴于律己,毅力、技術、體格、應變能力都凌駕于其他隊員。大吾太勇了,第4話,他為了能鉆進水泥板下面救人,竟然故意把自己的肩膀弄脫臼,看著就超疼!這真是應該至少記二等功。
想不到在救援隊的男人堆里居然還有個漂亮妹子雪,有這樣一個角色的出現真是挺令我欣喜的,可謂萬綠叢中一點紅。她還是佐倉大法配的,討我喜歡。雪雖然是妹子,但是毅力很強,有成為出色救護士的堅定不移的決心,是挺令人佩服和喜歡的姑娘。此作如果是以她作為唯一主角就好了,肯定會更有看點。
另一個男主駿的各方面能力比大吾就差多了。他從高中時就喜歡上了雪,因為看到她的手機背景是消防員的照片因此決定為了她而當上消防員,這小子為了追妹子展現的行動力真強。雖然他的最初動機不純,但他對救援倒真是表現得格外認真和拼命,做出了超越普通救援隊員的行為,也蠻不簡單的。不過他和雪交往的話我覺得還是不配,駿雖然一心一意喜歡雪,但是顯得缺乏內涵。大吾X雪還差不多。
有一話,開中華料理的店起火了,已經被老婆拽出來的男廚師為了救傳承了30年的醬汁,又沖進店里,結果此時店鋪爆炸,他被壓在倒塌的墻下差點掛了。看到這一幕我首先覺得有點好笑,居然有人為了救醬汁如此拼命,但后來我轉念一想,倒也有點能理解他了。換做是搞計算化學的人,如果記錄了積累自己30年科研成果的數據的硬盤落在了起火、可能倒塌的房子里,大概率也會冒生命危險沖進去救,畢竟這簡直像命根子。
Secret Mission~潛入捜査官絕對不會輸!~
總評:91.5
8話的泡面非表番。此作的設定和情節真是相當新穎、很有創意,令我眼前一亮。內容是一個女警察雷土吏子和一個男警察假扮戀人或夫妻作為臥底摸清販毒線索的故事,二人總是“不得已”地發生*行為,故事挺滑稽也很有趣,頗有看頭,是泡面非表番里的精品。
此作的內容真是挺奇思妙想,比如第一話內容是他們住到販毒份子租的公寓相鄰的房間,竊聽他們的販毒情況。由于他倆的房間長時間都沒有傳出令人害羞的聲音,令販毒分子產生了疑心。于是這一對男女警察為了更像夫妻、故意令隔壁的販毒分子能聽到他們奇怪的聲音,不得已只好做出情人間令人臉紅心跳的事。第1話劇情的畫面感營造得相當有趣,難以描述,墻的這邊倆人正在風生水起,墻的那一邊就是倆販毒分子靠在墻上饒有興致地偷聽,真是很好笑。毒販也等于在不知情的情況下給倆人做了助攻。
雷土吏子設定得真心挺不錯,顏值非常高(而且此作里她的作畫也好)、很豐滿,性格挺高傲和傲嬌,自尊心很強。明明她是個嚴肅正經的女警,但是為了工作,以及受到男主的慫恿,卻又不得不做奇怪的事、陷入不能自已的狀態并發出丟人的聲音,這顯得真是特別有矛盾感和羞恥的反差感。在做奇怪事情的時候她表現得格外瑟琴,顯得特別魅惑,和平時的樣子真是有天壤之別。再加上吏子很漂亮,身材完美,又害羞,此作又凸顯其內心的矛盾、復雜的心理,從各個角度來說此作真是很有看頭。吏子也真是令我喜歡,還令我感覺她真是蠻可愛的。
吏子的CV御苑生メイ表現得很好,配音非常出色,很有感覺,真不虧是配成人作品的大佬。
第3話真是夠險的,吏子差一點就被壞人給破了,好在男主及時救場。
男主也夠鬼畜的,看起來挺溫和的,一副人畜無害的樣子,但老是假戲真做、以公謀私,甚至還謊稱什么“我停不下來”于是毫不顧忌地做奇怪的事。如此冒犯前輩,吏子居然不抽他。
每一話的ED畫面都不同,吏子的姿勢都一樣但服裝不同,這服裝show挺有意思的,她穿某些服裝顯得真誘人。
地下忍者
總評:91.4
此作描繪的現代的日本世界里有很多忍者隱于民間,也有忍者組織,此作講述他們之間進行的不為大眾所知的爭斗,還涉及虛到構的講談高中(此作是講談社連載的,玩了梗)。
此作的風格極其特立獨行,劇情表現方式十分抽象且缺少連貫性,人物描寫顯得很跳躍和零碎,我完全搞不懂此作清奇的邏輯,我甚至看完了整部作品都沒理清楚人物關系和組織之間的關系。此作在我來看是那種一本正經胡說八道,但同時又故意給人以寫實感的作品,某種程度上刻意營造些反差感,帶給人強烈的不明覺厲的感覺。此作總是在看似挺正經、嚴肅的敘事中突然冒出來離譜的槽點、離奇的笑點,帶來強烈的反差。由于我看不懂此作劇情,老想棄掉,但是此作卻總是時不時出現一些蠻有看頭的橋段和戰斗,覺得棄了又可惜,令我挺矛盾的。此作的戰斗場面制作得蠻不錯的,是可圈可點之處。
男主云隱九郎顯得好老,一個30多歲胡茬男,居然還偽裝成講談高中的學生,很違和,他還始終不穿鞋。
佐佐魔的糟蹋流浪漢大叔的形象挺有意思的,深不可測,真是人不可貌相。居然他的真身是個妹子,套了個大叔的皮而已。
有個敵對忍者被干掉后變成貓,真離譜,居然還開摩托車,畫面感很樂。
帶粗框眼鏡男開的帶著“志能便”字樣的那個小客車頻繁出現,令我印象深刻。
忍者組織用的戰斗服叫摩利支天,我一直覺得這個詞很有神秘感,查了一下,原來是佛教用詞。
NIN組織的那個禿頭的平主任老是很自然地把手順著領口摸到那個中二女仆風格的妹子日比的衣服里,明明干著很猥瑣的事卻顯得非常淡定、繼續認真地談論工作。日比還老是不樂意,卻也不認真反抗,這關系挺樂的。但感覺這日比并沒什么可摸的,也就是A,我甚至都懷疑是不是偽娘。最后一話主任居然還把腦袋埋入日比的腿間借討論正事之機占她便宜,真是lsp了。
那個滿腿肌肉的高中妹子太厲害了,不愧是經過地獄般的歷練爬出來的女人。我本以為九郎的隱藏實力逆天,想不到連他都不是這妹子的對手,她最后被一刀口爆了,腦袋還被她切掉一半,這妹子夠狠的。不過九郎把她的鼻子給削下來了,倒也沒白死。削鼻子這一幕絕的,有點可惜一個女生就被這么毀容了。
最后一話曝光男主居然還有好多兄弟姐妹,雖然他掛了,但之后還有的是可以編的劇情。
此作雖然風格離奇,但ED卻顯得很正常。感覺近期動畫的主題曲整體質量有明顯下降,此作的ED歌曲就算不錯了。
鐵路浪漫譚 第二季
總評:90.3
第一季之前追過,是講美少女和日本列車相結合的泡面番,不同妹子對應不同列車,想不到此作能出第二季。這一季作畫不錯,少女們挺靚麗的。這一季的主題非常特別,講不同的列車妹子們去日本各處采集聲音、相互收聽錄音成果、互相討論的事情,劇情還挺舒緩的,但跟列車完全沒有直接關系,劇情給我留不下什么印象,只是感到不明覺厲,在錄音方面講得蠻專業,令我見識到了不少新奇的錄音設備和錄音技巧。
幻日的夜羽 -SUNSHINE in the MIRROR-
總評:爆表
此作的世界如同是lovelive sunshine故事的平行世界,雖然有和現實世界一樣的現代化的大城市,但那里也有著魔法。夜羽14歲就去大城市逐夢,想將自己的魅力展現給世界。但夜羽居然試鏡不合格,那些評委們真該去腦科眼科耳科好好治治了(PS:到最后一話,他們后悔自己有眼無珠,主動邀請夜羽,結果夜羽還不賞他們臉了)。此作講述夜羽在大城市里一直沒著落,只好回她看不上眼的故鄉努瑪梓后發生的事。通過一連串的經歷,夜羽和aqours另外的8個妹子逐漸結交成了親密的朋友,打開了心扉,同時也充分融入了努瑪梓,獲得了歸屬感。并且她最終決定留在努瑪梓和大家在一起尋找自我和夢想。在整部故事中,關鍵主線劇情是小鎮發生了異變,時而令動物發瘋、人類昏厥、天相異樣,導致努瑪梓籠罩在不祥的氣氛中。解除這個異變的唯一方法是靠心之共鳴。夜羽一度以為自己沒有魔法、無法幫助努瑪梓受到異變侵襲的命運,還一度為此自責,但實際上她的天籟之音正是解除異變的關鍵。最后在夜羽的帶領下,大家共同歌唱,終于讓努瑪梓回歸往日的生機和活力,夜羽成為了努瑪梓的救世主。
當我第一次看到此作動畫化的官宣時,我真是興奮極了!夜羽是我最最最喜歡的角色之一,我對夜羽的喜愛的感情真是如滔滔江水般連綿不絕,在我眼里夜羽可謂是不可思議的神奇的存在!論我對夜羽的愛,我不敢打包票說一定是世界第一(比如可能我比不過小林愛香對夜羽投入的感情),但我相信在全地球上絕對能排的上號。如果我有豐富的辭藻,我能夸夜羽一晚上。做一部以夜羽為唯一女主的動畫,這是我曾經難以想象的事,我覺得這是只有在同人動畫里才可能出現的、也是我有無限資金的情況下給動畫公司投資才可能實現的愿望。官方制作這么一部動畫,令我感覺此作就像是專門為我定制的似的,真的是令我覺得難以置信。看youtube上第一話先行版放送之前的CV的直播里,回顧了之前live演出時公布此作的場面,當時小林愛香都當場哭了。我要是在現場估計也得感動和激動得哭出來。
2023年6月25號,看youtube上官方發布第1話插入曲的時候,其水準就把我給震驚了,夜羽都不能用美若天仙來形容了,而絕對是天仙下凡!甚至說是美神的化身!這一段令我看得著實無比激動,令我的內心反復受到侵略。我感嘆,這一段動畫,絕對能令無數那些還不懂得夜羽無限魅力的人明白夜羽是何等難以置信的存在!不光夜羽的形象設計和整體畫面感覺做得超絕,夜羽的舞蹈設計還曼妙無比、輕盈灑脫,將夜羽內在和外在的美都淋漓盡致地展現了出來。我loop了無數遍,甚至還一幀幀看。后來,第12話的夜羽舞臺演出和第1話做了呼應,這一話的夜羽也是格外地美不勝收,驚艷萬分!舞蹈動作設計和鏡頭運用使演出充滿張力和躍動感(簡直是教科書級的完美),夜羽的神情、神態還特別鮮活,充分傳遞出她的心境和心聲,令我再一次感受到莫大的心動感,令心靈受到了強烈沖擊。第13話以可愛style出場的夜羽的舞臺演出再次令我迷醉不已。制作方真是竭盡全力釋放夜羽的魅力,GGGGJ!我真是想說,如果看了此作后還有人無法認識到夜羽的驚人的魅力的話,那他的審美真是無可救藥了。
說來,之前夜羽的魅力只是在關注Lovelive aqours sunshine動畫的群體中得到認知。難以想象,曾經的企劃中津島善子的自稱是夜羽的設定,竟然能延伸出這么一個新企劃,可謂是給夜羽提供了大放異彩、將其魅力無限釋放的舞臺!而且不僅有動畫,居然幻日的夜羽還出了兩部游戲《幻日夜羽 -湛海耀光-》(http://yohane-bid.com)和《蜃景努瑪梓》,真是太難得了!夜羽她一個人也帶給了其她8個水團成員新的繼續充分表現自己的舞臺,夜羽太棒了!以前也有很多動畫作品,其中某個配角非常出色、遠比第一主角閃耀得多,但可惜都被埋沒了,要是她們也能像夜羽一樣成為衍生企劃的center大展魅力就好了。
這動畫做得果然完全沒有令我失望,此作里的夜羽真是太可愛了,怎么看怎么可愛,美不勝收,平均每一話都截一百多張圖,記錄下來各種表情、各種角度、各種心態、各種情景下的夜羽,留住她的一顰一笑。夜羽真是怎么看都看不夠。我看此作的過程中,無數次地在心里深深感嘆,有這么一部作品作為夜羽大放異彩、展現無盡風采的專門舞臺,真是太不可思議了!終于令世界圍繞著夜羽轉了一次!
此作不僅把夜羽的外在魅力展現得淋漓盡致,還絕佳地塑造了她的內在,充分表現出了她的各方面優秀品質:內心特別美好善良,很為身邊的人著想,富有勇氣和毅力,還有知錯就改等各方面美德。她還有一些恰到好處的未熟的感覺,有著真實的青春期的女孩子的心理。感覺此作塑造的夜羽,真的就正好是我心目中的她最完美的形象。有這么一部作品專門刻畫和表現夜羽完美光輝的形象,真是太棒了!
此作里的夜羽起初執著于追求閃耀、戲劇化同時還快樂的人生,真是有她在sunshine作品里一貫的性格,真是完全繼承了那個世界的夜羽設定。而經歷了此作的故事后,她變得明顯更為成熟、穩重和有責任感了,內心得到了很大成長。
此作里出現了多次小時候的夜羽的樣子,真是好可愛!雖然我討厭小孩子,拒絕養小孩,但如果能養這樣的小夜羽,我絕對求之不得!
此作第8、13話里都出現了Aqours 9人的團體舞,果然夜羽成站在了center位,令我深感真是沒有誰比最閃耀的夜羽更適合C位,夜羽必須作為center!我真感覺之前sunshine作品里把center給了人設最不出眾的千歌真是太浪費了,這個位置絕對是應該屬于夜羽的。第13話大家穿的是那種比較清新可愛少女風的服裝,和夜羽特別搭,使我感嘆夜羽真是能完美駕馭任何風格的造型,她的美是無窮的,穿不同的服裝就能展現出不同側面的魅力!值得一提的是,此作的團體舞的制作質量真是比之前的lovelive作品有了飛躍性的進步,如今的演出已經看不到任何僵硬生澀感,動作已經相當灑脫自然,已經幾乎無可挑剔了。
夜羽是sunshine世界里津島善子的自稱,《幻日的夜羽》這部作品里其它Aqours的角色還是之前的名字,但津島善子這個名字則被完全舍棄了,夜羽在此作里真正的名字就是夜羽,這挺有趣。津島善子管自己叫夜羽是附帶著自己是墮天使的設定(這不是她有中二病!我相信這一點是真實存在的),而此作的故事則和墮天使沒什么聯系。如果能再做一部平行世界里講述津島善子作為墮天使大展風采的作品就好了,并且在里面梨子一定要作為夜羽的小惡魔出場!
看此作的過程中我反復深深感嘆夜羽的CV小林愛香配得太太太出色了,不僅聲線特別好聽,而且表現力超級好,充分展現出夜羽的人設特質,屬于是絕配中的絕配。而且唱歌唱得也是超好,此作里好多插入曲都是夜羽唱的,演唱可謂完美,而且就連最后一話里沒有伴奏的清唱都非常好聽。總之小林愛香作為CV的能力實在太杰出了!這部作品不僅令夜羽大放異彩,同時也令小林愛香大放異彩!PS:在U管上我訂閱的lovelive官方賬號上看到小林愛香頻繁做live、參加和此作有關的活動,此作也真是給了她絕佳的功成名就的契機。
夜羽從小就有一只母狗萊拉普斯陪伴,現在大到都可以當坐騎了。想不到這狗會說話,看PV時我本以為她是不會說話的。剛開始感覺配音略有點違和,是日笠陽子,稍微顯得聲音有點過于成熟了。但把此作看下去后,感覺日笠陽子的溫柔的恰到好處的聲線真是不錯,挺趁萊拉普斯的性格的。萊拉普斯的形象設定不錯,可愛和瀟灑兼具。萊拉普斯很看重夜羽,并且是一開始回到努瑪梓時高傲孤獨的夜羽唯一一個交心的伙伴,在此作中充分刻畫了夜羽和她的羈絆。萊拉普斯有個特別怕蟲子的設定,一聽到別人說有蟲子就瞬間竄到天上去,這設定有趣,也很像我。
雖然萊拉普斯被夜羽當做是妹妹,但她明顯比夜羽顯得成熟很多,完全沒有稚氣感,心態還有點像阿姨、保姆。此作中萊拉普斯總顯得心里隱藏著什么秘密,有什么想法,但始終不痛快說出來,是個伏筆。她時而顯得有點心事重重的樣子,這種感覺到了第9話尤為明顯。后來才明白,在小時候和夜羽特別要好、和夜羽一直是玩伴的萊拉普斯,似乎是覺得后來的夜羽長大了、變得獨立了,而且如今她又交了許多新的朋友、被其她小伙伴所喜愛著,懷疑夜羽不再需要自己、會逐漸忘了自己,然后開始心里就愈發鬧起了別扭,也和夜羽產生了隔閡。同時她還對于曾經那個充分依賴自己的夜羽抱有著一些留戀。后來萊拉普斯的這個心結總算解開了。第12話,夜羽曾經分給萊拉普斯能夠說人話的魔法貌似是被回收了,曾經被分離的夜羽的意念重新合二為一。雖然萊拉普斯此后仍在夜羽身邊,但沒法再和夜羽以人類的語言對話了,看得我有點心涼。不過好在她還能聽懂人話,就算是變成了啞巴了。PS:我其實沒搞懂如果夜羽不回收魔法會怎么樣,難道不能讓萊拉普斯一直說人話么?
第1話里夜羽的母親(俺的岳母)初次登場了,著實令我一驚,她也好漂亮,才直到原來夜羽的團子是繼承自母親的。夜羽媽第一話就出差去了,努瑪梓發生異變什么的她都完全沒經歷到,直到異變結束后才恰好回來。她要是知道夜羽拯救了努瑪梓,應該會相當為她自豪吧。此作里主要角色依舊沒有(討厭的)男性,夜羽的父親也完全沒出現過,我覺得夜羽的父親應該不是男的(我也無法想象夜羽和任何男性會存在聯系)。
最早我在youtube上看到官宣aqours的九人在此作的世界里的造型時我就覺得她們的設定都很出色,此作出色的絕不僅僅是夜羽。
黛雅在此作里是努瑪梓地區行政局的長官,她本身顏值就相當高,穿制服的形象更是顯得相當漂亮、帥氣、瀟灑,特別英姿颯爽,這設定真是好極了!這制服還是紅黑配色,正這是我大愛的超經典配色。估計她的CV小宮有紗第一次看到黛雅的這個出色的設定肯定非常高興。黛雅現在的工作的樣子真是像極了學生會長,每天指揮一大幫手下,顯得非常有才干,很靠得住,責任心很強。和sunshine里一樣,雖然她不說話的時候看起來是端莊穩重的美女,但其實骨子里有相當奔放灑脫的一面,經常管理不住自己的形象而失態。第3話以騎摩托的假面武士形象出場的黛雅更是相當帥氣,且身手出眾。感覺刀使巫女、女軍官之類的造型肯定也特別適合她。第13話還出現了單馬尾造型的黛雅,令我眼前一亮,這個形象也超棒!
黛雅的小跟班琥珀顯得有點小帥,比較中性化,這形象設定得不錯,不過此作里沒什么關于她的詳細描寫,也沒怎么參與到主線劇情里。我對她還挺好奇的,要是她有什么內心活動的描寫,比如暗戀黛雅什么的,就有意思了。
露比在此作以小精靈出場,她的這副形象和她的人設真是相當切合,顯得很可愛,感覺真是比她穿校服時候的樣子閃耀太多了。她作為學園偶像的時候我對她的感覺一般,但這個小精靈的樣子著實討我喜歡,我感覺之前的比較普通的校服的造型有點壓抑了她的人物魅力。居然露比可以進入黛雅的摩托成為控制面板的一部分,感覺這個設定真是很妙,而且顯得毫不違和。令我挺納悶的是,為啥黛雅是人類,她的妹妹露比卻是精靈?說不通啊。令我吃驚的是,第7話她居然變成了一般少女的樣子,搞不懂她是怎么在精靈和人類造型之間切換的,毫無交代,此作里也完全沒有她變身的畫面。
此作里鞠莉的形象設計真是太棒了,看此作時我反復被她的美貌和氣質所驚艷到!本來我之前就挺喜歡閃閃發亮的鞠莉,此作里鞠莉的形象更是令我拍案叫絕,很有獨特的美感。她的那對大大的角太適合她了,服裝的設計令她顯得很有范兒。她在此作里的設定名義上是魔王,但其實只不過是獨自住在一個小島上的大城堡里(跟私人別墅似的,她到了此作的世界里依然那么富有...),并且有千里耳的能力,還有一些看起來像可愛的軟體海洋生物的小跟班。
之前看官方人物介紹得知鞠莉在此作里是魔王,于是在本作中看到鎮子出現異變時,我起初還以為是鞠莉搗的鬼(原本sunshine的設定里鞠莉就比較喜歡搞怪,弄shiny煮什么的),后來才知道原來她是個正面角色,還靠自己的順風耳能力將小鎮上可能出現的危險傳遞給來訪的黛雅。此作第3、5話對她的刻畫甚好,不僅充分展現出她內在和外在的美,還展現出了她細膩的情感。她因為自己長著角,而且身邊總是有小魔物跟隨,于是覺得自己和鎮子上的人類格格不入,因此一直獨自呆在遙遠的城堡里,內心已經習慣了孤獨,而且很悲觀,最終成了自閉的家里蹲,還有嚴重的社恐。這點跟sunshine里特別外向、奔放的她的設定形成了截然的反差。不過到后來鞠莉的性格又一點點開放起來了,閃耀感逐漸恢復些了,最后一話看到美食的時候她兩眼放光,終于久違地說了句招牌式的shiny~
第3話,夜羽的前來,令鞠莉第一次聽到有人夸她的角,而且夜羽也是第一個主動和她親近的人。這令形象高冷、魔王設定的鞠莉感到了暖意并且臉紅了,害羞樣子的鞠莉真是美極了!原本我就很喜歡鞠莉的性格,此作看到這樣展現出另一番魅力的鞠莉令我著實很歡喜!之前我就覺得鞠莉和夜羽微妙地般配,我記得lovelive sif手游里還有這倆人一起做巧克力(也可能是其它食物類的)的劇情,在此作第3話里看到二人又有了交集,而且還是私密的接觸,而且在某種程度上還是夜羽攻略鞠莉并令她羞澀的劇情,真是叫我喜出望外!此外,鞠莉說夜羽是有能力改變鎮子的特殊的人的時候,我心里就想:鞠莉說得太對了!夜羽不用懷疑自己,你就是最最最特殊、最最最了不起的存在!
第5話真是令我這個夜羽X鞠莉的極小眾CP控一本滿足!!!!!!!!!感覺這一話就像是專為我做的似的。這次夜羽主動前來,努力打開了鞠莉的心扉,而且攻略有方,最后終于成功鼓勵了畏懼他人目光的鞠莉邁出新的一步,看到了與她以往想象中截然不同的世界,甚好。這一話有好多畫面令我很興奮,居然夜羽在把縮在橋洞的陰影里的鞠莉拉出來的時候都十指相扣了!如果再抱上去就更好了!之后倆人還各種同框一起露出幸福的表情,她倆真是太般配了!不知道此作會不會令夜羽X鞠莉的CP控一下子翻好幾倍(很可能,而且畢竟原先的基數太小)。原本sunshine的世界里,果南和鞠莉是官配,真想不到此作里居然是夜羽來攻略鞠莉,成了新的官配。在鞠莉有了夜羽這個真心朋友,并發現自己也被小鎮上的人接納和喜歡后,她露出的燦爛的笑容真是特別美,特別耀眼,令我感覺她遇到夜羽真是太幸運了,同時夜羽也真是太了不起了。要不是內心那么美麗、善良、真誠而且充滿智慧的她,誰攻略得了緊閉心門的鞠莉?在以前的sunshine中,夜羽并沒有太明確的官配,相對來說我對夜羽X梨子最有好感(夜羽還老叫梨子“梨梨”),而在看了此作之后,夜羽X鞠莉變成我首推的了。
第5話出現的鞠莉小時候的樣子超萌超可愛,我太喜歡啦!PS:居然在回憶中出現了鞠莉的父親,我本以為此作的世界里不需要男性也能生孩子。好在沒出現夜羽的父親的具體形象,就當夜羽的父親是女性好了。
梨子在此世界里是動物學者,服裝設計和她的氣質非常搭,整個人顯得很漂亮和優雅,同時也顯得很有知識的樣子。梨子在sunshine的世界里是個品學兼優的好學生的樣子,因而此作里學者的身份設定給她非常適合。梨子在此作里對于之前未見過的生物有種興奮和癡狂感,第6話她看到露比后那副癡態真是挺顛覆她矜持優雅的少女形象的,令露比感受到了危險,把可愛的露比都嚇跑了。梨子特別喜歡抱和蹭萊拉普斯(但明明在sunshine的劇情中她起初特別怕千歌家的狗,這回真是反差啊),每次萊拉普斯都被抱得一臉難受,真是身在福中不知福。要是我也能被梨子這么蹭就爽了!
第6話專門講述夜羽連同鞠莉一起攻略梨子的故事,并且結成了三人組,這組合我實在是超喜歡,超絕妙,正好是目前aqours里我最喜歡的三人(PS:鞠莉和黛雅在我心中平起平坐)。這一話里插入歌《Hey, dear my friends》真是把我美哭,三人的歌聲太動人了,傳遞出的情境太美好了,就像是美好又溫暖的夢境。這插入歌良心得沒法更良心,我真是太感謝官方了。這個插入歌里夜羽還把頭發綁在了兩側對稱較高的位置,這新造型激萌!
此作的梨子,和曾經的夜羽、鞠莉一樣,都是把內心鎖起來,和別人保持距離。在夜羽和鞠莉主動親近她、把她當做朋友后,終于梨子也打開了心扉,決定改變自己,傾訴了自己的過往和想法。梨子其實是害怕孤獨的少女,但可惜一直孤獨著,好在現在她總算遇到了和她最相配的人了。能看到夜羽、鞠莉和梨子仨人開心快樂地在一起的畫面真是太好了。
鞠莉和梨子在此作里也算是個CP了,特別是第7話她倆老在一起,似乎挺有共同語言的。畢竟也都是從長期的孤獨中走出來不久的二人。這CP挺奇妙的,在以前sunshine的故事里她倆交集不多。
小曜和果南還是那么要好,二人的人設也都很好。小曜的飛天郵差的設定真是很適合原本sunshine世界里就自由自在、喜歡倒處活動的她。果南除了繼續做sunshine世界里的潛水員外,同時還增加了非常厲害的機械達人的設定,居然還自己造了機器人,她穿工作服的樣子顯得挺協調自然的。她的性格依舊爽朗并且給人可靠感,還很親切,特別有貼心大姐姐的感覺,在此作里給我的印象很不錯。此作里果南和鞠莉之間倒是沒什么交集。
渡邊曜的表姐渡邊月在此作里出現了好幾次,雖然話不多、出現時間也很短暫,但人設還挺令我印象深刻的。她的頭發烏黑,個頭也大,整個人顯得挺厚實和敦實的,性格也挺穩重。居然最后一話她還開始調戲起梨子來,有趣。
千歌居然在此作里也是旅館家的女兒。在此作里她還和倆姐姐組成了假面三人組,顯得很中二,立馬就被夜羽敏銳地揭穿了。此作里看到了千歌她媽,釘宮給她配音的聲音真是令我一聽到就心癢癢,太好聽了。
我原本對sunshine世界里的花丸一點興趣也沒有,也一點都不覺得她漂亮,但也許是作畫或是服裝的原因,看第1話時我卻覺得相貌平平的花丸在此作的世界里也蠻漂亮的,令我有點刮目相看。此作里花丸是賣面包的,感覺這還挺適合她這樣比較慢性子的妹子干。花丸居然養了頭大肥豬來拉車,而不是常見的牛或馬,這創意挺奇特的。花丸的滋啦的口癖真是一點也沒變。
此作的各方面都相當優秀,包括原畫/作畫、色彩設計、舞臺動作和渲染、插入曲的創作等方面,還同時特別要夸一下的是BGM,真的level特別特別高,可以說是驚艷的級別,運用了豐富的民族樂器,旋律婉轉、悠揚、壯闊、宏大,聽感非常舒暢,營造出一定幻想系的色彩,極好地渲染出了此作的氣氛,此作也不愧是音樂題材的作品。負責此作音樂的是加藤達也,在這里要大加夸贊一下。
此作的人物畫風和之前的作品有明顯的不同,角色們比以往明顯顯得偏稚氣點。剛開始看此作時我心想如果是以前的畫風就好了,不過看完后覺得當前的畫風很適合此作,跟此作的全新的幻想世界的風格很搭(突然想起aqours的大家穿校服的樣子,令我突然發現一個槽點:難道努瑪梓的少女們都不上學么?豈不有很多文盲?)
我真是希望此作是半年番。看到第13話的劇情中大家合力圓滿解決了異變,令故事非常happy地end時,我非常依依不舍。我還特別期待在最后的最后宣告要做下一季,但可惜沒看到。我是真心希望此作能出下一季,這是多好的企劃啊!在最后有幾秒鐘的畫面里竟然看到了Saint snow的二人,令我有點小驚喜。她倆在此作里沒有正式出場,而在這幾秒鐘里露了一面,莫非是伏筆,預示著要有下一季、屆時她倆會正式出場?如果是這樣就太好了。
我之前一直不知道努瑪梓這個名字是怎么構思出來的,直到我在google上搜沼津,發現英文是numazu,才恍然大悟,原來努瑪梓就是沼津啊!我已決定在不遠的未來去努瑪梓旅游一趟了,努瑪梓的官方旅游網站是https://numazukanko.jp,看了內容后感覺真是很吸引我。離有從北京直飛的大阪和名古屋也就不到200 km,而且有JR能直接到,交通方便。
此作的周邊實在太多了,官網https://yohane.net上的各式各樣的周邊東西真叫我眼花繚亂,官方在吸金上也算是全力全開了。我在此作周邊上真是大出血,淘寶上代購的列表見http://bbs.keinsci.com/thread-38684-1-1.html,總花銷約6k軟妹幣,是我迄今在一部作品上開銷最大的,算是給摯愛的夜羽買禮物的付出吧!
我的幸福婚約
總評:94.8
此作是一部相當細膩、各方面制作十分精湛的很高水準的作品,我高度推薦觀賞。
此作時代背景差不多是大正時期。女主齋森美世是闊綽的齋森家的孩子,小時候母親去世,父親找的后媽對美世非常惡劣,導致美世在齋森家中完全失去了地位,天天干著傭人的活兒,極其可憐。后媽和美世父親生的女兒香耶還對美世天天冷嘲熱諷。這些導致美世每天都過得很憋屈,覺得自己很卑微,徹底放棄了對幸福的幻想。有一天,她被父親指定了婚約,嫁到了一個被他人形容得冷酷無情的男人久堂清霞家里,從此她展開了全新的人生。此作講述美世和久堂共同經歷的故事,期間還牽扯到通過血脈繼承的家族的異能、與鬼的戰斗等諸多元素。
剛到久堂家時候的美世像以前一樣自卑,唯唯諾諾,看不到任何笑容。由于之前和久堂相親的貴族妹子都是拜金大小姐、性格凈是毛病,令久堂很失望,也使得久堂一開始對這次新來到他身邊的美世態度冷淡。但很快,久堂就逐漸認識到了美世內在的美好,并了解到她過往的辛酸,開始對她展現出了充分的溫柔和關愛,他們的關系逐漸一點點拉進。然而,美世卻始終沒能完全打開心扉,她過于內向的性格導致她什么事情都憋在心里,害怕給久堂添麻煩,不好好吐露心聲,結果反倒造成了更多的麻煩,令我覺得郁悶。她還因為他人的閑言碎語導致胡思亂想,懷疑沒有獲得幸福的資格。直到最終弄清楚了美世的過往及她的異能,并且不再是久堂單方面地為美世付出,而美世最終也憑借自己的力量拯救了被惡魔擊傷的久堂之后,二人才終于徹底心意互通,并認清了自己對對方的重要性,從此成為了非常和睦美滿的夫妻,結局相當可喜可賀。
美世性格靦腆內向,是內心如同櫻花般美麗的女子,也是最完美的妻子,能將喜歡、重視的人照顧得無微不至。在齋森家時候的她的經歷真是太痛苦了,不過如果按人品守恒定律的來考慮的話,忍受這些痛苦而換來和久堂在一起美滿的未來,絕對非常值了。美世的CV是上田麗奈,配音配得相當好,把她心里一直以來悲涼、凄慘以至于麻木的感覺展現得很好。要是能登給她配音肯定也很合適。此作里能登配了美世在小時候很照顧她的傭人。
久堂真是很完美的男子,相貌俊秀,形象帥氣,責任心非常強,而且在此作里還具有最強的異能。一開始感覺他的性格有點aggressive,還覺得他和美世在一起不如幸次X美世來得般配,但看下去后發現從久堂身上真是找不出什么缺點,比起性格和能力平庸的幸次,如此閃耀的久堂和美世在一起明顯能令美世更加幸福。久堂對美世的重視、對她的執著,也比幸次有過之而無不及。這么出色的久堂在坊間全都是差評,都是因為那些渾身毛病的貴族大小姐沒被久堂看上,導致她們倒處說久堂的壞話。不過有這些惡評倒也幸運,美世作為齋森家的棄子被惡意送過去匹配差評如潮的久堂,反倒令他倆結為了最美滿、最令人羨慕的夫妻。
男二號辰石幸次對美世一直都心心念念,內心敦厚善良,是美世在齋森家經歷難過的歲月期間唯一能給她帶來慰藉、能令她流露出一絲笑容的人,他倆真是挺般配(雖然不如久堂X美世)。可惜啊,幸次被迫與壞女人香耶結婚,而只能看著自己深愛的美世嫁給了別人,真是挺不幸的。后來他為了救被綁走的美世,只能去找對于自己來說是情敵的久堂求助,肯定覺得自己很窩囊。
佐倉大法這回居然配了個壞女人香耶,這個聲線真是把她充滿傲慢的感覺表現得很鮮活。香耶顏值蠻不錯的,可惜內心太爛了,被慣壞了。不知道齋森家被燒沒后她到底去了哪里,如果她也能經歷一下美世受的苦就好了。等到時候她被勞動改造一番,重新做人、洗心革面,屆時如果能和幸次好好在一起,那倒也不錯。
久堂清霞的姐姐久堂葉月很漂亮,很精致和洋氣,對美世非常照顧,盡心盡力教她禮儀,是挺令我喜歡的角色。日笠陽子給她配音很不錯。
給久堂當傭人的由里江是個性格非常熱情和善良的老太太,這把年紀了性格還那么活潑真是挺討人喜歡。她積極給久堂X美世做助攻,極力幫助不解風情的久堂認識到美世美好的一面,真是很好的人。給由里江配音的著名的老牌CV桑島法子表現得真是相當出色,令我對這個角色印象深刻。
美世的哥哥一開始給人心機boy的感覺,很不討喜,陰陽怪氣,還有點妹控似的。第11話他瞬間換了發型,感覺一下子洗白了,成了正面角色,開始支持起美世來,這人格轉換得實在顯得過于突然。
原來陛下是驅使那些鬼的元兇,最后玩火自焚,活該,也沒想到最后美世在意識世界中的法力那么厲害,把陛下都給秒殺了。似乎陛下還沒完蛋,如果此作有第二季,沒準兒他會不甘心失敗、繼續搞事情。
久堂的父母從來沒出場過,令我挺好奇美世的岳父岳母會是什么樣,以及久堂的強大的異能是從什么樣的人那里繼承的。
租借女友 第三季
總評:94.55
《租借女友》前兩季我很喜歡,很有看頭,這個第三季真沒令我失望,令我對此作更欣賞了,更進一步覺得這個系列的劇情構思質量真是出彩。
這一季主要內容圍繞著和也當制作人發動眾籌給千鶴拍獨立電影的事展開。一直以來我都不怎么喜歡和也,感覺他的頭發亂糟糟也不好好梳梳,跟雜草似的,有點邋遢,性格也太不穩重,從始至終一直給我一種挫屌絲感。但經過這一季,我更多地認識到了和也的優點。和也對拍電影這事真是看得比什么都重,總算體現出了有責任心、有擔當、有毅力的稍微帥氣的一面,像個靠得住的男人而非幼稚的孩子。和也真是非常純情、執著,對千鶴毫無保留地全心全意付出,為了她拼盡自己全力,甚至哪怕千鶴最終不和自己交往,他都愿意繼續努力支持千鶴,心意堅定執著到這種程度真是很難得。第12話他倆一起放煙花時,和也在心中真情自白,結合著沉重的BGM以及畫面上千鶴偽裝的笑顏,這一幕真是令我感動,也把和也的形象拔高了。這一刻,我突然感覺給和也配音的堀江瞬表現得真好,感情演繹得真到位。
第9話非常好,講述千鶴在醫院陪伴她奶奶一之瀨小百合臨終的那一幕,情節和氣氛相當感人,充分展現了千鶴和奶奶對彼此的深愛的感情。真是可惜,奶奶最后還是去世了,救不回來了。千鶴的奶奶真是對和也X千鶴非常努力地助攻,努力讓千鶴意識到和也的優點、對她的重要。雖然千鶴和和也都以為奶奶是被蒙在鼓里,以為他倆真的在交往,其實我覺得奶奶早對真相有所察覺。這一話里和也將剛剛剪輯好的千鶴主演的電影拿到醫院的房間里播放,然后一言不發地離開,這個想法和舉動真是值得稱贊,也總算是勉強讓千鶴的奶奶的夢想實現了。
可惜在奶奶去世后,千鶴不僅沒有向和也敞開心扉,反倒是繼續努力演起自己完美的人設,給和也看自己一貫冰冷的面具。千鶴如此故作堅強,特別是在和也面前偽裝自己而強顏歡笑,真是很令我覺得心疼。本來她就很孤單,現在唯一的親人也去世了...看到孤零零一個人主持奶奶的悼念儀式的千鶴令我覺得她真可憐。
想不到第12話,在和也真情告白后,千鶴竟然徹底決堤了,我從沒見過有人眼淚的流量如此夸張。她有這么個機會大哭一場挺好,能釋放不少壓力。和也這段告白構思得真的是很出彩,說出了自己對心目中的理想女友的看法,并且充分映射出那個人正是千鶴。這種表達方式真是很高明,帥得都完全不像平日很挫的他了。
最后一話真是跌宕起伏。好在是在此話最末尾,千鶴的電影大獲成功,千鶴終于露出了不帶任何掩飾的真心的笑容,令和也以及我的心情都平復了,這個結局算是很理想了。
看了這一季我進一步感嘆千鶴不光是那種憑借美貌吸引人的妹子,而且還是令人尊敬的妹子,很有想法和原則,特別有職業素養,也很有內涵,非常不簡單。此作對她的設定和塑造真是太好了。千鶴最大的問題就是過于逞強了,太鉆牛角尖,對自己太嚴格,且內心防線高過頭了,導致本來她能獲得的開心和幸福得不到、內心有痛苦也無法和他人分擔,活得太辛苦和壓抑。千鶴對和也過度保持距離感,還各種回避和也對她付出的心意。她心里固執地認定和也對自己不是抱有戀愛對象的喜歡,而只是喜歡自己這種類型的人而已,明顯千鶴這是過于執拗地自我欺騙,好讓自己和和也劃清界限。和也和千鶴一直沒有能交往,一方面是和也絕大多數時間顯得都挺挫,另一方面是因為千鶴的攻略難度實在太高了,真是要用“千鶴心,海底針”來形容。
在這一季里看到千鶴是在路上走動都引人注目的妹子,居然沒有星探主動挖她,還得自己那么辛苦主動打拼,真不應該。我覺得千鶴就是缺少上鏡從而被關注的機會。如今網絡、直播這么發達,要是她當youtuber、帶貨、當模特之類的,經常有露面的機會,估計很快就能火,或者能被影視傳媒圈子里的人看中從而得到躍升的機會。她其實也完全可以多參加選秀類節目。
這一季還是令我覺得瑠夏喜歡和也實在不可思議。千鶴是99分的少女,瑠夏是90分的少女,和也也就是個60分的男性。60分想追99分是再自然不過的事,怎么會90分瘋狂追60分,而60分的還不稀罕90分、就只想著99分,難以置信。看了第31話,我進一步覺得瑠夏真是內心很純潔、純粹的妹子,內心挺美好的,一心喜歡和也,沒有夾雜其它感情,并且為愛情一直積極努力。可惜和也不好好珍惜她的感情,甚至避猶不及,真是挺不像話。既然和也鐵了心非千鶴不可,那就早點和瑠夏挑明了多好,要不然這三角戀得持續到什么時候,最后瑠夏肯定還是要受傷害,且愛得越深受傷害越多。第31話瑠夏的泳著裝真不錯。
小墨雖然不是我喜歡的類型,也不喜歡她的辮子和粗眉毛,但不得不說她的心靈真是很美好,也很可愛,非常樸實。之前她義務幫和也發電影眾籌的傳單,第10話看到和也因為千鶴奶奶去世而抑郁時,又努力地幫助和也找回快樂的感覺。小墨真是個能令人感到放松和安心的女孩。
這一季麻美完全沒出場,只存在于回憶中。但我覺得麻美不可能就這么偃旗息鼓了,可能未來還要攪局。果不其然,最后一話的尾聲居然她又出現了,看來她搞事還沒完。
這一季出現了新角色八重森米妮,是和也的大學學妹,居然就住在隔壁。我起初懷疑她會不會加入后宮,到后來發覺這個可能性為0,八重并沒有把和也當潛在交往對象看待,甚至和他都沒有和異性間的距離感。
剛看到八重這個角色時,她并沒有令我有任何興趣,也沒覺得她有什么魅力。但看下去后發現八重的人設真好,性格挺有意思,是個主播+coser,思想和打扮都挺新潮的,頭腦開放,好奇心強,性格積極,說話爽朗直白,還有點小萌,我還蠻喜歡這個個性鮮明又活潑的角色的。八重頭上的兩撮藍毛挺有亮點的,時而還展現出可愛的貓嘴。第2話她的警察制服裝不錯,歐派又圓又大,令我感嘆此作就沒個貧r。
八重聽了和也追千鶴的故事后極度支持和也,積極當助攻。第27話,看到八重很努力地給和也X千鶴牽線搭橋,令我感覺她很熱心的這一點蠻不錯。八重在第29話還使計令和也和千鶴有了二人單獨出去旅游+拍攝的機會,這舉動真是GJ。和也真應該好好感謝她。此外,八重令和也有了更多視角了解和認識千鶴,這一點上對二人的關系發展起到了有利的間接推動作用。
尼爾:機械紀元 Ver1.1a
總評:94.4
《尼爾:機械紀元》是個人氣相當高的游戲,雖然我沒玩過,但畢竟其女主2B人設外形實在過于出色,也是常被cos的角色,故我也略有一點了解Nier。這個動畫版可以算是萬眾期待的。整部作品看完后,我感覺此作整體做得很優秀,制作質量配得上游戲的人氣,能令觀眾充分感受到2B人設的杰出,并且能領會到此作獨特的世界觀和設定。此作的畫面和戰斗做得相當不錯,非常花本,人物動作相當犀利帥氣,觀賞性頗強。
此作主要內容是講地球基本上都被看上去像鐵皮桶一樣的機器人所占據,僅存少量人類與他們繼續戰斗,地球完全是一番末日景象。女主2B和男主9S都屬于寄葉部隊,是人造人,受地外空間站的人類指揮所控制。此作主要描寫2B和9S根據上級指示,在地球上不斷戰斗和偵查過程的經歷,最終干掉了作為機器人的意識中樞的亞當和夏娃。在這個過程中2B也逐漸誕生了越來越充盈的自己的情感,也見證了那些機器人的奇怪的自我意識的誕生和進化。
此作最大的亮點莫過于2B人物的塑造了。2B這人設真是絕了,可謂是神來之筆那種,魅力萬分。2B的外形真是特別瀟灑帥氣,極其吸引眼球,整個人物從上到下從里到外都充滿著美感,賞心悅目。她的面容格外精致,身材真是巨棒,細腰大長腿,蒙著眼睛還有種神秘感的加成。其服裝設計特別顯露身材的美感,展露出2B恰到好處的大腿根的肉感,胸口還能看到美好的弧線,一片白皙的美背特別漂亮,令人很想prpr。2B的身姿還顯得頗為犀利和挺拔,令人物整體顯得特別有氣質。2B的很多近距離特寫真是顯得格外漂亮,都引得我截圖保存。2B的戰斗力真是相當高強,看到2B如此美型的角色揮舞著長刀進行令人目眩的精彩戰斗真是享受。2B戰斗的時候偶爾還能看到一閃而過的她豐饒的大腿之間的白色胖次,真是美好的福利!此作里還出現了幾次2B揭開蒙眼布的樣子,眼睛特別美,令人不禁哦呼。總之,2B這個角色可以算是能滿足觀眾對最美最帥氣女戰士的一切的幻想!
我不了解Nier游戲里2B的CV情況,看第一話時,我覺得其CV石川由依表現得雖然也不錯,但聲音有點偏柔,覺得可能還有更適合她的CV,聲音的表現力和穿透力還可以更強。不過看完了此作之后,完整領略到2B的人設特質后,感覺石川由依配2B真心不錯。2B并不是那種很硬很剛的角色,也不是缺少感情的內心冰冷的機器人,其內心也有溫度和柔情。而且隨著故事的推進、2B的內心世界和情感不斷變得更豐富,這點愈發明顯。
此作還有其它寄葉部隊的女戰士,包括2A。不知道為啥人造人都做得那么漂亮或俊俏,明明只是戰斗或輔助用的啊!如果是玩賞用的,顏值做得那么高、身材做得那么好才說得過去。
9S這角色也很不錯,性格挺可愛的,估計能討不少女觀眾喜歡。他雖然是人造人,但跟一般的人類的頭腦沒啥本質區別,性格很活潑,想法也靈活,和一開始的性格冰冷固執的2B有截然的反差。9S作為2B長期的搭檔,倆人相處時間長了,2B多多少少也受到9S的影響,思想變得稍微“軟化”了,情感更像人類了,最后2B都不忍掐死被病毒感染的9S了,并且也開始稍微有了獨立的思想,最后開始思考起人造人與機器之間的差異。9S顯得挺喜歡2B的,不知道是否對她已經產生了微妙的愛意、想和她有更深入的接觸~ PS:我看到有4分多鐘的同人動畫里這倆人已經開始造人了。
輔助機真是不錯,非常萬能,又能射擊,又能偵查,又能提出建議,還能當飛行器和跳板用,還能變成大規模殺傷性武器(都不知道那么小的體積哪來的那么大能量)。既然輔助機那么好用,多生產一些多好。
此作的機器人幾乎都是鐵皮桶造型,不知道為啥這么設定,也不知道鐵皮桶里到底裝了什么東西、哪里制造出來的。始終我都沒看到它們制造的畫面。
鐵皮桶從人類文明的殘渣中學到了奇怪的東西,往奇怪的地方進化,甚至還ML!反倒是高等得多得多的2B和9S不懂這些東西。這些鐵皮桶有的進化出了原始又混亂的精神,有的還變得極度扭曲、偏執乃至瘋狂。此作里對一些“瘋狂的機器”的表現有的還挺有震撼力、令人膽寒的。看此作的過程中會令觀眾好奇到底這些機械生物是如何進化的、極限在哪里,是否真的有誕生類似人類的自我意識的可能性。
想不到,居然脫離戰斗的機器人還組成了村落,看到村子的欣欣向榮和和平,以及村長機器人的自述,令人不禁會思考暴力的戰爭有什么必要。既然機器人也能有產生自我思想的能力,交戰的雙方真應當通過對話和溝通來避免無謂的戰爭。
亞當和夏娃似乎是機器人意識的集合體的具象化,是無法理解不可描述的存在,一個兄控一個弟控,還光膀子露著肌肉,gay里gay氣,這形象令我很不適,他們還把自己當成神,我很討厭這倆角色。好在最后都被徹底消滅了,沒再節外生枝。居然他們之前把創造自己的外星人干掉了,這些外星人也太弱了。
也不知道潛藏在幕后的人類會議在搞什么鬼,似乎顯得對與機器人戰斗、拯救人類不那么積極似的,在最后一話更是展現出了一些不可告人的秘密,似乎要搞人類補完計劃似的在弄一些神秘的難以理解的事情。不知道還有沒有下一季,如果有的話,2B說不定就要和人類會議的大佬們為敵,揭開幕后的陰謀了。
起初女司令不給2B和抵抗軍他們派增援部隊,我還以為是女司令的鍋,看到后來發現這女司令其實也是一直在努力和爭取。最后她先斬后奏發動了軌道炮終結了BOSS這個舉動真是GJ!軌道炮這時候若不用什么時候用?
此作的ED很不錯,唱得賣力,聲音高亢,感情的穿透力很強,富有力量和激情。
銀砂糖師與黑妖精 Part 2
總評:93.75
此作的上一季做得很好,是我頗為欣賞的一部高水準作品。女主安又美麗又堅強,是相當令人欽佩的姑娘,令我一直很喜歡。極度美型帥氣的男主夏爾也塑造得非常出色。這一季和之前一樣做得很好,只不過赤色妖精拉法爾搞事的那幾話看得我有些郁悶。最后一話收尾很好,看到大堂里絢爛多彩旭旭發光的銀砂糖樹的絕美情景,著實令我覺得很舒服。
這一季主要內容是安當了佩基工房的總監,從心機婊那里奪回了夏爾,然后帶領佩基工房贏得了給圣堂制作銀砂糖裝飾的機會。他們在廢棄的城堡里制作銀砂糖的過程中遇到了一個對已故的人類主人忠心耿耿的小妖精,他對主人離別前的話語理解有偏差,導致極為執拗地一直絕食直到快要掛掉,最后被安的話語和她的銀砂糖點心安撫好了。再之后就是邪惡的赤色妖精胡作非為,打傷工房的工匠、擄走安、暴力地使役夏爾的情節。
第18話,安被拉法爾挑逗了,這令夏爾按捺不住了,于是夜里去占有安了。想不到夏爾如此會撩,這一話里夏爾嗦安的臉頰的那一幕太瑟琴、太高能了!(令我想起了當年高坂麗奈在夜里劃久美子的鼻子和嘴唇的那一幕)
拉法爾是和夏爾一樣從寶石中誕生的有特殊地位的妖精,有成為妖精王的資格。他小人得志的幾話看得我著實挺不爽。拉法爾真是惡劣,居然上來就弄傷了工房的職人一只眼睛,而且還把另一個職人的脖子劃得流了好多血,就算是作為威脅這也太過分了,真是一點都不懂下手的輕重。再之后他表現得更加肆無忌憚,揚言要成為妖精王,不以德服妖精們,卻沒收他們的一半翅膀去使役他們對人類搞各種恐怖襲擊,還把安也擄走強迫她給自己做銀砂糖點心,還抓著夏爾的翅膀逼迫他也一起干臟活兒,真是差勁歹毒至極。拉法爾卻還自我感覺良好,覺得自己有著正當的大義。他的結局真是又可悲又活該,之前被人類使役,最后因為自己的惡行糟至精靈同伴們的背叛。不知道最后拉法爾從城堡上墜落后成什么樣了,是摔得腦漿迸裂,還是逃之夭夭打算以后繼續搞事。
拉法爾一心要得到夏爾,想和他一起成為王。拉法爾跟夏爾說話時往往表現得像gay佬一樣,顯得很饞夏爾的樣子,相當扭曲。不知道有沒有這倆人的bl本子。我覺得要是有機會把夏爾拷起來,拉法爾可能真會對夏爾做“過分”的事。夏爾真是相當偉光正,對其誘惑完全不為所動,從未有絲毫合作的意愿,從最一開始就防著拉法爾,警覺性真是相當高。我剛一開始只覺得拉法爾是個性格惡心的家伙而已,都沒想象到其骨子里壞成那樣,起初我還甚至一度覺得夏爾對他的敵意太強而有點失禮呢。
那個據說是從植物中誕生的很小的綠毛女妖精的腦子就像壞掉了一樣,完全沒有基本的邏輯,我挺討厭她的。
喬納斯那個臭小子一直處于頹廢中,但安真是寬宏大量,跟圣母一樣,對這樣充滿罪過的人渣依然令其有機會發揮自己的價值,他在最后終于有點改邪歸正的樣子了。我感覺這小子應該把安視為救贖他的女神看待才對。
間諜教室 第二季
總評:93.75
之前追第一季時感覺不錯,這一季比之前甚至更出色,制作得相當精良。繼續講在克勞斯帶領下間諜妹子們完成各種任務以及日常故事,輕松有趣和驚心動魄的情節交替穿插。這一季最關鍵的是滅掉了大惡棍紫蟻,同時將克勞斯曾經的師傅,也是當年救了年幼的緹婭的紅爐的往事聯系了起來。
想不到這一季里我最愛的緹婭的戲份那么多,有點挺不可思議的,感覺她成了這一季的女一號,我對此十分滿足,截了大量緹婭的美圖。緹婭實在太美了,氣質太出色了。同時,這一季令我感到緹婭的內心真是善良,第14話她主動給阿妮特與偶遇的母親搭建溝通的橋梁,不顧隊友的反對。不過我稍微感覺她有點善良和天真過頭了,為了幫阿妮特她媽能夠回國甚至和隊友成為敵人,也不和克勞斯商量,并且還輕易相信了阿妮特她媽的謊言,疏于防備,結果在臨別前被她展露的丑惡的一面所傷害到。雖然緹婭的善良和天真從作為一個間諜角度來說有點不合格,但這是我喜歡緹婭的重要一部分。盡管緹婭的身材和相貌顯得很成熟,還有著像超能力般的讀心術以及完美誘惑男性的能力,但其實在很大程度上她在隊伍里屬于最不成熟的前幾位。令我一直有點納悶的是,緹婭也不像是有精妙心機的女人,她作為間諜時是怎么勾引男人為她服務的?這沒多少具體描寫。不過,第1話里倒是確實展現出了緹婭的魅惑能力,緹婭X艾露娜的畫面和對話居然如此橘里橘氣,令艾露娜都臉紅心跳神情迷離了!這一刻令我感覺緹婭不愧是專攻色誘之術,甚至連同性都可能攻略。似乎緹婭利用男人倒也不需要什么心機和語言技巧,對于一般男性,光是靠她那無法令人抗拒的美貌和肉體簡單直白地強攻就足夠了。
第22話出現了小時候的緹婭的樣子,真是相當可愛。那時候她似乎是棕皮,不知道怎么現在變成白皮了。
看完第16話,發覺阿妮特真是超級人才,真是很神奇的妹子。居然她在一開始就看破了假裝善良的實則極其險惡的她的養母的偽裝。我覺得她無與倫比的制作道具的能力其實只是她最杰出之處的1/3,剩下的2/3是她的洞察力。阿妮特還特別狠,最后分別時她對她媽設的陷阱真是毫不留情。阿尼特和緹婭可謂是兩個極端,要說阿妮特內心有多狠,緹婭就有多善良。但阿妮特表面上卻顯得一臉天真無邪樣子,令人覺得挺活潑可愛的,粉頭發的她真是教科書般的粉切黑,用一副可愛的樣子做出殘忍的事說出殘忍的話,同時還有著虐待狂的一面。也難怪她媽在蘑菇頭面前把她評價成妖孽。
阿妮特她媽可真是夠壞的,同時也夠會演的,明明殘忍陰險到不行,表面上卻裝得溫柔和善,令人不忍傷害,令緹婭之前完全沒察覺到她真實的一面。她肯定萬萬想不到自己撿回來的女兒遠遠高于自己一籌,預判了自己的預判,在她面前阿妮特可愛的樣子都是阿妮特在將計就計。有點可惜她媽后來直接就被炸死了,沒有受到什么折磨,沒令其認識到她自己才是小丑。
子安又配了個變態,紫蟻,專門折磨人將之訓練成對他完全順從的殺害間諜的殺手。紫蟻太變態了,子安作為變態的CV專業戶的普遍印象因此作而進一步加深。緹婭和紫蟻的對話,以及緹婭靠言語控制住紫蟻的手下的劇情,令我感覺邏輯太迷了。沒想到紫蟻是以挨一記克勞斯的經典的友情破顏拳來結束其間諜生涯,令我感到痛快和愉悅。
不知道當年頂尖的間諜紅爐是怎么被紫蟻給抓住的,感覺死得有點窩囊。
我本以為尸是沒有正常人思想的完全的神經病,因而靠色誘什么的對這種怪人完全無效。居然在第11話里,連他也被緹婭的話術和行為所迷惑&魅惑了。緹婭還用對待男寵的方式給聽話的他以獎勵,用自己的玉足摩挲他的面頰,使他甘愿作為奴仆服從緹婭。緹婭在控制人心、撩人方面可真夠能個兒的。可惜尸剛稍微洗白、表現出他并不是完全無可救藥,結果就在和紫蟻的戰斗中被紫蟻的兩個精英蟻兩肋插刀(那一幕可以作為“兩肋插刀”的標準配圖了),太慘了。我還以為他在未來會作為緹婭的pet、成為他的工具人呢。
莉莉這一季顯得挺傻呵呵的,最后執行完任務后她身敗名裂挺逗的,報紙頭條里的她都被ps成了窮兇極惡的面貌,樂。
吉薇婭和那個歐派很大的女人比賽飛鏢的劇情挺有觀賞性。想不到人家長期苦練的技能,吉薇婭直接就能戰平,真是天賦上的碾壓。
克勞斯繼續表現得帥氣+無敵,不過總算是在妹子們的圍攻下被戰勝了一次。這些妹子們如今的協同作戰能力真是得到了不小的提升。
現在剩下的伏筆是蛇這個搞事的組織,消滅蛇將是下一季的主線,期待下一季的播出。
無職轉生Ⅱ ~到了異世界就拿出真本事~
總評:93.45
之前追過此作第一季,是上檔次的話題性動畫。這一季主要內容是男主魯迪烏斯和冒險者團隊的人一起混,之后在和心儀的隊伍里的妹子薩拉ML時發現自己有ED,備受打擊,于是成為魔法學院的特招生,在那里試圖尋找ED的解決辦法。這一季的大部分劇情我感覺有些松散,有點東一榔頭西一棒子,沒有明顯主線,有些人物目前看來也有點多余,有的劇情還挺沙雕、無厘頭的。不過這一季的情節有兩個關鍵性進展,一個是魯迪在9話遇見了另一個和自己一樣被轉移到這個世界的妹子,于是開始積極試圖弄清楚大轉移的真相和神秘的人神的事,另一個重點是魯迪在魔法學院和他的青梅竹馬希露菲重新相逢,最后相互告白。
感覺這一季前一半里魯迪烏斯變得挺廢柴的,感覺他的實力不如之前,性格也嚴重走下坡路,有點墮落,還開始酗酒,玩女色,有點變得缺乏尊嚴感,在別人面前還經常顯得唯唯諾諾,跟小時候挺神氣的神童樣子的他相比差遠了。他居然還喜歡上隊伍里的薩拉,我覺得薩拉沒什么優點,明明魯迪烏斯救了她的命,就因為魯迪烏斯ED就把他拋棄,看到魯迪因為自己的X無能而難過時也不撫慰他一下(和后來的希露菲形成了鮮明的對比),顯得薩拉的性格真是挺爛的。明明魯迪的能力和實力跟薩拉不是一個level的,居然還受她的氣,真覺得魯迪很窩囊。
第3話看到魯迪ED時,我感覺真是報應,他那么快就把紅毛妹子艾莉絲拋在腦后,女隊友薩拉成了其新歡,結果最終被她和隊友拋棄,有點活該。這一話服侍魯迪的X工作者艾莉潔的人設蠻不錯,又漂亮又溫柔,而且線條極好,可惜這個角色后來就再也沒出場,我真心希望她也能成為一個主要配角。
魯迪烏斯經歷過的女子很多,但他似乎唯獨對洛希琪最情有獨鐘,始終念念不忘,居然第4話里還看到魯迪把她的胖次供起來。不知道魯迪最后和希露菲好上后,對洛希琪的想法會有什么改變,會不會還覬覦她。
第7話魯迪烏斯居然為了報復兩個獸族大小姐把洛琪希的手辦弄壞而將她倆監禁。這一段劇情真是相當高能,相當工口。魯迪烏斯也太狠了,居然一腳踩在一個獸族妹子的歐派上,把她倆拘束住之后居然還淡定地摸她們的歐派。令她們不吃不喝,還不給上廁所,導致其不得不在椅子上直接代謝,搞得濕漉漉一片,畫面不堪入目。官方和魯迪烏斯都真是惡趣味,太沒節操和下限了,魯迪烏斯已經完全不純潔了。這段內容可以出本子。很意外地是居然這倆獸娘還不記仇,被這么辱過之后居然還把魯迪烏斯認作老大了。
這一季剛一開始,會水魔法的希露菲突然被傳送到了一個國家的宮殿里,巧合地救了那個國家的公主愛麗兒一命,相互成為了對對方重要的人。當時我還以為這一季主角換人了,劇情要伴隨著公主經歷各種事端,擺脫暗殺、奪取王位之類,還以為多多少少有點要走百合路線的感覺。后來發現講公主X希露菲的劇情就僅僅這第0話而已。在魯迪進入魔法學院后希露菲才再次出現,此時的她是裝成進入魔法學院學習的公主的侍衛。
希露菲雖然不是什么大美女,身體幾乎沒什么線條,各處曲率接近于0,但顯得非常纖細和精致,性格靦腆,容易羞澀。她有些微妙的特寫和表現出的感情令我覺得她還挺可愛的,有一定獨特的個人魅力。她戴墨鏡的樣子還挺酷的,感覺這副形象作為刺客也挺合適。
這一季令我相當吃驚的是,居然魯迪烏斯見到戴著墨鏡出場的希露菲,一直都沒認出是她來。更難以置信的是居然還一直把她當成男性,在魯迪對她有生理反應時居然還自言自語地說“我喜歡上男人了”,直到第10話魯迪才開始懷疑化名“菲茲”的她是女的。難帶希露菲戴上墨鏡、沒有歐派,就不像女性么?明明她的面龐那么細膩,個子也矮,沒有喉結,聲線也是妹子,魯迪能把她視為男人真是夠奇葩的。
魯迪這ED還真是來得挺邪乎,是很奇怪的心理引起的問題而非生理問題。在不經歷頭腦思考、理性這道關卡的情況下,靠條件反射倒是可以支棱起來。第10話居然在意外的推倒事件后起立了。還以為他的ED竟然就這么被菲茲無意間給治好了,還在想這倆人身體的相性不錯,但結果他高興得太早了。
希露菲對魯迪是真心喜歡,越來越壓抑不住對他的感情了。第11話,居然希露菲主動倒貼,喚雨把自己淋濕,然后還謊稱衣服靠自己脫不下來(巨嬰么!?),讓魯迪烏斯脫,脫到只剩眼鏡和內衣時居然說還有一件(原來那一件是指眼鏡,我還以為是指胖次或胸口的布),令我感嘆顯得挺簡單天真的她竟然夠會倒貼的。總算在摘了眼鏡后,魯迪烏斯認出她是兒時的青梅竹馬希露菲了。她這時候闖入魯迪烏斯的感情世界倒真是恰到好處,正好他身邊沒其他女人。看到這里我也有點可惜希露菲完全是BG,第0話她和公主之間綻放出的百合僅僅綻放了一瞬。
最后一話信息量夠大的。希露菲帶著公主給她的spring藥主動給魯迪送上門。這藥效真靈,不枉賣100金幣,令魯迪完全把持不住、喪失理性,甚至第二天都有點失憶了。可惜翻云覆雨的過程一筆帶過了,一瞬間就把場景切換到天亮了,真是遺憾。魯迪摸著床上的一灘血漬,這畫面真是很有深意,希露菲的重要的第一次就這么給了他...第二天醒來魯迪看到希露菲已經不在了,這一刻不禁令人想起當年他和愛麗絲運動后的第二天早上的情景,那時候愛麗絲已經遠去了。好在是沒一會兒希露菲就回來了,還笑著看著魯迪,一下子令魯迪懸著的心放下來了。似乎這一次盡情的發泄把魯迪心理性ED的病根洗刷掉了。最后看到他和希露菲求婚,最終二人開心地笑著站在一起,挺可喜可賀的。希露菲的性格也真是很好,特別遷就魯迪,特別為他著想,得知他ED的時候沒有不開心而是去撫慰他,充分接受他的缺點,在魯迪對自己索求的時候又全身心奉獻自我,沒有任何猶豫。希露菲的性格也沒什么復雜、扭曲、任性、算計、難懂的地方,和她交往挺安心的。希露菲真是可遇而不可求的好妹子,希望魯迪之后別胡搞瞎搞而傷了她的心。我總覺得魯迪未來的感情世界大概率不會只被希露菲獨占,可能還要有其他妹子侵入,畢竟從過往經歷來看魯迪在情感方面不容易長時間地專一(他爸就是,和那么好的老婆在一起居然還和女仆出軌),而且此作里還有那么多和魯迪有過瓜葛的妹子也不會和他再也沒有情感上的交錯。
這一季里畫伯居然配了個正經的女戰士,真是難得。以前畫伯總是配性格奇怪的女人。
第4話出場的艾莉娜麗杰居然是個極其無節操的沉迷于**的女人,倒處亂吃男人,居然和魯迪臨時加入的小隊里所有男性都云雨過,而居然至今也沒懷孕或染病。她竟然還逆sell春,主動把錢給男人令其和自己一起舒服。而且她還對男人不挑,居然老男人、丑男人她都纏上,似乎也不嫌對方有沒有味道、臟不臟。這女人太奇怪了,感覺她就像得了怪病,不**就會死一樣。結果,第5話還真出現了這個設定,說是她受到了詛咒,不**就得掛。但我感覺她這是在開玩笑,畢竟她那么主動、樂在其中,完全不像是把這詛咒當包袱。
這一季基列奴一次都沒出場,可惜。真希望在之后的動畫里早日再看到她。
Lv1魔王與獨居廢勇者
總評:93.45
此作里原本的大魔王被勇者麥克斯為首的隊伍擊敗,過了很長時間后,魔王復活成為了一個嬰兒肥體態的看起來傻乎乎的小女孩樣子的魔王,絲毫沒有之前的威武和霸氣,而只像是個自大的傻瓜。她對之前擊敗她的勇者念念不忘,找到勇者后發現如今的他已經從當年的帥小伙變成了糟蹋的獨居廢大叔。魔王看不過去曾經的勇者自暴自棄,決定住在他那里照顧他,試圖令他振作。此作就是講述在這之后倆人在一起共同經歷的各種各樣的事,前一半主要是日常,之后勇者曾經的隊友在故事中依次出場,勇者還卷入了王國和他之前隊伍里雷歐新建立的國家之間的紛爭中。在整個劇情中,勇者和魔王對彼此的認識和感情不斷加深,起初勇者對擅自跑來的魔王沒什么好感,之后慢慢地接受了她,最后還變得對她重視了起來。
此作整體制作質量上佳,內容蠻有創意和樂趣。我雖然對勇者的興趣不大,但這魔王的設定挺可圈可點。此作挺花本,原畫和動畫都很好,特別是戰斗做得頗為出色(尤其是第11話)。
魔王或其手下轉生成妹子的動畫不少,比如之前《迦希女王不會氣餒》就是,此作也有相似性。令我挺納悶的是,明明此作的魔王之前看起來兇神惡煞,怎么看都應該是雄性,咋轉生后就成了女的了?
魔王的形象設定挺有意思,嬰兒肥且咋咋呼呼的樣子還有點可愛。她經常顯得傻了吧唧,十分天真,缺乏正確的自我定位,喜歡蠻干。魔王是大空直美配的,她可謂是配這種沙雕妹的專業戶,此作她給這魔王配音真是恰到好處。如今的這個魔王一點也不像魔王,沒有什么野心,也沒有什么惡意,甚至顯得心腸挺好,第10話她不想看到勇者以前的隊友雷歐和弗雷德之間的戰斗,還努力把勇者引過去解決紛爭,真是已經完全忘記了自己原本是制造混沌的魔王了。
魔王可以變身成體態豐饒的女學生妹的形象,這形象很不錯,十分青春靚麗,而且水手服還特別短,大面積展露的腰腿令她顯得頗有視覺誘惑。可惜這種狀態出場的戲份只是占少數。
不知道曾經和魔王戰斗的帥小伙勇者經歷了什么現在才變得如此萎靡不振。只是看到了勇者之前的負面新聞真不少,特別是亂搞男女關系方面,如今變得這么廢柴一定程度上也是不少人的惡意所害。他居然如今還看瑟琴的網站,還用plane杯,亂七八糟的屋里的一團團廢紙估計有不少他的遺傳物質。按說他作為拯救世界的前勇者應該不缺女人才對,墮落到這種地步真是悲哀。
明顯勇者是對女性還是有興趣的,但魔王的Q版的樣子無疑令他不會產生任何興趣和幻想。不過在魔王偶爾變成學生妹的狀態后,勇者對她的態度往往就展現出了一定程度的改變,會在一定程度上把她當異性看待。令我有點意外的是,勇者在第2話看到了身材性感至極的剛出浴的潔妮亞,而且喝酒時和她離得那么近,甚至還發生了梨斗摔,居然勇者也沒有什么很明顯的觸動,都沒刻意壓槍,我感覺很不合邏輯。
魔王的女下屬潔妮亞的形象很是驚艷,身著死庫水,整個人顯得特別豐腴飽滿,歐派、大腿格外有肉感,光澤圓潤的尻還經常在鏡頭里出現特寫,死庫水的質感也都做出來了,真是賞心悅目,給觀眾帶來了巨大的眼福。第2話她揍勇者的時候還有r搖,觀賞性極好。第2話她在大街上果奔甩著歐派追逐勇者的那一段真是太高能了。第7話出現的潔妮亞的漆皮裝真是極好,顯得非常飽滿和性感,這個造型也很適合作為SM女王出場。
勇者的破公寓里藏在壁櫥中的那個女鬼不知道是什么來頭,一直沒有正經戲份,但老是時不時露一面。直到最后一話她才終于說了一句話,我立馬聽出來是我大愛的小林優。希望下一季里神秘的她能開始作為有臺詞的正式角色出場。
曾經勇者小隊里的尤利婭的粗壯結實的腹肌真是性感,但可惜最后和普通人結婚生娃了,完全沒了當年的魅力。
公司的小小前輩
總評:93.25
此作主要講述剛入職的男主篠崎拓馬和他的女前輩片瀨詩織里之間的故事,內容主要是公司里和私下里他們之間的故事,還有不少同事出場,這種講述職場戀愛故事的動畫不少。此作風格輕松舒緩,畫面明亮,作畫上佳,觀感挺好,是第一印象就很討人喜歡的作品,光是看到OP就令我決定追了。
此作最出色的地方就是詩織里的人設了,非常有亮點。她的個子小小的卻是巨r,歐派和身長呈嚴重反差,長個子的營養真是都用在發育歐派上了。歐派大到上樓梯都看不到腳下。此作第8話是泳著回,真是經典的7、8話定律。這兩話里前輩的泳著甚好,真大!特別是第10話的一個中場泳著插圖,真是大到離譜!
詩織里內在和外在一樣美好。她的性格特別可愛,非常愛笑,笑容特別燦爛和養眼。她總是非常溫柔和善,整個人暖洋洋的,很明媚。剛看此作時我感覺她的人設有那么一丁點乃木坂春香的感覺,都是眼睛都特別大,而且發色都一樣。
詩織里有一點點貓屬性,還經常變成Q版貓娘,這副樣子總是有各種可愛或者有趣的表情,很討人喜歡,這個設定進一步給她增添了萌點。第10話在幼兒園她還被小孩要求扮演貓,那個樣子真是特別誘人,同時又很羞恥。
此作有顯著福利向,有時不很純潔,出現了許多不可描述的令人浮想聯翩的臺詞和畫面。頭腦顯得很純潔的詩織里也真是毫不自覺,總無意識地說出令男性興奮的言語、做出令他們把持不住的行為,時而顯得很有色氣。她還挺能幻想,比如第1話她在胡思亂想中莫名其妙就滿臉通紅陷入絕頂了,第3話她看到兩個毛絨玩具貼在一起時還腦補成自己和男主在床上的事前準備。此作也因為這些描寫顯得比較偏男性向。
男主沒有詩織里的人設特點那么鮮明,相貌平平,人設整體還挺不錯,比較踏實穩重,品行很好,能挺好地烘托耀眼的詩織里。雖然他看起來挺正經的,但終究還是個普通男性,腦子里時而對詩織里產生各種幻想,包括夾雜著工口的。畢竟詩織里體態那么夸張,還頻繁給男主各種甜頭,他能不YY、不想入非非才怪。
詩織里的母性很強,很關照男主。劇情中藉由各種事件,詩織里給男主的視覺、觸覺、聽覺都奉獻了極大的福利,真是令人很羨慕男主,實在太幸福。詩織里雖然名義上是男主的上司,但在男主入職沒多久就對他產生了上下級、同事間以外的好感,對他很上心也挺積極的,男主生個病她都主動跑到他家給做飯。不過此作的戀愛發展比較磨嘰,倆人總是處于各種曖昧的關系,到了12話,這倆人才剛剛名義上成為了私下的朋友,有點急人。明顯這倆人都相互喜歡,關系都那么親昵了,也沒有任何其他可能性,就早點告白了唄!這倆人都太害羞了,要是其中一方是比較主動的,事早就成了。值得一提的是,本來詩織里就很可愛,在羞于表達好感時她還會臉紅,有羞澀、害羞感加成時她更是顯得可愛得不得了。此外,她的真心想法每次被戳穿的時候流露出驚訝的小神情也特可愛。
男主的青梅竹馬早川千夏看起來不怎么少女,稍微有點中性的感覺(但第8話她泡澡時我發現她的歐派也得有F,之前真是小看了她),對男主也沒有任何面對異性的好感或心動。沒想到第4話居然她一個人cosplay去會場,真是令我吃驚,想不到她還有這樣的趣味。她的這一面被公司的主任秋那千尋看到時真是蠻羞恥和尷尬的。她臉紅的樣子我感覺還蠻可愛的,也是第一次令我覺得她的人設有了可愛這方面的分量。早川剛進入公司時就因為主任對她展現的溫柔而心動過,不過進入公司后看到他奇怪的行為,很快幻想就破滅了。自從早川cos時被主任看到這件事,二人之間有了共同的秘密,關系明顯拉近了。想不到早川居然還自己畫漫畫,明明看起來是個標準的OL,竟然有宅女的一面。第6話,居然她讓主任幫自己畫R18 bl本子,還一起出攤,真是不可思議,哪有讓公司領導干這種事的。
主任是個蠻神奇、很特別也挺有趣的角色,令人琢磨不透。他老是顯得一副樂呵呵的樣子,非常天然,對人親善,從不嚴肅。盡管他是個正經的會社員,而且還是小領導,卻對男主和詩織里倆人的感情發展抱有超濃厚興趣,各種偷窺,想各種辦法助攻,以作為吃瓜群眾觀察二人的感情發展為樂,這趣味和他的身份真是有極大的反差。
主任對早川表現得沒有任何距離感,諸如很自然地就摸她腦袋、緊挨著她坐,顯得好像沒有常識一樣,令人弄不清楚他對早川是like還是love,亦或是只是對男女關系沒有自覺、腦子缺根筋,真是挺奇怪的人。得虧早川拿他沒辦法,若是早川對主任沒有任何好感,說不定就舉報他職場性騷了。目前他倆的戀愛關系到底有沒有還很含糊,不知道什么時候這倆人才能開始真正認真地挑明感情、發展戀愛關系。從后來的劇情中知道,主任居然對別人產生不了戀愛感情,以至于從小到大有很多妹子心儀他卻最終也沒能交往。希望早川能是他生命里的唯一例外,我感覺這倆人還蠻般配的。
第10話出場的男主的同輩妹子來棲和主任的同輩的女課長鷹司顯得關系很親密,有點橘氣,身形矮小的來棲都沖著有點御姐氣質的鷹司臉紅,還被她溫柔地摸頭,關系明顯不一般。
第6話出場的男主他姐真夠豐饒的,不比詩織里小多少,希望以后能看到二人共同入浴時的球賽。姐姐很喜歡詩織里,積極為弟弟和她的發展助攻,覺得弟弟和她結婚了之后自己就可以像吸貓一樣吸詩織里了,想法有點hentai。雖然弟弟還沒和詩織里結婚呢,姐姐就已經各種對她親親抱抱了,比如最后一話在電車上就看到男主姐摟著詩織里。姐姐真是占了弟弟的大便宜,免費爽詩織里。男主姐居然有時還會在一大清早就騎在男主身上(雖然隔著被子),雖然二人是姐弟關系,但這也顯得太沒羞恥心了,如果碰到了男主的帳篷就尷尬了。
此作每話一半進度時都會出現兩幅插畫,很多都非常出彩,甚至令人眼前一亮。比如第3話的插圖中女主霸氣側漏夾雜著微妙性感的樣子真是魅力萬分。
我喜歡的女孩忘記戴眼鏡
總評:93.2
此作主要是以一個男初中生小村楓為視角講述他和一個令他心儀的同桌女生三重愛之間的故事。三重的視力差到離譜,幾乎殘疾,不戴眼鏡的時候看書都得貼著看、不貼到對方臉上都分不清對方是誰,也因此總做出各種離譜的行為。這樣明顯不能離開眼鏡的妹子居然還老忘記帶眼鏡,或者由于意外的原因沒法戴眼鏡。此作著重描寫小村楓和這個非常冒失的三重之間一步步的感情發展。小村在剛入學見到三重的一開始就著迷于她,由于小村對三重不斷極力付出溫柔和關懷,三重也一點點認識到小村的特殊性,發現自己不想離開他,也不愿他離開自己。最后二人雖然還沒有正式告白,但在彼此都互相喜歡對方的這一點上已經基本心意相通了。此作算是個頗有個性的純愛番,總的來說此作制作質量不錯,不少劇情挺有愛也挺有趣,特別是非常與眾不同的三重愛的人物的成功塑造絕對是此作極大的亮點。
三重愛的人設非常有個性,令人印象深刻。她不戴眼鏡時只能使勁瞇著眼睛以盡可能獲得多一點的視力,這使得她的眼神看起來特別可怕,這是其人設非常大的亮點和特點。一個可愛女孩子竟然有這樣兇惡的眼神,這是我看過的海量動畫里前所未有的設定,她的這種眼神和她自身的可愛真是有巨大的反差感。我想那些喜歡被妹子以惡狠狠的表情嫌棄的抖M觀眾肯定會很喜歡她的這種眼神,會令他們興奮起來。而個別時候三重又戴著眼鏡出場,眼神和不戴眼鏡時截然不同,有時神情柔美,有時眼睛看起來又圓又亮,顯得又可愛又靈巧,這種樣子的她也令我很喜歡。
三重的視力真是差的離譜,居然能以為近在眼前的塑料袋是貓,甚至就連眼前的人是誰都分不清。真好奇三重的眼睛里看到的是什么,想多看看她的第一人稱視角。我心想這要是測視力還不得0.1?失去眼鏡的她實在過于辛苦和危險,難以想象她是怎么能避免事故和意外發生的。她忘戴眼鏡的幾率實在太高了,我感覺應該放一個備用眼鏡在學校,要不然考試那天沒眼鏡就麻煩了。她還老是弄壞和弄丟眼鏡,真是太費眼鏡了。
三重不僅視力極差,腦子還挺迷糊,也挺沒常識,居然完全不知道保持和異性間的距離,竟毫不顧忌地把自己的臉往小村的臉上緊貼,絲毫羞澀都沒有,令小村多次感受到天降大福利。
越往后看此作,我越來越覺得三重的性格真是挺萌的,而且萌得有個性,十分與眾不同,經常還有點呆萌(更準確來說是糊涂萌),顯得有種頗為神奇、神秘和捉摸不定的感覺。她還動不動就一驚一乍,常人難以理解她獨特的腦回路。她還經常表現出由于眼睛看不清楚而對當前情況的疑惑、好奇的樣子,此時老是伴隨著發出“嗚嗚嗚”、“嗯嗯嗯”、“呃呃呃”以及“哎”的聲音。她的這些怪怪的反應更增添了其萌點。
三重的頭發的細節真多,而且多次出現純手繪的頭發大幅飄動的畫面,我感覺真是太費工了,不知道畫師畫這種場面時作何感想...值得一提的是,三重的發色偏紅,再加上忘戴眼睛時總露出不爽&危險的眼神,令我想起來周防尊,她當周防尊的女兒我不覺得違和。
再說說小村。第一話剛開場,看到畫面中央有個走路的小個子男生,我心想千萬別是男主,然后發現很不幸,還真是。當時更令我大失所望的是,近距離畫面里看到他長得特別丑,缺乏氣質,就是個純情小正太的模樣,令我沒有追的想法。但看下去發現,男主小村楓這角色其實還可以,其內在性格遠沒有一開始他的外在形象令我失望。小村的性格踏實、不張揚,全力以赴幫助和保護三重,對她的感情一心一意,100%付出自己的真心,這些方面挺可圈可點的,也難怪最后三重能喜歡上他。
小村看上去是個純潔&純情小正太,經常因為缺乏常識的三重的一些不自覺的帶有撩撥的行為和話語而心動、滿臉通紅。但他其實也沒那么單純和簡單,還挺自作多情的,經常因為自我腦補而導致心里泛起波濤。他還挺有想法,心里還盼著三重能忘戴眼鏡,這樣她就會依賴自己、能有和她近距離接觸的機會。小村也不是完全純潔的孩子,骨子里其實也有點紳士,也抱有一些雄性的妄想,其心理活動能引起男性觀眾的一定共鳴,這些方面也蠻有趣的。
此作對小村和三重的關系刻畫得不錯,著實挺有愛,隨著劇情發展他們的關系越來越近,周圍的人都看得出他倆特別相互在意。隨著小村在三重心中的好感度不斷攀升,三重對他也是越來越沒隔閡,還要求他在教室里給自己梳頭,還每天都要近距離看他的臉。小村還怕被周圍的人以為他在和三重交往而導致被說閑話,實際上他倆關系那么親密,還凈做出臉紅心跳的事,不被同學們當成情侶才怪,他倆早點光明正大正式交往了多好。無奈三重的腦子發育異于常人,我懷疑目前的她是否懂得什么叫戀愛,她對戀愛這個概念可能認識得還非常模糊。在第11話里,居然三重覺得小村有父親的感覺,對他發了父親卡,對小村來說這可真是悲傷的事情。不知道啥時候小村在三重心里從目前大約70%父親+30%喜歡的人的狀態轉變成100%喜歡的人。
此作幾乎完全是在講述三重和小村的故事,沒有副CP,也極少出現其他角色,其它角色即便出場也沒多少戲份,這一點和大多數戀愛動畫截然不同。
我一直懷疑那個帥哥東同學是不是其實對三重是有好感的,從第12話才得知原來他是喜歡隔壁的上大學的姐姐。這不禁令我想起來《夫婦以上,戀人未滿》里面的大帥哥天神,他也是喜歡年上的而對同齡妹子沒有興趣。這可真是有共性。
此作的ED構思真是挺奇特的,很少見,沒有任何場景切換,始終就只有一個鏡頭,三重從左往右走。
我剛看此作的時候對此作的畫面印象很差,第1話一開場是上學路一直到教室里的連續畫面,又一次看到了鈴木信吾作為監督的那種標志性的光污染+畸變嚴重的透鏡效果+繁多的動作連貫的路人,再加上室內視角的大幅運動,真是看得我頭暈,對此作的好感度立刻降低。當時我心吐槽難道真有人喜歡鈴木信吾這種風格么?好在是一直看下去此作后逐漸適應了這種畫面風格,這風格也不算明顯減分項。
能干貓貓今天也憂郁
總評:93.25
女主福澤幸來是個未婚的年輕女上班族,幾年前撿了只貓,結果長大后這貓像黑熊那么大,平時都是站立姿態,有人類的頭腦,非常能干。被取名為諭吉(湊福澤諭吉這個梗)的它擔負起了照顧忙碌的福澤的飲食起居的家庭主夫的角色。此作講述這一女一貓共同的生活,還有不少和福澤的職場及其同事相關的劇情。此作是一部內容有趣的作品,制作得挺不錯,福澤和諭吉的人設都挺有意思,喜歡有趣的日常動畫的人很值得一看此作。
福澤和諭吉在一起真是挺有愛的,這個CP非常好。福澤在職場上是個行事干練的女白領,但私生活非常糟蹋,生活作風散漫,把家里弄得跟垃圾場一樣,也完全不會做飯,不懂得照顧自己,職場以外的她像如同半個廢人。而貓貓諭吉則家務萬能,非常愛干凈,完美地承擔起了福澤的保姆的角色。福澤的生活真是少不了諭吉的照顧,而諭吉也離不開福澤賺錢給它買貓罐頭,他倆真是特別適合在一起。
由于福澤在家里過于懶散和缺乏常識,在此作的劇情中她老是被福澤嫌棄,甚至惹它生氣,挺有趣的。諭吉倒也真是很寬容和大度,每次到最后都原諒它不懂事的主人。雖然福澤名義上是諭吉的主人,但有時候她甚至像個大孩子一樣還會向諭吉撒嬌。我感覺有了萬能的諭吉,福澤就不需要任何人類伴侶了,有諭吉寵著她、包容她就完全夠了。而且諭吉又軟又大又溫乎,換做是人類伴侶抱起來也遠沒這么舒服。福澤還能把它當做大型毛絨玩具抱著睡,挺享受的。
第12、13話對諭吉與福澤之間的關系描寫得很好,特別有愛。在這段情節里看到原本福澤的日子過得非常辛苦,每天回家之后都累得幾乎暈倒,家里完全亂得一團糟,都幾乎沒有下腳的地方。但即便如此,福澤對撿來的諭吉還努力關照,這令諭吉決心要為她報恩,于是努力學會倒垃圾、做飯,最終成長為了能干的優秀的大貓,把家整理得干干凈凈,把福澤照料得舒舒服服,令每天辛苦工作一天的福澤在到家時都高高興興的,并且期待吃到諭吉做的美味。自從有了諭吉,福澤的精神狀態有了極大的改觀,全公司的人都看得出來她發生了巨大變化,和剛進入公司時判若兩人。
起初我并不算多喜歡諭吉的形象,感覺沒啥亮點,覺得若是它再可愛一些或更美型一些就好了。不過看下去后覺得這大貓的設定很不錯,挺契合它內在的踏實穩重認真的形象的。此作里的鄰居看到諭吉的這種姿態居然不覺得意外,諭吉還經常光明正大去超市,按理說它應該會被捉去研究所研究才對。現在諭吉的形態真是很不像貓,故意裝做貓也裝不像,令福澤都有種自己沒有在養貓的感覺,都快忘了正常的貓是什么樣子了,有趣。此作一直沒交代諭吉的成長過程,到底是怎么從小貓一點點變得那么大的,真令人好奇。不知道諭吉是公的還是母的,從它說話的沉重聲線上來看似乎是公的。
第8話福澤居然展現出了非凡的體術,將男混混迅速制服,之后回頭的那一刻瀟灑極了!(這個畫面在之后幾話里還重放過兩次)不知道她哪來的如此強的戰斗力,從她的性格和外表上真是完全想象不到她還有這么特別御氣的一面。這一幕英雄救美,令那個便利店店員在瞬間都對她心動了。第9話,還出現了福澤在電車上制服癡漢的回憶,而且她居然還能在電車上不扶任何東西站著睡覺,真是異于常人。
第9話講述福澤的公司的后輩柴咲由里與福澤之間的關系,這段真是令我小興奮,橘里橘氣。柴咲很早之前就關注起經常在電車上巧遇的福澤來,被她的氣質所吸引,還對她臉紅。然后有一次她被福澤英雄救美,福澤抓住了摸她的hentai,導致倆人的人生完全交錯上了。進入同一家公司后福澤成了柴咲一直憧憬的對象,在柴咲的眼里福澤有點像超人。公司里福澤的同輩尾代聽了柴咲講述自己與福澤的邂逅時說“你們干脆結婚算了”,我心想這話說得太好了!我強烈支持她倆結婚!我本身也感覺福澤那樣比較隨性且糟蹋的女人不太像是能和一般男性交往的樣子。順帶一提,柴咲的顏值真是很高,一些角度的特寫很有美感的,希望她的戲份能再多一些,特別是能多一些和福澤的百合劇情那就太好了。
第11話柴咲和她表妹仁科理央聊到喜歡的人的那段對話真是超高能,令我好興奮!沒想到第8話被福澤救的便利店店員竟然是仁科。仁科因為那件事開始憧憬和思慕起福澤,想到福澤的時候她就像思春期的少女一樣羞澀臉紅。她對福澤當時救她的英姿的描述令柴咲立刻想起了福澤,柴咲肯定萬萬想不到自己表妹喜歡上的正是自己喜歡的人、自己和妹妹都被同一人救過。這姐妹都算是未來的情敵關系了。柴咲聽表妹對那名女俠的描述時還感嘆“我都開始想干脆找同性當對象好了”,然后妹妹心想“我能說...我也在想同樣的事嗎”。哎呀哎呀,福澤以其魅力同時掰彎了兩個妹子,改變了她們的擇偶性取向,她倆還正是姐妹,真是不可思議的事!真心希望福澤最后能和柴咲或仁科其中之一交往上(同時交往更好)。
福澤的公司里有個挺帥氣的男性小領導,他之前和福澤有過近距離接觸,還曾經抱過喝醉的她。起初我還以為這倆人會有些BG發展,后來發現此作完全沒有他倆的BG線,這點很好。我可不希望福澤和男性交往。
值得一提的是石川由依給福澤幸來配音很好,很能展現角色自己的個性,那聲線我原以為那是小松未可子給配的。
說來,此作剛一開場,看到很長一段的意義不明的街道景觀圖,里面有過曝和經常整體偏藍的色彩、光污染、流暢的鏡頭運動、嚴重視角畸變、密集的流云且帶濃厚的陰影和倒映效果、密集且走動的路人,令我感覺這真是再典型不過的鈴木信吾的畫面風格。我起初覺得此作的內容真不適合這種風格,后來看下去倒也習慣了。同一季的兩部動畫,此作和《我喜歡的女孩忘記戴眼鏡》都是這樣風格的畫面,真是巧合。
堀與宮村 -piece-
總評:93
《堀與宮村》之前的動畫我都追過。這一季是講述堀和宮村以及他們的同學在不同時期經歷的一些零散故事,貫穿高一到高三畢業,故名piece。這一季沒什么可以特別評價的,該說的上一季都說了。這一季內容基本上還是延續前作的人物關系,以戀情發展以及有趣的日常故事所組成,給之前人物的形象設定和人物關系進一步添磚加瓦。
第1話宮村逃避和大家一起洗澡、上游泳課用自己生理期為理由不裸露身子下水很有意思,虧他好意思用這種理由。
第12話講述了堀的父母年輕時候在高中時相識的故事,很有意思。堀媽夠厲害的,是不好惹的瞇瞇眼。有個年下的女生招惹她,一開始堀媽看起來還挺受氣的,居然突然給了那個叫囂的女生一個大逼兜。然后堀媽微微開眼了,氣場令人不寒而栗,真是帶刺的玫瑰。不知道是不是堀爸京介有抖M的一面,就好這一口,居然因為這個突然愛上她了。京介到現在看起來還是挺散漫不正經的,和當年的風格沒怎么變。堀媽當年顏值也蠻不錯,如果平時睜著眼睛,顏值也不輸于堀。如今的堀還真是一定程度上遺傳了父母的性格,有一些父親的大大咧咧的豪放感,也有母親的好勝和進攻性,只不過不是藏在里面而是直接展露出來。
堀真是挺適合男裝的,第2話她穿黑色的男子應援服的樣子相當有范兒,收到女生們的極大歡迎。最后一話堀居然被理發師誤剪成了短發,感覺這副造型真是顯得更帥氣了,更能吸引妹子了。
看這一季我再次感覺堀和宮村之間著實很般配。特別是最后一話,令我進一步意識到雖然堀的性格挺強勢、剛烈、獨立,但她也不想孤獨一人,很渴望有宮村這樣的人來陪伴,不想離開宮村,也擔心有一天他會離自己而去,這展現了她內心意外的細膩和柔軟的一面。
妖幻三重奏
總評:92.95
此作里有各種各樣的妖怪,有的邪惡有的無害。花奏鈴是妖巫女,從小就吸引身邊的妖怪。她的青梅竹馬風卷祭里繼承家族使命,一直在消滅邪惡妖怪,也一心保護鈴。有一天出現個強大的貓妖白金,在要吃掉鈴的時候被祭里封印住力量,而不爽于他倆戀愛情感的白金在最后一刻發動了技能,使得祭里從男孩子性轉成了妹子,妄圖以此終結他倆的戀愛關系。此作就是講述在此之后的故事,祭里逐漸適應自己的女體,和鈴不斷加深了感情,也逐漸和白金從敵人成為了朋友,最后還一起努力消滅了強大的能吃魂魄的邪惡妖怪。
看第一話的時候其實我是矛盾的,在追和不追之間橫跳了幾次。一開始看到此作是妖幻題材,覺得不是我喜歡的類型,沒打算追。然后看到OP里閃耀的妹子以及充滿百合氣息的畫面,又有了追的動力。正片開始后,我發現那個挺秀氣的紅毛主角祭里是男性,鈴還喜歡他,這BG關系又令我沒興趣了,還心想男主要是妹子、此作全是女性角色就好了,比如《閃亂神樂》。到了這一話末尾,看到白金令祭里性轉時,我按捺不住喜悅,還叫出了一聲“好!”,心想白金真是GJ,干了件大好事!看到祭里被性轉后也沒有復原方法,貌似在此作中他會一直處于性轉狀態,而且預感之后鈴會和TA親密無間、場面會橘里橘氣,再加上此作還很有福利,我就愉快地決定追了。
此作的作者是著名的ToLoveRu的作者矢吹健太郎,自不必說,此作必定福利滿滿。此作里日常、搞笑、驚險的劇情都有,令人賞心悅目的各種福利有機地穿插在其中,不愧是矢吹健太郎,非常在行。此作盡管也有驚心動魄以及一些認真描寫感情的劇情,但總的來說輕松歡樂的劇情還是占大多數。
我真是蠻喜歡祭里的女體化的形象的,面容姣好,清秀可愛,身材太頂了,穿著忍者服的時候更凸顯曲線的美感,整個人的形象真是賞心悅目,鈴作為女性的魅力都完全比不過原本是男性的祭里。剛變成女生的祭里居然跑去男廁所,還蹺二郎腿,各種走光,很有趣,這種充滿反差感的福利真是棒棒的。祭里的戰斗力相當強,這美少女忍者戰斗的樣子挺有觀賞性,給祭里漂亮的形象之上又增添了勇敢帥氣的加成。祭里的性格也很不錯,溫柔又開朗,還把保護心愛的鈴當做最最重要的事,非常有責任心。這角色設定真心很好。
祭里起初對于自己變成妹子這事還挺抗拒的,我心想:不要繼續掙扎了,好好當美少女吧,明明是別人求之不得的美事!后來祭里逐漸適應了女體,開始忘了要變回男兒身的事了,似乎對自己的現狀還挺滿意的(祭里這樣的“不幸”真是令人羨慕)。
此作對祭里和鈴的感情和羈絆描寫得不錯,二人真是特別相互重視。鈴對于怎么看待變成女生的祭里比較糾結,我心想就當百合不就好了!表面看上去,這倆人在一起真是很不錯的百合CP。此作里還有一些二人做臉紅心跳的事的鏡頭,比如最后一話祭里中了幻術后,在幻想的空間里祭里竟然在澡堂里赤果果地被鈴所摩挲,這畫面感真是美好啊!鈴一直試圖尋找讓祭里變回男性的方法,但我是真心真心真心希望祭里永遠變不回男性,變成BG的話就無聊了。祭里是女生也并不影響鈴在靈魂上對TA喜歡啊,何況祭里的女體化的形象如此之漂亮,同為女生也可以毫無距離感地接觸,有啥意義變回男性去(除非是鈴想和祭里制造后代)。
鈴的相貌不像祭里那么令人眼前一亮、那么閃耀,但外形也有亮點,此作多次描寫了鈴的潔白嫩滑的大腿,真是誘人。
剛開始看此作時我覺得鈴的聲線很像花澤香菜,還驚異于香菜的高產,后來才發現不是香菜。鈴的發型和《怕痛的我,把防御力點滿就對了。》的女主真是挺像的。
作為妖怪王的白金這角色設定得不錯,看到后面它還挺討我喜歡的,是個樣子挺可愛的懶惰的長壽的胖貓,性格蠻有趣,既有人性的理性的一面也很有頭腦單純的動物的性格。雖然它是妖怪之王,但并沒有真正的壞心思,第11話還挺身而出保護鈴,不惜自己差點掛掉。這一話也揭示了白金的過往,原來它本身并沒有主動傷害人類的意愿,只不過是幾百年來人類的一系列行為侵擾到他,所以曾經才厭惡人類。其內在其實挺好的,還主動保護其它無害的妖怪們。白金雖然是妖怪,但是對男女戀愛之事卻非常敏銳,非常懂。但它卻以為祭里性轉了就不會和鈴戀愛了,這點真是有夠無知,難道它不知道在二次元還有百合以及偽百合么???
祭里的爺爺也挺有趣,是個lsp,看樣子80多歲了,還在饒有趣味地看青春少女雜志。祭里被性轉成美少女,我估計他其實是暗喜的,能有眼福了。
第8話出場的給祭里制造道具的妹子香爐木戀緒的造型挺有亮點的,里面穿黑色比基尼,外面罩一個圍裙,露出度很高,有微妙的性感。她給鈴制造的那個性感忍者裝真是很有情趣,要是祭里平時穿著這身戰斗就養眼了。香爐木相貌不錯,性格開朗活潑,制造能力極強,是個不錯的妹子。她挺心儀祭里的,要是以后成為姬友就好了。第9話還出現了她對氣味極其敏感的設定,居然湊近了聞祭里腋下的香氣,場面真是很令人臉紅心跳。第9話她試圖令祭里恢復男兒身的過程很有趣,一開始祭里的氣球泄了氣,似乎有點變回去的希望,但大根還是沒能出來。結果沒過一會兒,突然歐派開始反彈,氣球突然膨脹起來,最后衣服都包不住了直接彈了出來,這一段真是令人喜聞樂見!帶著大瓜回家的祭里把爺爺都看傻了。戀緒在最后一話居然還貼近了聞鈴,真是橘里橘氣的,還令祭里有點警覺了起來。
祭里的那個忍者男前輩二之曲的相貌設定挺有點像《獸娘動物園》里的一只大鳥,也是頭發長度正好到上眼皮,目光十分銳利,還有種陰暗的氣息。他到后來都開始對美少女版祭里有點臉紅了,看到她可愛的樣子都心動了,他似乎快忘了祭里里面的靈魂其實是男性了。
此作的大福利部位總是被白金以一臉吃驚的臉擋住,令我想起了《宇崎學妹想要玩》里神經兮兮總在奇怪的地方出現的貓。不知道BD里完全福利解放的畫面是什么樣,我挺好奇。
此作中途間隔了很久才繼續播放,一開始和《不當哥哥了!》是同期播放的,令我當時覺得挺有意思,一季竟然有倆性轉動畫。
轉生成自動販賣機的我今天也在迷宮徘徊
總評:92.7
此作男主開場就掛掉,而且是開場30秒就因為在山上開車時撞上了飛來的自動販賣機而噶了,然后轉生成了自動販賣機。說來轉生成非人的作品不算多,上一個這種作品是《轉生就是劍》。男主變成異世界的自動販賣機后,利用體內有限的可選的技能,絞盡腦汁使自己在異世界能生存下去,并盡可能多地銷售商品。他還遇到了一個對他非常感興趣、表現得很親熱的可愛+怪力妹子拉密絲,和她成為了要好的朋友。之后的劇情主要就是講述他倆和其他一些人共同冒險、攻略迷宮的經歷,一起經歷了各種麻煩事和險境。在這個過程中拉密絲和被她稱為阿箱的男主的關系越來越親近、越來越信賴彼此。阿箱雖然外表上只是個冰冷的機器,但他為大家的不斷努力付出最終贏得了很多人的喜愛,其樂融融。此作很大一部分內容都是描寫男主的內心活動。
總的來說此作挺有意思的,挺富有創意,阿箱和拉密絲的設定都不錯。戰斗過程做得略微粗糙,但無關大礙。
男主作為自動販賣機沒法說話,僅能靠著非常有限的提示語音盡最大努力和他人進行交流,實在很不容易。基本上只能靠別人提問,他再用兩種不同的提示音回答來表達看法。好在拉密絲和他心靈比較相通,越往后越容易明白男主的想法。看此作前期的過程中,我有點納悶怎么大家似乎都能猜出男主的提示語的意思并且進行溝通,感覺那些人的理解能力也太強了,還以為這個異世界的人都這樣。沒想到在第7話,男主和那幾個袋熊始終無法建立對話,始終被他們認為是在說無意義的提示語,交流老費勁了。
男主的腦子很靈,非常有經營頭腦,應變能力極強。他的內心挺善良也是個優點,比如第3話小姑娘買東西后搖號抽獎,男主故意讓她中,令她很開心,而好賭的老頭子就沒這待遇了。以男主的品行和才能,如果在異世界是真人的話估計能受很多妹子喜歡。男主目前的自動販賣機的形象還挺可愛的,長著兩個大眼睛,不知道什么時候能變回人類。
男主雖然一開始只是個普通的自動販賣機,但隨著劇情進展、不斷升級,到最后越來越逆天。他不止販賣飲料和零食,居然還能賣洗發液、condom、曼妥思、姨媽巾、氣球、簡易廁所,第4話居然賣工口書,甚至還能輸氧,真是令觀眾開了眼界了,也令我感嘆作者對自動販賣機了解得還真是很深,連昭和40年存在的東西作者都知道。后來男主甚至還加裝了太陽能電池板,賣的東西還能彈射,還能有吸塵器吸入功能,還能隱身,還能變成洗車機、音箱...最后一話還賣了AED救了倆妹子。總之,除了不能移動外,男主簡直無敵了。在滿足自動販賣機的設定的前提下真是能隨心所欲地進化,不過奇怪的是非常有限的提示語音始終沒法擴展。
男主真是特別能活用自己作為自動販賣機擁有的技能,尤其是第6話的行為相當不可思議。他被BOSS吞掉后,居然彈出能釋放瓦斯的氣體充滿BOSS的肚子,然后通過微波爐加熱紙和罐頭產生火星,引爆BOSS的肚子,這想法真是人才。
福山潤給男主配音配得著實很不錯,把情感表現得特別豐富,令外表只是個自動販賣機的他的性格表現得格外鮮活。
女主拉密絲設定得不錯,很有元氣和精神,天真無邪,挺可愛的。她的歐派真是豐碩,小腹顯得很結實,是個很“飽滿”的妹子。她有著筋肉怪力的設定,能輕輕松松般起男主,這點和她的陽光且頭腦簡單的形象挺搭的。
那個很懂道具的拉密絲的朋友休爾米人設也不錯。她雖然沒有拉密絲那么出色的身材,但比拉密絲更有女人味,盡管她像個工科女,不怎么修邊幅,但眼神和氣質顯得有微妙的性感,我挺喜歡這形象的。第4話,阿箱給她輸氧后,她帶著興奮又認真地看著阿箱說:“你要是人類男性的話,我可能就淪陷了”,這一幕的休爾米真是蠻令人心動的,展現了和平時截然不同的一面。阿箱雖然目前的形態是個機器,但這番話也令他心中興奮起來。
想不到最后一話拉密絲在意外之下對男主親親了,男主還被休爾米和一個弓箭手美少女親了,居然一個自動販賣機能享受到這樣的福利真是不可思議,鐵皮箱子男主這甚至都算是有點開后宮了,作為機器來說真是沒有比這更高的幸福了。
廟不可言
總評:92.6
是個風格主流的福利后宮番。男主赤神明光的父親非常風流,借了寺廟的錢不還。赤神從小就拒絕變得像他父親一樣渣,堅持不沾女色。但有一次他偶然邂逅了女主蒼葉結月,導致他一直自我克制的對異性的興趣覺醒了,而且還把持不住。他為了能鎮定下來,決定去三日月寺廟修行,不料這寺廟實為尼姑庵,蒼葉等幾個妙齡女子就住在那里。經歷一連串事件后,原本被排斥的赤神被寺廟的妹子們逐漸接納,關系越來越親近。赤神既幫助寺廟干活,又打開了一些妹子們的心扉,和她們變得像家人一樣。從設定上來看此作似乎應該是后宮番,但到這一季結束時后宮的感覺目前還不算明顯。雖然已經有幾個妹子對赤神總是表現出主動白給,但赤神還只是停留在不斷享受突如其來的梨斗般的福利的階段。
此作不少劇情蠻歡樂,福利滿滿,對赤神的塑造不錯,是個很有個性的人。但此作對妹子們的情感和與赤神之間的關系的刻畫一般般。此作作畫較粗糙,不怎么講究。總的來說此作制作不怎么精良、有點放飛自我,但由于設定新穎、內容風格挺夸張,也是個能令人感受到不少樂趣的作品。
赤神的人設真是很有創意。他本身對歐派特別沒有抵抗力,為了能夠消除雜念,主動去寺廟里努力修行、力圖開悟,真是特別自律。不幸的是,他努力回避工口的東西試圖變得心神清凈,但是工口的東西、離奇的福利事件卻又總是接連出現在眼前,也導致他不斷受傷害,諸如被妹子暴揍、自插雙眼來消除雜念什么的,真是辛苦呢~ 他還顯得相當純潔和純情,努力不產生非分之想,是個很簡單、耿直、一根筋的男子。雖然赤神顯得有點傻氣,但人品很好,也表現得挺溫柔、有責任心。再加上他長得也還行,體格也好,被妹子們主動喜歡上是理所應當的。
此作里有很多奇怪的關于寺廟的設定,居然寺廟還有專用的記賬電腦程序,挺樂。此作里有各種奇怪的苦行方式,充滿了許多工口+荒誕的奇妙元素,是此作挺獨特的一個地方。此作里還出現了一本《苦行全集》,令我蠻有興趣的。赤神對苦行表現得挺有癮的。
這寺廟里祭拜的佛像的腦袋一直都沒修好,始終歪著、用膠布纏著,真是出戲。有這樣的佛像,難怪三日月這么冷清、沒香客呢。我感覺這佛像靠自己手工都能修好,至少看上去不至于那么夸張。
此作第一女主算是蒼葉結月。這姑娘蠻不錯的,認真努力,溫柔可愛,對赤神挺包容,在未來和赤神結婚后應該會是個賢妻良母。最后一話將她和赤神之間的關系提升到了新的高度,來了個收尾呼應。最初是結月把無處可去、在雨中縮成一團的可憐兮兮的赤神撿走,而最后一話在同一個地方,赤神的一個擁抱給予了失意的結月充分的溫暖。
此作里有兩個黃毛妹子,蒼葉月夜和美亞,以及和一個橙毛角色神樂,令我一度覺得有點臉盲。我感覺這是此作一個有點失敗的地方。而且這仨妹子的歐派也沒有明顯區分度,更容易分不清角色。
月夜第2話色誘赤神引誘他犯錯誤的劇情很逗。一開始各種看他不順眼,后來發現她真是個能干的男人而且本性很好,之后順利加入后宮,還讓赤神對對她的初吻負起責任。
神樂居然想讓美亞按照父母的要求成為工口女王,和盡可能多的男性交往,這家長太變態了。
作為蒼月家三女、是個loli樣子的海月雖然年幼,但是level很高,姐姐們想什么她心里都很清楚,還語言玩弄赤神。她還經常一臉平靜地說一些工口的玩笑,第9話她把搗年糕形容成0和1的碰撞,還把姐姐的歐派形容成年糕,導致赤神產生雜念。她還經常仗著自己年幼,“表面上”顯得天然和不懂事,伺機和赤神搞親密接觸,真是挺有想法和心機的。
嬉嬉姐挺有意思的,是我挺喜歡的角色。她顯得不怎么正經,喜歡戲弄人,性格蠻有趣,還經常坦胸露r,挺有色氣的。第6話她終于穿了次其它的衣服,事業線巨突出,脖子上的飾物給其進一步增添了性感。雖然名義上嬉嬉姐是個成熟的大人,但給人感覺也不怎么靠譜,要不然怎么三日月寺廟那么破敗?嬉嬉姐的發型很有個性,擋住一半臉,這造型不錯。我估計嬉嬉姐得三十多歲了,顯得挺有風韻感,是個吸引人的地方。她明明身材和顏值都很好,性格也開放,居然一直還沒結婚,挺怪。感覺她在情感方面已經變得挺佛系、躺平了,已經沒有任何戀愛情感了。
此作ED歌曲平庸,但妹子們的福利圖不錯。
藍色管弦樂(第15話之后沒繼續追)
總評:91.4
男主青野一的父親原本是個知名小提琴家,但后來出軌了,從小練小提琴、如今上初中的青野也由此放棄了小提琴。青野的性格一直比較孤僻,沒有朋友,離開小提琴的世界后他的人生更加落寞了。有一天他偶遇了學校拉小提琴很爛的女孩秋音律子,體育老師給他倆牽線搭橋,讓青野教她小提琴。之后的故事就是講青野和律子之間逐漸變得熟絡,一起升上了管弦樂社很強的高中,然后講他倆和其他人在管弦樂社的故事。
本以為此作將會是講男主女主從小提琴方面的師生關系開始,逐漸成為朋友,再然后開始產生喜歡的感情。但看下去發現不是,在上了高中之后他倆之間并沒什么戀愛感情的發展,又加上出現了許多其他角色,導致他倆的交集不算很多。
此作前期的觀感不錯,感覺是一部踏實認真制作的作品。一個小提琴高手但是如今已經放棄小提琴的男主,遇到了小提琴菜鳥但是對小提琴有極大熱忱的女主,這種相遇挺特別的。青野在第5話心里還說,律子的琴聲帶走了他的痛苦,感覺這倆人之間真是蠻有愛的。
律子的性格一開始令我覺得不討喜,但顏值不錯,稍微有一點點EVA的明日香的感覺。隨著劇情進行,她的優點逐漸顯露出來。她很正義凜然,看到被欺凌的同學不肯袖手旁觀,甚至不惜自己受到傷害。她雖然只是個小提琴菜鳥,但很勇于表現自己,也很努力上進,面對挫折也不氣餒,是心態積極的妹子。在她的影響下,青野逐漸走出了陰霾,重新開始他在小提琴方面的人生。另外,看律子這樣的美少女拉小提琴本身也是件挺享受的事。
青野的小提琴拉得是真好,屬于天才級別。其性格也不錯,長大后的他比較內斂,對律子上心,對周圍人也挺溫柔,但要說在性格上有特別出彩的地方我倒也沒覺得。青野他媽顯得真年輕漂亮。
第7話回憶了青野在小學二年級時參加完大賽后撩海帶頭女小櫻春的事,沒想到那時候的青野那么攻,顯得那么閃閃發光,跟現在陰沉的他判若兩人。那時候的青野拉得就那么好了,感覺過了7、8年,似乎青野也沒多大長進似的。小春真是很純情,對青野充分傾注了感情,可惜青野把她徹底忘光了,我還以為青野能回憶起來呢。小春長相不好看,但面對青野時表現得青澀感倒是挺可愛的。她充滿內向和怯懦感的配音配得挺不錯的。她很想交朋友,但又不敢開口,真是挺自我矛盾的人,還自我厭惡,好別扭。其實她完全沒必要去強迫自己交朋友,順其自然就好,更何況交朋友又不是必需的事。
第8話的夜晚空地的對話場景中,小春和律子之間的關系顯得橘里橘氣,二人對彼此來說都是特殊的人。小春性格內向,沒有朋友也就罷了,連律子這么開朗容易交很多朋友的人,心里也把律律放在最重要的位置上,說“是你給了我一片港灣”、“對我而言,你是獨一無二的”。此作要是從BG線變成百合線就好了,本身青野對她倆也都沒有戀愛的想法。
大約10話左右往后的劇情,雖然有的也可圈可點,但我感覺劇情逐漸變得平淡,沒什么有看頭的戀愛內容,律子的戲份也少了,而且很大一部分內容都是講拉小提琴也很好的一個白毛佐伯直和青野等人之間的事。我實在不喜歡(甚至反感)佐伯,性格冷淡,樣子呆呆的,也沒什么閃光點,實在缺乏人物魅力。而且管弦樂隊演出時3D人物渲染得太生硬,不自然,曲目老是演奏那幾個,令我覺得乏味。出場的角色也沒什么養眼的,對青野的描寫也沒令我感到出眾。由于愈發覺得內容味同嚼蠟,看完15話后就決定棄了。此作要是好好描寫青野和律子的感情發展,去掉佐伯這個無聊的角色,觀感肯定會好很多。
夫妻交* ~一旦做過就回不去了
總評:90.8
泡面非表番,只有8話,是一個知名漫畫的動畫版。此作是個小制作,staff非常少,一人兼多職。雖然此作不算上檔次,但畫面表現力不錯,女主也蠻有魅惑力,觀賞性可圈可點。
此作故事中有兩對已婚夫婦,倆男主是哥們,倆女主是閨蜜。有一次他們四人一起喝酒吃飯后,居然正大光明進行換wife play。倆男主都同意讓自己的妻子和自己的哥們暫時在一起住幾天,結果果然出現了不得了的雙重出軌。此作極力渲染背德感,老是表現出自己在和小伙伴的配偶摩擦時,心里卻還惦念著自己配偶的矛盾心情。他們逐漸地發現,自己和對方的配偶相性更好,更能獲得興奮的精神上和生理上的體驗,因而逐漸地動搖了他們與原配偶的感情關系。我看,既然互換對象之后更happy,索性就都光明正大離婚然后重新組合完了,從故事里我能明顯感覺到兩對夫妻在交換伴侶后更相配。交換后,一對是男方比較主動,另一對是女方比較主動,這比起一對都是比較內斂、一對都是比較aggresive的組合明顯更好,生活更有情趣也更少有矛盾。
明日歌真是不錯的妹子,我挺喜歡她的,很可愛,也很積極主動,對禮司顯得非常渴求。比起原來的妻子,禮司明顯和明日歌更相配。而且之前二人在大學期間就在一起,似乎有點像《宇崎學妹想要玩!》里宇崎和櫻井之間的關系似的。禮司戴著眼鏡,性格穩重,看起來文質彬彬的,沒想到肌肉挺發達,活兒很不錯,令明日歌非常享受。他倆要是不能在一起就太可惜了。而禮司的原妻子雖然相貌和身材極好,但性格太悶太無趣,雖然表面上和禮司在一起挺協調,但生活長期缺乏激情、情趣的話,感情還是容易被動搖。
幼女社長R
總評:90.7
此作的上一季是兩年前的事了。難得能出新的一季。這次還是泡面番,而且依然是OP+ED長度比實際內容都長。此作每一話的故事都短小有趣,構思精良,想象力天馬行空,繼續充分展現了這幼女社長以及她的奇怪的公司成員的獨特個性,是一部很不錯的小品動畫。
Dr. STONE 石紀元 第三季 前半
總評:95.7
此作的前兩季我都追了,這真是創意絕佳的作品。這一季依舊做得非常出色,主要內容是龍水加入科學王國后,大家一起造船、出海,尋找石化光線的源頭。期間發現了一個有人存在的小島,那里有當年千空他爸等宇航員乘坐的返回艙,但也有掌握石化光線的敵人,最后憑借千空等人卓越的智謀和琥珀杰出的肉體能力奪去了返回艙中的鉑,由此有了能夠無限制作硝酸的催化劑,隨意復活石化的人指日可待了。
這一季里千空繼續大放異彩,不斷有令人嘖嘖稱奇的神級表現,就連無線通訊耳機、遙控四驅車、鐵甲船什么的都造出來了(感覺未來20-30年內科學王國造出80486級別的芯片都有戲)。千空他們居然還比較輕易地獲得了大量的油和金屬礦石的,這小島附近的資源真是夠豐富的。
琥珀在這一季有著杰出的表現。她的卓越身材令人贊嘆,第8話穿上開胸衣服的時候真是又圓又大。她的兜襠布還特別性感。第9話的時候被那個島上的妹子化妝后,顏值真是驚人,美麗程度絕對是此作的天花板,她的美女姐姐都完全比不過她。我早就覺得琥珀要是換掉原始人的打扮肯定能非常漂亮,果不其然。第9話出現的千空和欺詐師的女裝造型蠻有趣的,很有想象力,可惜只是一帶而過,要是能看到女裝千空的正經劇情就有意思了。
銀狼在這一季顯得真是挺卑微的,害怕和千空他們出海冒險,但又想要得到村子里的人的稱贊和尊敬,假裝奮勇游泳去追已駛出的船,結果不幸假戲成真。用卡牌稀有度來形容的那個畫面里,銀狼的價值連西瓜都不如,真是毫無地位。他的女裝還算說得過去,不過還是怪怪的。不過說不定那個島上的首領其實還就是喜歡這種偽娘。
島上的首領在這一季始終沒露面,甚至是男是女都不知道,說不定是個戰斗力強大的LES,這樣的話之前送過去給她當后宮的女孩子們就...
龍水、卡瑟吉、克羅姆等船上的大家都被石化了,慘,特別是看到有的人還掉落了碎塊。這要是某些關鍵部位掉落而且接不回去那就太糟糕了。西瓜真是立了大功。
弗朗索瓦這個新角色不錯,是龍水家的女廚師&管家,一心一意服侍龍水。她的存在解決了沒有專業廚師的出海的關鍵性難題,也是科學帝國子民獲得飲食方面的幸福的關鍵存在。她的性格嚴肅認真,能力非常強,在作為廚師以外也是龍水和千空重要的助力。
最后一話講述千空他爸在這個新發現的小島的事。他爸雖然智力和知識不足千空的1/10(果然最后得知不是親生的,千空的親爸的腦力應該更出色),但他還真是很有想法,居然從河流里花幾十年收集鉑粒(令我想到《黃金神威》里在北海道河里淘金時收集到的白貨),蠻不可思議的。感覺他就像是和千空能相隔千年進行對話似的,就明確知道現在的千空一定會來取鉑,所以才如此拼盡全力收集直到最終去世為止。他的這個愚公移山般的毅力真是令人贊嘆。而按照常理來說,千空他爸做這種事>99.99999%的概率完全是浪費一生做無用功。如果我是千空他爸,我則會盡可能把腦子里的知識先全都復寫出來(寫教材等),以成為未來人類復興文明的關鍵助力,這樣才肯定是最高效率地利用生命做有用功(反倒是,千空他爸等宇航員的后代真就成了原始人了,完全沒有文化。要是盡可能在傳授知識給下一代上竭盡全力花功夫,絕不至于退化成這樣)
此作重視科學,但從科學的角度來說,我感覺那個石化光線實在是沒法解釋,現實中完全不存在類似物。或許只能說是完全超越人類理解能力的極高強度輻射的隕石。倒要看看最終作者怎么圓這個設定。
海盜戰記 第二季
總評:95
之前19年看第一季的時候,我對此作評價超級高,可以算神作檔次了,是一部宏偉、蕩氣回腸的史詩級作品,充分描寫了北歐那邊戰爭的殘酷,震撼了我很多次,也感動了我很多次,給我留下了深刻印象。沒看過的話強烈建議補補。隔了那么多年,看到了此作第二季,我是相當欣慰的。
這一季依舊是非常高水準、有深度的作品,令我非常滿意。此作真的是很有吸引力,看大多數其它作品時都感受不到作品對我的吸引力有那么強,看這種精彩的作品過程中經常感覺時間過得特別快。此作有很多地方表現力、藝術性相當出彩,很多地方BGM令我感到驚艷。
此作極其真實地描寫了英格蘭和北歐地區在充滿野蠻的戰爭時代的情景,人民實在生活得過于悲慘,家園一次又一次被殘暴的軍隊蹂躪,想獲得一個能安穩生活的家園是如此渴望而不可及的事。活在這個時代真是太可悲了,要么被蹂躪,要么成為幫兇蹂躪別人。此作從始至終突出戰爭帶來的悲劇,以及對權力、征服感的渴求所帶來的慘像。在那個缺乏秩序和普適價值的時代,想好好努力、靠辛勤的勞動安穩地過活,就這么平凡且簡單的愿望卻都難以被滿足。我真是挺好奇作者作為日本人,是怎么對北歐古時候有那么充分的了解、在作品中將那個時代的人文和環境描寫得那么真實,真是令人佩服。
這一季的主角換人了,從之前的克努特換成了托爾芬。上一季的劇情和這一季的劇情沒有緊密的銜接,之間經歷的故事沒有明確交代,這個跳躍很突然,只知道之前丹麥國王被殺了之后,克努特繼位,托爾芬被流放成為了奴隸。這一季主要講述托爾芬從當年馳騁戰場的厲害的騷年,如今變成了一個老實本分干農活的奴隸。他可謂是重新做人,有點主動參加勞改的感覺。男二艾納爾之前家園被掠奪、自己成了奴隸,被地主凱迪爾買來后,托爾芬和艾納爾成了搭檔,一起種田。在此期間,托爾芬從原本戰士+掠奪者視角,切換成了生產者的視角,令他對世界的認識煥然一新,成長了非常多。本來眼看再過不久他倆就要滿足地主的條件擺脫奴隸身份了,而且現在日子過得也不錯,可慘劇卻連續發生。先是地主心愛的女奴隸阿爾涅斯的如今已經是奴隸老公格魯薩爾跑來奪取她,在農場大鬧一場,最后格魯薩爾身亡;然后又因為地主家的傻兒子闖禍,導致如今已坐上國王寶座的克努特找到由頭,帶著一批厲害戰士殺到農場試圖奪去之。經過一場大戰后,地主被干掉,阿爾涅斯也沒了,失意的托爾芬和艾納爾一起回到了托爾芬的故鄉,冰島,并決心在vinland建立新的王國,能令來到此地的人們都不受戰爭的摧殘,成為真正的樂園。
如今的托爾芬和當年真是判若兩人,一開始我都完全認不出他來。看第一話時我納悶怎么這一季的托爾芬從當年勇不可擋、犀利的戰士變成了落魄的奴隸,而且整個人木呆呆的,難道是平行世界?從形象和心態上真是一丁點看不出他曾經的影子。不過托爾芬雖然很多年沒再上過戰場,武藝倒是一點也沒退化,和實力深不可測的蛇打得不分勝負。
如今的托爾芬真是特別正直特別善良的人,對曾經殺害的無數人誠心懺悔,哪怕那些人基本都是不得不殺的敵人。第15話托爾芬開始思考如何消除戰爭和奴隸制度,這兩樣真是此作所展現的各種罪惡和慘劇的最本源。此作描寫的時代的大多數人都向往著沒有戰爭和奴隸的世界,而托爾芬作為曾經殺人無數然后又醒悟的人,他對于和平的渴望、對戰爭的憎恨真是遠比一般人更深,也因此更有志向去改變這樣的世界。
我覺得此作的一個敗筆是第21、22話,真是非常莫名其妙,明明托爾芬戰斗力那么強,干嘛非得故意被約姆戰士團的大個子打來獲得與國王見面的權利?難道把大個子干掉難道來證明自己不是一般人就不能與國王相見?這邏輯真是很迷,看著托爾芬明明比大個子更強還要故意挨他的揍,真是令我不爽。雖然后來托爾芬也解釋了自己這么做的原因,但我感覺還是牽強。解決戰爭并不能光靠打不還手,否則施暴方往往會變本加厲,并以同樣的手段侵害更多的人,也不能單純靠以暴制暴,而是要靠相互理解、建立良好的秩序。而且也很迷的是,怎么大個子打他100拳,居然就打到太陽落山了?時間也太長了,而且打了100拳之后他累得氣喘吁吁,哪像是身經百戰的戰士,這段劇情真是太奇怪了。
男二號艾納爾是個標準的好人,雖然也沒有什么特突出的亮點,但性格挺不錯,是最真心關心托爾芬的人。每次托爾芬做殺人的噩夢的時候艾納爾都將他喚醒,艾納爾對他的關懷也令托爾芬一點點從麻木中蘇醒。二人關系越來越好,相處得越來越融洽,曾經有一次都有點令我的腐魂覺醒了。
艾納爾第一次見到坐在地主的車上的阿爾涅斯時對她一見傾心,感覺看到了自己生命中的女神。她為阿爾涅斯的美麗所著迷可以理解,但我有點不理解為啥艾納爾在知道她是地主的情婦、已懷了地主的孩子,而且她還有前老公格魯薩爾的前提下還對阿爾涅斯那么上心,畢竟阿爾涅斯無論如何也不會成為自己的戀人。尤其是艾納爾為了格魯薩爾能脫身,居然肯冒那么大危險、可能把自己都搭進去的時候,真是令我覺得其行為不可思議。只能說艾納爾是全心全意為阿爾涅斯著想,只要她能過得好,寧可冒著危險犧牲自己,這種覺悟雖然顯得有點糊涂和頑固,但也相當不簡單。
格魯薩爾的生命力真夠頑強的,被蛇插了一刀戳中要害,居然還能偷襲蛇把他勒個半死,死前走馬燈能挺那么長時間。本來格魯薩爾殺了不少人,令我覺得他也不是什么善人,沒什么必要去救,但看到最后,他說不想讓兒子成為維京人,這一刻我對他的印象有了極大的轉變。維京人在此作里就是窮兇極惡的反派,破壞和平,踐踏他人,以殺戮和掠奪獲得成就感和滿足感。
阿爾涅斯真是很可憐,是此作最悲情的角色。她本是個美麗的良家婦女,結果流離失所,被迫和尚未成年的兒子分離,并成為了地主的女人,最后還眼睜睜看著心愛的前丈夫流血死去,命真是太苦了。第18話,阿爾涅斯還被凱迪爾打得半死不活,流產了,最后整個人還沒了,真是令人非常心疼。她的一連串痛苦的經歷真是充分體現出消除戰爭、消滅殘酷的奴隸制度,以及保護人權、守護和平的絕對必要性。
艾納爾也真是令人感到遺憾,他在他真愛的阿爾涅斯活著的時候沒表白,結果對她說出喜歡的時候她已經離世了,這真是他一生永遠的悲痛記憶。
買下艾納爾的地主老爺凱迪爾真是相當了不起的一個人,可以用首善來形容。他給艾納爾開出非常好的條件,不是將他作為奴隸看待,而是把一片私有林子承包給他讓他開墾,并且從他那里收購糧食。當艾納爾賺的錢足夠贖回自己時就成了自由身。而且老爺還親自下田干農活,自己像個雇傭似地賣力。明明艾納爾名義上是他的奴隸,但他對艾納爾卻挺和氣,顯得禮貌和尊重,完全看不出是主奴的關系。老爺的性格相當和善和豁達,唯一對他人生氣時也是教訓自己的蠢兒子的時候。有這么善良開明的地主老爺真是不可思議,也太理想化了,現實中我覺得不可能有。這也令我不禁懷疑此作是不是故意先把他描述得特別完美,令人喜歡,然后再講他的悲慘遭遇,通過反差來渲染殘酷性。
意想不到的是,第18話令凱迪爾徹底人設崩塌!!!果然有個180度的轉折!!!原本我覺得他是圣人,而這一話他居然表現得像殘暴的野獸。雖然表面上和善的人有另一幅面目不稀奇,但我萬萬沒想到凱迪爾的這兩面居然如此極端。也難以想象這樣的人曾經經歷了什么才變成了以前善良的狀態。感覺凱迪爾并不是兩幅面孔同時存在、表里不一的人,而是人格有一個開關,一個極善一個極惡,在他看到阿爾涅斯背叛他時開關一下子就撥到極惡的一頭且回不去了。
凱迪爾的父親,也即此作的大老爺,雖然性情古怪,待人嚴苛,帶其實內心十分善良,是個值得尊敬的人。他認為奴隸并不是下等人,只不過是運氣不好。這說得很對。奴隸的智力、體格完全不輸于其他人,如果有公平競爭的機會,誰最后成奴隸誰成為地主還不一定的。他還勸人向善,居然愿意把自己的土地給蛇,讓他丟掉劍,并且還幫助男主帶著格魯薩爾逃脫,做法著實挺了不起的。整部作品看完,我感覺大老爺才是真正的圣人。而且他是純粹的、刻在骨子里的善,而不像凱迪爾那樣有兩種極端人格。
流浪戰士團伙的團長蛇的形象設定得不錯,相貌帥氣,挺有男性的性感,有勇有謀,既理性也懂人情,頭腦很清醒,行事老練。我覺得此作可以給蛇出個OVA或者外傳什么的,因為他的形象挺有主角范兒。想不到在戰爭之后,蛇和他的小弟們居然成了農民了,反差真大,算是解甲歸田了。蛇這么好的人設如果以后不再出鏡我覺得就有點可惜了。
流浪戰士團伙里那個賊眉鼠眼的金發長臉的家伙真是太壞了,性格跟狐貍一樣,居然慫恿地主家的蠢兒子殺托爾芬當成人禮,看到第3話末尾他被團長一拳打得臉癟進去而且牙還崩飛了真解氣!托爾芬的耳朵邊緣被砍掉了一截,真是可惜,不知道當時接上去的話還能不能長好,至少應該試一下。
地主家的蠢兒子真是太蠢了,滑稽得不可思議,能力低下卻自信心爆棚,整個一二愣子,什么本事都沒有,但是心高氣傲,不肯干農活,還要參軍立戰功,真是幼稚得很。第11話他在殿下面前切豬肉失敗,總算因為愚行令自己的自尊心被徹底擊潰了。到了第23話,很難得地看到他終于變得成熟了,認識到托爾芬是真正值得他學習的人,還好好去勞作去了,能改過自新很難得。
地主的另一個兒子托吉爾強得離譜,在此作里是top級的,而且很有頭腦。都是一樣的父親,怎么這倆兒子差別就那么大呢!不知道托吉爾之后去哪兒了,是此作未來的伏筆,說不定以后變成個反派BOSS。我感覺托吉爾可能比蛇還要強,托爾芬都未必打得過他。
克努特原本是個清秀、有點偽娘感的少年,沒想到這一季長了胡子的他開始變得狠毒、陰險,居然給哥哥下毒,簡直成了和斯韋恩王一樣的險惡家伙。權力、地位真是能極大地改變人,克努特也沒能脫離這一點。更難以想象的是,居然他陷害老好人凱迪爾一家,做得太過分了,惡行過頭必會糟至反噬。斯韋恩王在這一季里一直陰魂不散,老在克努特旁邊叨逼叨,真煩。不知道克努特老了會不會也變成斯韋恩王那副丑陋的樣子。
克努特在第22話里說的那些話真是超級中二&腦殘,說是要集結維京人,與上帝反抗,真是什么鬼邏輯,上帝都沒有個實體,他們要反抗個啥?還揚言要建立樂土,可分明自己就是在破壞樂土,說一套做一套。他明明就是追求無限的權利,卻給自己找個大義。真是壞蛋不可怕,就怕壞蛋有理想。
看完第23話,令我很是納悶怎么托爾芬那么容易就讓克努特放棄征收農場了,我覺得托爾芬的話語的力量按理說也不至于能改變克努特的統治思想和價值觀才對。此作這個部分著實有點怪。
最后一話,托爾芬剪了頭發,新造型著實很不錯,令我有種驚喜感,眼睛顯得有神了。他整個人不再顯得蹉跎,而是有未來的王者風范了,肯定能在下一季里闖出一片天地,令我很期待下一季。很有可能屆時他和克努特因為價值觀的差異要成為在戰場上相見的對手。
此作OP1不錯。雖然歌很一般,但是氣氛感做得出色,畫面設計的藝術性很強。
我推的孩子
總評:95
這部作品是2023年4月開播的動畫里最強之一,是超人氣大制作,特別推薦一看。此作內容的創作水準相當高,劇情超一流,極其有看頭,不斷有驚人展開,后續發展令人意想不到,對人物塑造和人物關系的描寫極好。我感覺作者特別懂信息傳播和公眾心理,還很懂營銷、影視、偶像,這絕不是普通作家能創作出的作品。此作動畫整體制作得特別精良,絕對配得上原作水準,所有地方都做得非常用心和考究,尤其是原畫質量極佳,仿佛制作方有近乎無限的制作經費。我非常期待此作能出第二季。
此作第1話居然長達82 min,這是故事的前情提要部分,可以視為一個獨立的劇場版,完整地交代了正篇劇情之前的故事,這部分做得實在是特別精彩。我先說這部分我當時的觀感:
男主原本是個小鎮的醫生,曾經和他關系很好的一個身體虛弱的小女孩最終因病去世,妹子生前對一個叫愛的偶像極度執著,這醫生后來也狂推這偶像。沒想到竟然有一天,懷了龍鳳胎大著肚子的愛竟然來這里生產。男主被愛的狂熱粉不講武德地推下山崖掛了之后,竟轉生成愛生出來的龍鳳胎中的男孩,被取名為阿庫亞;而龍鳳胎中的女孩恰好是因病去世的女孩的轉生,被取名為露比(阿庫亞這名字總令我想起《為美好的世界獻上祝福》里的阿庫婭,而露比則令我想起黑澤露比)。倆人都保留了前世的記憶。第1話主要就是講之后愛的演藝道路,以及阿庫亞和露比自幼對自己母親小愛的狂熱支持。
第1話的視角頗為新穎,劇情展開相當離奇,槽點滿滿,真可謂是超級神奇的劇情。居然16歲產子,這設定也太高能了,也不知道愛是因為什么原因懷上的孩子,真令人好奇。而這背后的故事,正是正片劇情里男主要揭開的。喂奶的劇情真是不得了,居然露比真的就吸了。男主居然相當有節操,不肯從愛的歐派吃奶。我想,要是讓其他粉絲都有機會轉生成愛的孩子,我不信有其他人能做出和男主一樣的選擇。但我相信他不可能在出生后完全沒吃奶,說不定什么時候模糊的記憶會被喚醒。
愛的設定真是絕佳,外表極度閃亮和閃耀,顏值驚艷,絲毫不亞于水原千鶴,養眼極了。而且她的笑容甜美無比,還有天才般的舞臺表現力。雖然她在演出時展現的笑容是純商業笑容,但也沒有其他人的商業笑容能超過愛,愛已經做到了滿分。當她在一次演出時看到她的倆寶寶在現場揮舞熒光棒,那一刻她興奮的神情真是美到如夢似幻,那一幕畫得實在太好了。
此作揭露了很多偶像、演藝領域的殘酷現實,顯得特別真實,真是充滿了各種心機和潛規則,非常冷血和無情,也特別不公,真正出類拔萃的藝人得不到重用、得不到機會、收獲不到人氣,與此同時藝人還要面對腦殘粉絲們惡毒的攻擊。愛雖然年紀輕輕,但也早已經深刻懂得這個大染缸的陰暗,顯得非常早熟,她非常明白在偶像領域必須靠欺騙觀眾來生存。愛從小沒有父親,母親因偷盜而判刑后進入了孤兒院,從小她就缺愛,在演藝界她一直表演著虛偽的自己,對粉絲們展現虛假的愛意,對他們訴說著愛的謊言。不過盡管如此,她在心底里是真心渴望著愛,希望愛的謊言最終能成為真正的愛,這與其名字正好呼應。在她生下寶寶后,對他倆全力傾注了自己的愛,也從他們身上感受到了被愛。可惜,當愛正值人生巔峰,有了名望、有了家人時,卻慘遭毒手,真是令人心痛。
愛的眼睛有兩個星星,就像夢幻斑斕的宇宙,極其閃亮耀眼,這個設定很棒。而阿庫亞和露比的眼睛則一人繼承了一個星星,這個設定挺有意思。
從第2話開始就是正片的劇情了,愛就只存在于回憶中了。正片的劇情主要講阿庫亞成為高中生之后,為了尋找殺死愛的兇手而踏入演藝圈經歷的各種故事,劇情同時描寫了好幾個妹子的經歷以及她們與阿庫亞的關系。
阿庫亞這角色在此作中展現的人格魅力真是太棒了,很超凡脫俗,肯定在女性觀眾心中人氣相當高。他的形象很帥氣,不愧是母親的高顏值遺傳。他的頭腦極其睿智和理性,思維縝密,擅長推理,性格冷靜,行動力還特強。他總是表面上顯得冷言冷語,有時還顯得對別人不屑一顧,實則他的內心非常善良而且熱心。哪怕是和他關系并不近的人,如果他認為值得去幫,在其遇到困難時他也會積極伸出援手,甚至不惜自己為此辛苦一番。他的為人處世的方式真是很出色,同時很有他自己的個性,在故事中好多次都立了功,甚至還救人一命,真是令人佩服。也由于他卓越的性格魅力,令佳奈和黑川茜都為他所吸引,最后愛上了他。
第一次我看到阿庫亞長大后,感覺他真是個帥哥。以這樣的顏值按理來說絕對能作為偶像出道,哪怕演技一般般。但一開始雖然他的演技在線,卻沒能踏上演藝的道路,令我覺得真是可惜了。好在是通過一些契機,再加上他自身是閃閃發光的金子,他被動地被挖掘了出來。阿庫亞雖然并不指望也并不主動希望自己出名,但這一季末尾,看到他接到了舞臺劇重要男角色的工作,再加上他的極品相貌和杰出的才能,我估計在下一季他肯定能變成受關注度很高的業界新星。
在此作里阿庫亞身邊雖然圍繞著出類拔萃的美少女,但他卻沒有對哪個妹子產生出過真正喜歡的感情,始終顯得特別超脫。這倒也能理解,一方面是他有自己的使命,另一方面他的心理年齡比身邊的妹子實際上大二十多歲,存在代溝,本質上有種俯視的感覺;還有一方面在于阿庫亞真心喜歡的小愛的level實在太高了,現在接觸到的妹子都沒有能達到那樣高度的。不過,按常理來說,他面對那些美少女,甚至還有對他倒貼的,其心理和生理一點沒有響應按說也不應該。
正由于阿庫亞總是顯得太認真、太理性、太無懈可擊、太高冷,因此此作里出現一些他破防,或者一些出戲的行為的時候會感覺得格外有反差感,這很有趣。比如最后一話他在新生的B小町的live時大甩熒光棒的樣子真是巨樂。
要是男主轉生成了小愛生出來的女孩露比就有意思了(與此同時生病去世的妹子轉生成阿庫亞)。男性轉生成女性的作品也不少。要是真這樣的話,劇情肯定有很多能大加發揮的地方,出現很多喜聞樂見的槽點,甚至會有不少不可描述之處。
露比小時候很可愛,畢竟遺傳了母親超優秀的基因。她在長大了之后雖然也蠻漂亮,但感覺只是個標準的美少女而已,跟愛相比就顯得太平凡了,沒有美到那種能令人哦呼出來的閃耀感覺。在性格上長大了的露比也沒什么突出的地方,遠沒有阿庫亞的光環閃耀。感覺長大的露比的那個黃色側馬尾造型真是特別像《孤獨搖滾!》里的虹夏。不過她在最后一話倒是出彩了,能令如今已經是大叔的B小町的老粉絲看到了當年小愛的魅力,令其被圈粉。希望在下一季能看到露比有出彩的表現。
有馬佳奈是個令人印象深刻的妹子。當初當童星的時候性格不討喜,令我印象不佳,沒想到如今長大后和那時有了截然的反差。從第3話開始,我發現長大后的佳奈表現出的性格很不錯,她開始變得特別照顧起別人的感受,甚至能為了一部戲的成功一定程度犧牲自己,跟小時候以自我為中心的她截然相反,這樣的佳奈很討廣大觀眾喜愛。佳奈由于從童星畢業后遭遇一次次連續失敗的經歷,導致她如今完全失去了自信,總表現出消極的心態,一直沒有自己的忠實粉絲支持她是她心中最大的傷痛。她總在自我貶低自己,覺得自己注定要繼續經歷失敗。這么美麗又努力的小姑娘笑著說出那樣殘忍的話,看得人真是挺心疼的。可惜演藝圈的黑暗和殘酷,令她的努力和善意得不到回報,真是相當可憐。
第9話出現了佳奈唱歌極佳的設定,我之前真是小看她了。明明她長相不錯,演技一流,唱歌還非常厲害,長大后也一直在努力工作,居然一直沒紅,挺不可思議的,令我懷疑難道在此作世界的設定里她長大后顏值變殘了?值得一提的是,她小時候穿青椒的布偶裝唱唱跳跳的那一幕真是像極了平安名堇小時候扮演具足蟲,都是紅極一時,并且成為了長大后自己心中的黑歷史。
佳奈在心里很喜歡阿庫亞,但是又礙于面子和過往,始終假裝作為普通朋友相處,不肯表露真心。而當阿庫亞主動約她、主動為她做些什么的時候她會顯得特別開心,這時顯得真是蠻可愛的。可惜阿庫亞骨子里不把她當異性、可交往對象來對待。此作的故事中,當佳奈看到阿庫亞和黑川在節目里接吻時感到心里很難受,這時的她更令人覺得可憐,不僅演藝事業不順利,從小就開始在意的人還被后來出現的女人搶走。
一直很悲觀也確實很悲情的佳奈,總算是在最后遇到了可能令她人生和事業發生轉變的重要契機。她加入了新生的B小町,在露比和MEM的鼓勵下,可能她將有機會能夠把一直蘊含在體內得不到釋放的偶像潛力噴薄出來,從而很快走紅。希望在下一季能看到佳奈重新變得閃耀和自信。
黑川茜在演出中不慎把有希的臉劃破后,那些流言蜚語和網暴真是極盡惡毒,活生生把她逼得跳橋。好在阿庫亞立了大功,在她跳橋的一刻將她突然抱住,這一幕真是經典,阿庫亞GJ!看到這里令我深感阿庫亞真是很正義和善良,不僅是他自己母親的事,哪怕他看到一起演真人戀愛秀的和他沒什么關系的妹子在網上受到霸凌,他也決意去反擊那些隱藏在屏幕后面撒播惡意言論的bad people,試圖通過自己的努力去懲罰邪惡,其舉動真令人欽佩。
黑川茜為了報答救她一命的阿庫亞,決定化作阿庫亞最喜歡的少女的模樣,也就是小愛。我本以為茜就是個很平庸只不過相貌不錯因而能得到演出機會的妹子,沒想到她居然不是等閑之輩,竟然能復刻小愛的星星眼,令我著實對她刮目相看。令我覺得納悶的是,既然她如此能耐,肯定也經歷過很多世面,怎么還會因為網暴而跳橋,不應該啊。沒想到茜扮演愛的時候能令那么理性冷靜的阿庫亞臉紅起來,真是令我相當意外,看來阿庫亞對小愛真是有著男女之間的那種喜歡的感情。茜抓住人物內心和性格的能力是真強,這使得阿庫亞決定利用她來回溯小愛的過往來尋找兇手,這個劇情構思真是相當可圈可點,厲害了。
我對茜總體感覺一般,之前感覺她的長相也只是中上,但看到阿庫亞吻她時她緋紅嫵媚的面容,突然感覺她的顏值也蠻高的。
想不到最后一話出現了茜和佳奈小時候結了梁子的設定,真是很有趣,以前是演藝圈的死敵,現在更加對立了,還增加了情敵的關系了,樂~
此作的還活著的女角色里,我目前最喜歡的是MEM 啾。MEM 啾無論是頭腦還是相貌都很像鬼塚夏美,非常懂得通過網絡經營自己的關注度并從中獲利,很能說會道,深諳公眾的心理,很有一手。同時這一季開播的《偶像大師 灰姑娘女孩 U149》里的米莉亞也是這種youtuber設定的妹子,真是巧合,也傳遞出未來這種設定的角色可能會越來越多的信號。
想不到小愛去世后過了十幾年,MEM 啾居然能根據阿庫亞的泛泛的描述而一次就說中小愛,這妹子也真不簡單,真是非常懂偶像。后來才知道MEM 啾原來已經25歲了,難怪能知道B小町,B小町火的時候她還是個孩子。
我是越往后看越喜歡MEM 啾,她的相貌和性格真是很可愛,顏值又高又有個性,整個人有點萌萌的貓屬性,挺俏皮,性格非常討人喜歡,對周圍的人和氣友善,沒有任何壞心思和陰暗負面想法,挺純潔、純粹的。最了不起的是,她有著成為偶像的執著夢想,居然不惜為此謊報年齡多達7歲,這種執著挺令人尊敬的。光從相貌上看真是看不出MEM 啾居然比周圍其她妹子大那么多。倒也因為她的歲數更大,經歷更豐富,因此比同齡人的直覺更為敏銳、看問題更透。之前我還心想,要是MEM 啾在戀愛真人秀的節目結束之后在此作中就不再出現的話就可惜了這個人設了,當我看到她被邀請加入新生B小町的時候我真是蠻開心的,說明從此她將成為一個有固定戲份的角色了。希望在未來能看到她在舞臺上、作為偶像的道路上進化、變得更加閃耀。
想不到那個成熟的導演大叔居然是啃老族,明明看上去是個業內的獨立工作的有身份的人,竟然還和老媽住在一起。在阿庫亞小時候他就已經有所小成,沒想到現在都40多歲了,還天天受媽媽照顧,真是...
起初社長的老婆和社長結婚的目的是為了之后能蹭上小鮮肉,這想法真是夠爛的。她那時候特別討厭愛的兩個小孩,但在愛去世后,卻認真地擔當起了母親的角色,真是比曾經成熟了非常多,心態也轉變了非常多,現在成了一個比較可敬的角色。她的顏值和身材都很不錯,只在幕后工作真是可惜了她出眾的外貌,以她的相貌在以前不當個模特真是太可惜了。希望在下一季能看到她也能有機會展現一下自己的風采。
此作的OP我雖然說不上多喜歡,但風格很有個性,尤其是一開始的說唱的那幾句令人印象深刻,總的來說可圈可點。
此作ED一般,但前奏那一小段很不錯,特別有振奮感。每當一話末尾突然出現驚人展開時且這一段音樂一響起時,就特別能把觀眾的情緒和對下一話的期待感給吊起來。
此作里說道一個月能掙100萬日元的偶像都不多,甚至小偶像一個月只有20萬,令我覺得挺吃驚,不知道是真假。100萬日元也才5萬軟妹幣,比大廠的碼農也強不了太多。感覺此作對試圖嘗試成為偶像的年輕人有一定勸退作用。
我家的英雄
總評:94.8
此作剛開始看時,會令人感覺這是一部非常樸素的動畫,似乎沒啥亮點。但只要看完兩話,就會覺得此作非常吸引人,再繼續看下去會覺得此作的劇情真是超絕,是一部相當精彩的懸疑推理類作品。此作能給這么高分不在于制作質量,主要在于劇情構思真是特別厲害,高潮迭起,故事展開環環相扣,劇情發展特別有懸念,是特別吸引人一口氣看完的作品。此作是我強烈推薦一看的,絕對不要錯過!此作之前還有真人版,不過我沒看過,鑒于此作的故事本身那么出色,估計再差也差不到哪去吧。
此作的男主鳥棲哲雄是一名47歲的父親,女兒被黑社會的渣男欺騙了感情。哲雄發現那個渣男不僅對她施加暴力,甚至還要謀財害命。機智的哲雄在危急時刻把那個渣男延人打死了,結果也因此攤上事了,開始被黑社會團伙的人糾纏。特別是黑社會派的追查此事的恭一特別謹慎和難搞,令哲雄吃了大苦頭。此作內容是講述哲雄、他的老婆鳥棲歌仙以及女兒鳥棲零花這一家子經歷重重險境的故事。哲雄想方設法給自己開脫,在最后一話終于算是脫身了,把事情了結了。
鳥棲哲雄真稱得上是奇人,超級了不起,機智得不得了。雖然他明明只是個平凡的上班族,外貌顯得非常普通,是個老實巴交的中年人,不像是有什么過人之處的樣子,也不容易令人產生戒心,但實際上其推理能力、反偵察能力、隨機應變等方面的能力都超強,絕對是個當頂尖間諜的料。他居然還懂得如何處理尸體,手段極其專業,令我吃驚不小(這樣的處理技術要是真被現實世界的bad people學了去就糟糕了)。他還很懂得利用對方心理給自己創造機會,甚至還試圖令黑社會組織內部互相猜疑實現自己的脫身。哲雄真是扮豬吃老虎的那種人,誰也想象不到這個看起來形象瘦弱、性格軟弱順從的男子居然不是等閑之輩。此作對他的形象設計和刻畫真優秀,諏訪部順一的配音也恰到好處,用的聲線和表演方式與其形象太搭了。
哲雄除了自己超凡的能力外,另一個特別令人欽佩之處是他對女兒的父愛,不惜一切代價也要從惡勢力手中保護女兒和整個家庭。當他陷入困境和絕望時,想起自己的女兒,又會繼續拼盡全力地努力,很了不起。
哲雄最大的失誤就是沒有早點報警,當初他監視黑社會團伙時被拽進巷子里毆打,在此之后就應當馬上報警,這樣的話也就沒有之后的事端了,也能完全保護好女兒。而且其實早在那個黑社會男的試圖把他拉到巷子里時他就應該意識到之后會發生什么,應當馬上暴擊其要害,使之站立不能,并大聲呼救。
哲雄的老婆也同樣是個不得了的人才。她明明看上去只是個普通家庭婦女,但得知哲雄滅了渣男后還挺淡定,然后還積極配合他的行動,包括毀尸滅跡以及反制黑社會團伙,隨機應變能力也同樣特別強,非常冷靜且富有勇氣,還特別會演戲。第10話,居然她還一個人入侵恭一的房間成功把嫁禍包塞到了他的保險柜里,其能力和膽量更是令我驚嘆和贊嘆。哲雄能跟她結婚真是太幸運了,這兩個外貌平庸而內在超牛的人能結合在一起也真是很神奇的事。但也不知道這倆人怎么才生出了那么糊涂笨拙的女兒。
哲雄的女兒真是全劇最蠢角色,甚至給哲雄的計劃幫倒忙。她父母都那么機智,竟然她會被延人那廝欺騙感情,太蠢了,看此作時看到她犯蠢時挺容易來氣。家里出了這么大的事,女兒始終被蒙在鼓里,哲雄和他老婆也真是夠能掩蓋的。
給黑社會賣命的恭一真是極難對付的家伙,整部作品里他對哲雄死纏爛打非要找出他殺死延人的證據,要是沒了他哲雄一家就輕松了。雖然哲雄超厲害,但偏偏碰上了這么個警惕性非常高、洞察力特別敏銳、難以糊弄的人,真是倒了大霉。本以為最后哲雄把恭一陷害成功后就事情就了了,居然又出現了反轉,恭一去給麻取打小報告,當時令我覺得哲雄真是太難了,什么時候才能熬出頭啊!當時看到恭一受傷,在大街上流著血,心想他趕緊掛了,或者被黑社會干掉就完了,居然血槽很長。我當時也還一度猜想會不會哲雄不忍看到他的那副下場而心軟,把他救了,令其成了友軍,結果恭一對哲雄也挺絕的,到最后也不放過哲雄。恭一的身世挺慘,但并不值得同情。他明明是個很有能力的人,卻要加入黑社會團伙,給這些人干事導致自己落得悲慘下場完全是活該。最后他不僅受了重傷,被黑社會追殺,之前好不容易攢的錢大部分還都被沒收了。看此作時我有時還覺得,趁恭一一個人背對著哲雄時,哲雄干脆也把他干掉多好,然后也讓他和延人一樣化歸土壤中。
最后哲雄居然跟麻取肉搏,這個展開著實驚人,倆父親竟然直接干上了,場面很激烈。我原本還心想會不會看到哲雄這么坦誠,最后兒子控麻取選擇放下仇恨,看來對他沒法報以幻想。不知道最后麻取為什么自殺了,沒搞懂他是什么心態。哲雄收拾麻取的尸體真夠利落的,在黑社會小弟進入房間之前那么短時間內就收拾得干干凈凈,而且居然還僅憑自己一人之力就把他的尸體在不被任何人察覺的情況下運上了貨車并且在山溝埋掉,真是不可思議。這段居然沒有任何描寫,我感覺挺不應該(亦或是最后一話時間有限只能略過?)。
總之最后的結局意外的順利,恭一消失了,非要搞清楚兒子下落的麻取沒了,黑社會對哲雄也沒有興趣了,也沒驚動警察,結局比我想象的好得多。
我意外地發現此作漫畫還沒完結,不知道在TV版之后又有什么事端。不知道會不會恭一要報復哲雄對他的陷害,或是警察發現了什么線索。最后哲雄看到電視上播報山上有泥石流的畫面的那個鏡頭耐人尋味,莫不成后來麻取的尸體被沖出來,或者在搜救隊找人的時候翻出來了?雖然此作非常好看,但我希望故事到此就結束了,不怎么想看到此作再有第二季,因為不忍心看到哲雄再被折磨。
小鳥之翼 BIRDIE WING -Golf Girls’Story- 第二季
總評:94.8
此作的第一季我超級喜歡,是百合度極高的少女高爾夫球動畫,比賽過程驚心動魄,人設絕贊,極有看頭,是我吐血推薦的動畫,絕對稱得上是寶藏。這新的一季我盼了好久了,繼續做得特別出色,看點滿滿,觀感極佳,情節緊湊,很多比賽過程驚心動魄,我太喜歡這番了,對我的吸引力極強。強烈推薦沒看過的人看看。
這一季把兩位女主伊芙和葵的身世都交代清楚了。難以想象,一個表面看上去是一個運動類的作品,居然有那么離奇、曲折、糾纏的故事線,人物背后故事如此復雜的作品在運動番里真是極為罕見。如果你以為此作就是僅僅講妹子打高爾夫球的動畫,那就大錯特錯了。
這一季多次令我感覺到人物關系真混亂。第16話,伊芙和葵在雨中的對話中,伊芙有一種惡魔的感覺,往日很自信的葵在此刻咄咄逼人的伊芙面前顯得特別脆弱,因自己的生父問題而茫然。而伊芙不僅不安慰她,還無情地說要和她比賽一決高下,之后就斷絕和她的關系,令葵更是失魂落魄。這一幕里的伊芙真是有點太狠心太絕情了,令我覺得葵很可憐。
我原本以為伊芙和葵是同父異母的關系,算是親姐妹,想不到伊芙通過擊球找回了自己關于親生父親的記憶,原來伊芙是葵的名義上的父親一彥和一個洋人大小姐私奔生的孩子,而葵的父親居然是學校里高爾夫球部的老師亞室。難怪亞室之前顯得好像是個深藏不露的不平凡的家伙,原來曾經是那么厲害的球手,他和一彥的競爭關系都有點像如今伊芙和葵之間的關系。這樣一來,高爾夫球部的女部長神宮寺在名義上成了葵的后媽了,想來真是離譜。
這一季的比賽過程繼續非常扣人心弦,在擊球的表現力上的表現比第一季更上一層樓,伊芙掌握了新的武器,將師傅雷歐的子彈擊球、親生父親一彥的彩虹擊球融合到了一起,還加入了艾莎擊球的爆炸效果,在最后還把葵的球桿的加成也融入了進去,可謂達到了人類史上最強擊球的境界,擊球時就像施法一樣,真是嘆為觀止。
這一季又冒出些不自量力試圖挑戰伊芙和葵的人,果然都被二人碾壓了,挺好。我之前懷疑伊芙和葵畢竟還只是少女,進入職業界的話說不定會遇到很多遠超過自己之前遇到過的強敵,結果就連日本女子高爾夫球獎金第一的選手都被葵給完虐了。雷歐新培養的徒弟艾莎我還以為有多厲害呢,原來就是個準星一般的重炮手而已,也就是至多1.5流的水準。她的助跑擊球挺有意思的,不知道現實中有沒有人這么打球。
想不到十六七歲的伊芙和葵,居然真的已經在世界女子高爾夫球領域中實力登頂了,我還以為這是她們需要進一步成長之后才能做到的事。最后一話看得我很滿意。想不到居然能看到葵給伊芙當桿妹這么一天,令我著實吃驚。伊芙的擊球結合葵的能令實力加成的球桿,真把那個特別自負的號稱女帝的尤哈·哈米萊爾給贏了,實在很爽。雖然最后葵因為曾經牽扯黑社會的事被挖出來而被剝奪了獎金,但這都不重要了,高爾夫球屆都知道了伊芙這個名字,那個尤哈雖然拿了冠軍,但在領獎臺上肯定心里覺得以這種方式奪冠對自己來說是種恥辱。
伊芙被禁賽三年,此期間葵也在養病,時間一轉眼就過去了。看到這倆人重新健健康康地在球場上成為對手,感覺這個結局不錯。三年前她倆就已經是最強了,又經歷了三年的修煉,未來的高爾夫球界豈不得被這倆人長期稱霸了。
在這一季里伊芙和葵之間還是沒能分出勝負,最后一球葵差一丁點沒進真是太可惜了,不過好在沒影響伊芙實質性獲得了比賽的冠軍。我感覺葵最感興趣的是伊芙這個人,而伊芙對葵感興趣的主要是她的球。雖然我更想看到更為閃耀的伊芙戰勝葵,但如果伊芙如果不能徹底贏的話,她就會始終和葵競爭,就總會和葵在一起,這樣想來不讓伊芙贏倒也好。伊芙對葵和她的約定真是太執著了,最后一話里葵的遺傳病發作導致無法繼續比賽,伊芙不把女帝放在眼里,卻把葵在這個球場上的最佳成績當做虛擬的競爭對手。
最后一話里伊芙用大力發球導致手疼得不得了,而葵因為舊病導致頭疼得不得了,但此時作為球員和桿妹的二人為了共同戰勝女帝而互相支持和鼓勵,有種同甘共苦的感覺,再次令我感到這倆人真般配、在一起真有愛。等以后倆人都從高爾夫屆隱退了,就結婚吧!
沒想到那個毒蝎女維佩爾如今有了正經的丈夫,開始變得稍微穩重踏實了,成了正面角色。現在她的形象有點像歐巴桑,成了太太的樣子了,沒以前的姿色好了。
可惜那個斷手的白發女羅茲在這一季再也沒出場,看來是真中槍了,徹底涼了。她的形象令人印象深刻,有不少喜歡她的觀眾,要是有機會復活就好了。
伊芙的桿妹早乙女一奈真不錯。起初我不怎么在意她,畢竟長相就是路人水平,但到后來我越來越欣賞這個角色,也覺得她挺有趣的。她對伊芙忠心耿耿,為了當她的桿妹竟離開學校一個人跑到它國,成了伊芙重要的助力。雖然伊芙或許只是把她當工具人,但一奈真是心甘情愿地成為伊芙的工具人。我還逐漸發現一奈的性格也蠻有意思的,對堅持的事情非常認真和執著,而且在高爾夫球上的判斷和分析能力相當卓越,是作為桿妹的天才。
一開始伊芙還獨斷專行,聽不進去一奈的建議。而到第22話,居然伊芙意外地變乖了。一奈不讓她用爆炸擊球,她還真的老老實實就封印住,最后想要用爆炸擊球還事先向一奈申請許可。看來伊芙也是確信了一奈的水平,把自將交給她管理了(同時也有那么非常微妙的妻管嚴的感覺)。
在最后,伊芙被禁賽三年,突然消失了。居然一奈一直等著伊芙的回歸,哪怕日本頂尖的球手邀請她當桿妹她都不答應,她對伊芙真是太忠實了。明明伊芙是否會回來都是未知數,如果永遠都不回歸,或者找了別的桿妹,那一奈的人生就要浪費掉了。可是一奈就是像忠犬一樣等著她期待的人回來,真是不容易,這執著也很了不起。
一奈對伊芙沒有那么明顯的像雨音對葵那么特殊的球員-桿妹以外之間的感情,她對伊芙的桿妹的位置是超級執著,不肯讓給任何人的。最后一話看到葵臨時給伊芙當了桿妹,我就在想一奈此時是什么心情。結果還真給了個畫面,這一幕里一奈觀看比賽時心想:“天鷲同學,只有今天,我會把伊芙同學桿妹的位置讓給你”,一奈果然特別不甘心自己最重視的伊芙以這種方式被占有。
我還感覺藤田咲給一奈配音配得蠻不錯的,那種有輕微的粗、莽的感覺很適合不修邊幅、大大咧咧的她。
雨音一直是葵的專屬桿妹。我之前對雨音也不怎么關注,想不到她在第20話里大放異彩,是其人生高光時刻,對葵奪冠有關鍵性貢獻。居然她還有氣象預報員的證,簡直有呼風喚雨的能力,真是邪乎、離奇,令人嘖嘖驚奇!然后,這倆人在賓館里坐在床上正對著對方的場面真是橘里橘氣,雨音露出了一種奇妙的神情,眼神和嘴角的弧度流露出對葵的愛戀、愛慕又復雜的感情,還有種微妙的瑟琴的感覺。之后她對葵說“如果你覺得自己攪亂了我的人生,那就負起責任到最后吧”,這話太高能了,絕對是發自真心的愛的告白!雨音已經決定把自己的人生都托付給了葵。然后葵猛然抱住雨音說“雨音 我愛你!”,這一刻二人的百合怒放,真是令我激動!沒想到這倆人之間有如此直球的發言,哎呀哎呀~ 官方也太會了。看到這里,我感覺伊芙股徹底跌停,伊芙都被此刻的葵拋在腦后了。看了第23話后更感覺雨音對葵真是盡最大全力去照顧和呵護。雨音肯定沒有期待過葵對她有任何戀愛感情,但如果葵真的是以這種感情喜歡她,她肯定是求之不得的。
給伊芙當輔助的老頭兒的為人真不錯,努力在背后支持伊芙,居然還有針灸執照。沒想到之前他在打球方面是雷歐認同的厲害角色,此人真不簡單。要是此作里出現一次他曾經打球的畫面就好了。
葵她媽也挺不容易,一心為女兒的前途著想,看到自己女兒也患上不治之癥,心都碎了。不知道葵她媽看到曾經的情人亞實如今和女學生神宮寺交往、親密地在一起的時候作何感想,會不會有吃醋感。我感覺葵她媽一個人真是挺孤獨、挺累的,難過時需要有人撫慰一下,她身邊那個黑衣人我覺得和她有發展潛力。
感覺有的場景里亞實和亞彥之間、亞彥和雷夫之間顯得gay里gay氣的。
這一季里居然出現了球打到旗子的布上掉下來進洞的畫面,令人嘖嘖稱奇。球打到果嶺上經過旋轉進入球洞看起來是極其罕見、純靠運氣的事,到后來似乎這種進洞方式都已經是伊芙可控的了,經常出現,顯得是故意這么打的,伊芙真是神乎其技,后期的實力明顯比之前上了一個臺階(但再離譜也沒有此作最開始那樣直接從疾馳的列車的縫隙中打過去夸張)。第22話,居然伊芙還把球桿打斷了,真是不得了,伊芙用的球桿廠商的股價可能因此下跌。
這一季居然沒換OP歌曲,還是那個我不挺喜歡的雖然風格鮮明但表現方式很張揚鬧騰的歌曲。
鬼滅之刃 刀匠村篇
總評:94.5
鬼滅之刃之前的動畫我全都看過,這一季依然制作得相當精良,畫面表現力登峰造極。這一季劇情是炭治郎、霞柱時透、戀柱甘露寺蜜璃都去了給殺鬼隊鍛刀的村子,村子遭到上弦伍玉壺和上弦肆半天狗的入侵,這幾個人展開了和這倆上弦的激戰,苦戰之后在殺鬼隊沒有傷亡的情況下干掉了兩個上弦,并且彌豆子還恢復成了人形,可喜可賀。
首次出場的那個眼睛和嘴調換位置的上弦伍的形象設計真夠惡心的,令人看著非常不適。感覺這個家伙相對于其它上弦來說比較弱,時透一個人就將之干掉。
上弦肆和上弦伍之間雖然只差了一位,但前者比后者厲害太多了,感覺一個上弦肆能頂三個上弦伍。上弦肆實在太難對付了,能分身,每個分身戰斗力還特強,還有多個形態,本體還特別小、特別能跑和能藏,脖子還賊硬,干他真是費老勁了。上弦肆本體的性格和形象巨惡心,是個內心卑鄙齷齪的小老頭兒,還總裝弱小,明明吃了很多人卻還顯得自己是受害者,真令我討厭。
想不到彌豆子竟然那么容易就變回人了。不知道太陽把她曬到什么程度突然就變了,畫面上也沒個交代,令我覺得奇怪。早知道這樣就能變回去,早點曬太陽多好。而且既然她本來不怕太陽,為啥起初被太陽一照就像被炭火烤一樣?挺說不通。變回人后鬼頭明里總算有臺詞了,不用純靠哼哼來領工資了。
在此作中對甘露寺第一次進行了充分的描寫。她真是又大又白,感覺過多的脂肪明顯會影響戰斗,如果像傳說的亞馬遜女戰士一樣割掉的話可能戰斗力會進一步增強。她過于豐碩飽滿,衣服都完全包不住,也沒法怪上弦肆稱之為蕩婦。她得早點重做一身衣服了,現在的衣服太危險,萬一什么時候沒兜住就糟了。甘露寺的吃貨設定在這一季里還專門做了描寫,感覺她有可能把吃的巨量食物轉化的能量都儲存在了那個部位。
甘露寺的戰斗身姿很有觀賞性,就像藝術體操表演一樣。不知道她的刀怎么造出來的,和彩帶一樣,和其他人的刀完全不是同一種概念。她的性格挺有趣的,總顯得沒什么緊張感、不怎么認真,甚至往往帶著一點玩的心態來戰斗。香菜給她配音配得真是相當好,聲線聽著舒服,把她的自由和有趣的性格表現得很到位。
居然釘宮配了只烏鴉。
把兩個上弦滅了之后,居然炭治郎出村時還得蒙著眼睛,我感覺完全多此一舉,畢竟村子的位置都被上弦探出來了,隱藏村子還有何必要?應該常駐一個柱在這里。
輝夜姬想讓人告白-永不結束的初吻
總評:93.9
我本以為在上一季所有的故事就都結束了,畢竟白銀和四宮倆人都深吻了,沒想到還有這么一季,只有4話。白銀和四宮這倆人都有里外兩面,尤其是輝夜,更是有著雙重人格。二人之前都在使用著表層人設,因而上一季的接吻屬于是倆人都帶著面具的狀態,不算把自己的最真實一面展露出來。而他倆非要達到以真實一面再次接吻和確認心意才肯罷休,而眾所周知這倆人的關系發展極其磨嘰,結果白銀愣是又花了四話才達成二度攻略(相當于之前攻略的僅僅是輝夜的一半)。這個過程真是苦了白銀,戀愛談得真夠辛苦的。不過好在努力沒白費,終于最終二人對彼此不再有任何隱瞞,心意真正徹底相通了。
輝夜的雙重性格真是挺不可思議的,給人感覺冰冷的輝夜的那個人格和平時的有點傻氣的她的形象完全是兩個極端,和之前輝夜給人的印象完全判若兩人。之前動畫里冰輝夜只是在回憶中短暫出現了一次,當時我還覺得那個狀態的輝夜挺驚艷的,結果這個輝夜成了這一季的女主。這兩種狀態的輝夜貌似可以通過戴和不戴頭巾進行切換,有意思。
冰輝夜可真是超級傲嬌,比平時的輝夜還要傲嬌,而且她還特別執拗,還一個勁固執地認定自己的性格是被別人討厭的那種,老是覺得白銀不是真正喜歡她,總是不斷在意念中誕生自我煩惱。這性格可真是超級難對付,白銀可真夠難的。
第2話里冰輝夜躲避白銀的那一段的身手絕了,簡直是約爾靈魂附體,真是夠科幻的。這是什么情況完全沒個解釋。
那個在此作里時不時冒出來的一對情侶,居然最終在賓館里一起穿著浴衣出現,看來是100%上壘了。不知道白銀和四宮什么時候也干他們干的事。
百合是我的工作!
總評:93.85
我雖然沒看過此作的漫畫,但之前就在百合會上聽聞此作大名,對動畫還是挺期待的,同時又覺得此作名字怪怪的。此作雖然不算制作得特別精甚細膩、經費特別足的作品,但觀感真的很好,風格獨特,故事、人物關系的發展很有看頭,人物塑造很好,整部作品看完后我覺得很舒服。這是一部我很喜歡,也非常推薦的百合番,而且是還是硬核的百合作品。
此作的女一號白鷺陽芽是個心機少女,表面上裝成討所有人喜歡的可愛閃耀的乖乖女,目的是被有錢人看上然后嫁給他。有一天她在路上撞到了名為“莉貝女子學園”的咖啡店的店長御子柴舞,被她訛詐(我覺得是訛詐)要求去她的店里打工。那個店的設定是貴族大小姐學校,店員都扮作學院生,舉止高貴優雅,店員之間還時不時上演橘里橘氣的舞臺表演,令來客們感到非常享受。白鷺想扮演好學院里的新生,卻由于稱呼店里一位相貌美麗身材高挑性格溫柔的店員美月為姐姐而將其激怒,并令其非常抗拒,這令白鷺非常納悶,但也因此勾起了她靠自己的實力攻略美月、令其認自己作為妹妹的動機。此作前一多半主要就是圍繞她倆關系的發展展開劇情。在這倆人互通心意,成了真真正正的莉貝學園的姐妹并關系穩定后,后1/3主要是講新加入咖啡店并長期暗戀白鷺的雨宮果乃子和一直在咖啡店工作的橘純加之間的關系發展。
此作的咖啡店的設定真是很新穎,可以一邊喝咖啡、感受貴族學園的氣氛,還有機會欣賞店員之間的百合劇情。店里的客人們也真是上道,特別關注店員們的互動,我感覺大部分人來這里不是為了喝咖啡而是來看戲,TA們還對店員關系的發展積極加上各種評論和解說,真夠來勁和八卦的。我估計來客絕大多數都是百合眾,再加上少部分是來看美女店員的。這種設定的店在現實中肯定開不起來。白鷺X美月以及純加X果乃子間的“演戲”總是半真半假,看上去是在演,但實際上反映出真正的人物關系的變化,這個設定很有意思。有時候為了不破壞咖啡店氣氛的和諧,她們還試圖通過天衣無縫的表演來傳遞出自己激烈的心理活動,很樂。
白鷺這妹子人設很棒。她的心靈真是很不錯,雖然她很會掩飾和偽裝自己,甚至把自己說成是極盡追求利益的人,但其實內心很是善良。雖然她對幾乎所有人都展示著自己虛假的一面,但對于她真正在意的人,她很會為對方著想,表現得特別真誠,甚至會去多管閑事。看到后來,我還愈發覺得白鷺真是挺閃光的妹子,其內心以及很多特寫著實顯得非常可愛。
此作剛一開場,聽到小倉唯配白鷺的聲音就令我耳前一亮。小倉唯這聲線配白鷺真心很合適,越往后看越覺得配得真是好,又符合角色形象,又特別有亮點,表現力又強。我之前對小倉唯的聲音其實沒啥興趣,她對白鷺的演繹真是給我留下了深刻印象。
美月是個身材高挑凹凸有致的偶像級大美人,難以想象她竟然和身材矮小的白鷺是同級生。美月在前期劇情中給我的印象相當差,雖然她在店里表演得看起來像高雅淑女似的,想不到性格居然那么惡劣,都不肯和白鷺好好說話,當時她的怒點到底在哪真是很令我好奇。第4話講述小學時候白鷺和美月的關系的劇情真是扎心,看了這話才明白為啥美月會對如今重新相見白鷺那么厭惡。原來,從小學時白鷺就八面玲瓏,努力和所有人搞好關系,對于她并不看重的同學也笑臉相迎,但實則只對班級里孤獨一人的美月付出真心,試圖打開她的心扉。可性格別扭的美月實在不知趣,把白鷺善意的偽裝和謊言揭穿,真是對自己、白鷺,以及那些無聊散漫的同學都沒好處。對鋼琴態度認真的美月和那些玩樂主義的同學志不合不相為謀、不去迎合她們這一點很正常也很正確,但因為自己的話語弄得白鷺風評被害,她的情商真是太低了,不懂得白鷺對她有多好。看完這話我還是沒太明白,為啥小時候白鷺突然就不和美月一起練琴了,這么唐突也難怪美月生氣,令她感到遭到了背叛。美月的另一個怒點是白鷺居然認不出自己就是她當年的同學,這也不怪白鷺,誰讓她發育得如此夸張,顯得像比白鷺大兩三歲的姐姐似的。好在是后來在白鷺和美月關系徹底鬧僵后,一直誤會白鷺的美月終于有機會認識到了那時白鷺對她的真心和好意,成了美月對她轉變態度的契機,最終在眾人面前二人lovelove放閃起來。白鷺對美月的喜歡是毫不掩飾地直白表達出來,但美月還是太傲嬌,在最后一話時又開始紅著臉回避起來,還挺可愛的。
美月的歐派實在太夸張了,特別是最后一話將這個設定凸顯得尤為充分、惡意炒作,真是喜聞樂見。她穿夏季制服的時候轉身的畫面展現的回轉半徑真是太大了!本以為美月是那種對穿著比較保守的人,居然在最后一話里,眾人都提示她穿夏季校服那個樣子過于工口、不宜在客人面前展露,她還不聽,這令我挺意外的,難道她對工口的概念沒有任何認識么?在動畫里就已經巨大了,在一張官圖里其單邊歐派甚至比白鷺的腦袋都大了,可怕。
此作里我最喜歡的是純加,她真是個非常出色的角色,越往后我越欣賞她。
純加的性格和內在很優秀。她表面看起來是輕浮辣妹,有時還像惡脾氣不良少女,但其實挺好相處的,內心很溫柔,想法很成熟,有大局觀,也有內涵,居然還看世界名著。她把所有事情都做得很好,為了咖啡廳一直在背后默默努力,比其他人付出得更多,為了保持咖啡廳的和諧、維護好大家的關系也是煞費苦心。她的洞察力很敏銳,準確地意識到果乃子對白鷺戀愛的想法,由于不希望這份執著的戀情破壞咖啡廳的人際關系而努力好心說服果乃子放棄戀情。寧寧說純加這是煩人的溫柔,確實如此。純加有時候顯得挺隨性的,比如在后臺時坐姿往往特別瀟灑(不雅),但在店里卻扮演成毫無破綻的品行優秀溫文爾雅的貴族少女,這反差挺有意思的。隨著此作的觀看過程,純加在我眼里的顏值和氣質真是越來越好,越來越令我欣賞,甚至一些特寫令我還覺得賞心悅目,引得我給她截了不少圖。總之純加的人設真是極好的!
純加的CV小市真琴配得很好,那種穩重優雅又帶一些御氣的姐姐大人的感覺表現得很出色。剛開始看此作,聽到純加的聲音,我還瞬間以為是畫伯配的,因為有一些畫伯的標志性的濃厚的鼻音的味道。如果畫伯配純加也一定會很好,不過在某些場景里畫伯如果太過于放飛自我,那可能會引發笑場并成為NG。
果乃子這個名字令我想起穗乃果,名字太怪了,而且nai如果換個字就顯得很無法描述。我不怎么喜歡果乃子的相貌,毫無亮點,她的性格也沒什么吸引我的地方,顯得扭扭捏捏,過于內向,往往還顯得挺陰暗,在我眼里沒有任何閃光點。她極其癡迷于白鷺,各種偷拍白鷺,滿手機都是她的偷拍照,真是夠變態的。她十分不適合當服務員,只是因為追求著白鷺所以也來當服務員。不幸的是,她看到了白鷺和美月結為姐妹,更糟糕的是,這姐妹關系不是演戲,在后來百合還綻放了,令果乃子極度吃醋。她對白鷺的獨占欲實在太強,一心只想著她而不顧及周圍,為了自己的私利甚至不惜破壞白鷺X美月的關系,一度令我覺得是挺反面的角色,后來還表現出黑化和病嬌感,令人覺得可怕。
第9話講述了果乃子在初中時和白鷺的過往,這一話劇情不錯,終于讓觀眾明白了果乃子為什么那么癡迷和鐘情于白鷺。原來,果乃子在初中時是個非常不合群、非常內向,對他人完全沒興趣的孩子,不喜歡班級里那些彼此間有說有笑的“好孩子”。但由于她被其他同學安排了做慶賀老師結婚的展板的任務,內心一直不爽,在壓抑不住郁火時為了發泄而把展板給破壞掉了,在她面對班里的大家、自身面臨形象危機的時候,被白鷺所救場,進而白鷺還打開了她的心扉,令白鷺有了唯一一個朋友,而且也是唯一一個知道白鷺秘密的人。不僅白鷺令她得到了救贖,看到了光明,再加上近距離欣賞到白鷺的美貌為她所吸引,果乃子深深愛上了白鷺,乃至無法自拔。這段劇情真是顯得挺真實和自然的。果乃子曾經以為白鷺是那種特別受大家喜歡的光彩照人的好孩子,經歷這件事情后才知道原來白鷺是裝出來的,她有著自己的想法,有著自己的秘密,有著自己的動機,不僅她不是果乃子不怎么喜歡的“好孩子”,反倒是挺有心機的圓滑的“壞孩子”。這顯得白鷺很真實,也令果乃子覺得她和自己有了某種交集。說起來,我在高中時也特別不喜歡班級里那些合群又沒思想的“好孩子”,感覺他們太膚淺。通過這事,我進一步覺得白鷺的心地真是很好,為了幫助被孤立、陷入危機的果乃子,不惜向她曝光自己的偽裝。白鷺在小學時候“多管閑事”幫助了美月,結果令自己受了傷害,令偽裝破滅,徹底失去了班級里的地位,沒想到初中時候她還是繼續“多管閑事”,或許她已經知道了這樣做的危險,但還是選擇幫助并信任果乃子,白鷺還真挺了不起的,是真正的很好心的人。由于美月和果乃子曾經都是特立獨行、很孤獨的人,也都曾經被白鷺打開心扉成了交心的朋友,白鷺覺得她倆挺像,但其實這倆人有50%不像,美月有不爽的地方就直接說出來,也不怕被因此被其他同學敵視,而果乃子則是一直憋在心里,忍受委屈和不悅。在果乃子的眼里,美月就是自己的情敵,尤其是看到如今白鷺又和美月這么親密,心里更是難受極了。畢竟白鷺是果乃子心中的唯一的朋友,而且是最特殊的朋友,我也能明白為什么果乃子對白鷺執著成這樣。看過第9話之后,雖然我依然不怎么喜歡果乃子,但從此我也覺得果乃子的形象沒有那么負面了,至少覺得果乃子那么喜歡白鷺是理所當然的事了。
其實我覺得白鷺和美月還是和果乃子最終在一起都可以,都挺有緣分的,作為CP都挺適合的。但是白鷺明顯對美月更為看重,美月在她心中的優先級更高,是最最發自真心重視的人。感覺在白鷺眼里,果乃子是好朋友+幫扶對象以及閨蜜,但友情以上的感情只給了美月,并且美月對白鷺有著吸引力。雖然相對于美月來說,果乃子更鐘情于白鷺,是離不開她、喜歡她到無法自拔的那種,但可惜白鷺卻給不了果乃子相應程度的感情。可以說果乃子對白鷺是單箭頭,而白鷺和美月之間是雙箭頭。上天對果乃子無疑有點殘忍,感情得不到回報,只能眼睜睜看著深愛的人的心被別人奪走。
看了第11話才知道為什么果乃子那么堅持只是和白鷺做朋友而不告白,不是因為怕告白失敗這種一般性原因,而是她知道白鷺從不會對誰產生戀愛的感情,對所有向她告白的人她都會拒絕。因此自己如果向白鷺告白的話,就做不成特殊的朋友了,而是會被她視作其他那些普普通通的接近她的人。果乃子的這么不可告人的內心想法居然那么痛快地向純加訴說了出來,令我挺意外的,不過倒也能理解,畢竟這種別扭的情感一直壓抑在心里無疑很難受,肯定希望能有個人分擔。她倒也真是碰上了正確的聆聽她心聲的人。純加真的是非常好心,不忍看到果乃子一個人因為戀情而流淚,第11話她緊緊抱住果乃子撫慰她的畫面把我都感動了。
沒想到后來純加對果乃子那么上心,特別親熱,居然結成了姐妹,還當著所有客人的面撩她,這真是特別出乎我的意料,純加對果乃子的攻勢太猛了。純加的行為至少有一半是因為她過于善良,想努力撫慰果乃子,不希望看到好好一個妹子因為竭盡全力也得不到的感情而那么悲傷,但我感覺可能也在一定程度上果乃子對純加的相性就是比較好,要不然也不至于純加能做到那種能令人看了臉紅心跳的份兒上。純加X果乃子是此作的官方副CP,雖然我不算喜歡果乃子,但由于純加在我眼里實在太出色了,所以我也控這對CP,希望能花開結果,能將果乃子對白鷺死心塌地的感情轉移方向。目前果乃子還是心心念念白鷺,但對純加的攻勢已經露出些臉紅了,說不定以后也會愛上純加。我感覺純加真是超有魅力,如果純加以同樣方式攻略別的妹子,對方的心早就被她俘獲了。
那個在后廚的寧寧一直沒怎么描寫,令我有點好奇。在第10話我才知道原來她是之前扮演服務員西園寺的人,她曾經也扮演純加的妹妹,這令我一下子豁然開朗了。她對純加對果乃子看法的開導還挺及時的。目前的寧寧完全是個橘外人,不知道未來會不會也有她的百合劇情。
五影堂葉子真是個壞女人,居然玩弄西園寺的感情,還騙了她的吻(估計還是她的初吻),玩兒完了就把她無情地拋棄掉。被她玩弄的妹子肯定還有很多。伊藤靜給她配音真是很適合,那種成熟感非常強。本來純加之前就從來沒愛上過誰,看到了五影堂X西園寺這個反面教材后估計更對戀愛有抵觸了。不知道五影堂會不會以后再在此作中出場。
店長御子柴是個有點神奇的人,也不知道多大歲數,表面上看是loli,不禁令我想起《魔禁》里的小萌老師。她也是個橘外人,估計不會看到她會和誰在感情上有所發展。
我很希望能看到此作第二季,不過鑒于這種硬核百合作品的受眾人群少、收益很低,像是《終將成為你》、《Citrus》都沒有續作,我對此作出第二季很不看好。
黃金神威 第四季
總評:93.75
這個極具個性的作品終于又出新作了。此作的故事線太長,人物和設定又非常多,而且動畫播放時間跨度又特別長,導致很多劇情我都會想不起來,一些人物關系都有點糊涂。
看此作時我多次感嘆作者真是創作鬼才,比如第40話鶴見和鯉登他爸一起救鯉登的過程逗死了,我笑點那么高的人都被逗樂了,一個破三輪車居然還漂移,車炸了后老爹居然赤膊拿著車把在飛奔,行為太迷惑了。居然最后還來了個皇后樂隊佛萊迪的梗。再比如第48話杉元都快淹死了,白石給他用嘴輸送空氣,杉元直接把他pia走,樂極了。此作總是突然出現出乎意料、沒有邏輯卻特別高能的笑點,令人措手不及,像彩蛋一樣,真是此作的一大特色和亮點。
第41話,阿西麗帕當導演記錄寓言故事的那一段真是超級惡趣味,拍攝的東西全是和歐金金有關的,包括伸到冰窟窿里、無限伸長當晾衣桿,甚至被切斷,令此作污出了新境界。不知道阿西麗帕對歐金金是怎么理解的,說出那么低俗的相關寓言居然都沒有一絲害羞(我印象中都沒見她害羞過)。
第46話出現的美女醫生家永挺漂亮養眼的,沒想到居然被月島一槍崩死了,這角色真可惜了。這一話里月島的戰斗力強得離譜,令我挺吃驚的。這一話茵卡拉瑪生產過程描寫得不錯,所有人一起為孩子的降生齊心協力,也令被憤怒沖昏頭腦的月島放下了殺意,生孩子過程用到的工具和技術挺新鮮的,不知道是瞎編出來的還是確實存在。
第47話出現的那個郵遞員真是搶眼,自稱地獄郵遞員,沒啥戰斗力,卻想大出風頭,希望體驗一回人生巔峰,真是很有性格的老同志。結果沒帥多久,就被阿西麗帕一腳踢到屁股上跌入河里,好在最后順利退出戰場,沒被子彈崩死。
第48話的“槍”戰真是樂死了,宇佐美和殺人犯倆人相見的一刻,立馬瘋狂擦“槍”,居然用自己的液體對射,一個表番居然有這么大場面真是令我著實大吃一驚!作者的想象力真是天馬行空,制作方也真是有勇氣,11區的審查也真是很open。
看了第45話我終于明白為啥有好多人對鶴見篤四郎死心塌地(像是鯉登、月島、宇佐美),甚至把他當做心目中的唯一、像男神一樣,原來他就是在培養對自己忠心耿耿的人的方面極其有心計,這陰險的家伙還真是有頭腦。比如第40話鶴見救鯉登感覺就像英雄救美一樣,無疑把他的心都俘獲了。鶴見沒毀容時候意外地還挺帥的。
不過鯉登現在也有了自己的想法,不是宇佐美那樣就是希望自己完全成為鶴見的棋子。鯉登看到谷垣源次郎偷偷把茵卡拉瑪帶走的時候網開了一面,這令我對他的印象加分了。
雖然阿西麗帕并沒有明確表達過對杉元的喜歡,但是她已經明確在心里訴說出不想讓杉元離開自己,也因此一直不說出破解金子藏放地點的暗號。估計早晚他倆之間要出現明確的戀情。這倆人真是很相配。
既然阿西麗帕腦子里有暗號,感覺之前大費周章收集人皮都成無用功了。每一季看完時我都在想不知道此作到底出到多少季的時候才能找到金子,感覺離找到金子仍遙遙無期。這一季最后居然說確定制作最終章了,終于要有個結果了。別到時候發現金子其實是坑爹的。
這一季的OP歌曲和畫面都很有氣勢,很可圈可點。ED的歌曲一般,但是畫面設計真是很有亮點,是畫面在報紙上來回掃動,報紙上有各個人物的介紹,風格獨特。
總之就是非常可愛 第二季
總評:93.7
此作是出了名的純愛作品,第一季是很久以前的事了,最近很多作品都出了第二季,此作能出第二季真不錯。這一季繼續大撒狗糧,第一話剛開場女主司就對男主星空親了上去。這一季主要內容依然是這對年輕夫妻lovelove的日常劇情,甜得發齁,二人經常膩歪,星空每一話都要夸司可愛好多遍。
司實在是非常溫柔可愛的少女,而且對星空特別體貼,有一種令人覺得有了她夫復何求的感覺。她也繼續像以前那樣表現得很容易害羞,楚楚動人的樣子真是很可愛。司的一些特寫真是很美麗,還有的微妙畫面令我感到她頗為曼妙誘人,比如第7話她睡覺嫌熱時無意地在睡衣中露出小腹的那一幕。
當年我看第一季的時候還沒怎么注意到司的CV鬼頭明里,現在覺得她配得真是超出色,用的聲線頗有她配近江彼方的感覺,和司的形象真是絕配,稍稍偏沉的聲線有恰到好處的成熟和溫柔感,同時又不失少女的可愛、活潑和嬌羞,有時候聲音還很甜美。這個聲線真是絕好地塑造出了司獨有的性格和形象,我挺喜歡這個聲線的。
星空總是直白地說出害羞的話,開心地向別人介紹自己老婆的美好和結婚后的幸福,總是弄得司非常不好意思,這樣的橋段是此作的獨具特色的場面。
星空X司的關系其實在這一季有明顯深化,更加了解了對方,但在生理方面似乎沒特別實質性的進展,還沒互相看過果體。在第1話末尾看到星空和司在被窩里蹭時,我覺得這倆人不可能還沒上壘吧,不可能沒有生理沖動吧,結果似乎還真就是進展到僅限于隔著衣服的前戲階段。第6話,倆人果著一起入浴居然那么費勁,都心理斗爭半天,總算最后是把燈關了后司才肯一起泡,真是太見外了。不過第8話的時候,在桑拿房里司倒是表示愿意讓星空扒下她的浴巾,可惜就差一點,被攪局了。
第11話,二人結婚后入進入了婚帳篷,最后一帶而過,伴隨著司的一聲嬌喘,畫面拉到遠景,不知道進展到什么進度了。按理說都這個場合了,不搞點事說不過去啊!別這倆人結婚一年的時候還沒做過認識一個月的情侶就普遍做過的事。值得一提的是,這一話二人在湖邊看著月亮時司的那一段自白做得很可圈可點,司把對愛情和婚姻的看法訴說了出來,令二人的愛情的深度升華了。
第7話出現的百鬼這妹子很有個性,我喜歡。紅色的瞳孔+黑色馬尾有種nico的姐姐的感覺。她是星空的初中同學,現在貌似是在五金店工作,顯得是個典型的工科女,沒什么少女的感覺,氣質挺直挺剛硬的,完全沒有女孩子的柔美。司還擔心星空和她有一腿,真是想多了,怎么看他倆也不像是有交往可能的樣子。百鬼聽到星空得意地講述司的那些典型女孩子的特征時(只喝溫水、買餅干看重外包裝等等),以她的近乎男性思維方式的頭腦感到完全難以理解。看星空在那里夸贊結婚和妹子的各種好處時,最后百鬼感嘆說她也想找個妹子結婚。這句話可真高能,我很支持她和妹子交往!
這一季新出現的黑皮小孩球馬挺有意思,完完全全就是個笨蛋,此作把她的這個形象表現得非常突出。
星空的弟弟在這一季也出現了,明明是個高中生,居然是個黑社會人設,行事作風令我想起了《極主夫道》里的龍。他和有棲川綾意外地出現了一些交集,不知道以后能否有所發展。
星空的中學女老師是個大齡單身,她那么長時間都一個人,從性格來看感覺也不像是有結婚希望的樣子,居然在這一季里完成了從交往到結婚的全過程,這進度還真夠快的。想不到在學校有個男老師一直喜歡她,而且她那么容易就接受了。
這一季出現了時子婆婆,是曾經司和星空的證婚人,也是曾經司名義上的監護人。她雖然是個老奶奶,但性格挺俏皮的,愛開玩笑,跟司和星空關系很好,居然還會python,真不簡單。時子還巨有錢,居然能把山買下來,挺神奇。
時子和司的關系相當深遠,似乎都要追溯到半個多世紀以前,這一季里非常隱晦地描寫了她倆之間的過往和感情,到最后我也沒搞懂到底這倆人是什么關系、對彼此真正的想法是什么、時子隱藏的愿望是什么,也不知道她怎么那么有錢。最后一話時子身體有恙,似乎時日無多,可能在下一季會出現一些略虐的設定,把時子和司的身世過往揭開。背景神秘的司到底是什么來頭,這真是此作這部溫馨有趣的日常動畫里令人有所不安的貫穿始終的重要隱藏線。
賽馬娘 Pretty Derby ROAD TO THE TOP
總評:93.7
我挺喜歡賽馬娘系列的,之前的動畫一直都追了。這一季的畫面水準真是意外的高,令我眼前一亮。這一季的畫面看著真是舒服,色彩、線條、畫風都特別好,制作得相當用心。賽馬過程做得驚心動魄,跑到最高潮的時候馬娘們的面容都變得扭曲甚至猙獰了。同時我能體會到計算機技術的發展給作品表現力帶來的不可忽視的提升,運動感比之前更強了。而且這一季還把高速運動帶來的視覺模糊感給刻意做出來了,進一步增強了表現力。可惜這一季只有4話。
這一季的主角又變了。成田路是女一號,織姬是女二號,女三號的好歌劇也是個重要的角色。此作主要講述她們訓練和比賽的經歷。
成田路在此作中被描寫得很正面。她真是非常努力,失敗之后會失意、會難過,但最后還是會繼續充滿斗志去挑戰。她的性格很好,也因此收獲了大量極力支持她的人。最后一站她總算贏了,不懈的付出獲得了應有的回報,挺勵志的,也令人欣慰。她的教練員真是不錯,為了她的成長和成績用心良苦,而且非常考慮她的心情,甚至有點像她的老父親的感覺。成田路和教練員的關系顯得太親密了,總會令我懷疑會不會越界,但事實上倆人彼此完全沒這種出格的想法。
織姬的相貌真是漂亮可愛,絕對是當idol的料。但在此作的故事中她想得太多了,老想著本應一同出生的妹妹,老覺得虧欠她,覺得是自己奪走了本該屬于她的人生和幸福。性格比較內向和孤獨的織姬選擇一個人負擔壓力,總是陰著臉,甚至后來還黑化,真是有很嚴重的心病,早該去醫院看看了。她的這些內心活動著實令我我不怎么喜歡,令我看得不舒服。
好歌劇這角色設定得很有個性,總是氣勢昂揚,姿態非常高傲和張揚,就像時時刻刻都在演出歌劇一樣,表演欲極其旺盛,這個角色令我感覺挺有意思。她的心態很好,即便全力拼搏后輸了比賽也不會顯得消極,眼睛總是看著前方。在之前的賽馬娘動畫里我就對她這個個性的名字很有印象,但之前一直都沒具體描寫過,這回總算知道她也是個厲害角色。德井青空給她配音配得相當好,把她那股獨特的勁兒演繹得滿分。
這一季稍有點百合,但不多,跟上一季的東海帝王X目白麥昆的段位比差很遠。這一季主要是成田路對織姬挺積極主動,很是跟她湊近乎。織姬對成田路倒是沒什么特別的想法。
女神的露天咖啡廳
總評:93.6
此作的男主粕壁隼從小父母因事故雙亡,由奶奶帶大,奶奶以前是開名叫Familia咖啡廳的。奶奶去世后,剛考上東大、時隔多年未見奶奶的隼回到Familia,準備將之拆除并改建成收費停車場,卻意外地發現奶奶的房子里住著5個曾經給這咖啡廳打工的只比他大一點的妙齡美女。隼想把她們趕走,她們卻死賴著不走。后來隼懷念起奶奶的往事、對咖啡廳的回憶,決定雇傭這5個妹子重新令咖啡廳開張。之后的劇情就是講他們一起努力經營咖啡廳,并且彼此間不斷加深感情,最后有三個妹子還正式對隼產生了戀情。
我對此作評價很好,人設、劇情、風格真是很不錯,越往后我越喜歡此作,真的是每一話都看得很開心,看點和樂趣滿滿。一個年輕男子的周圍圍著五個妹子,無疑令人本能地覺得這是后宮福利番,確實此作有這樣的元素,但此作絕非是簡單粗暴的賣肉作品,故事里對人物的刻畫和他們的關系的描寫有很多可圈可點之處,不少劇情挺有愛、挺有溫情的,是一部有一定內涵的上檔次的作品。
不得不說隼真是挺令人羨慕的,頭腦好,相貌也不錯,還很有*福,這五個妹子經常以各種形式主動或被動地獻出的福利(尤其是喝醉后的白菊)。起初這五人把隼稍微有一點當做弟弟看待,而不怎么把他認真地當做異性,對他也不是很害羞,令我起初感覺這幾個妹子沒一個正常的。看起來好像比較正常的流星,在第2話還直接掀起裙子給隼看胖次,真是難以置信!隼似乎對異性的理解完全為0,居然他能面不改色地拿著妹子的bra,真是沒有和異性相處的基本常識,也是難以置信!
后來以紅葉為首,一些妹子愈發地在意起了隼,對他產生了心動,進而誕生了面對異性的喜歡的感覺,甚至還開始輪番攻略,不惜靠賣肉達到目的。可惜啊,隼對于5個漂亮的大姐姐居然沒有任何心動和色心,也不知道他的男性的欲望在哪里,對她們的示愛也顯得無動于衷,實在是身在福中不知福。此作里我感覺隼比五個妹子們都成熟很多,有的時候顯得他是成年人在看一些小孩子似的,可能也因此由于不在一個頻道上,再加上隼的自律性太強、始終想著經營店鋪,導致隼對她們難以產生想法和沖動。
隼的人設著實很不錯。他的頭腦聰明,做事和對待感情都認真負責,很可靠,非常有理性,作風正直,自律性很強。他對5個妹子雖然在嘴上往往不饒人,但對她們實際上很寬容和溫柔,把她們都當家人看待。而且他知錯就改這點不錯,盡管有的妹子有時說話難聽,但他認識到自己有不足的時候會很快悔改并承認錯誤,有這樣的心胸挺不簡單的。再加上隼長得也不差,身材也挺好,我覺得隼這個角色還挺完美的,同時又不缺乏個性。
所有妹子里我最欣賞的是紅葉。紅葉可謂是集美麗、帥氣、才華橫溢于一身的真正的女神,她的人設真是神來之筆,超超超級符合我的審美!!!光是從外貌上我就非常欣賞紅葉了,再加上其超出色的氣質和內在的美好,以及玩音樂時閃閃發光的形象,在我的心中真是滿分的極品人設。從此作剛一開始我就關注起紅葉,往后真是令我越看越喜歡、越看越覺得漂亮和出彩。我感覺此作其她四個妹子加起來魅力都賽不過紅葉。
紅葉雖然平時的外表和氣質顯得有點高冷,但笑起來的時候真是挺迷人的,有時還帶有些甜美的感覺,養眼極了!到后來,一點點變得溫柔、一點點變得愛笑的紅葉更是越來越吸引我,每一話我都給她截不少圖。
紅葉的身材也超一流,第6話男主看到了紅葉換衣服的場面,她穿的黑色內衣和她白皙妖嬈的胴體相映襯真是顯得特別性感!第7話,紅葉洗澡的時候她的又大又白真是令人噴血!我覺得紅葉穿貼身的跨欄背心的造型很不錯,顯得瀟灑自在,而且又能勾勒出弧線,顯得性感。我發現官方設定紅葉才79cm,這也太假了,我覺得起碼得86啊,明明入浴時顯得那么大。
第5話紅葉的live真不錯,氣場特別帥,而且唱得挺好,歌聲有沖擊力并且富有感情,這個部分做得挺花本的,幀數很足,在一個戀愛喜劇番里能有這樣水準的live很難得。看了這一段令我更喜歡紅葉了。紅葉的歌聲和形象那么出色,在音樂方面沒出道真是可惜了,如果能堅持下去我估計早晚能闖出一片天地。她平時酷酷的,沒想到她發現隼看到了她的live后變得很害羞和難為情,真是很有反差感,這一幕的她顯得挺可愛、挺萌,很有趣,之前真沒想到紅葉也有這樣一面,令我更喜歡她了。
沒想到紅葉居然還是個名門望族家的孩子,第7話她媽坐著勞斯萊斯來的。居然她媽對她從小那么嚴苛,令她一直不敢反抗母親。她本來打算離開咖啡廳,回家接受母親強行安排給她的相親、生子的無聊的人生和惡心的命運。好在是隼站了出來,幫助她擺脫了她母親的控制。沒想到紅葉媽其實也是知書達理、尊重兒女意愿的母親,洗白得還挺快。在最后她竟然還督促紅葉攻略隼,令我很意外,想不到她居然如此開明。
此作里紅葉是和隼走得最近的。在第7話之前她就已經對頭腦機智又本性善良的隼逐漸開始在意起來,誕生了一定好感。第7話居然她還拽下來跨欄背心主動讓隼揉歐派,真不可思議!(但和腦殘的秋水讓隼揉歐派不同的是,紅葉知道這意味著什么,這都已經表達出以身相許的意愿了)。經歷第7話隼為紅葉在其母親面前出頭,紅葉更是打算全身心奉獻給隼了,她感到自己的命運被隼所拯救。在這一話她最后還突然抱住了隼,說會給他泡一輩子咖啡。這高能真是突如其來,想不到紅葉真是一點也沒有傲嬌、一點也沒有強撐著矜持、一點也沒有糾結,居然直接就這么敞開胸懷主動A了上去。我要是隼就太好了!能被女神般的紅葉倒貼,隼也太幸福了!可惜耿直的隼完全不知道欣賞紅葉的美,居然對她并沒產生出心動。紅葉由此也把其他四個妹子遠遠甩在了身后。
紅葉對隼好幾次都特別撩,比如第11話開頭,真是嫵媚萬分,我真是羨慕死隼了,換了我肯定把持不住。第11話紅葉居然對隼都說“月色真美”這樣的示愛老梗了。可惜隼太不解風情,是死板一塊,浪費了紅葉的感情。此作里紅葉多次顯得對隼特別體貼,給他端水什么的,想必如果結婚后肯定會很幸福。再考慮到紅葉本身是個白富美,家里是土豪,以后讓頭腦聰明的隼經營資產,肯定人生會非常發達。我真是完全想不到能有任何理由去拒絕紅葉這位真正的女神。
一開始我還以為五個女角色是平起平坐的,后來感覺紅葉在五個人里面明顯有特殊的位置,她是第一個偷跑的,第8話拍合照時她還是在正中間,感覺作者似乎有意讓她成為最特別的一位。我最喜歡的角色能有這樣的待遇真是令我挺意外也挺開心的。
紅葉這么又美又帥又玩音樂的妹子在此作里有不少女粉,要是她和誰有百合展開就好了,或者當個女牛郎,絕對能把普通妹子迷得不要不要的。
看到第8話五個人一起在海邊拍合影的時候,我突然覺得這五個人組成偶像組合絕對能火,并且一定要讓紅葉當C位。
雙馬尾妹子流星這角色也不錯,紅葉是那種略帶帥氣、充滿氣質的美,而流星則是那種形象偏可愛、很容易討人喜歡的美,是標準的“可愛的女孩子”。雖然她也有點傲嬌,有時難對付,但性格和心靈很好,在工作上也很努力。第9話,隼在她最需要安慰的時候安慰了她,使其好感度突然大增,納入后宮了,還主動抱著隼哭唧唧說要永遠在一起,不錯。向隼變相告白的那個畫面里的流星顯得真是挺楚楚動人的。之前沒想到她居然是童星,難怪交際能力那么強。說來童星這設定如今不少,平安名堇也是,《我推的孩子》里佳奈也是,童星長大后的普遍命運都不好。
白菊存在兩種相對立的性格,平時是顯得最保守和檢點的,穿露肉的衣服都不愿意,但聞了酒精之后,就變得比誰都奔放,這設定真是離譜。酒精對她來說就是*藥,哪怕只是吸入了消毒濕巾上的酒精,都會露出不可描述的一面,變得工口起來,主動自己解衣,然后使勁蹭隼,還扒他衣服,這副樣子的白菊真是令人把持不住,這個設定真是有趣。可居然隼對她沒感覺,還避猶不及,真是不可理解(PS:OP里看到隼似乎生理上缺失一塊,不知道是不是由于這個原因導致隼缺乏正常的生理反射)。
白菊在此作出現很多次大福利表演。第4話,隼把花朵頭飾戴在了白菊巨大的歐派上,那一幕白菊的造型和姿態真是令人興奮!第8話,以她這副姿態,把又白又軟的東西放在隼的頭上(經典的腦墊波),真令人超級羨慕他!可惜隼身在福中不知福!
白菊的設定如此特殊,但一直也沒什么感情上的描寫,始終有點神秘。直到最后一話,才終于把白菊的過往、和隼及其奶奶的羈絆展露出來,并且吐露了自己對隼喜歡的心聲。看來她非常看重隼,真心想和隼永遠在一起,這一對也不錯。不知道白菊攝入酒精后人設就切換開關的這個設定會不會在未來的劇情里有個明確的交代,我很想知道這怪毛病是什么引起的。
此作對秋水沒什么細膩的內心的描寫,但她令我印象深刻。秋水的頭腦格外簡單粗暴直白,傻乎乎的,是絕對的無頭腦少女,精力特別旺盛,就喜歡吃和玩。她還很喜歡搞怪,經常帶著奇怪的面具,感覺就像小學男生一樣喜歡做傻事,很有趣,這妹子實在很有個性。秋水還是武斗派妹子,貌似還很喜歡特攝劇。第4話她居然真的使出了旋風腳,像直升機一樣旋到天上去了,真是太厲害了!她的武打能力真不是花拳繡腿,第4話一個人就干掉仨痞子,了不起。秋水還是健身狂,居然茶杯和啞鈴同時拿著,結果掉落的啞鈴把隼的筆記本砸壞,真夠逗的,而且居然砸壞了兩次,不可思議。秋水的身材又緊實又豐腴,第3話她穿粉色背心的樣子太工口了,衣服顯得特別緊身,歐派的渾圓輪廓一覽無余。秋水是真正的腦殘,也自然沒什么節操,不知道一般男女之間什么能做什么不能做,居然她在闖禍之后還主動提出讓隼捏她柔軟的地方來試圖贖罪,這舉動真是難以置信,完全不知道節操為何物。我真是無法想象和秋水交往會是什么感覺,似乎會經常和她不在一個頻道上雞同鴨講,但肯定生活會挺有樂趣。
相對來說,櫻花這個角色令我不算太感興趣,性格和相貌給我的印象中規中矩。在這一季里她尚未對隼有明確的表示,但我估計早晚也得成為后宮。櫻花居然有個雙胞胎妹妹菊花,雖然是雙胞胎,但感覺櫻花真是顯得像她的姐姐。菊花有那么點姐控的感覺。
此作OP不錯,風格清新,妹子們的動作設計不錯,特別是開頭和結尾的TuruTuru很有魔性感,令我印象深刻。
最后一話居然出現了大概十幾年后,已成為大叔的隼和女兒在一起的畫面,這場面來得非常突然,我還以為要完結了呢,沒想到最后突然宣布決定制作第二期,甚好!這個女兒是隼跟誰生的,實在令人極其好奇,看起來那女兒似乎頭腦挺苯,而且還是藍頭發,令我懷疑是跟秋水生的,但是秋水是所有人里最不可能和隼發生戀愛感情的。如果說是和紅葉或流星生的,又不是很像。我還以為最后男主能跟不止一個妹子結婚生娃呢,看來還是只能選擇一個而放棄其他的,蠻可惜的(令我想起了《五等分的新娘》)。
和山田談場Lv999的戀愛
總評:93.5
這是一部很有趣而且制作精良的戀愛番,我挺推薦一看的。
此作里上大學的女主木之下茜是個網游玩家,男朋友和網游里另外一個女的交往上了。因為男友劈腿而感到不爽的她,偶然在網游線下活動中遇到了知名的FPS游戲玩家山田秋斗。山田看起來是個特別冷淡的帥哥,但其實內在溫柔,在短暫的邂逅中茜成了令他在意的對象。此作之后就是講述二人的關系發展,通過各種各樣的故事一點點加深了相互了解,愈發相互吸引,最終在最后一話時彼此確認了戀愛感情。剛開始看此作時不禁令我想起另一部也是跟網游有關的戀愛番《網絡勝利組》,在此順便吐血安利一下,真是好看極了,給我留下了美好的回憶。
茜的人設很出色,挺討我喜歡。她的顏值挺高的,顯得挺精致的,但在此作里居然沒人稱贊過她漂亮,令我覺得有點不應該。她的性格挺有趣可愛,平時的樣子顯得挺天然、簡單、單純的,不怎么穩重,有時候顯得傻傻的,沒有女神的氣質。不過第9話她發燒時朦朧迷幻虛脫的樣子倒顯得是個大美人,很是迷人。茜的一個很大優點是待人真誠友善,有著美好的心靈。第5話著實不錯,展現出了她內心多么善良,面對害她的瑠奈,很大度地原諒了她的行為,并充分表達出自己希望和她做朋友的真心,最終將她打動,從此成了要好的朋友。這也令山田更進一步認識到了茜的優點,很大幅度提升了對她的好感。
水瀨祈給茜配音配得挺不錯,雖然聲線不算多有個性,但適合演繹茜,表現力挺好。
茜在游戲世界里的形象蠻有趣可愛的,這個設定我挺喜歡,而且挺有她本人的風格的,能明顯給人感覺這就是茜玩的游戲角色。不知道山田怎么在網游里選了那么傻愣愣的爆炸頭角色,懷疑他是不是故意弄個丑角色的,和他現實中的形象有極大反差。
茜的前男友的那個新歡長得好丑,他真是眼瞎,明明有茜這么漂亮而且對他很上心的女友居然不珍惜,難以置信。
山田這角色設定非常出彩,非常帥氣而且很有氣質,話不多,性格穩重,相對于同齡的男生絕對是鶴立雞群,有那么多女生追他迷戀他真是理所應當的事。山田的內在和他的外在同樣出色,雖然表面上他顯得是性格冷淡的人,但實則非常溫柔,重視他人的感情,尤其是特別重視茜。之前山田對任何追他的妹子都沒有敞開自己的情感,令我感覺山田這么帥又這么溫柔簡直是種罪孽,令無數妹子傾心于他,而他卻又不能給出任何回應。并不是山田看不上她們,而是山田在遇到茜之前,他的戀愛感情從來就沒有萌發過,都不知道喜歡是一種什么樣的感覺。
此作對山田,以及他和茜之間的升溫刻畫得很自然,也很有看頭。第10話尤其出色,是非常經典的女主生病然后男主立功照顧她的劇情。這一話把倆人的關系提升到了新的高度,之前表現得性冷淡的山田終于充分釋放出了暖意和愛意。
剛看此作時得知山田竟然是個高三生,比女主還小,令我挺吃驚的。他真夠自由的,都高三學生了,居然還能花那么多時間玩網游,還能獨住,真是超級自在。令我甚至懷疑他已經考上或保送了大學所以才這么閑。我也懷疑山田是頭腦好到用50%的考試時間都能考上東大的人,所以都不用用功了。山田雖然比茜要小至少一歲,但茜并不比山田更成熟,明顯更成熟的反倒是山田。山田真是絲毫都沒有高中生的稚氣感。
此作里極少見到不茍言笑的山田露出笑容。而最后一話,山田在表達出自己對茜的喜歡后,居然咧著嘴得意地笑了,這一幕真是令他的形象出現了極大的反差,挺令我吃驚的,看到了山田的另一面。這個極其難得的表情也只有茜能看得到。
山田的班長椿也是FPS玩家,我感覺明顯她曾經是為了更接近山田才玩的。椿表面上顯得平靜冷淡、對戀愛毫無興趣似的,但心里卻暗戀著山田,始終將感情藏在心底,真是挺累的。她顯得是班級里對山田最沒興趣的女生,但其實她是比任何女生對山田都愛得更深的人,第13話她的傾訴充分體現了這一點。當她看到山田手上茜畫的某種意義上像是宣誓主權的小圖案的時候,椿明白了一切,這時候她哭的畫面真是挺扎心的。山田又弄哭女孩子了...哎呀哎呀。雖然我希望椿以后也能找到和她般配的人,但她真沒有可能找到比山田更完美的戀愛對象了,若是拿山田當參照的話,她估計就沒有能真心喜歡上的人了。山田對椿完全沒喜歡的感情,她的感情得不到回報著實挺令人遺憾的,要是此作沒有茜的話,我倒是挺推山田X椿的CP。椿平時的發型太難看了,太正經了,毫無少女的美感,第9話她在家的時候把頭發解開后顏值高多了,能有茜80%的水平。
游戲中看起來非常可愛能和女孩子聊得非常自然的公會會長琉璃姬居然是個叫瑛太的男的玩的角色,真令我意外,他居然還把上廁所說成是摘花,真是挺妖的。此作里對瑛太的內心情感沒有什么描寫,他也只是作為玩家和瑠奈的哥哥登場。給我感覺瑛太是肚子里挺有想法的家伙,無疑他是個好人,但總覺得有點令人猜不透,有點神秘。如果有下一季,說不定會對他有更多深入的描寫。
茜的閨蜜桃子挺不走運的,不斷參加聯誼想找男朋友,但是始終沒見到一個靠譜的。桃子這么努力卻沒有收獲,而茜則和內外都滿分的山田交往起來,有沒有主角光環真是差距太大了。瑛太和小桃之間在后來出現了一些交集,小桃那么想找男朋友,瑛太在她眼里那么帥,這倆人又都是單身,不湊一對可惜了。不知道什么時候能開始交往,挺想看他倆處對象的。
瑠奈這個上初三的小姑娘也是個不錯的角色。山田給她一直當家教,能和大帥哥經常獨處,她喜歡山田是理所應當的。但實際上她似乎對山田并不是有戀愛感情,也沒有因為山田和茜關系的拉進而吃醋。她起初對茜很抗拒,我看主要是單純的排外、想維持自己的小圈子所致。
隊伍里的鴨田老頭實在太違和了,畫風都和別人完全不一樣。他的人真是很好,是個很和善樂于交流的大叔,感覺這種形象的人理應不會熱衷于玩游戲才對。他的游戲世界里的形象居然是微生物,真是很新鮮,這么微小的角色用物理攻擊應該很難打中,對于弓箭手來說更是災難。
熊熊勇闖異世界 PUNCH!
總評:92.85
此作能出第二季令我感到很欣慰,也很難得。此作的第一季給我的印象很好,是一部有愛又很純真的作品,觀感舒服,女主優奈給我留下了深刻印象,而且還有不少百合元素。這一季的劇情令我覺得此作真是挺有童話色彩,故事內容很簡單純粹。優奈這個角色依舊是此作的最大閃光點,漂亮可愛,內心溫柔善良,正義凜然,還有點小性格,是很討我喜歡的角色。剛開始看這一季的時候再次感嘆優奈的發型和瞳色真是挺有點像夜羽的,正好以夜羽為女主的lovelive外傳動畫《幻日的夜羽》也快要出了,挺巧的。
感覺這一季的經費非常吃緊,很多畫面顯得極其廉價,本來兇猛的怪獸畫得跟吉祥物似的,許多戰斗完全是ppt,演出很粗糙。有些劇情也挺套路,明顯感覺就是純粹為了表現優奈的強大而創造的設定,明顯作者刻意制造那些問題就是等著優奈來解決,而且也只有優奈能解決。不過優奈的人設實在好,能令我愿意追下去,這些缺點在我眼里可以接受。
第11話,居然出現了優奈的新衣服造型,真是令我哦呼!優奈是絕對的美少女,可惜美麗都被熊熊裝嚴嚴實實包起來了,穿一些漂亮衣服必定能給人非常驚艷的感覺(而且還有與平時印象的反差感的加成)。優奈穿其它服裝的時候她會感覺心里不安,害怕別人的視線,害羞的樣子令優奈顯得更萌了。
此作里每次優奈在沒見過她的人面前出場時,迎來的反應總是:“kuma?”,難道就沒人能給點其它反應么?
感覺優奈在這一季里面子更大了,見了國王都不用行禮,而且貴族乃至國王都欠了她的情,真是“上面有人”。
感覺優奈召喚的兩只大熊熊緩和熊急真是挺可愛的,又毛茸茸又乖巧,特別是縮小到大型玩具熊的那種大小時,真是挺令我想抱一抱它們的。此作的妹子們對這兩只熊也都沒抵抗力,看到它們就被征服了。
這一季里菲娜居然開始教訓優奈了,第7話她對優奈說教時優奈居然跪在地上,感覺就像妻管嚴的老公被老婆教訓似的,樂~
此作依然算是很輕很輕的百合番。優奈在此作里有三個小迷妹,算是后宮了。有一話還出現優奈同時被這三個小loli抱著,真爽!
最后幾話里那個反派貴族真是作死,沒想到最后被判了死刑,下場可真夠慘的,還以為此作的世界里不會有死刑。比他更壞的是他那個傻兒子,這才真應該被好好收拾,比如每兩天打10大板,連著打一個月,然后再去踩縫紉機直到18歲。
反派貴族兒子找的那個看起來像吸血鬼似的前冒險者的戰斗力居然很強。當然,不用交手也顯然知道肯定不是戰斗力天花板的優奈的對手。我還以為他會不會是像優奈一樣的真實的游戲玩家,后來發現是我想多了。
國王的御廚是新出現的角色,我覺得這個胡子胖大叔的人設挺不錯,性格開朗,對美食的烹飪非常執著,而且也挺有風度。
此作有好幾個經典的BGM,有的活潑,有的悠揚,有的激昂,當初看第一季的時候就覺得很不錯而且印象挺深的,聽感很好,這一季又反復復用了。這些BGM我是真心喜歡,是此作另一個非常大的閃光點。
其實我感覺優奈的配音不是很理想,聲音有點渾濁,有時候發音有點怪、有點莽,要是讓夜羽的CV小林愛香配肯定好得多。
放學后失眠的你
總評:92.65
此作的男主中見丸太是個高一學生,有失眠癥,夜里睡不著覺,而白天又特別困乏。有一日他在學校的天文臺偶遇了有同樣失眠癥的女同學曲伊咲,二人結成了伙伴,不僅在學校里二人一起睡覺,半夜還一起出去浪。后來被老師發現,于是只好組成了天文部,以此名義使得二人繼續能膩歪在一起。此作的劇情就是講他們一起經歷許多夾雜著浪漫和感動的日常,關系越來越近,最后相互喜歡上,彼此坦白了心意。
整部作品制作質量不錯,二人的關系挺有創意,整部作品結束時觀感很好。打分不是很高主要是期間有些劇情有點零散,節奏偏慢,情節的連貫性和緊湊性不足,但故事中有很多次描寫男主女主之間浪漫的情節著實非常出彩,把那種少男少女真實的心境表現得很好。此作中出現的漫天的星軌很漂亮,在斑斕的星空下二人享受著和對方在一起的時光、坦白著自己的感情,那種場面著實相當浪漫。
這倆孩子看起來都是好學生,中見還是戴眼鏡的看起來是那種性格比較沉悶老實的少年,但很意外的是他們在一開始卻完全不懂得保持異性間應有的距離,居然二人還不熟的時候就在天文臺靠在一起睡了,然后很快就相約在深夜里一起出去耍直到天亮,真是不可思議。他們的肉體接觸真是比感情發展早得多,但是他倆卻又不是做壞壞的不純潔的事。他倆之間真是早早地就出現了不可告人的秘密,要是讓其他人知道了絕對會有不好的聯想。
中見發現貼著曲睡就能很快睡著,曲聽著中見的心跳聲就能睡著,這真是專門為了讓這倆人能膩歪在一起的神奇設定,很是科幻。第5話居然倆人在深夜偷偷溜出去在海邊相互交纏著睡在一起,不是男女朋友卻如此大面積肉體接觸不是可思議。這要是被捉奸了真是跳到黃河里也洗不清,大抵會以為這是事后。
第7話末尾部分真不錯,很浪漫。中見在深夜里通過手機app弄個人電臺,自說自話,讓曲收聽,然后曲也弄個人電臺,自說自話,讓中見收聽。曲睡著后,中見把音量放到最大,聽著她的呼吸聲入睡,仿佛曲就在他身邊,這構思真是甜極了!這部分有好幾個畫面的構圖我也很喜歡。
雖然中間和曲早的相處方式早就像情侶一樣了,但是始終沒有表白的意思,二人也都拿不準對方在戀愛感情上是怎么想的。還稍微令我覺得說不定曲在戀愛感情的發展上少根筋,并不懂戀愛是啥。在倒數第二話,居然曲主動親了中見,著實令我很意外,竟然第一個主動表白心意的是她,這進展真夠突然的。在最后一話終于迎來了大高潮,中見在星空下認真地說出了對曲的喜歡。這個結尾非常美滿。以后他們睡不著覺時再也不會寂寞了,他們心里都知道自己所愛的人正在和自己一起共度寂靜的深夜,真好。
曲的性格不錯,天真開朗,很好相處。曲的眉毛太粗,這點起初我不太喜歡,感覺明明把眉毛畫細點能令顏值提升很多,何不設定成細眉毛?不過看習慣了,倒也覺得這粗眉毛配合著她的大眼睛顯得挺有獨特的可愛,很能凸顯曲淳樸的內心。雖然曲沒什么身材,但是穿手工耿那種牛仔吊帶褲的時候有種很微妙、獨特的性感,不知道官方是不是拿捏住了一些人的癖好故意設計的這身服裝。在ED里,曲穿死庫水的畫面顯得她還挺結實的,蠻有槽點。雖然此作里曲小時候由于心臟病導致發育慢,但我感覺她的身體素質一點也不差,也不怎么需要擔心以后會有慘痛的情節出現(本作是4月開播的,容易令人想起當年虐心的《四月是你的謊言》)。
中見的性格特點總的來說不算非常突出,相貌不算帥,但也有特點。他是個表面上看老實巴交的充滿書生氣的男孩子,但也有著與同齡人不一樣的想法和煩惱,并不平庸。中見挺能給人安穩感的,很踏實,他對曲的喜歡發自真心,曲應該能和他一直順利走下去。
此作的女老師戲份不多,但她的形象設定真不錯,算是個御姐。她看到中間和曲在學校里霸占天文臺秘密偷情,并沒把這事捅出去讓他們受處分,而是幫助了他們使得他倆可以光明正大地在一起,真是好老師。
曲的姐姐人設不錯,她的相貌和曲完全一致,只不過是棕色長發,但整體看上去卻比曲的整體顏值高不少。第10話曲洗完澡頭發披下來的樣子也比平時的發型好不少。曲真是應該好好考慮弄個新發型,能令氣質和顏值增加至少50%。
白毛學姐雖然已經畢業了,但還好心義務給中間指導攝影,蠻不錯。她對中間臉紅過,可能會令人以為她對中間有好感,但果然這不是什么官方設定,這倆人并沒有在感情上出現什么交集。
此作里除了學姐、曲的姐姐、老師外,還有中間班里的其他小伙伴,但沒給我留下什么印象。不過有個高個子黑皮女角色的姓是“穴水”,這令我記憶深刻。后來發現11區有個穴水町,姓穴水的人大部分都在那。這個姓真是會令人有奇怪的聯想。
此作令我聯想起《徹夜之歌》,也是講述男中學生半夜不睡覺,出來和女人游手好閑的故事。那部作品很優秀,順帶推薦一下,風格和此作完全不同。評論《徹夜之歌》的時候我就說到,深夜對于絕大多數中學生來說是個神秘的世界,現實當中只有極少數中學生才能有幸接觸到深夜,能有機會在深夜游蕩更是太難得了。曲和中間可謂提前踏上了成人的階梯。
說來我如今沒有失眠癥的煩惱,不是因為我不失眠,而是因為絕大多數情況下我根本沒有需要強行滿足的作息,也因此作息大約20~30天轉一個周期。像我這么自由且規律性變化的作息,100萬人里都未必找得著第二個。看到那些有失眠癥的人發彈幕訴苦,我心里只有呵呵倆字。
第11話中間說他睡不著,其實是因為他害怕明天的到來,睡著后一睜眼就是充滿壓力的明天。這種感覺很多人都有,我也是,一醒來就要處理新的一天的各種事,因此希望延遲睡眠來讓今天更長一些,更多地享受時光。以前我上中學的時候更有強烈的這種感覺,一醒來就要上該死的學。
此作男主用的相機是佳能的,只不過商標改成了canan。“佳能,感動常在”這個廣告語真是和這部作品非常搭。感覺佳能公司真應該和此作搞個聯動。
此作ED相當出色,風格、編曲、人聲都很棒,又有強烈的韻律感,聽著又舒服,挺抒情、浪漫和舒暢的,ED畫面做得也不錯,其中白色背景下曲的回眸一笑還真是挺美的。ED真是值得反復欣賞。相比之下OP歌曲感覺就比較俗氣了。
雖然我早已經完全不在B站看動畫了(至少首次看任何番都不在惡心的B站看,頂多偶爾看個彈幕),但還是偶然地發現B站繼續做極度齷齪的事。比如第11話,居然把曲和中間接吻那么關鍵的鏡頭給刪了,真是罪大惡極!未成年人之間就連接個吻都不行!?比Iran甚至有過之而無不及吧!這樣搞只會助長少子化問題(少子化都是后事了,結婚率現在都不行了)!若要希望未來能有更多勞動力,必須先從不反對青少年戀愛做起才行(并且必須從詞典里刪除“早戀”一詞)!
江戶前精靈
總評:92.5
此作的名字起初令我沒什么興趣,原本不抱期望,沒想到做得還蠻不錯的,是個內容輕松有趣的日常番。此作里的女主小金井小糸是個中學生,家里是開神社的,400年前她的祖先從異世界召喚了一名叫艾爾達的女精靈,她不老不死,一直作為神社的神明至今,如今輪到小金井小糸擔任她的巫女職責。此作主要講述小糸和艾爾達經歷的日常故事。此作做得很精細,作畫非常用心,人物畫得很有美感,而且色彩看著也很舒服
艾爾達的人設相當不錯,非常有個性,像個活寶。她雖然是活了至少400多歲的精靈,但是性情和習慣顯得就像小孩子一樣,沒自制力,貪吃貪玩,往往還任性。她還特別會撒嬌,經常惡意賣萌、犯了錯誤試圖萌混過關,絲毫沒有神明該有的嚴肅正經的樣子。而且艾爾達嚴重社恐,是個家里蹲,天天呆在神社里不出門,害怕出門見陌生人。她還是個狂熱的游戲宅,天天看漫畫、玩游戲機,每天還要喝一罐紅牛。她厭煩神事工作,經常犯懶擺爛,也因此一直被小系嫌棄。雖然艾爾達始終表現出一副好逸惡勞的廢宅形象,但顏值相當高,形象有成熟的韻味,性格挺有趣可愛,所以很討人喜歡,她也被周圍居民們從小到大一直愛戴著。居然還有給她上供VR游戲機的,真是很懂她。
此作里艾爾達的表情挺獨特,經常看到她歪著嘴笑或者作汗顏狀。她的Q版的樣子很萌,而且表情極其豐富,出現過的表情都可以湊表情包了。
艾爾達真是個大美人,一些特寫相當漂亮,有時引得我忍不住截圖。第5話出現的艾爾達穿風衣造型真美麗帥氣,魅力萬分,不去當模特都可惜了。她好好打扮一下真是巨美,不小心把小系壁咚時令她都動心了(要是不小心親上去就好了)。
雖然艾爾達看起來頭腦挺簡單的,但其實也有心事和想法。她不肯出神社去見大家,一方面是社恐,另一方面也是因為人類的壽命和她相比太短暫,她怕她所喜愛、重視的人類在去世時會令她感到悲傷。
艾爾達有很多江戶時期的古老的回憶,訴說了許多那時候的見聞,使得在此作里觀眾能領略到好多江戶時期的風貌,挺有意思的。
不知道艾爾達有什么和人類不同的能力,似乎除了不老不死以及會意念傳話外也沒啥,她照樣會得感冒、照樣會倒霉,也沒什么特別的身體能力。
不知道艾爾達的零花錢每月有多少,香油錢是怎么分配的,感覺她過得很滋潤,有不少錢能拿來揮霍買各種漫畫、游戲、模型,而且又漂亮又不會衰老,要做的工作簡簡單單,這樣的日子真是甚爽。
艾爾達的CV是小清水亞美,她給艾爾達配音配得很出色,把她演繹得活靈活現,性格特征凸顯得非常好,而且聲線聽著很舒服、很有個性。她把艾爾達整個人顯得挺軟弱、怯懦、陰柔、有點發虛的感覺表現得恰到好處。
16歲的女高中生小糸雖然年紀只有艾爾達的一個零頭,但比艾爾達成熟踏實得多,對神事工作上心,也對艾爾達非常重視和照顧。她非常想變得成熟,她的那個紅色的手提包太違和了,明顯看上去是20多歲女性才提的包,卻被一個穿著校服的中學生拿著。她很看重名牌,居然香奈兒的紙袋她都一直不舍得扔,被反復使用。
雖然此作算不上明顯的百合,但說是準百合我認為夠格,有些橋段里小系和艾爾達之間的關系顯得真是挺有愛的。比如第6話艾爾達拉著小糸鉆過晴空塔上的圍欄的一幕就是,這個場景給了二人特寫,感覺有點像二人一起跨越禁忌,共同去往美好的彼岸的感覺,這一刻令我感到挺欣喜的。另外,第9話出現的小糸腦補的艾爾達對她的公主抱的畫面很美好。最后一話射箭的劇情挺好,最后一箭時小糸怕自己射到艾爾達而故意瞎射,艾爾達的表現驚人,飛身躍起,用手里的箭靶去接小糸射的箭,感覺這倆人真是很有羈絆。還有一話,小糸和艾爾達在深夜里提著燈籠在熄燈的街道按照特定路線行走做神事,這一段氣氛感也很好,寂靜漆黑的夜里倆人并肩同行顯得挺有愛的。
小糸終究會長大、老去,最后與艾爾達分別,壽命的差異總是想來會令人憂傷。分別是免不了的,但至少小糸以后別和男性結婚就好,最好能一直陪在艾爾達身邊直到老去。
不知道小糸她爸媽在哪,也可能我忽略了,感覺好像已經去世了似的,印象中都沒出場過。當第9話小糸看到她媽的曾經的錄像后樣子還變得有點沉重和憂郁。
622歲的幼兒體神明約爾黛是釘宮配的,聲線和角色很搭,難得又看到了釘宮。本來艾爾達就夠像小孩子了,而約爾黛幾乎完全就是兒童。
第8話出現的比艾爾達年長200歲的精靈赫伊菈的相貌和打扮真不錯,顯得很摩登,很閃亮,那個頭飾挺別致和優雅的。看上去她光彩照人,但沒想到是個賭徒,比艾爾達的作風還要糟糕、比她還墮落。居然赫伊菈的巫女把她的照片做成了卡牌,還真有想法。
此作出現多次紅牛,不知道是紅牛給了錢還是官方拿了授權,在動畫里看到真正的紅牛感覺挺新鮮的。
此作ED設計非常有創意,我相當欣賞,畫面是緩慢運動的鏡頭,空間感和3D渲染很出色,令人眼前一亮。而且歌聲挺溫和,ED的整體觀感非常舒服,令人享受。
偶像大師 灰姑娘女孩 U149
總評:92.5
我很喜歡《偶像大師》,這次是以一批幼女為主角的新作。此作講述經營偶像大師那些偶像的公司有了個新企劃,招了一批小學生loli作為偶像,由一個男制作人帶領。劇情主要是講這個制作人和loli們一起經歷的故事,他們一點點共同成長,最后終于得到了登上萬人矚目的舞臺的機會。整部作品12話,每個妹子都有專門的一話進行描寫。幼女當偶像真是很有新鮮感,剛開始看的時候有種很奇妙的感覺,有的地方甚至令人覺得有點犯罪感,此作是loli控們的福音。此作的作畫很精良,制作得挺用心的。不過我不算是lolicon,并非此作的loli角色令我都有興趣,她們的魅力和養眼程度和>=16歲的妹子比還是有一定差距的。所以總的來說,我還是希望能把同等資源用于制作初代的偶像大師或者灰姑娘女孩的續作上。我也挺期待看到U149的loli們長大后的樣子,其中有的loli真是好苗子,長大后可能顏值或氣質挺驚艷的。
穿布偶裝女孩仁奈很天真、元氣、活潑、可愛,頭腦發育比其他loli慢得多,還挺討我喜歡。
橘愛麗絲的顏值真高,她要是不當偶像絕對是可惜了。起初我感覺她的性格有那么點像真姬,挺傲嬌的。她一開始的性格比較高傲,不像多數其他loli那么簡單純真,這點稍微有些不討喜。果然之后發生了改變,逐漸打開心扉。她想得真多,第11話自我鬧別扭可真麻煩,好在最后和父母溝通化解開了心結。愛麗絲她媽顏值蠻高的,愛麗絲真是遺傳了好基因。
第3話講米莉亞和佐藤心的直播的劇情蠻有趣。自稱heart的佐藤心這個妹子真是挺有意思的,我喜歡,有點鬼塚夏美和MEM 啾的感覺,經常直播,能說會道,懂得營銷套路和取悅觀眾,而且特別財迷、特別重視漲粉,看到有人打賞、看到米莉亞能給她帶來收益時,就立馬失去原則了,樂。米莉亞則是個天真無邪的妹子,還很有做直播的天賦,誤入heart的直播間后很快就被觀眾追捧了。heart和大家都特別害怕這些孩子們過早接觸直播、看到成年人的世界,諸如金錢、沒素質的噴子,結果米莉亞這么可愛的孩子居然也被垃圾腦殘的閑言碎語噴了,好在是米莉亞非常簡單和單純,完全沒因此產生負面消極的想法、也沒往心里去。米莉亞在未來很有做直播的才能,但其實我并不想讓她像heart一樣專門做直播,要不然等以后她逐漸成長、內心變得敏感后,難免會因為滿嘴噴糞的觀眾的一些話語變得有點消極。世界上的垃圾人實在太多,比如我認識的一個人在抖音不求回報地免費教日語,教得很好,卻招來不少類人(而不是人類!)說各種惡心話。
濃妝艷抹的的場梨莎顯得真是早熟,比所有人都急著出道。她非常努力,但太急功近利,脾氣大,心氣太高,還傲嬌過頭,容易弄得周圍人不開心,這不太好。梨莎的顏值是真高,經常引得我截圖。她不管穿什么都顯得很漂亮,好看的不僅僅是其平時辣妹風格的造型。她居然是個嚴重父控,只喜歡父親一個,而且居然很認真地說想和父親結婚,有這樣的女兒真幸福,loli還是二次元的好!此作里梨莎的父親并沒露面,令我很好奇,不知道他是怎么被梨莎那么喜歡的,是很優秀還是有什么特別原因(諸如用了什么調教女兒的方式)。
踢足球的妹子結城晴挺有個性,令我想起了橘地真。感覺她可能是因為成為偶像而耽誤的天才足球少女。她不喜歡打扮得可愛,死活不穿偶像的衣服,真是挺怪的。后來她穿偶像服裝時我感覺還是有那么點微妙的違和,有點運動員當偶像的感覺。居然她最后也開始享受成為偶像了,她把演出場館想象成球場,這種視角蠻有意思。
千枝這小姑娘的裁縫技能真是厲害,在這方面堪稱天才,還發揮想象力和運用自己卓越的技能給高中社長偶像的被咖啡弄臟的衣服救場。我覺得她要是跟著高中社長偶像干肯定在未來事業會很成功,但她還是選擇當偶像,我感覺這大概率不是最好的選擇。畢竟她的性格內向,也缺乏閃耀照人的感覺,當偶像肯定不是她的長項,起碼比起她在裁縫方面展現出的天賦來說差得多。
櫻井桃華的劇情沒給我留下什么印象,但這角色的外貌設定著實不錯,顯得很高貴端莊優雅。
此作里能看到一之瀨志希,難得。這角色挺可愛,性格有點貓屬性,挺俏皮,還挺有偶像風采,演出的時候蠻養眼的。能看到高垣楓也令我欣慰,真是挺漂亮的,早見紗織的聲音很好聽。要是給她們專門出動畫就好了。有一話也難得地看到了凜和卯月,真是令我懷念,希望以后她們能再次成為動畫的主角。
剛開始看此作的時候,我感覺此作最大的敗筆是制作人的設定,比歷屆制作人的形象都要糟糕,居然是個矮矬子,神情充滿稚氣,頭發居然還往上梳露出額頭,不知道是誰設計的,太糟糕了。我原本還以為他是初中生當制作人,沒想到是個成年人。再過幾年,此作的妹子們都比他高了,得尷尬死。居然在第10話里,他還沒高垣楓高,真是太丟人了。好在如我希望的,此作里沒有loli喜歡上制作人的情節,要不然得把我惡心到。
雖然制作人很丑,不過他倒是挺稱職的,非常為U149的loli們著想。這些孩子凈是非常有個性、難懂的,制作人耐心地試圖了解和理解她們,有時有點像他們的哥哥,并且在為了她們能獲得成功上也挺拼盡全力的。他自己也有點不成熟,把他的形象設定成那樣可能就是想突出這一點,免得制作人和孩子們之間的顯得代溝太大。
我心里危險的東西
總評:92.25
這是個初中男女戀愛喜劇番。男主市川京太郎是個不起眼的矮矬子,而且還很中二,經常胡思亂想。因為他有一次偶然地看到女主山田杏奈一個人邊吃邊笑邊犯蠢的一面,開始在意起了她,并開始對她有了喜歡的感覺。此作之后就是講述二人日常一起經歷的事情。隨著共同的經歷越來越多,他們的關系越來越近,到最后雖然沒告白,但是已經明顯像男女朋友一樣了。此作整體還蠻有意思的,有些劇情很有看點。作畫蠻不錯,絲毫沒有崩壞,而且許多地方做得很精致細膩,女主非常養眼。
此作的最大亮點是山田的人設。外形很驚艷,個頭明顯比同齡人高一大截,人設居然達到了172 cm(長大了肯定得175以上),而且歐派甚巨。這樣的身材居然是初二,說是高三或者大一我還信,二次元的設定就是隨心所欲。山田特別能吃,而且酷愛零食,兜里總是藏一堆零食,經常吃得一臉幸福。她的基因真好,靶向能量轉化效率極高,一般人吃那么多都得橫著長,她不僅豎著長,而且肉還長在了最該長的地方,簡直就像能操控生長一般。山田是給雜志當模特的,以她的身材,再加上能和古見硝子相媲美的極品容貌,在作為模特或者偶像的道路上肯定以后能很火。山田在此作里偶有穿時尚衣服出場,有時令我覺得很能哦呼出來。比如第4話她穿便服的樣子就巨漂亮,有種不似這個年紀女孩的成熟美感。
山田的性格也挺討人喜歡的,挺隨性、豁達、歡樂的,絕大多數場合下都顯得不是心思細膩的少女,還經常賣蠢干傻事,有時還有點缺乏常識似的,整個角色顯得挺有趣和可愛。她的一些想法有點令人捉摸不透,經常顯得有點飄忽的感覺,難以憑常識來理解她。
男主京太郎人設一般般。他襯托山田的閃耀是夠格的,但和山田交往在我來看完全不夠格,實在不覺得他倆哪里相配。京太郎跟山田相比感覺完全不像是同一個年齡的,也不是同一個世界的,說山田是他姐姐都毫不違和。從形象上看,一個靚麗的高挑大美女搭配一個相貌平凡的矮矬子,怎么看都不搭;從性格、喜好等方面看,二人也沒啥交集。按理來說他倆不可能有什么發展,但很奇怪的是,山田就是對京太郎有濃厚興趣,特別在意他。她真是對京太郎格外地重視,每當看到他眼里有自己的時候就會開心起來。隨著劇情發展山田倒貼得越來越厲害,對京太郎越來越不設防,毫無避諱地做出各種親密舉動,這真是令我看不懂。到底山田對京太郎是抱著什么樣的想法接觸的,這令我感到疑惑,如果說是沒常識的山田不懂得保持與人之間的距離,這有點牽強,如果說山田對京太郎是戀人那樣的喜歡,我又不懂京太郎哪里那么吸引山田,而且顯得很大條的初二的山田在戀愛感情方面是否已經發育了還是回事。反正京太郎實在太幸福了,一個陰暗boy,沒什么閃光點(雖說對山田比較溫柔),就能有山田這樣極品大美女不知由于腦筋怎么回事就主動貼自己了,真是羨煞死人。他得極度珍惜這個機遇和山田的感情才行,并且鼓足勇氣好好面對山田的感情,不能老是顯得扭扭捏捏的。
不知道怎么會有動畫故意把男性設定得那么矮,而且女方還特別高,這樣的組合顯得太殘念了,《古見同學有交流障礙癥》也是如此。如果是身高差不多也就罷了(比如《總之就是非常可愛》、《式守同學不止可愛而已》),而男的比女的還矮一頭,這顯得也太違和了。這樣的搭配,如果能令我感覺這樣的倆人是相配的話,那男主的內在必須相當出色才行,顯得形象高大,比如像《石紀元》里的千空那樣,那樣我會覺得男主的成長點數是點在了內在上、配得上女主,而并不是相比女主顯得像殘疾人。
此作的一些劇情挺開放的,有不少工口的畫面、特寫。男生之間有不少大膽、下流的對話和想法,發言尺度甚大,顯得太早熟了,居然還在教室里直白地亂講,真是完全不介意自己的低俗被女生們鄙視。男主也不怎么純潔,居然第1話還拿著里面山田當模特的雜志“用”了。雖然他也凈胡思亂想,并且有非分的想法,但相對于那些品行糟蹋的屌絲男同學來說還是有節操和下限的。
我與機器子
總評:90.8
2022年12月開播的泡面番,總共28話。此作主要講述一個叫“機器子”的女仆機器人和他的小男孩主人以及其它小伙伴的事。
此作是個極度無厘頭純搞笑沙雕作品,創意天馬行空,高能展開不斷,槽點滿滿。雖然有些劇情挺迷,但也有很多劇情相當歡樂,構思很有趣。里面的梗多得很,機器貓、龍珠、咒術回戰、鬼滅之刃等作品的梗都出現了。本身我是不怎么喜歡周星馳之類無厘頭搞笑作品的,但此作著實有些可圈可點的地方,于是還是追了下去。
故事一開始相貌挫得像野比一樣的男主想買個漂亮女仆機器人,結果買的廉價女機器人居然是個只會幫倒忙的胖妞,也就是機器子了,這令男主大失所望。此作對機器子的描寫和刻畫蠻不錯的,形象十分鮮活生動令人印象深刻。她身寬體胖,完全沒有女仆該有的樣子,感覺以其形象當胖虎家的女仆還算勉強說得過去。這機器子長得傻里傻氣,外表粗壯內心魯莽,腦回路奇葩,又完全不會干家務,總是幫倒忙,各種添亂,還胡搞瞎搞,而且自我意識嚴重過剩。但她也并不只有缺點。她相當重視主人以及與主人相遇的緣分,對女仆的工作很有使命感,對保護主人盡心盡力,在關鍵時刻也有出人意料、令人刮目相看的表現。所以機器子給人的感覺挺矛盾,雖然她這樣又丑又蠢又亂來的家伙給自己當女仆避猶不及,但相處時間長了的話也會發現其優點,也有那么一丟丟很微妙的可愛的地方,把她趕走會令人割舍不下。
機器子的戰斗力真是逆天,始終自詡為女仆,實則等同于最強兵器,身體暗藏各種武器。終于在第26話曝光了機器子的過去,以前的戰友來找她,原來她是最強兵器還真不是亂講的,還真是作為戰斗女仆創造出來的。那時候的機器子和如今真是有了180度的反差,就如同《極主夫道》的男主似的。曾經機器子嗜戰斗如命,而如今的她卻追求極致的可愛,顯得頗有女子力(但極度做作+反胃),居然還當偶像在電視上蹦蹦跳跳。此作就是要刻意塑造這么一個極其矛盾的個體。
機器子的外在形象實在令人愛不起來,聲音也很粗獷。然而在第5話,粗壯的機器子在和熊搏斗后消耗太大,里里外外都變成了美少女模樣,令男主開始心動了起來。要是這副模樣能一直保持就好了。結果果然好景不長,吃了一頓肉又回去了。看來還是得餓著她。
此作的男主看上去像極了野比,他的兩個小伙伴我知大猩猩和持男完全就是胖虎和小夫的樣子。持男和小夫一樣家庭非常富有,我知大猩猩的周長很大(但整體尺寸則比胖虎明顯小一號,而且眼睫毛長得離譜,挺違和的)。三人之間的人設關系幾乎就是《機器貓》的翻版,但也有一定不同。《機器貓》里野比總是受胖虎的欺負和小夫的嘲諷,但此作里男主雖然也受大猩猩和持男的嘲笑,但關系好得多,三個人彼此之間都很重視友誼,有時也挺為對方考慮,沒有對立感。
第15話給持男加分不少。雖然他家極其闊綽,也時不時嘲諷男主,沒想到他卻那么看重和男主和大猩猩在一起的時光。他從小就被父親強行當做精英培養,生活非常緊湊,沒有喘息的時間,但即便如此也不肯離開他們倆而順從父親的安排去別的地方上學,其人格蠻不錯的。
總評:94.6
此作是個很有創意的硬核搞笑作品,劇情和設定的創意相當別出心裁,槽點滿滿,人氣相當高,是個極為熱門話題作。此作的播出過程也真是命途多舛,到了2023年3月末才播了最后一話,本來半年前就該播完的。
此作男二號高丘敬文的舅舅島?陽介從17歲開始昏睡不起,過了17年,終于從病床上醒來了,并自稱是從異世界歸來。他不是神經病,而是真的從異世界反向穿越回來,擁有異世界的記憶,掌握異世界的魔法,還帶回來了異世界的裝備。此作講述脫離現實社會甚久、呆慣了異世界,性格和腦回路奇特而且又有魔法的舅舅回歸現實后與敬文以及暗戀她的藤宮一起經歷的日常故事,以及三個人一起通過屏幕回味舅舅曾經在異世界的奇幻經歷。
此作最大亮點就是舅舅這個角色的塑造,令人印象非常深刻,特別有個性。要說舅舅也不至于多丑(雖然有點猥瑣怪大叔的感覺),也就是平均相貌水平,五官也端正,但在異世界的人眼里他的長相大抵是癩蛤蟆亞人那種形象,被嫌棄厭惡,甚至莫名其妙就遭到圍攻。大多數穿越到異世界的作品里主角都過得瀟灑自在,而這舅舅所穿越的異世界對他真是很不友好,完全沒穿越對地方。不過好在他在異世界倒還有著逆天的戰斗力,也算是以特殊方式走了龍傲天+開后宮的路線。
舅舅非常守舊,觀念特別落后,特別執著于去異世界之前所癡迷和留戀的老事物。他還是個SEGA的骨灰粉,我估計此作的作者肯定對SEGA特別有情懷,也借著舅舅這角色抒發自己對SEGA的懷念。此作里有很多老物的梗,估計多數95后都不知道在說什么,卻能引起80后們的廣泛回憶。雖然舅舅才34歲,也不算多大,但表現得真像是個老古董,就如同MP2似的。
舅舅是個真正的死宅,鐘情于游戲里的角色。他從未有和真人交往的經歷,不僅是因為他的性格和相貌毫無女人緣,而且是即便有美少女倒貼,他都不來電,完全意識不到對方的動機,在和異性相處方面明顯缺根筋,木訥至極,完全不懂浪漫、完全不解風情。舅舅真是對妹子完全不感興趣,連一絲都沒有。美少女白給他都沒任何想法,更別提他能主動和誰告白了,我完全想象不到他在未來能和什么樣的女性結婚。不過舅舅卻對敬文和藤宮之間積極助攻,敏銳地意識到藤宮暗戀敬文,還讓藤宮和敬文住一個房間之類的。要是舅舅的這份敏銳用到暗戀他自己的妹子們上就好了。
舅舅有時候對妹子倒也表現出極度的溫柔,令本身就暗戀他的妹子淪陷,做足了前戲。但其實舅舅卻完全沒有攻略對方的意思,因而總是最后令妹子們極度失望。他老是撩完了就跑,并且說特別不解風情的話,令妹子空歡喜。舅舅雖然熱衷于電玩,但對galgame倒是沒什么興趣。他要是真心喜歡galgame的話,肯定會有戀愛腦和細膩的情感,絕對不會表現得直成這樣。
舅舅有時候的行為很沒常識,甚至變態,毫無自覺地做出赤果果的耍流氓行為,這是其招牌式的性格特點之一。比如第5話把梅貝爾的衣服扒了,近距離聞她的味道,還把她束縛住抓住她的腿,那副丑陋的面容再加上子安變態的聲音,真是活生生的癡漢。第4話末尾太高能了,舅舅給舅媽吸出毒,然后要脫她盔甲,舅媽使勁抗拒。這一刻真是重演了須鄉當年對亞絲娜干的鬼畜之事。又是子安,又是肉松遙,這重現真是絕了!不知道制作方是否有意搞這個梗,這可真是神梗!
舅舅是子安配音,可謂超絕配,CV界不可能有第二個人比子安更適合配舅舅。子安的那種聲線把舅舅那種蠢兮兮、呆愣、單純、刻板、(在外人看來)變態、經常一驚一乍的怪人的形象演繹得絕佳。
舅舅第3話變成舅媽的模樣真可愛。youtube播放量大漲,反倒令他備受打擊,清醒地意識到如今的人對熱血的東西遠沒有對青春可愛的妹子大。確實如此,youtube上cosplay布料少的角色彈鋼琴很容易大火,而上傳玩游戲視頻的up主的人氣就低多了(我關注的“82電玩大叔”就是這種感覺,即便玩得很好,解說也很風趣,但畢竟是大叔,人氣跟賣萌賣可愛賣顏值賣福利的妹子沒法比)。
敬文這角色也不錯,很關愛舅舅。腦子相對正常的敬文給奇葩的舅舅當陪襯,就像說雙口相聲的二人,這搭配很不錯。敬文特別喜歡聽舅舅講述異世界的故事,特別是那邊的妹子和舅舅之間發生的往事,那個世界令敬文充滿了好奇。
舅舅對于異世界喜歡自己的人毫無察覺,敬文對于這個世界里喜歡自己的人毫無察覺,但彼此都非常關注對方的潛在交往對象的事,這真是很有趣的關系。
暗戀敬文的那個同齡妹子藤宮的人設我挺喜歡。她挺傲嬌的,不坦誠,看她害羞時慌亂的樣子挺萌挺可愛的,我很喜歡看她這副樣子。她還有挺暴力的一面,對于危害敬文的事物都想抹殺,對敬文的獨占欲相當強。她真是對敬文太倒貼了,居然還想讓他看自己的果體。可敬文對她太直了,就如同異世界里舅舅對妹子們的態度,真是令人著急。舅舅本身是個怪人也就罷了,明明敬文是個普通男青年,也沒女友,按理說不可能對異性沒有想法才對,可他對那么主動又那么美好的藤宮卻絲毫不來電,真是離譜。藤宮的身材真是絕贊,太飽滿了,特別是第4話洗澡的時候福利很贊。藤宮就是欠缺打扮,好好打扮一下絕對相當有風采(但可惜敬文總也不懂得欣賞)。
異世界里有一個相貌可謂極品的傲嬌女精靈蘇翠路奇拉澤加魯涅爾魯蕾婭阿格蘭澤爾加·艾爾加,對舅舅特別好,起初對他是在意,在后來對他產生了熾熱的戀愛感情。但她盡管反復向舅舅倒貼和暗示,告白的話語卻始終憋在心里。本來舅舅對感情的遲鈍程度就是∞,艾爾加就永遠別指望舅舅能主動意識到自己的新意了。
艾爾加的很多特寫很漂亮,跟仙女一樣。平時她很嚴肅,但是有時卻會意外地展露出驚慌害羞的表情,挺可愛的。她羞紅了臉傲嬌的樣子也特別萌,這點和藤宮有點相似。肉松遙給艾爾加配音配得很不錯,充分表現出了她傲嬌和可愛兼并的人物形象,以及內心的敏感之處。
舅媽是個大醋壇子,且獨占欲極強,看到舅舅和梅貝爾、艾麗西亞有近距離接觸時都要攪局,跟她成為情敵的人真是危險了。
最后一話舅舅和艾爾加從天上降落的場面挺美好,艾爾加在implicit方式的示愛上已經盡全力了,可是舅舅到最后對她也沒產生出任何男性對女性的感情,真是大殘念。舅舅從異世界離開后就只剩艾爾加自己了,我感覺她有點可憐,要是她也能穿越到舅舅的世界,繼續發展二人的關系就好了。
藤宮和舅媽相當相似,都是挺令人心疼的,都那么主動了,除了說出告白的話語外的事都做了,可長久以來的心意和努力卻沒換來任何回報,唉~
藍發的梅貝爾的外形設定真是很驚艷,顏值極高,外表冰清玉潔,有時候又顯得楚楚動人,跟仙女下凡似的。可是她里外完全不一。第6話表現出了她宅女、家里蹲、廢柴、強烈不愿工作的一面。甚至只要不工作,讓她當狗都愿意(我很想飼養之),內在挺沒羞恥心的,和冰霜美女的外表形成了鮮明的反差。之后的各種事情也都進一步玷污了她看似神圣、只可遠觀而不可褻玩的形象,完全成了一個腦筋挺蠢的沙雕角色。梅貝爾和舅舅之間也有很多顯得非常曖昧甚至不可描述的高能情節出現,這令艾爾加非常警惕。
除了艾爾加和梅貝爾這兩個舅舅的后宮外,舅舅還有個準后宮女神官艾麗西亞。我對這個角色感覺一般,但她的size真是碩大,令人印象深刻,實在太圓潤飽滿了,入浴畫面真是大福利。舅舅第11話看到她的果體后居然立馬自我消除記憶,難以置信怎么會有腦筋這么正、如此刻板的男性存在。豐崎愛生給她配音的那種純潔和柔軟的感覺不錯。
此作OP的像素畫風格不錯,還有把3D畫面搞得有像素感的濾鏡效果挺好。
最后一話的最后出現了好幾副誘人的香艷的畫面,似乎是給下一季留出伏筆,希望此作能出下一季。這一季最后也沒交代舅舅怎么穿越回去的,感覺還有很多可編的。要是舅舅的后宮們也能穿越來這個世界就好了,我挺想看到舅舅最終能和艾爾加結出點實實在在的果實來的。
小智是女孩啦!
總評:94.45
這真是一部極其有趣的戀愛喜劇作品,人設新穎,樂趣滿滿,槽點眾多,很多情節相當高能。這是我一部極力推薦一看的相當上佳之作。
很多作品都講述男女之間各種各樣的不尋常的關系和相處模式,而此作這種關系是前所未有的。此作的女主相澤智長得五大三粗,又壯又結實,上了高中還沒什么女子力,但是戀愛之心卻萌發了。她終于向從小就和她在一起玩鬧的此作的男主久保田淳一郎在櫻花樹下告白了,結果淳還是像曾經一樣把智當成一起打打鬧鬧、對練空手道的玩伴,沒把她當異性看待,更完全沒意識到她這是在愛的告白。此作主要講述這倆人的奇葩關系的發展,小智想被淳當女孩子看待并和她發展戀愛關系,但淳卻始終不開竅,始終把她當成老伙計對待、一點也不懂保持異性間的距離。
小智的人設真是相當有趣。她真是開口脆,第一話一開場,看到一個妹子在櫻花樹下向男生告白時,她居然突然發出那么粗獷的聲音,真是令我一驚。雖然小時候的智和男孩子沒區別,也成天混在男孩子堆里打鬧,但上了高中后的她卻想讓自己被他人當做女性、希望他人覺得自己有女人味。這點真是挺有點像《式守同學不只可愛而已》里的女主式守,二人還都是空手道頂級高手、戰斗力爆表,同齡男生都完全不是其對手。但與式守同學不同的是,智如今還是跟以前一樣不修邊幅,行為處事上粗獷得不得了,沒主動試圖去扮演成正常妹子。而式守則在上了高中后積極改變自己,盡管內在的強大沒有改變,但至少表面上已經是個像偶像一樣的可愛女孩子了。所以淳一直沒能把智當異性和潛在的交往對象看待,一半是淳確實太遲鈍,另一半也是智自己的問題,形象和性格還那么man,一點都沒有作為女孩子的自覺,與此同時頭腦還極其簡單和粗暴,甚至挺有點蠢。不過,智的歐派卻挺宏偉,唯獨這點她確實是100%的女子特征,當淳隱約地看到她歐派的時候才會猛然間意識到她其實是異性。上高中后的淳還是像以前一樣對智經常勾肩搭背,完全意識不到她的內心和對他的看法已經發生了明顯的變化,總弄得在意他的智很不好意思,淳這樣的行為在外人來看甚至有點像在耍流氓。
雖然受到性格以及過往相處方式的阻礙,小智和淳的戀愛關系發展得總是不順利、總是錯過良機,但好在最終在小伙伴美鈴的助攻下,終于二人確立了戀愛關系。在第12話,他們的戀情終于開花結果了,真是可喜可賀。他倆的關系真是非常獨特,摯友+對手+戀人,這樣的關系在整個世界上可能都找不出第二個,這是獨屬于他倆的戀愛,此作的這個構思真是絕妙。
雖然小智平時都是女漢子的感覺,但第3話美鈴和卡洛爾給她搭配打扮一下后,和淳約會時的小智的外貌意外地很不錯,氣質跟平時判若兩人,居然還蠻可愛的。第6話,她被美鈴和卡洛爾打扮并穿上女裝后,有種微妙的男性易裝的感覺,小智還都羞恥得不好意思出門,簡直就像原本是男孩子變裝一樣。雖然漂亮是漂亮,但是她胳膊稍微有點粗,略有違和。這一話小智遇到淳后的劇情真是尬死了,居然淳還真沒認出是她來,不可思議。我覺得小智要是想變得像女孩子,還是得把頭發留長了才行,要不然哪怕性格變得(裝得)像女孩子,還是頗有些違和感。值得一提的是,此作中小智有時候露出的小害羞模樣令我感覺挺有魅力的,有著和平時截然不同的反差萌,這個時候的她確實展現出了一定女子力。
小智這么猛,不知道她和淳在做愛做的事的時候會是什么場面,說不定小智一個害羞就把淳打殘,甚至一下子踢到他要害。考慮到這點,得先把小智拘束起來才行。
此作起初有兩個小太妹跟小智找茬,最后成了她的小迷妹。第4話小智英雄救美太帥了,一腳把流氓高三學生踹飛,然后摸了摸她倆的頭,那一刻小智在她倆眼里肯定是完美男友的感覺,又威風凜凜又帶給她倆安全感。我也挺想看小智和她倆交往,那樣的話小智就徹底拋棄女孩子的設定完了。小智到后來迷妹越來越多,第12話她以男型造型登臺時迎來一群小女生的歡呼,要是此作有百合劇情就好咯!
高橋李依給小智配得實在太好了,特別要大力夸贊一下。難以想象曾經配溫柔如水的高木的CV也能把這么有野性和爆發力的女猩猩的聲音表現得這么好,高橋李依真是實力派。
此作里我最最最喜歡的就是女二號群堂美鈴,其外在形象、性格、內心,都特別令我著迷,對我有巨大吸引力,在我眼里她魅力無窮。這角色設定真是絕了,比小智的人設還絕。我已經將她納入后宮列表了。
雖然此作對人物的作畫不是那種特別精細的,但美玲很多時候的特寫真是顯得顏值賊高!而且還有那種神秘、暗黑、性感甚至妖艷氣質的加成,甚至能把我驚艷到,令我為之傾倒!
美鈴的性格設定極其有趣。她的外形陰郁,說話聲音暗沉,雖然長得很不錯但面癱,表面氣質沉穩高冷,內在性格狡猾老練,特別腹黑和毒舌,整個人陰氣特別重。她還總想搞點事,弄點惡作劇,肚子里常備壞水。美鈴說話時的用詞還經常很有槽點,有時候令人感覺邪惡,比如什么“黏膜水平的接觸”。她從小就跟智和淳在一起,對他倆再熟悉不過,如今美鈴不斷給他倆的關系添油加醋,經常旁敲側擊,甚至搞點小陰謀,試圖加速二人交往。美鈴不僅是吃瓜群眾,還插足了進去。比如一開始雖然淳完全不把智當異性看,也完全沒有和她戀愛的想法,但是美鈴一提到如果智和其他男生交往了怎么辦,淳就會立刻陷入沉默,然后再欺負一下美鈴,這情節很逗。
美鈴和小智的人設挺有互補性的,陰沉的美鈴總是靠語言解決問題,嘴巴總顯得挺壞,還有很多陰險的想法,整個人感覺有點像女巫、巫婆。而陽剛豪放的相澤總是靠武力解決問題,像狂暴女戰士。這倆性格迥異的人能玩到一起、關系這么融洽,真是很有緣分。
美鈴雖然顯得特別老謀深算、深思熟慮,但實際上她往往想得太多,把問題反倒弄復雜了,尤其是自己與小智之間的關系。美鈴其實完全不是無懈可擊、心靈特別強大的妹子,她也會難過、困惑、憂傷,害怕孤獨,還特別不想失去小智。美鈴的心靈上有這樣的弱點,顯得她更像真實的女高中生,也更令我喜歡和心動,并令我感到攻略她也是有可能、有機會的。
小智對美鈴來說是非常特殊的人,美鈴特別希望能和她在一起。美鈴對淳毫無興趣,她撮合小智和淳的動機純粹是在于和淳交往能令小智變得更像女孩子一些,從而令自己和小智能有更多的交集。我總在懷疑會不會美鈴其實心里對小智是愛的感覺,但由于是同性,因而一直深深隱藏著。第12話,扮演灰姑娘的美鈴和扮演王子的小智在臺上跳舞的時候美鈴笑得很開心,感覺這是她感到至上幸福的時刻,可能她心底里也希望能把這一刻的美好永遠延續下去。我一度有點害怕,別此作最后真的曝光美鈴其實就是真心愛著小智,而由于此作是BG番,無疑會拆散這美好的百合戀情。好在是到了最后,并沒有出現這個設定,美鈴對于小智和淳正式相戀也沒有誕生什么負面想法。我覺得,美鈴肯定還是最希望獨占小智。
美鈴最大的黑歷史就是初二時,淳居然主動和她交往,結果才兩三天,淳就感覺和她不合跟她提分手。明明本來應該是受他的氣的美鈴先開口才對,竟然被淳很無恥地(雖然是沒自覺地)搶先了。這是美鈴在語言對抗上的最大慘敗,也等于遭到了極大的羞辱。看到這里我都替美鈴生氣!美鈴這個失算太糟糕、太可憐了!
美鈴在此作里的表情真是豐富至極,都夠一個表情包了,官方GJ!很多她的經典表情都被我截了下來當做QQ表情用。
剛聽到美鈴的聲線時令我感到特別有既視感,其CV日高里菜配過八滿結、伊理戶結女,她們的聲線和腔調著實很有共性。特別是美玲毒舌吐槽的屬性,真是像極了高中時的結女。日高里菜給美鈴配音用的暗沉的聲線真是太棒了,我真的超喜歡,可謂絕妙至極!
美鈴她媽是個外貌特別標志也特別標準的女白領,和美鈴有一定相似之處,但感覺她媽相對來說還算正常一些,陰暗程度遠不及美鈴。真不知道美玲的腹黑性格是怎么形成的,在劇情里看到美鈴從小就已經有點像邪魔一樣了。貌似美鈴她媽和小智她媽曾經有過什么過節,但此作里沒明確交代,這令我好奇。美鈴她爸始終沒有出現,也令我好奇。
金發洋人卡洛爾是個像暖陽一樣的妹子,身材真是絕倫,做體育運動,諸如跳繩、仰臥起坐,都令男生不好意思去看。她的性格挺逗,看起來天真無邪,經常主動貼和粘別人,里里外外像棉花糖一樣的軟萌天真可愛。然而她心里不僅不糊涂,嗅覺還相當敏銳,腦子很靈,對于男女之事相當懂,還說美玲的性格惡劣。卡洛爾喜歡搞事,絕對不是什么善茬,經常很討打,總是一副皮笑肉不笑的樣子,顯得是真明白卻裝糊涂,用奇葩、妖孽來形容卡洛爾非常貼切。這種有點粉切黑的妹子就得靠美玲來治,在美玲面前卡洛爾是小巫見大巫。卡洛爾和美鈴見面不久就結成了奇怪的CP,亦敵亦友,她倆有時候則互掐,但對于小智和淳交往的事方面卻在同一戰線上,并且經常看到卡洛爾主動向美鈴表示親熱,這相處的關系很有意思。這倆人都是內在“惡劣”而且特別機智的女子,只不過卡洛爾是表面上顯得(裝得)呆萌清純,而美鈴是在表面上就能令人察覺到有暗戳戳的深藏不露的想法,是心機少女的兩種極端形式。
看到第9話,果然確認了卡洛爾一直是帶著偽笑的面具,一直在裝。美鈴扮黑臉給御崎光助X卡洛爾做助攻沒想到效果拔群,令卡洛爾從陷入極度悲傷再轉變到極度幸福,終于被一直喜歡的御崎告白了,二人的心意終于徹底相通了。美鈴雖然總是陰著臉,但本性真是善良。這一話卡洛爾居然跑到淳的房間里還把他推倒,還親了上去,令我頗感意外。卡洛爾搞事也搞過頭了,不過倒還沒有主動把衣服解了貼上去,否則如果再恰好被小智看到,那可真就是糟糕透頂了。看完這一話我也終于確認了卡洛爾是BG,雖然很粘美鈴,但對她并沒有實質上的戀愛感情。
第12話,在學園祭結束后男女情侶都在一起跳舞,連小智都有淳作為舞伴,看美鈴孤身一個人落寞,我挺心疼的。好在是卡洛爾這時候暖洋洋地貼了上來,令美鈴空虛的內心充盈了起來。雖然不管卡洛爾對美鈴怎么主動,百合關系都不可能有(畢竟有多余的男性御崎光助的存在),但卡洛爾如果能在美鈴找到真愛(最好是同性)之前能經常親親抱抱貼貼作為友人來撫慰美鈴孤寂的心靈,那也是挺好的。雖然田邊總是想伺機追求美鈴,但美鈴哪怕孤身一人也絕對不搭理膚淺&傻兮兮的松崗配音的田邊,這令我在看到時挺愉悅。否則要是萬一美鈴忍耐不住孤寂,一時做了傻事,給了田邊好臉色,那美鈴的形象就崩塌了。到現在卡洛爾和小智都有戀愛對象了,唯獨美鈴還沒有,我真心希望她能找到與她這個異類完美般配的人并獲得幸福。
卡洛爾的CV是個新人,天城莎莉,她給卡洛爾配音配得相當不錯,能令人印象深刻,也給這奇特的角色的形象加分很多,在此夸贊一下。
卡洛爾她媽和卡洛爾相似度極高,歐派都很大,都有種天真+迷糊感,這種膠皮般的外表讓人捉摸不透里面到底是什么樣。她和美鈴之間也是不對付。卡洛爾在家里居然穿得軟乎乎,跟幼女一樣還和她媽還親親抱抱,這畫面真是難以形容,顯得很降智。小智她媽和小智的相似度也極高,樣子顯得豪放灑脫,真像是個大號的小智。相對來說,美玲她媽和美鈴的相似度就沒那么高了(倒也好,要不然兩個大腹黑在一起生活,相處得恐怕會超別扭。)
7、8定律對此作生效了,第7話的泳著回真是賞心悅目,卡洛爾大到離譜,而小智也展現出似乎不該屬于她的東西,令淳無法直視。這一話還曝光淳居然從小就控歐派,居然小時候對小智她媽瞬間都有了幻想,初戀是其碩大的柔軟。這一話小智擁有的作為女人的屬性給淳留下了極其深刻印象、對他有巨大的殺傷力,一定程度上顛覆了他對小智的認知。美鈴則是絕對的平板,在女性的一面上完敗給女漢子小智,和卡羅爾比更是兩個極端,真是殘念~ 偏偏美鈴自己還對這方面很在意。要是小智的料分給美鈴2/3就好了,就合適了(i.e. 把r量分給真正需要的人)。
藍色監獄
總評:94.4
此作是一部很優秀的男子足球作品。內容是一個足球監督繪心甚八組織了一個叫藍色監獄的企劃,從全日本高中生中召集了300名有潛力的前鋒,通過在藍色監獄這個設施里組織各種選拔,以最終挑選出能登上世界舞臺的頂尖攻擊手,試圖顛覆日本足球在世界的地位,改寫日本足球的歷史。在藍色監獄中,繪心甚八極力給選手們灌輸要成為自私的最強大的攻擊手的理念、讓他們放棄把團結合作放在第一位的日本足球的傳統,甚至慫恿他們為了自己進球可以犧牲他人、不擇手段。此作的內容就是講述少年們經歷變著花樣的一次次選拔的過程,其中以男主潔世一在這個過程中的不斷成長作為主線。
此作的觀賞性非常強,氣氛渲染得很好,比賽過程跌宕起伏,各種出乎意料,總是特別吊人胃口,看完一話忍不住馬上看下一話。此作在畫面制作上相當精良和花本,屬于是高成本大制作的精品。而且此作真是越往后越好看,中途把此作棄掉的人真是糊涂和沒眼光。我相當期待此作第二季,看看主角們怎么對付日本U20代表隊。好在看官網上說第二季已經定下來了。
此作對男主潔世一的塑造真的是極其出色,對他的成長、進化歷程描寫得相當好。他的身體素質并不出眾,也沒什么自信,一開始只能算是個平凡的球員。但他真是特別有腦子,很有想法,總是在絕境中拼盡全力地思考,尋找屬于自己的武器、補完自己的缺陷,同時還嘗試將他人的長處和能力融合進自己體內,在和強敵交手中不斷地試圖自我進化,最后逐漸進化成了能主宰比賽的球員。他老是覺得自己是凡人,實際上他在進化能力上是天才,初期屬性值很低,但有近乎無限的成長潛力。在此作所有角色里,論極限條件下展現出的智力值,潔世一絕對是天花板,是個稱得上神奇的角色。我感覺還真得感謝藍色監獄這企劃,要不然潔世一的才能就完全被埋沒了,藍色監獄的選拔過程也真是把看起來溫順的他的內心里的虎狼般的野性激發出來了。原本的他沒有什么大志,但如今他對于成為世界第一攻擊手已經有了執著和野心。
不光是潔世一,藍色監獄里很多有潛能的角色在絕境的比賽中也突破了自己的極限,完成了自我進化。藍色監獄的企劃一開始看起來有點不靠譜,甚至有些荒唐,但現在來看搞得還是挺成功的,不過日本球協對此還是持高度懷疑態度,希望能看到下一季里經歷殘酷選拔活到最后的藍色監獄的選手能虐一虐在溫室里成長的U20代表隊。
蜂樂這小伙子不錯,性格挺好的,傳球和過人能力突出,相貌秀氣,整個人還有種可愛的小受感覺,挺容易令別人愿意與他合作,他在隊伍里作為助攻也相當有用。男主和他在一起挺般配的,他也非常看重男主,可能會有一些他倆的bl本子出現吧。不過我感覺此作里蜂樂的內心戲稍微有點太多了,其心理疾病塑造出的那個莫名其妙的怪物的戲份比很多配角都多。
千切豹馬的外形真是出眾,像一個高挑的大美女,周圍人也都把他稱為公主,有些他的特寫畫面還挺養眼的。他在生活上也具有一些妹子的元素,比如特別看重保養秀發。他的速度驚人,跑得飛快,甚至能把球傳給未來的自己。我感覺他跑得那么快不如也參加短跑比賽,很有可能會有挺大發展。他的腿的舊傷是個隱患,但好在在一整季里舊傷一直沒發作,不過這也可能會是定時炸彈,以后在關鍵時刻掉鏈子。
久遠奸詐得不得了,居然比賽到一半成了對方球員,真是離譜。最后改邪歸正了,難得,令我刮目相看,看來他還挺有良心。本來他自己什么都不做就能順利在藍色監獄里留下來,但被主角一方隊伍的眾人極力拼搏的精神所感染,最后還是選擇冒著風險和他們一起繼續為了登上頂點而奮斗。不過最后他還是被刷了,畢竟實力很有限。
松崗配的雷市陣吾充滿野性和獸性,和《鬼滅之刃》里豬突猛進的感覺簡直如出一轍。不過目前來看他并沒有某些方面特別出眾的才能。雖然這一季最后他留了下來,但未必能繼續走多遠。
紫毛的玲王一直以為自己挖掘出的天才呆萌球員凪要一直和自己踢球,而且這倆人關系挺基的。沒想到第12話凪居然加入了男主的隊伍,這時候的玲王的表情就像自己的老情人跟別的男人跑了似的。雖然我不怎么喜歡玲王,但這劇情對玲王也太殘忍了。凪也真是太冰冷和絕情了,不顧舊情,如今覺得誰更有能力和才華就和誰在一起。雖然后來凪也和玲王示好,結果玲王卻不接受了,像極了被丈夫拋棄的充滿怨氣和仇恨的寡婦。玲王有種如果得不到對方,就要通過粉碎對方令對方屈服的扭曲心態。不知道以后玲王和凪還有沒有機會重新交心。雖然玲王排名挺高,但我真沒覺得他有什么特別突出之處,頂多是各方面屬性都較高,但高得很平均。
馬狼挺有個性的,是個絕對的硬派猛男,體格高大,射門和突破能力強悍,自詡為國王,看不起其他人。起初的他絕對不給別人傳球,執拗得很,是自負的天花板。雖然他很強,但也沒強到光靠一個人就能進球的程度,第17話看到他吃癟的表情我感覺很逗。他在生活上居然特別自律和計較,對很多雞毛蒜皮的事要求得格外苛刻,完全沒有魯莽漢子的感覺,這導致此作里出現了他被幻想穿女仆裝,真是瞎眼。第16話末尾的小劇場居然還專門玩馬狼的女仆咖啡廳的梗,更瞎眼了。
第18話對馬狼的描寫甚佳。潔對他采取放棄play,令他從失敗中徹底認識到自己的無力,高傲的自尊心被潔的閃耀的表現所碾壓,現實迫使他重新徹底審視自我。然后馬狼在崩潰的邊緣突然覺醒,重拾自我,并爆發出驚人的能力,瘋狂過人后大力射門,震驚全場。這一連串的劇情的構思真是相當精彩,對馬狼的心理斗爭和劇變表現得特別好。他經歷了心態的徹底的粉碎和重組,脫胎換骨。此作里我最喜歡的是潔世一,其次就是馬狼了,其個性真是相當突出,形象留給人的印象特別深刻。
國神煉介算是個不錯的球員,身體素質強,技術也還行,居然最后被刷下去了,感覺有點可惜。而看起來整部作品里實力墊底的南無三居然沒被淘汰,真是不可思議,也不知道怎么活下來的,作品里沒個具體交代,不知道是不是他有什么還沒展現出來的潛能,估計這是個伏筆。
凜是此作的BOSS,本以為他是綜合實力的天花板,但也一點點被極速進化的潔世一所觸及到,甚至在某些時刻超越了。感覺潔世一完全超越凜的交點早晚會到來。雖然凜強到像bug,但出乎我意料的是他居然在世界級選手的5人隊伍面前顯得那么弱,表現得毫無辦法。雖然我知道肯定此作要給主角和凜的隊伍潑潑涼水,讓他們知道天外有天,但也沒想到凜會無力到那種程度,還以為起碼也能和對手稍微過過招。本以為這場比賽里男主最后也會像之前一樣面對逆境可以小宇宙爆發,但卻完全沒派上用場。我真覺得有點像主角一方放了水似的。說來,很多游戲、動畫里都是某個角色作為敵方BOSS時強得離譜,但加入自己隊伍后就變弱了,此作也沒免俗。凪、馬狼、凜曾經作為敵人時雕得不得了,結果變成己方之后表現得也就那么回事,貌似之前的光環都消失了似的。和世界top選手的比賽讓自負得不得了的凜重新審視自己,充分挫了挫他的銳氣,這倒是令我覺得挺好,他之前高傲、目中無人的樣子令我挺不順眼的。
繪心甚八的相貌、身材、性格和說話風格真是像極了《飆速宅男》里的御堂筋翔,非常激進,性格狡詐,很會煽動。他是神谷浩史配的,感覺和配御堂筋的游佐浩二的聲線相當像,我起初還以為是同一個CV。繪心甚八和御堂筋互相cos對方肯定沒有絲毫違和感。
一開始和潔世一結伴的才華橫溢的白毛居然就那么被一腳球悶到臉上被淘汰了,還展現出了極致的顏藝,有點可惜,這也充分展現了藍色監獄規則的殘酷性和戲劇性。他如果沒那么早就被淘汰,很可能最后還能留下來。
故事的中期進行的換人賽挺有意思的,三個人一組對決,贏了的一方可以從另一隊里搶走一個人,當一個隊伍里輸到只剩一個人的時候這個人就被淘汰了。在這個過程中原先的敵人可以變成隊友,原本的隊友可以變成敵人。我覺得國足也可以試試這么搞,說不定能靠優勝劣汰篩選出幾個真正像樣的,別以后再到處丟人了。
此作里梅西、C羅的名字出現了N次,感覺作者好像挺崇拜他們似的。
藍色監獄英文叫做blue lock,此作里被簡寫為BL,每個人還有“BL排名”,不結合語境的話立馬就想歪了。
不要欺負我,長瀞同學 2nd Attack
總評:94.4
我很喜歡此作的第一季,女主長瀞的人設實在太好了,性格超級皮,特別雞賊,特別歡脫,顏藝滿分。這新的一季制作質量和劇情令我十分滿意,延續了之前的高水準和極高的歡樂度,觀賞中令我屢次覺得男主女主之間獨特的相處方式真是又有趣又很有愛。
這一季里聒噪調皮的長瀞繼續瘋狂挑逗純情害羞老實憨厚的前輩,沒完沒了地笑嘻嘻地念叨他惡心,同時故事也充分表現出她對前輩的重視和真心,而前輩也明顯心里承認自己已經喜歡長瀞了,在最后一話二人都親密地抱在了一起(看二人腿部的特寫我一瞬間還以為二人接吻了,令我吃了一驚,看來接吻還得留到下一季)。雖然傲嬌的長瀞還是始終不肯承認交往,別人一提到他倆在交往她就糊弄過去,但無疑他倆的關系已經和情侶沒什么兩樣了,到最后都已經在實質上約會了,二人最終也都支支吾吾地坦白這對于自己來說不是約會演習。他倆多喜歡對方都已經不言而喻了,但以戲弄前輩為樂(并以此暗中表達愛意)的長瀞就是不肯坦誠、放下架子告白,而前輩則始終缺乏勇氣,太懦弱,一直也沒明確說出對對方的喜歡。
這一季里男主開始戴隱形眼鏡,感覺戴了之后帥了不少,但還是顯得比較弱雞的樣子。其實男主顏值還可以,要是好好打扮打扮,提升自己的氣質、氣度的話,會更能配得上長瀞,但這樣的他倒未必是長瀞喜歡的類型了。長瀞就是喜歡平時牽著他的鼻子走,而極個別時候男主的主動又會給期待特殊獎勵的長瀞帶來驚喜。
長瀞真是太討我喜歡了。她的眼神太傳神了,表情設計絕妙,鮮活靈動得不得了。她的帶著各種壞笑的面孔真是令我印象深刻。這一季里長瀞還頻繁化身為黑色圓柱,只剩兩個眼睛,這個極度抽象的形象設計得頗為有趣,也有特殊的萌點。長瀞的身材真是令我不由得屢次贊嘆,雖然不是凹凸有致的那種,但真是超級曼妙。看這一季第一話時就再次令我感嘆起來,長瀞的裸足+黑皮細腿+mini裙的組合真是太棒了,賞心悅目,并且這一話里居然還有男主給她給穿絲襪的劇情,特寫鏡頭更是充分展現了長瀞的妙處。長瀞穿死庫水和搏擊運動服的樣子也特棒,小麥色的皮膚再帶上曬痕,真是很性感。這一季里經常有給長瀞加了濾鏡的特寫,有的畫面里長瀞真是養眼極了,特別是長瀞想要親親的樣子的特寫里她真是可愛到爆!
第5話長瀞去男主家照顧發燒的男主,這段真是挺有愛。長瀞雖然平時很調皮,但在照顧前輩這方面一點都不含糊,居然還試圖趁他燒迷糊時親上去,可見對他的重視和喜歡的程度。長瀞給前輩喂粥時,糊涂狀態的前輩眼里看到的是OL裝的長瀞,這可真是看到了夢幻的景象(其實是看到了長瀞作為自己妻子的未來),男主真令人羨慕。OL裝的長瀞真棒,有種別致、反差的美感,也令我感謝男主的腦補帶給觀眾的福利。
這一季里長瀞的幾個小伙伴繼續顯得元氣滿滿,給長瀞和前輩的關系添油加醋、調皮搗蛋,給故事增加了很多樂子。那個長頭發的不良少女居然家里是開健身房的,她在傳單上的造型令我感覺真是很有她的風格。以她的身材和野性,在健身房里教散打真是毫無違和感。
第3話長瀞的姐姐出現了,紅瞳黑發是很迎合我審美的人設。她外貌上看似乎應該是個正經的女人,沒想到性格和長瀞頗有相似之處,喜歡戲弄男主、和他開玩笑,只不過不像長瀞顯得那么歡脫,而是更有想法,更會打算盤,還陰陽怪氣地試探男主,內在很腹黑的樣子。想不到長瀞這么能鬧騰的孩子,唯獨面對她姐的時候顯得被壓制著,在她面前嘚瑟不起來,真是一物降一物的感覺。長瀞姐和長瀞一樣眼睛總能彎出奇怪的弧度,而且有連著上嘴唇的虎牙。
作為美術部原社長的學姐在這一季里再次令我感到她真是神奇的角色,還是那么極度隨性、顯得沒有世俗的常識,并經常提供巨大福利。她穿兔女郎裝跑步的樣子很銷魂。第6話居然她藏在柜子里,突然一絲不掛地出來,真是超奇葩的女人。第8話里她和男主練柔道時她的歐派實在太壯觀了,差點釀成胸殺案,或者給男主造成歐派恐懼癥。好在是長瀞突然出現救場。第11話出現的社長的坦胸的騎行服真是驚艷,巨大的白皙和黑色衣服的映襯極富視覺沖擊力!太頂了!如果在真實世界中出現,估計得把周圍雄性駕駛員的視線都吸引過去并釀成嚴重車禍。水樹給社長配音太蘇了,深沉的聲線真迷人,能令人耳朵懷孕。
這一季出場了不少新角色,社長的表妹也是其一。此角色真是和她的表姐很有相似度,性格極度簡單直率。她明明才高一,居然已經是爆炸極的歐派了,而且完全沒有羞恥心和戒備心,第一次出場就面不改色淡定地說可以給男主當果模,自己立刻就把衣服脫了,真是太科幻。這表妹現在是美術部的一員,將男主作為前輩而主動照顧,不知道保持男女間的距離感,甚至給他喂食,但并沒有任何戀愛的想法。雖然長瀞或許也知道其實沒必要吃她的醋,但還是會忍不住吃醋,挺有意思。
弦音 -連結的一射-
總評:94.2
這是《弦音》的第二季了,距離上一季好幾年了。上一季的觀感很好,新的一季果然沒令我失望。這一話剛開始的超級精致細膩的驚艷的射箭特寫一下子就把我再次拉入了此作的世界。真不愧是京阿姨,水準就是絕,色調、光澤、構圖、動感和氣氛的表現等等都超講究,令觀眾在一開場就能立馬感受到大作的范兒。觀看這一季過程中令我N次贊嘆效果做得可謂嘆為觀止,將很多大片級的運鏡納入到弓道的表現中,還加入了魔法效果和肉眼不可見的效果,比如氣流、聲浪、波紋,還有飄散的櫻花和綠葉,再加上此作非常出色的配樂,這些元素令弓道競賽的觀賞性太強了。此作里射箭的場景我感覺被京阿尼做得像藝術品一樣。此作里還有很多細節,諸如地板的反光、光暈、鏡頭拉伸等效果都做得特別出眾。
這一季主要是講風舞高中弓道部大家的訓練、人物之間的關系、隊伍之間的接觸,以及最后參加全國大賽的事。
上一季我就留意了妹尾,第一話她剛一出場時再次令我感覺真帥,個子高挑白凈瘦削,眉目清秀。雖然她不是賞心悅目的大美女那種角色,但她那種帥女的氣質真是特別有風采,還有一些御姐的感覺,帥氣感完爆除小雅哥和愁以外的男角色。而且妹尾的箭術高超,第6話沒有一次脫靶,真是太贊了。弓道部的另外兩個妹子挺漂亮,特別是其中深紫發色的妹子顯得又漂亮又優雅,是大家閨秀的感覺,像是輝夜姬的那種人物風格。這三個女角色真是養眼,看第6話的時候我還不禁發出聲贊嘆了一句:“京阿尼畫妹子真是一絕!”。第6話,她們三人射箭時還施展了魔法,箭矢的軌跡劃出了一片花瓣雨,射箭聚力時還產生了氣流,觀感真是領我吃驚。
妹尾甚至都受到了其它學校女生的關注,感覺她有成為萬雌王的潛力。連桐先的女生都覺得妹尾很帥,還主動找她交往。第11話,參加全國大賽時,很多其它學校的女生看到妹尾出現時立刻簇擁上去,還說想晚上去她的房間,令我感嘆妹尾居然都有這么多迷妹了,真是看得我很開心!我以為帥氣的妹尾只會受女生歡迎,沒想到第9話,辻峰隊伍里有個大塊頭男子居然也迷上她了。此作里其她妹子都沒有妹尾的待遇,我感覺京阿尼的人似乎也知道妹尾這個角色的人設招人喜歡,故意給她多做了劇情。此作要是出一個外傳,以妹尾作為女主,并且是百合作,那就絕好了!
小雅哥還是那么牛,那肌肉那氣場,真是能令很多女觀眾迷醉。居然他說練習時連中211箭,感覺他已經出神入化了。雖然他年紀輕輕,但無疑已經是大師級了。
第5話專門講七緒和海斗斗氣的事,海斗過于沖動暴躁的性格令我不怎么喜歡,而七緒說的刺激海斗的話我也不喜歡。這倆人真是基友,還說要比拼一輩子,明顯是官方賣腐。這倆人外形還蠻般配的,一個粗獷,一個秀氣。
愁的顏值真是高,第7話里有好多愁的特寫,真是顯得格外精致俊俏。愁可真是土豪,居然還有自己的弓道場,令人羨慕。愁的妹妹令我覺得有點成熟過頭了,特別是說話的聲線和語氣,感覺不像是那個年紀的小女孩該有的天真的樣子。她和遼平有一定感情基礎,以后交往的話挺不錯。
辻峰高中弓道部真是太寒酸了,5個人連個練習場地都沒有,只能在野外射箭(射野箭),還得考慮風的影響。他們也沒有指導,純粹自己瞎摸索,真是個很有個性的隊伍。此作里有人評價他們是野武士,真是恰如其分。隊伍中還有個戴黑口罩的,有點像忍者似的,挺神秘的。白發的二階堂的射姿和射箭時候的氣場挺帥的,不過水平也就那么回事,只能算1.5流。他堅持非主流的斜面射法,極其特立獨行。我沒搞懂為啥他如此執著于此,貌似是受到了叔叔的重要影響。他對叔叔的感情和重視程度真是夠夸張的,都有點叔控感覺。二階堂的性格挺別扭的,對小雅哥擺出臭臉,在第9、10話的表現有點討人厭。不過在小雅哥幫了他后,在第10話的最后他居然傲嬌了,挺樂。
辻峰和風舞的全國大賽對決挺有戲劇性的,辻峰前一半狀態極佳,居然第一輪全中(五連發五連中的效果相當震撼),后一半卻一塌糊涂;而舞風恰恰相反,最后離奇地贏了比賽,真夠邪乎的。我都已經做好了風舞失利的心理準備了。
最后風舞碰上了桐先。按照之前場次的中靶率,桐先輕松獲勝是鐵定的,除非風舞再次運氣爆表且桐先不在狀態,才可能扳回絕對實力的差距。沒想到風舞表現得意外出色,再次超水平發揮。但果然,人品還是守恒的,上次對決用掉了太多。這場比賽居然打到了第二場加賽,能跟桐先膠著到這種程度已經很不得了了。但沒想到最后是湊脫靶而輸掉,我還以為要輸也是因為其他人脫靶。這場比賽做得真是驚心動魄,感覺就像是看點球大戰一樣。雖然我是愿意讓風舞贏的,但是如果真贏了也不好,畢竟以風舞的水平理應無法在全國大賽奪冠,他們贏了的話對桐先也挺不公平的。桐先不僅贏了這場,而且還最后奪了冠,這很是理所應當,桐先也配得上成為冠軍。
話說愁真是厲害,感覺此作的高中生里最穩的就是愁了,我心想是不是他在全國大賽上一箭未失,我真是很難想象愁脫靶的樣子。湊和他比果然還是有差距,還是不夠穩。桐先的隊伍里那兩個小黃毛有點拉胯,要是換上更穩的,桐先平均每場比賽估計能中>=18靶。
此作的弓道比賽里只有中和不中之分,感覺多多少少不科學。射中中央和射中外環總應該在計分上有點差別(哪怕只相差30%)才對啊。至少是在戰平的時候,不應該靠加賽,而應該看根據環數算的計分。
雖然我對弓道知之甚少,但還是多多少少覺得此作把弓道描寫得有點玄化了,說什么弓道沒有盡頭。小雅哥的程度在我看來已經站在了弓道的頂點,但他似乎還想要在弓道的路上再去探索更多的可能...
這一季沒有專門的ED畫面,每一話都是ED歌曲伴著劇情內容出現。這一季的ED不錯,女聲挺抒情、溫婉和高亢的。
轉生王女與天才千金的魔法革命
總評:93.75
此作真是特別硬核的百合作品,之前早就在百合會上聽說了,果然名不虛傳。此作的制作頗為精良,整部作品觀感很飽滿,是百合愛好者絕對不容錯過的作品。此作整體制作精良程度很高,不僅人物畫得精致,在戰斗方面制作的水準也真是相當高。特別是第5話艾妮絲和尤菲莉亞共同和黑龍戰斗以及在最后一話這倆人間的大戰,視覺表現力真贊,做得相當用心,很有沖擊力。
女主艾妮絲菲亞是從現代社會轉生到這個世界的公主。她的戰斗力超強,從轉生前就有使用魔法的(中二)夢想,但可惜轉生到這世界后,雖然身為王族,卻沒有魔法能力。她很有個性,身份高貴,但是跟平民冒險者一樣,到處打怪采集素材,也缺乏作為公主的自覺。另一個女主尤菲莉亞是公爵家大小姐,有著極強的魔法天賦,本來和王子有婚約,結果王子在畢業前喜歡上了個歐派很大的女生蕾妮,并為了解除和尤菲莉亞的婚約將她污名化。正在尤菲莉亞當眾被侮辱,感到絕望時,自稱掠奪公主的艾妮絲菲亞突然騎著掃帚從窗戶闖入,將她擄走。自此尤菲莉亞就成了艾妮絲的后宮,此作劇情就是講述這倆人在一起經歷的各種故事,包括一起研究魔法,跟龍戰斗,揭開蕾妮背后故事,跟愚蠢的弟弟的戰斗,繼承王位,等等,內容挺跌宕起伏和曲折的。
艾妮絲很有個性,真是奇葩,自由自在無拘無束,她的發明創造在那個世界的人眼里都是些天馬行空的東西。她的性取向很明確,第2話里她小時候就直言說“我不想和男性結婚!”、“我更想跟女性親熱!”。這種腦筋令百合眾很喜聞樂見。
此作里有很多精彩直球場面,令人很興奮。第一話艾妮絲把尤菲莉亞擄走的這一幕真是太GJ了(雖然有點違和,畢竟之前她倆的交情也沒那么深厚)。看到艾妮絲菲亞扛著一臉慌亂的尤菲莉亞在夜空中揚長而去的場面,我感到很開心,感覺艾妮絲菲亞挺有點大盜魔理沙的感覺。艾妮絲攻略神速,第1話末尾綁了尤菲莉亞后,第2話見到岳父直接就讓他把尤菲莉亞許配給自己,還一通夸尤菲莉亞漂亮可愛和完美,并向她展現自己的簡單純潔和真誠,僅僅一話就把尤菲莉亞給收服了,在床上心滿意足地抱上了她。很多作品里性格細膩敏銳的角色往往一季還沒啥進展,令人著急,而艾妮絲這個簡單粗暴的家伙一話就把她癡迷的對象給搞定了,真厲害,看著痛快。
第5話出現了艾妮絲的禮服裝,雖然挺漂亮,但我感覺還是挺違和的,畢竟明明之前她都是給人假小子的感覺。沒想到她居然那么大,比尤菲莉亞的似乎還大。
此作對艾妮絲和尤菲莉亞倆人的關系描寫得不錯,一開始尤菲莉亞是被擄走,完全沒做好準備,隨著對艾妮絲認識的加深,開始感謝她闖入了自己的世界,把自己帶離了被束縛的無聊人生。再之后,二人一起與惡龍戰斗,拼盡全力拯救了國家,也令二人的關系更加緊密。再到了后來,艾妮絲自己開始鬧別扭,尤菲莉亞逐漸轉受為攻,主動撫慰和關愛艾妮絲。在最終,為了艾妮絲的幸福,還不惜與固執的她一戰,表現出的勇氣很不錯。我一直覺得艾妮絲是奇才,戰斗力是此作的頂峰,真沒想到最后尤菲莉亞化身魔炮少女,把艾妮絲給虐了,真是顛覆了我對二人實力的認識。
最后一話二人戰斗結束之后的內容太高能了,瘋狂秀恩愛,閃得不行。看到倆人在床上居然嘴對嘴真的親了上去,令我大吃一驚,雖然對于這么硬核的百合番,出現接吻并不意外,但看到這倆人真的就那么直白地親了上去,還是令我感到沒有防備的猛然興奮,這也太直了!尤菲莉亞在這一季的最后完全是攻,在床上壓在艾妮絲上面,平時還試圖主動要求和艾妮絲親嘴,而此作前期顯得特別豪放的艾妮絲反倒現在是害羞的一方了,樂~
此作最大失分點在于艾妮絲繼承王位的劇情,感覺作者像是刻意編出個事來,制造矛盾點,結果從故事邏輯角度以及從艾妮絲的人物性格角度都顯得挺牽強。這一段里艾妮絲表現出來的想法和姿態令我感到挺莫名其妙甚至有點不爽,一點都不像她自己,非要強行接受自己根本沒必要去承擔的義務委屈自己,還得看惡心大叔們的臉色、和不認識也不喜歡的男性生孩子,我感覺她真是很蠢。她還非要堅持履行自己作為王女責任,好像非得這樣自己才能夠被認同、有自己的存在感似的,都不知道在想啥。之前她顯得頭腦很清醒、明事理,結果卻在這方面上如此糊涂,令我挺失望的。明明她之前那么奔放,不受世俗約束,結果這段故事里卻顯得那么固執和矯情,難以理解。雖然后來她也坦白了為什么非要執著于王女的身份,但我感覺還是不太說得通。但這段劇情里尤菲莉亞表現得蠻不錯,做了自己該做的事,盡自己最大所能做了自己能做的事,也展現出艾妮絲在她心中的無可取代的最關鍵的位置。
PS:關于繼承王位,明明有很多選擇。比如艾妮絲父親繼續執政15年,然后讓發配邊疆的傻弟弟回來執政。或者和顯得年輕漂亮的艾妮絲的母親再生一個,然后在艾妮絲父親繼續執政20年后,再讓自己的兒子執政(或者如果是女兒就讓女兒執政,基本也不可能再是艾妮絲這樣的奇葩,又喜歡瘋又是彎的)。反正不管怎么著都比著急忙慌地讓艾妮絲成為女王好得多。也不理解為啥艾妮絲的父母那么著急退位,似乎也沒好好想過艾妮絲的感受。
王子實在是神經病,性格太惡劣了,好在是解除了婚約,要不然尤菲莉亞在未來就一點也獲得不了幸福了。后來知道他居然是個姐控,而且還內心自卑。他殺了蕾妮(好在沒死),還跟艾妮絲大戰一場,最后居然強行洗白了。我真是很全方位討厭這個愚蠢的歐豆豆。艾妮絲居然還覺得虧欠他,我挺不理解,明明是歐豆豆自己腦子有問題、自作虐。和艾妮絲一戰失敗后王子竟然變得一下子老實了起來,令我感覺有點太突然。要是他內在性格沒那么壞的話,哪干得出把蕾妮掏心這么毒辣的事情。
我挺喜歡紫毛緹爾蒂·庫拉雷特這角色的,她和艾妮絲是老交情,既是朋友也是魔法上的合作對象。她深居簡出,人物形象和氣質像極了群堂美玲,都是暗色系帶有神秘感的妹子,整個人形象和性格都暗沉沉的,還都特有想法。她是研究詛咒方面的專家,其研究興趣和整個人展現出的形象真是極度一致。她和美鈴雖然都顯得都挺陰森、挺怪的,但其實內在性格都挺好,都有自己重視的人并愿意為其無私付出。第11話她和艾妮絲并排坐在一起安慰她,還提出愿意和她私奔、周游世界,去研究各自喜歡的魔學和詛咒,我覺得這個提議很好啊!這倆人挺配的,在一起挺有愛的,我蠻希望她倆深入發展的。
艾妮絲的女仆伊莉雅·克拉爾是個很出色的配角,顏值很高,外在和內在都顯得很有氣質,端莊穩重,還挺帥氣,是了不起的女仆。伊莉雅X蕾妮這一對CP我愛了。伊莉雅第一次露出白皙的脖子讓蕾妮吸血的時候我就感覺這一對CP很奇妙,第8話又看見伊莉雅對蕾妮公主抱,令我興奮了一下,第9話伊莉雅嘴對嘴給蕾妮喂自己的血,我覺得這絕對是官方暗示這是CP了。最后蕾妮成了伊莉雅帶的新人女仆,二人在一起的機會更多了。雖然此作里二人還沒展現出相互喜歡的關系,但我感覺以后很可能會有些發展,她倆的關系給二次創作留出的余地很大。
尤菲莉亞的父親蠻不錯,長得挺帥(沒有典型的老父親的感覺),想法真是開明,能坦然接受自己的女兒成為艾妮絲的玩物,居然沒因此受什么刺激。
旁白和琉米是釘宮配音。釘宮的鼻音還是那么好聽,給琉米的配音特別酥、柔和銷魂,令人釘宮病又要發作了。
此作是個超級無比happy的end,二人比翼雙飛,并且攜手創造出了幸福的國度,這也令我感覺出第二季的可能性微乎其微,本身硬核百合番受眾群體就少,這一季又花了那么大本,收回成本可能都難。而且感覺劇情繼續編下去也沒什么余地了。
尤菲莉亞簽了精靈契約,可以永葆青春長生不老,宿命論使得她早晚會與漸漸老去的艾妮絲離別,這令人想來挺惋惜的。但聽有人劇透說艾妮絲因為龍的刻印什么的好像也能壽命無限,這倒不錯,這倆人能永遠在一起真好。不過如此想來,國王若永遠都不換,也顯得有點離譜(我覺得可以等歐豆豆發配邊疆改過自新后回來繼承王位,然后從此這倆人與王國政務不再產生任何瓜葛,可以痛快游遍天涯海角)。
雖然艾妮絲菲亞的CV千本木彩花配的也不錯,但是個性感還是有進一步提升的空間,如果讓UMB來配或許更好。
銀砂糖師與黑妖精~sugar apple fairy tale~
總評:93.75
此作里銀砂糖是用砂糖制作的工藝品。女主安是個銀砂糖師,母親去世后自己一直一個人生活。有一天她在精靈奴隸商人那里偶遇一名叫做夏爾的男精靈戰士,為他的容貌所驚艷到,然后買下了他作為之后旅途的保鏢。此作就是講述安和夏爾在一起的故事。起初夏爾對安極其冷淡,不斷諷刺和挖苦她,但安始終用自己的真心去面對夏爾。在此作中,二人一起經歷過風風雨雨后,彼此建立了親密的牢不可分的感情。
此作整體水準很高,各方面都非常精良,制作用心,令我感覺很有愛,是一部我很欣賞的作品,喜歡少女向作品的人更是絕對不容錯過。安和夏爾的人設都非常出色,作品中對他們的描寫和二人的情感刻畫很細膩。此作的整體劇情很棒,既有輕松有趣的故事,又有非常溫婉感人的橋段,而且越往后越好看,隨著劇情的進行能令人越來越喜歡上安和夏爾。此作的音樂水準也很突出,令此作表現力非常飽滿。
此作里的銀砂糖工藝品挺有意思的,故事中出現的不少銀砂糖作品都相當驚艷,設計構思、工藝、色澤都絕妙。而最出色的那一件,莫過于安在最后一話拿出來的那一件,精美絕倫,難以想象安是怎么做出來的,簡直神乎其技。現實世界中要是有賣水晶重現出的那件作品就好了,再打上燈光,作為擺件肯定賞心悅目。
此作里一直都沒有出現關于吃銀砂糖作品的描寫,令我很好奇是怎么吃的。直到最后一話才出現了國王手下的妖精吃安的作品的一幕,但居然不是用嘴吃(我以為他會像吃棒棒糖一樣舔),而是把銀砂糖一下變沒了,這吃法真是無語。
安的人設極贊,既完美,又有個性,是令我很喜歡的少女。她給人的外在形象只是一介普通的弱女子,但實則有著非常堅強的內心,特別獨立和要強。她善良、純真、樸實,而且很溫柔,面對起初夏爾的百般挑釁,總是在生氣后依然繼續寬容地對待他,不懈地努力和他成為交心的朋友而不是主人和奴隸的關系。她還很勇敢、正義凜然,面對奴隸販子對精靈的暴行敢于挺身而出。安在此作中的經歷真是特別艱辛,明明她才華橫溢,但老是得不到公平競爭的機會展現其才華,還總是被惡人陷害。此作里總有人以為她是溫室里的花朵,卻不知她是飽經風霜的野花。好在在最后一話的品評會上,她的能力總算在公眾面前得到了承認,成了被國王認可的銀砂糖師,可喜可賀。
雖然安非常獨立和自立,不愿意依靠別人,對一開始喬納斯的求婚毫不理睬,但她也有著很少女心的一面,在面對夏爾的撩撥時會臉紅心跳,顯得真是挺可愛的。
第8話做得相當好,充分表現出了安作為銀砂糖師的尊嚴和執著,無論怎么失敗都不氣餒,而且能夠從不同角度看待問題,不僅技術卓越而且非常聰明,主動了解公爵對他深愛但已經消逝的女精靈的印象和思念,從而終于制作出了這變態的公爵真心想要的銀砂糖塑像。一直苦著臉的公爵最終得到了個極品的私人定制手辦,從而露出了笑顏,這結局真是挺好的,如果他對這作品的長期凝視真的能令他摯愛的精靈戀人再次重現就好了。安和夏爾一度失去了彼此,但在這一話里終于再次重逢,二人都更加清晰地感受到了對方對自己的重要,情感關系出現了極大的升華,擁抱在一起的畫面充滿了美好、幸福和愛意,此作對這一段的表現很優秀。安也真是奇人,居然搓出來那么漂亮且透亮的眼珠子,里面仿佛能看到星云,這成為女精靈砂糖塑像的點睛之筆。
夏爾是那種少女漫畫里典型的完美化、理想化的二次元男主,外表帥氣,相貌極度俊美,戰斗力無比強大,同時又很有性格。起初的他相當毒舌、高傲、倔強,對安的溫柔和誠意顯得不屑一顧,還挺壞壞的,看女主身形嬌弱、性格柔弱,就各種撩她,好像不欺負她、令她受辱都難受似的,非要扮黑臉,這性格挺扭曲和病態的。他還甚至有些誘受,有時非要勾引安以命令的方式使役他。安要是個脾氣差的妹子的話早就抓他翅膀讓他閉嘴了。夏爾這么扭曲倒也能理解,畢竟他被作為奴隸使喚久了,對人類嚴重缺乏信任。好在是在旅途中安始終對調教夏爾非常有耐心,表現得非常寬容大度,并且付出誠意和真心。逐漸地,當夏爾理解了安的所思所想,充分明白了她是位多么美好和充滿魅力的少女后,與她的隔閡一點點消失了。在最后夏爾甚至都決心為了安而付出自己的一切,居然為了能讓安成為銀砂糖師而將自己的翅膀交給了饞他身子的貴族女人,成為了她的奴隸。最終這個劇情挺虐的,安要是知道夏爾會這么做,就1000%不會當銀砂糖師了。我很希望此作出第二季,看看夏爾何去何從,應該最后還是happy end。PS:我估計那個饑渴的貴族女人回去肯定會把夏爾全身舔個遍。
我感覺此有點bug的地方是一開始安買夏爾也太便宜了。夏爾這么強大,為了捉他還死了幾個人,按理說應該貴得離譜,怎么貧窮的安會有那么多錢買他?而且就算安買他花了自己攢了好幾年的大筆積蓄,那也不應該在不算很長的旅途結束后就立馬把夏爾的另一半翅膀交給他、令他獲得自由才對,至少也得讓夏爾給自己當一兩年保鏢才能回本啊。
沒想到喬納斯那么壞,本來起初他看起來挺溫和的,也沒什么壞心眼,居然第3話被安拒了之后妄圖害她。原本還以為喬納斯會洗白,沒想到真的就是超級厚顏無恥,居然自己還真以為從安那里搶來的作品是他自己做的,真是徹頭徹尾的神經病,腦子徹底壞掉了,有這樣的人真是不可思議。想不到后來在變態公爵那里,喬納斯再次給安使壞。感覺安的脾氣真是太好了,早該狠狠地給他個大逼兜。雖然在最后喬納斯還算給安做了點好事,立了點功,但還是完全不足以扭轉他的丑惡下賤的形象,他還是沒能好好審視自己、重新做人。
第5話出場的男銀砂糖師凱特不錯,很有自己的堅守,只把作品賣給他愿意賣的人,而且充分考慮對方的經濟承受能力,而性格惡劣的貴族無論開多高的價格也買不到他的作品。凱特真是很有骨氣,也很有良心和善心,我挺欣賞這樣的人的。
在地下城尋求邂逅是否搞錯了什么 第四季(迷宮篇)
總評:93.65
之前每一季我都追了,這一季主要是講貝爾等人連同其他眷族的隊伍一起探索深層迷宮的經歷。后來貝爾掉到更底層和大伙失散,大伙、貝爾和琉小姐都經過無數次絕處逢生,最終大伙總算找到了奄奄一息的貝爾和琉小姐,總算是活著從地獄里出來了。總的來說這一季觀感很好,其實前一半做得相對中規中矩,最精彩的是后半,劇情特別有吸引力。
貝爾出息了,成了level 4,有點脫胎換骨的感覺了,fire bolt的威力已經是個大招級別了,令所有人都刮目相看。這一季里他很多次單挑BOSS相當帥,身手極其敏銳流暢,成長驚人,感覺貝爾得有level 4.5了。以前他給人一種小白兔的感覺,而現在卻成了隊伍里的靠山了,仿佛只要有他就有希望、就有奇跡。不知道啥時候貝爾能到level 5。其實我覺得以這一季里他的算得上傳奇的經歷,再加上如此出色的身手,理應到level 5了,至少我不覺得其他level 5的戰士能比現在的貝爾強多少。
工會接待女艾伊娜對貝爾越來越動心了,還沖著他臉紅。在她眼里貝爾不再只是個boy了,已經把他當可靠的男性看待了。貝爾的后宮又隱性地增加了,這引起了赫斯緹雅的隔空警覺。
豪放的耍大刀的黑皮女阿伊莎在這一季之前就是我印象挺深的角色,其人設相當有特點,性格剛毅豪放,其肉體給觀眾的視覺帶來極大福利。看到她這一季也出現,而且還是個活躍的角色,令我欣慰,要不然她這么出眾的人設如果只是作為個不起眼的小配角就太可惜了。她的歐派實在太大了,此作里還故意給了些特寫,感覺可能比赫斯提亞都大一號。第12話她居然還試圖單挑樓層主,戰斗的身姿很是帥氣,相當勇敢無畏。第14話她更是在春姬的加成下奮勇躍起砍死了大BOSS,帥得不行,這應該算是她在此作里人生最高光的時刻了。在隊伍里立功最大的除了貝爾就是她了。在失去貝爾后,阿伊莎完全挑起了隊伍的大梁,不僅戰斗力強大,而且頭腦冷靜,判斷力強,始終士氣高昂。阿伊莎平時稍微有點高傲,但對隊友真是很重視,是個很有血有肉的角色。第13話時她看到春姬被雙頭龍的嘴炮轟沒,表現出了出離的憤怒。之前給劍姬專門做過外傳動畫《劍姬神圣譚》,要是給人設這么出眾的阿伊莎也做個番外篇就好了。
第6話莉莉和貝爾在晶體的窯洞里的對話做得不錯。要說貝爾和此作哪個女角色最般配,我覺得還是莉莉,莉莉對貝爾是純純的真愛,莉莉也特別需要貝爾作為自己的依靠的陪伴。
卡珊德拉是這一季最令人來氣和火大的角色。這女人莫名其妙,太耽誤事了。她特別墨跡和窩囊,也不把自己的預知向大家說清楚,就光一個勁讓大家別去,根本說服不了大家,也沒法讓大家為此做好準備、事先擬定作戰策略。而且她還老說喪氣話拉低士氣,大家奮勇戰斗時也派不上用場,總算第12話的時候在達芙妮對她臭罵后覺醒了,想起來干自己該干的事了。然而第13話,她面對雙頭龍的嘴炮傻愣著不動,春姬為了救她送命了,此時我真是氣啊,恨死了卡珊德拉,為了這么個垃圾角色,喪失了我挺喜歡的春姬(好在,第14話里春姬不知道爆發出了什么潛能,居然啥事都沒有)。
達芙妮這個角色中規中矩,能力一般般,外形也沒什么特殊的。她和卡珊德拉有點橘,倆人認識很久了,第14話卡珊德拉突然向達芙妮抱上去、弄得達芙妮很害羞的那一幕挺橘的。
我越來越喜歡命小姐,很帥氣,在面對雙頭龍的時候她真是拼盡全力,非常奮勇。她還主動擔當起春姬的護衛,第2話顯得橘里橘氣的,看得我挺高興。我真心希望她倆能發展出感情,這倆人很配,比起貝爾X春姬好多了。第13話,看到落水后的命小姐被三個魚咬出血,我真是心疼她啊!我一度特別擔心命小姐真的掛了,而且第14話使出大招壓制雙頭龍的時候感覺她就是決意豁出性命了,看到雙頭龍被滅后我心里一個勁兒著急:大家趕快去救水里的命小姐啊!好在命小姐只是受了外傷,命挺大,要是食人魚在水里咬斷了昏厥狀態的命小姐的喉嚨就慘了。在此之后命小姐貌似一直昏厥中,不知道為啥卡珊德拉不趕快把她治愈。
我不僅磕命小姐X春姬,還很磕阿伊莎X春姬。特別是第14話,渾身是傷的阿伊莎抱著使完大招虛弱狀態的命小姐時,那畫面感真有愛,春姬表現出了被戀人抱在懷里的那種嬌弱感。
第11話,貝爾怎么看都已經沒救了,胳膊也斷了,人魚瑪麗的血也太好用了,居然胳膊那么容易就接上了,也不知道這人魚是個什么來頭,此作都沒交代。人魚不惜自己放血也要救貝爾的表現不錯。不過人魚怎么就喜歡上貝爾了,這點描寫的不夠,應該把他們的邂逅再多展現一些。瑪麗也算是后宮王貝爾的準后宮了。
之前韋爾夫沒派上太大用場,作為一個鍛造師,武力平平。沒想到第15話他居然想到了就地取材在地下城打造新的魔劍的想法,還真是一位內心烈火熊熊燃燒的鍛造師呢,給此角色的形象加分了。第16話我對韋爾夫更是刮目相看了,居然光用錘子愣是敲出了一把不朽的魔劍,跟彈藥無限的火箭筒似的,這也太夸張了。韋爾夫在這一話簡直是神乎其技,這理應不可能是一個普通鍛造師光憑借意志力就能做出來的壯舉。
那個邪惡的馴獸師馴服BOSS沒成功,反倒被BOSS秒殺令我挺開心的。這個馴獸師的長相很像我小學一個姓侯的同學。
這一季的后半里關于貝爾和琉小姐結伴的旅程做得真好,劇情緊湊、扣人心弦。他倆在深層陷入的絕望感在此作里描寫得很出色,無數次死里逃生,卻總也看不見深淵的盡頭,如同踏入了地獄卻始終尋覓不到出口。好在這倆人在一起,能相互鼓勵走過人生幾乎最黑暗和絕望的關頭,用相濡以沫來形容毫不為過。這些劇情里貝爾和琉小姐也真是超超超超超命大!基本沒食物沒藥品,經歷無數次戰斗,受過無數次致命傷,可是靠意志和相互加buff最后居然都挺下來了,最后和終極BOSS決戰時這倆人好像之前都沒受過傷似的,生龍活虎,真是不可思議的二人(以及不可思議的劇情)。
第19話挺莫名其妙的,按說琉小姐和貝爾明明能一起逃出競技場,居然琉小姐把橋炸了,并把貝爾弄到橋的另一頭令他能夠脫身,然后琉小姐就被怪物虐。琉小姐的這個行為很多余啊,顯得編劇強行催淚。我還真以為琉小姐就要永遠地離開我們,我都做好心理準備了。貝爾還真是勇猛,對琉小姐特別有情有意,似乎都沒想過自己的生存幾率、沒做任何心理斗爭,就立馬跑去救琉小姐了。在琉小姐眼里貝爾已經成了能創造奇跡的英雄般的存在,真是沒法不對他心動。
之前就覺得琉小姐很不簡單,看了第20話,沒想到她是那么有故事的人。當年眷族被全滅令她受的打擊真是太沉重了,失去了活下去的目標。她居然一個人能把暗黑眷族全滅,真是超厲害。這一話的BGM整體很出色,特別是前半部分出現的女聲歌曲水準相當高,非常可圈可點。
本來我覺得琉小姐這么獨立、自強、勇猛的人應該不會對貝爾誕生喜歡的情愫,但二人共同出生入死、經歷過無數次絕境后,關系已經緊密得牢不可分。在這一季中途琉小姐就已經明顯對貝爾有了一些喜歡的感覺,對貝爾臉紅了,對半果著的他羞澀了。二人還肌膚緊貼在一起取暖,那畫面里二人顯得很是“綿密”。估計琉小姐以前從來不知道喜歡的感覺是什么,這是她的初體驗。
二人合力干掉大BOSS,并最終被救回到地面上后,琉小姐醒來后不顧自己的著裝,第一件事就是飛奔去找貝爾,可見貝爾在她心里已經重要到什么位置了,說是她如今最重要的人都毫不為過。傷愈后,倆人出來約會,琉小姐居然穿上了女孩子的白色連衣裙,這真不像她的風格。琉小姐居然還對貝爾臉紅了,還害羞了,還逃跑了,真想不到她這么英姿颯爽強大的女戰士,終有一天會對某個人露出那么羞澀和嫵媚的表情、會變得那么可愛!貝爾幸福地后宮+1。說來貝爾還真是徹底改變了琉小姐的角色形象,似乎從她心中在意起貝爾開始,琉小姐才慢慢地有了作為一名少女的意識,而以前她只把自己作為冒險者看待。
琉小姐經歷無數次慘烈的戰斗,我一度覺得她的衣服撐不住了,還心想如果其衣服徹底破了,難道要果著?豈不會嚴重分散貝爾的注意力?居然第16話,似乎琉小姐是拿了女性尸體的衣服后,重新給自己裹了一次,總算不用擔心她果體的危險了(如果真是尸體上的,豈不會已經很臭了?)。貝爾看到她纏胸還羞澀了一下,逗。阿伊莎的纏胸布到后來也是越來越破爛,都搖搖欲墜了,這要是完全破了那就糟糕了,以她的size,戰斗時候的甩動可能會造成離心力嚴重妨礙戰斗,并且令隊友們都無法直視。不過莉莉的背包那么大,我估計總應該有阿伊莎備用的纏胸布吧(或者直接用紗布代替,紗布不可能沒有備著)。
這一季的ED1構思不錯,赫斯緹雅在眼前甩動著歐派輕盈地舞動,氣氛甚是愜意,感覺就像面對著和自己戀愛中的少女,令人春心蕩漾。
再得一勝!
總評:93.65
這是以女子高中生的柔道為題材的動畫。以柔道為題材的動畫作品真不多,我有印象的動畫里這還是第一個。像這樣的題材,除非做得很爛,否則我基本都是會追的。此作的制作高于我的預期,十分值得推薦,是一部用心制作的佳作。整部作品很有愛,觀感很好,劇情有趣,在柔道方面表現得很硬核,關鍵場景的畫面表現力頗為出色,而且主角青葉西隊伍的大家都挺令我喜歡的。此作是認真做的一部女子柔道作品,完全沒有任何福利,人物的畫風也并不主流,單純指望看美少女在柔道過程中獻福利的膚淺觀眾就要失望了。
此作主要講述園田未知、瀧川早苗、冰浦永遠,以及后來加入的姬野?和南云安奈,她們共同組成的青葉西柔道部。劇情中刻畫了她們的性格、內心,描寫了彼此間感情的加深,并講述了她們平日的練習和參加比賽的過程。
冰浦永遠是此作所有角色里我最喜歡的。雖然由于此作畫風原因她不可能顯得有多漂亮多養眼,但她的氣質和眼神特別好,在我眼里還有種微妙的性感,她在場時總令我側目。她在柔道方面展現出的那種認真、好強、執著,以及平日顯得羞答答的感覺,都令我相當喜歡。再加上她在柔道方面極其強大的實力,顯得特別閃耀,這令她整個角色在我眼里特別有魅力!這個角色設定太棒了!
冰浦的性格挺萌的,容易害羞和臉紅,挺內向和怯懦,人多的時候甚至買個東西都不太敢上前,和不太熟的人說話都有點結巴。這跟比賽時大顯身手的她的感覺截然不同。她一穿上柔道服就變得十分有勇氣,和平日判若兩人,這個反差感真是很有趣。此作里還出現了冰浦很能吃的可愛的設定,和她冰清玉潔的形象也很有反差。其實倒未必是她本身很能吃,而是柔道對體力消耗太大。
我很喜歡看平時純潔樣子的冰浦在獲勝后展露出的微妙的邪性般的笑容,甚至有種嘲諷感,真是與平時判若兩人。我希望她最終變成此作世界里的女子高中柔道界的大魔王,對各種強敵最終都能露出這個表情,由此展現出對手遙不可及的壓倒性的強大。
最后冰浦和最終BOSS隊伍立川里的bug級強人洋人艾瑪的一戰真是驚心動魄。果不其然,最后輸了。就算贏了也沒意義,冰浦再厲害也不可能再把剩下三個人都贏下來。輸給艾瑪也是沒辦法的事,盡管冰浦的柔道技術巔峰造極,但畢竟對手又有技術又人高馬大,體重大、力量強,手長腿長,冰浦實在無計可施。此作里柔道比賽不分公斤級,這令冰浦面對高出幾檔公斤級的對手吃虧太大。對手要是純外行也罷,卻碰上了個技術也是top級的,目前的冰浦實在拿她沒辦法。不過此作在未來,估計冰浦終究還是要和她交手,不知道屆時冰浦會用什么秘密武器。
園田未知不怎么漂亮,外形看起來普通得不能再普通。其性格非常隨性、天然,極其陽光開朗,總是顯得活力滿滿,是個挺有趣的孩子。雖然她在柔道方面目前還沒什么成就,目前實力只是中等稍微偏上,但能感覺到她骨子里有柔道的天賦,而且柔道獲勝時喊出的“一勝!”是柔道令她難以抗拒的巨大誘惑。她對柔道的極度熱忱是這個角色最大的魅力,挺討人喜歡的。
我一開始懷疑此作會在一定程度上是百合番,發現主角之間并沒有什么百合元素。不過此作一開始著實無法不令人腦補冰浦對園田有特別的好感。冰浦在初三和園田初次交手后開始在意她了,當時比賽完就想和她搭話但是退縮了。然后就為了和她能一起練柔道,而去考了她要上的高中,這實在顯得對她太有愛了。
早苗是個文靜的文學少女的感覺,雖然也認真地練柔道,但水平實在不怎么樣。本以為她去參加比賽也就是走個過場,沒想到第10話,她和大胖妹的比賽意外地膠著,消耗了胖妹一半的體力,令我對她刮目相看。早苗在隊伍里總的來說是拖油瓶,從文靜的外貌和平庸的身材來看就不像是練柔道的料,但她對柔道是發自真心的重視,不懈地付出汗水,能認清自己的不足,這些也挺了不起的。希望這些努力不會白費。
由于柔道部人數不足,曾經的柔道部的高三社員姬野?一度離開了柔道部,但后來看到高一的充滿活力和斗志的新人們,被她們所感染,終于在經歷很長空窗期后重新加入了柔道部。姬野?的顏值真不錯,僅次于冰浦,是個有成熟氣質的美女。如果此作是主流的畫風,學姐應該顯得是個靚麗的大美人。
南云安奈是個有點神秘和神奇的妹子。她有著卓越的運動天賦,在劍道方面是全國top水準,居然她為了和園田她們一起練柔道,放棄了劍道,感覺真有些可惜。她在柔道方面大概率不會像她在劍道方面取得那么大成就。很期待下一季里南云能有出彩的表現。在此作的大賽中老師沒讓她上場真是挺可惜的,就算她剛開始練柔道一兩個月可能派不上用場,但畢竟多一次實戰鍛煉對于提升她的比賽經驗還是很有好處的。
青葉西的柔道部顧問紫乃老師真不錯。在第2話末尾一閃而過時我還以為她是男的,好在是女的。第3話她把粗暴且粗壯的男老師一下子摔到地上,解救了園田和南云,真是帥得不行。紫乃的外表形象特別帥,性格顯得中性偏男,氣質很有魅力。紫乃對待學生們還非常認真和上心,盡自己所能支持她們,同時她自己的柔道實力也格外強大。大家能遇到這么好的顧問真是很幸運、很難得。
我本以為此作沒有百合,后來發現此作的百合的點是在紫乃身上,令我既意外又小愉悅。之前除姬野外其他柔道部社員都退部后,只有紫乃和她在一起,和她對練,還經常開車帶著她,感覺挺有CP感的。第12話,居然紫乃還拍姬野的屁股,一點見外的感覺都沒有,感覺這倆人的關系真是非同尋常呢!我希望此作在未來能有更直球的描寫,哪怕紫乃對姬野沒有師生以外的關系,只是描寫出姬野其實對紫乃在心里有特殊的好感也好。
第11話居然出現了紫乃學生時代的迷妹犬威凜架,當年她還給紫乃送過巧克力,現在她成了高中柔道界的名教練。二人再次相見,犬威還是那么愛慕著紫乃,看得我挺開心。紫乃這么帥的女人,要是沒有迷妹才顯得不正常。這番的主要角色里沒有什么直球的百合,但從這倆人的關系可見作者絕對是有一定百合偏好的。我真是挺希望看到紫乃最終和某個同性交往的,我也完全無法想象男性和她交往的景象。
總的來說主角這一方的隊伍整體實力目前來說還是比較弱。但提升潛力還是挺大的。園田未知對柔道的熱忱,早晚會轉化成為她的實力。瀧川早苗現在顯得派不上什么用場,但如果能把短板補上,并且專攻寢技,在未來應該能有所作為。冰浦永遠的技術已經無可挑剔,隨著年齡增長、力量和體力進一步加強,戰斗力無疑還會進一步提升。姬野?空窗期太長,等恢復熟練度,估計也能當個小號冰浦來用。南云安奈還處于雪藏狀態,說不定會以后會很耀眼。可以預期在未來,主角一方團體戰就算仍打不贏此作最強的立川,但至少也能打到對方第4人。
我是真心希望此作能出第二季,我對此作很有好感,也很希望青葉西在以后的全國規模的大賽上能有好成績。
傲嬌反派千金莉潔洛特與實況主遠藤同學及解說員小林同學
總評:93.55
高二男生遠藤和高二女生小林都是高中廣播社成員。有一天小林玩一款女性向游戲《與你墜入魔法的珍愛》時拉著遠藤當解說,居然他們的解說直接傳到了游戲世界里男主齊格瓦托的頭腦里,被他當做神諭。而能直接和游戲里的角色交互、改變游戲情節的發展,也令遠藤和小林吃驚不已。此作主要要講述對戀愛比較木訥、不怎么開竅的王子齊格瓦托在這倆人解說的引導下和他在同一個學園上學的未婚妻莉澤洛特真心相愛,一點點打開她的心扉,最后消滅了試圖控制她的古老魔女,拯救她脫離游戲劇情中她原本的悲慘宿命,最終迎來比happy end更幸福美滿的end。
此作的劇情真是很會編,很有創意,也很有看頭和樂趣,視角非常新穎,之前沒有過創意雷同的作品。此作許多角色設定不錯,人物形象豐滿,齊格瓦托X莉澤洛特和遠藤X小林這兩對都非常有愛,很討人喜歡。此作的BGM也很不錯,非常豐富,很能襯托氣氛、增加場面表現力。此作的畫面倒不怎么精良,staff名單里動畫部分全是國人的名字。不過這和此作的優點相比倒也不是什么值得一提的。
作者還真是有想象力,居然游戲世界的神明久遠最后跑到現實世界來了,附身在男青年上,然后他又想重新進入游戲世界里占有夏娃轉生的菲妮,劇情設定真是夠奇葩、夠大膽的,最后的部分信息量相當大。久遠這個神渣來搗蛋倒也好,他最后促成了遠藤和小林之間告白。要是他不出現,還不知道遠藤要憋到什么時候才會和小林告白,說不定到畢業前還沒告白,最后就錯過了人生中最相配的人。
此作的結局做得真是相當優秀,美好得不得了,看得我很滿足。小林和莉澤洛特哭著分別時真是挺令人傷心的(PS:看到小林那么喜歡莉澤洛特,我還感覺她倆要是有百合關系也一定很美好)。我心想要是小林和遠藤也能穿越到游戲世界就好了,結果還真被我料到了,果然來了這么一出,最終他倆出現在了齊格瓦托和莉澤洛特的婚禮上。我想,要是來個反穿越,齊格瓦托和莉澤洛特也能有一天出現在遠藤和小林的婚禮上就更好了。
莉澤洛特這個傲嬌的反派大小姐設定得真是很不錯,氣質高貴,相貌絕佳。她外在顯得好強、倔強、盛氣凌人,但內在有非常可愛柔美的一面,真是越往后越令我喜歡這個角色。齊格瓦托起初并沒受到她的吸引,直到聽了遠藤和小林的解說才開了竅,總算認識到她的美好了。被齊格瓦托視為神明的遠藤和小林真是嚴格意義的“神助攻”。
莉澤洛特真是教科書般的滿分傲嬌,這是她最大的性格特點。她傲嬌和扮黑臉的時候每次都被解說的遠藤和小林戳破,搞明白實情的齊格瓦托表達出對她的善意夸贊時,她總會滿臉羞紅,就像紅蘋果一樣,這時候總顯得格外可愛!莉澤洛特對齊格瓦托在表面上老是一副冷淡和說教模樣,但是是真心喜歡著他。要是沒有遠藤和小林的牽線搭橋,相互喜歡彼此的二人真的就要因為誤解、誤會而錯過了。齊格瓦托和莉澤洛特倆人間的情感經歷一波三折,最終收斂到一起,到后期表現得甜膩膩,真是令我看得挺開心的。
莉澤洛特可謂是有著被動的傲嬌技能,甚至她明明都不想傲嬌,但回過神來發現自己已經傲嬌過了,她還曾自言自語說“我怎么又在說這種話,總是辜負殿下的一片好心”。她也知道自己的性格有多執拗,還說“我爭強好勝,絲毫沒有可愛之處”。可惜她總意識不到自己的內心其實是多么的可愛、多么地吸引齊格瓦托。
第6話情節真好,把我都看哭了。這一話把莉澤洛特從小對齊格瓦托的喜歡、對他的真摯感情做了充分的描寫,能令人體會到她對齊格瓦托的愛意是多么深厚,但又因為刻意與他保持距離,而導致自己多么壓抑和難過。這倆人不能相愛、不能最終在一起的話真是天理難容。小林和遠藤給她和齊格瓦托之間牽線搭橋真是立了大功。他倆做解說也真是很高明,刻意不讓齊格瓦托過早知道莉澤洛特要被魔女附身的真相而因為責任感去愛她,而是引導他逐漸認識到莉澤洛特的苦心和真心并發自真心地愛上她,從而令二人間的感情達到高潮。這解說真是GJ,絕了!
莉澤洛特對菲妮挺關照,都明顯超過了女性朋友的關系,我都覺得有一點百合味道了,第3話居然還真來了個官方吐槽“看來同學們以為這是百合情節啊”,這令我歡喜。第5話她倆認了姐妹,又一次場面顯得特別橘里橘氣,還令小林稍微有點疑似進入了游戲的百合路線。可惜這倆人走的都是BG路線,此作并沒任何真正的百合。
菲妮也是個此作里重要的角色,是在標準版的游戲世界里被莉澤洛特欺負的妹子。我對她的外貌和性格倒是沒什么興趣,不過菲妮倒真是很有特點,戰斗力強得離譜,她老媽也是個狠角色,不愧是親生的。沒想到沒多少少女魅力的菲妮最后會被巴爾杜爾所赤誠地追求,但我感覺這倆人倒算不上多般配。
小林這妹子的設定真不錯。其實她并不怎么漂亮,但性格很不錯,非常簡單和純真,并且有獨屬于她的可愛和魅力。她對游戲里的戀愛很懂,但是對自己身邊的人卻非常大條,殊不知遠藤一直在暗戀她,對他也完全不知道要保持異性間的距離。小林玩游戲時容易激動,甚至會興奮地甩起自己的馬尾,這形象令我印象十分深刻。小林是香菜配音真是太好了,感覺有一陣子沒怎么見到香菜主役了。配小林這種角色特別適合香菜這樣表現能力極強而且還能恰到好處地放飛自我的CV,香菜把她的開心和激情感覺表現得極其生動,尤其是把她看到齊格瓦托和莉澤洛特間的關系有關鍵性進展時表現的那股亢奮的勁兒表現得絕了,給這個角色加分太多了!香菜配小林真是要用絕配來形容!
最后一話,居然久遠讓小林陷入黑化的手段是激發她心中對姐姐的嫉妒,沒想到小林這么容易就被刺激到了,有點不像小林的性格。我之前也沒意識到小林對她姐姐有嫉妒心,感覺這一點的鋪墊稍顯不夠(小林她姐真漂亮,和小林的氣質截然不同,不怎么像姐妹,小林她姐確實也有值得小林嫉妒的地方)。曾經小林拯救了莉澤洛特的內心,而現在莉澤洛特反過來去拯救小林的內心,這個相互呼應的劇情構思相當可圈可點,充分體現了二人對對方來說是多么重要、羈絆多么深厚。
話說,剛開始看此作時,我感覺小林玩的這游戲的展開真是太俗套老套了:王子在學園里是被眾人關注的對象,后來喜歡上了一個不起眼的妹子,這令其原本的性格惡劣的婚約者貴族大小姐非常不爽,于是給那妹子下絆子,最終反派的大小姐自食惡果bad end。當時我覺得這種展開簡直太有點模板化,我最早接觸的這種展開的作品是《轉生成為了只有乙女游戲破滅Flag的邪惡大小姐》。不過雖然一開始展開相似,但后續發展倒真是截然不同。
間諜教室
總評:93.5
此作講述一個世界最強的20歲男間諜克勞斯培養8個妹子成為超強間諜團隊的故事,在故事中他們經歷了各種事件,期間將每個妹子的個性和特點都做了充分展現。
此作的整體素質很不錯,是一部優秀的作品,值得推薦。此作的劇情構思挺好的,緊湊,有看頭,不空洞。有的劇情輕松幽默,有的又扣人心弦、跌宕起伏,還有很多令人意想不到的超展開。此作的男主以及許多妹子的人設都挺出色。
男主克勞斯真可謂是奇人,自稱是世界最強間諜真是沒有任何水分。他的實力深不可測,就像開掛一樣,都不需要主動去做什么,僅靠被動技能就能完勝任何人、令他人甘拜下風。他總是顯得很優雅、從容,情緒冰冷波瀾不驚,表情幾乎都沒變過,任何時刻都毫無破綻,一切盡在掌控,經常覺得他事先拿了劇本,或者有預知未來的能力似的。
雖然克勞斯令人遙不可及,也仿佛并不會對誰付出感情,但他并不冷血,而是本性善良,對妹子們很關照,還挺有愛心。第6話他在小姑娘的裙子上繡了只貓,令我被萌到了。
克勞斯的人設特別英俊帥氣,且透露著高雅、神秘的氣息,長發又令他增添了藝術家的元素。再加上其性格極度理性穩重,以及有bug級的實力,令他真可謂幾乎完美的存在。這個角色的設計真是頗為優秀。
克勞斯訓練妹子們的手段挺高明。他和8個妹子們平日都住在一個大宅子里,他讓妹子們可以隨時憑借各種手段試圖制服自己,可她們即便合力無數次也尚未成功一次,而且感覺距離成功遙不可及。在一次次的和這位世界最強間諜不斷過招的過程中,她們的能力得到了極大的提升。
8個妹子各具特色。其中莉莉中規中矩。一開始她看起來比較弱雞,在間諜培養學校是吊車尾,沒想到她會主動找克勞斯杠上,自我能動性大大超乎我最初的想象。起初我本來覺得她啥也不會,沒想到在使毒方面能力超一流,自己還有超乎尋常的毒抗性。莉莉的size真是夸張,說不定比緹婭還要大。在泡澡的時候,以及第5話她試圖色誘克勞斯的時候穿的那身衣服,顯得真是大極了!莉莉被團隊里所有妹子認為是最傻的,她那種有時顯得略帶傻氣的樣子挺有意思。
此作里我最喜歡的是緹婭,真是美絕了!外形實在太符合我的審美了,深藍色長發齊劉海+白皙的皮膚+深色瞳孔+高貴美艷的氣質+絕倫的身材,真是無敵了,每當有她出場時我的目光都被她吸走了。她偶爾穿上凸顯身材的服裝時更是令我內心哦呼起來。緹婭在間諜團隊里扮演的是色誘他人套取情報的角色,很難想象除了性冷淡的異類男主克勞斯外有哪個男性能過她的美人關。不知道她在學校學色誘的時候都學過什么,是否有過什么不便描述的技巧?也不知道她是否真的有過某方面經驗。
艾露娜的特征挺新鮮的,是帶來不幸和預知不幸的能力,利用這個特點可以殺人于無形。她的相貌真不錯,就像洋娃娃一樣特別精致可愛,可惜她由于自己的能力,一直以來都被周圍的人懼怕和議論,這令她內心孤獨、悲傷,始終表現得很消極和憂郁,并且對與人交往表現出恐懼。最后她被團隊的大家友好、親切地接納,找到了屬于自己的歸宿,重新綻放了笑容,對她來說真是難得。
一開始我一直都沒注意格蕾特,感覺她的人設不怎么漂亮、沒啥個性。到了后幾話有了對她的專門描寫后,我感覺這妹子真是挺不錯的,能力出眾,真心實意愛著克勞斯并主動追求他,并極力希望為他減輕負擔,克勞斯也唯有在她面前能放下一切戒備。克勞斯真是需要有這樣一個人作為助手和分擔壓力。此作關于格雷特對克勞斯的感情描寫得挺不錯,令人感覺她真是個好女人,而且有著纖細敏銳的內心世界。不過果然克勞斯對她還是產生不了戀情(要不然無懈可擊的他就相當于有了破綻,人設就崩塌了),只想將她作為家人去好好愛護。雖然克勞斯極其優秀,但此作里其他妹子倒是沒有愛上克勞斯的,只是尊重和敬仰他,而唯有格蕾特努力爭取踏入他的世界,格蕾特也因此沒有情敵,在克勞斯眼里無疑格蕾特是最特殊的女性。感覺克勞斯多多少少有種情感缺失的感覺,不知道以后在格蕾特的關懷下能不能將克勞斯的情感補完,然后最終克勞斯也對格蕾特產生出對異性而非家人的愛。格蕾特的變裝太神了,變裝成克勞斯沒有絲毫破綻,居然連聲音和身形都能完美重現,不可思議,不過克勞斯的開掛般的能力她是完全復制不來的。
白色短發的吉薇婭的人設也挺好,性格有點男孩子般的爽朗特征,穿偏中性的黑色衣服時顯得是個小帥。其身手敏捷,戰斗力很不錯,很能派上用場。
最后一話的最后,居然緹婭似乎成了叛徒,令我很好奇之后的劇情和真相,希望此作能出第二季。但愿我大愛的緹婭不要在第二季人設崩塌,她目前的設定在我心目里很完美。
擁有超常技能的異世界流浪美食家
總評:93.3
這是一部趣味盎然的異世界美食番,構思很有創意。此作的男主穆寇達原本生活在現代世界,被異世界國王當勇者召喚到了異世界,結果到了那里的他被鑒定毫無戰斗技能,但有個獨特的技能是用異世界的貨幣從原先世界網購,這使得他在異世界也能享受到現代世界的便利,并且能充分發揮他在現代世界擁有的各種生活技能,特別是其非凡的烹飪能力。此作主要講述他靠美食誘惑使得外貌看著像巨狼的超強神獸芬里爾成了他的伙伴,然后共同在異世界倒處闖蕩。每一話男主都在烹飪方面大顯身手,令芬里爾吃得滿臉幸福陶醉,每次的烹飪還都不重樣,男主真是超級能耐。芬里爾的超強的戰斗力也不斷刷新其他人的三觀,總是捕到各式各樣的罕見的怪物和猛獸,令收購這些東西的公會的人一次又一次汗顏。
此作做得很不錯,每一話觀感都輕松有趣。此作元素相當豐富,除了美食外,還有冒險、戰斗、養成、經營等,對角色間的關系描寫也挺不錯,充分展現了穆寇達和芬里爾彼此的相互信賴、關照和依靠。此作的美食挺硬核,烹飪過程制作得挺講究,而且食材還都是在異世界就地取材,很有新鮮感,諸如烹飪豬頭人頭領什么的。此作里男主什么都吃,芬里爾干掉什么就吃什么,但唯獨沒見他吃哥布林,令我好奇哥布林的肉是什么味道。
穆寇達這角色不錯,性格開朗和善,烹飪技能超絕,換成其他人很難和高傲的芬里爾相處得那么融洽。他還挺有腦子,還知道拿網購的現代世界的廉價商品諸如鹽、胡椒、洗發液等在異世界售賣,這肯定不少賺。芬里爾每次捕到珍奇異獸就能讓男主大賣一筆,不知道此作里男主都得有多少金幣了,我感覺換成rmb的話至少也賺了好幾百萬了。男主雖然完全不會為了私利而利用芬里爾,但客觀地說芬里爾無疑是男主的搖錢樹,令他能白躺著賺錢。
芬里爾的設定非常出色,很有個性,形象相當生動。它極其強大,形象威風凜凜,性格穩重高傲,有很強的自尊心,但卻頗為貪吃,面對美食完全經不住誘惑,一看到男主烹飪就立馬精神起來并露出充滿期待的神情,吃的時候完全不顧顏面,真是和平時的氣質很有反差感。男主對它的脾氣和性格也拿捏得挺準確的,能令它始終老老實實心甘情愿地跟自己在一起。有的時候它還顯得挺萌挺可愛,就像男主養的大犬似的。男主對它也很關懷,最后一話還給它渾身洗毛。這一人一犬的關系在此作里刻畫得著實很有愛。日野聰給芬里爾配音配得很不錯,氣質非常對味兒。
男主后來收的使魔史伊的設定也不錯,性格軟萌萌、慢吞吞、肉肉的,很有史萊姆的感覺。木野日菜給史伊配音能令人耳前一亮,表現得相當好。芬里爾和男主之間是老伙計的感覺,而男主對史伊則是當成寶寶來看,對史伊關愛有加。男主、芬里爾、史伊在一起有點一家三口的感覺。史伊雖然只是個史萊姆,但卻是個超凡的個體,不知道是什么來頭,居然意外地厲害,等級和戰斗力隨著和男主與芬里爾的冒險一路蹭蹭上漲,到最后愈發強大,似乎戰斗力都有芬里爾的1/10了,并誕生了五花八門的能力和戰斗方式,男主真是撿到寶了。但無論史伊變得如何強大,始終都還是男主的頭腦單純簡單的乖寶寶,其性格挺討人喜歡。PS:突然想起來此作里沒有吃史萊姆的,不知道如果真出現吃史萊姆的劇情時史伊會如何表現。
此作里給男主庇護的女神也挺有意思,看到男主和芬里爾大吃特吃,饞得她讓男主供奉異世界食物,然后也吃得一臉幸福。向男主討食的她真是一點女神的尊嚴都沒有,有那么點智障阿庫婭的感覺。我也好奇她在天界都吃什么東西,那邊在食物方面肯定特別蠻荒。
感覺此作接下來還很有得編,希望此作能出第二季。
東京復仇者 圣夜決戰篇
總評:93.15
這是《東京復仇者》第二季。我曾經追過此作第一季,劇情很有懸念,但男主實在是太窩囊了,很多次令我看得挺來氣。當時最后一話來了個巨大的伏筆,這回可算是迎來了第二季。這一季主要是講東卍與另一個強勢不良少年團伙黑龍爭斗的事,男主武道對于滅掉黑龍立了功,但回到未來后,還是沒能改寫女友死亡的命運,甚至東卍的重要伙計們全被滅,比原來的結局甚至還慘,這給下一季留足了懸念。不知道是不是下一季就能徹底真相大白了,引出導致男主始終沒能避免的bad end的最關鍵因素。
最后一話真夠夸張的,甚至顯得滑稽,居然一群不良少年大鬧火車站,這令我再次認定了此作是不存在保安和警察的世界。此作的場景真是一個架空的超離譜的世界。
這一季對于男主意識不在線(行為不受控)的期間發生的事還是沒有任何交代。男主無法控制這段期間自己無意識的行為的話,無論自己意識在線期間付出多少努力都是白搭,這其實是此作最bug或者說最變態的地方之一。男主還不如就不回到原來的世界,就一直在12年前的副本世界里活下去,始終主動避免事態惡化。若這樣活12年之后,如果橘日向沒死,那么回到原先的世界(此時等同于回到過去)自然也就沒任何問題了,或者根本沒必要回去了。令我好奇的是,如果男主在未來的世界里死了,那么從過去穿越回未來的時候會不會瞬間掛掉,然后就再也沒有然后了。
這一季里男主依然總是一臉吃驚、驚慌的廢柴樣,經常哭哭啼啼,思考方式和行為完全不具有成年人的特點。不過他在教堂和大壽打的時候的表現倒還可圈可點,豁出性命單挑大壽,死活都不屈服。男主的戰斗力始終沒有提升,感覺到現在也就是小混混戰斗力的平均值,不過長處是血特別厚,怎么打也不掛。
這一季的反派BOSS是黑龍的老大大壽。其體格是此作目前出現的角色里最強壯的,性格真是惡劣至極,居然還打妹妹柚葉,欺負女人真不是東西。第6話居然還重重的一拳打到柚葉的正臉,我都擔心她毀容了。大壽還不是個簡單的莽夫,蠻有管理能力和頭腦。他居然還信基督教,但我感覺他純粹是在以某種方式給自己的暴行開脫。
mikey的戰斗力真是∞,第33話居然一腳秒殺大壽,太離譜了。我還以為即便是mikey,面對目前出現過的最強對手大壽也得陷入苦戰。到現我在完全不知道mikey的極限在哪里。男主穿越回未來后,在廢墟上的mikey和之前的形象判若兩人,真是令我很意外,那個黑頭發的mikey還有那么一點別樣的性感的感覺。
這一季另一個關鍵新角色是東卍副隊長的柴八戒,這名字令我不由地想起豬八戒。他從小開始就屈服于哥哥的淫威之下,明明戰斗力挺強的,令我感覺很窩囊。本來八戒的人性不錯,卻出生在不幸的環境里,因為大壽的暴力一度變得扭曲了,挺可悲。
大壽的妹妹、八戒的姐姐柚葉這個角色令我很喜歡,顏值和身材相當不錯,特別是腿。她的外形很有經典的青春JK特征,ED里她穿JK服裝和泡泡襪露著美腿跑步的樣子真是挺養眼的。我起初看到她和八戒在保齡球館,形象顯得挺配,還以為她是八戒的女友。居然柚葉也挺能打,第26話飛起一腳把黑龍的混混的腦袋踹到真帥(瞬間還看到了白色的胖次)!柚葉雖然表面上顯得是混社會的不良,后來我發現她的內在也挺不錯,內心還挺端正的,對日向還挺好的。最后,居然柚葉喜歡上了男主,真是令我大吃一驚,感覺這嚴重有損柚葉帥氣的形象。雖然男主面對大壽寧死不屈倒是挺有點型,但她喜歡上那么個戰五渣又挫的男生,還是令我感覺眼光差了些。
我本以為日向的內在是個完全溫柔的妹子,沒想到第5話她騎在男主身上把甩了她的男主胖揍一頓,完全不留余力,顯得簡直不是同一個人,真是驚到我了。挺奇怪的是,第36話男主穿越回未來后,日向又死了。mikey把東卍的人都殺了就完了,干嘛還殺了日向,不可思議,也許是還有隱情。想來如果日向的弟弟在未來也死了,就成了死局了,男主再也沒機會改變過去了,那就有趣了。
稀咲還是那么壞,是東卍變質的最大毒瘤,我很希望這一季能看到這廝被好好教訓,看到他被mikey驅逐出東卍的時候他的那副吃了翔的模樣令我挺開心。感覺明顯他是在針對日向,否則怎么可能日向死N遍。我很希望知道背后的緣由。看到稀咲被逐出東卍時,我就心想他絕對不會就此收手,果不其然。必須得想辦法把這個陰險狡詐一肚子壞水的bad people逼上絕路、弄得他永世不得翻身才行。
千冬真是挺不錯的,他戰斗力不錯,腦子也比較靈,還是個小帥,是個令我覺得很順眼的角色。千冬非常靠得住,很真誠,也很有勇氣,和男主完全一條心,共同出生入死。他居然真心相信男主是從26歲穿越來的,還挺有眼光和洞察力的。廢柴男主能有這么個貼心的搭檔愿意全力以赴地幫忙真難得。在下一季,能否拯救東卍和mikey,就看他倆了的合作。
飆速宅男 LIMIT BREAK
總評:93.1
《飆速宅男》是我一直追的作品,很久都沒出新作了,突然出了這新一季令我挺意外的。記得上一季總北在失去原高三的三名大將后,又加上了拖油瓶,表現得挺窩囊的,看得我比較郁悶。這一季繼續講總北、箱學、京都三支隊伍在高中聯賽中角逐的故事。此作依舊非常熱血,人物感情起伏和劇情轉折總是相當夸張。
此作的拖戲真是老毛病了,這一季也尤為明顯。一會兒回憶一會兒心理活動一會兒說話說半天,一定程度降低了劇情的緊湊性和連貫性。比如第10話,用了一整話各種心理描寫、各種回憶殺、各種二人對話,水了一話居然還沒把手島和葦木場的山岳獎沖刺這點事講完。然后第11話一開始又是回憶,我干脆用兩倍速看回憶部分了。好在是凡人手島贏了,沒白看那么多鋪墊(不過感覺不科學,手島哪是葦木場的對手,哪怕他全力全開。不過若是說手島有潛在的未被發覺的技能,是把觀眾的喝彩轉化為自己的力量,倒也說得通)。我感覺此作可以壓縮到一半(13話)長度,更突出重點,這樣會更好。最后的決戰做得很精彩,十分扣人心弦,令看到最后的我感覺挺痛快和暢快。
這次的高中聯賽里總北總是顯得很被動,好像總是對手技高一籌而且智高一籌似的。好在是最終小野田險勝,令我很happy,一掃之前的憋屈感。小野田獲勝時我感覺小野田真是神奇的存在,真心超級了不起,不愧是此作的主角。在最后和真波決勝之前,小野田在這次聯賽中感覺沒太多存在感,都不像是男主。而最終奪冠的小野田真是出盡了風頭。憑借他總北連續兩度奪冠,小野田山王的稱號必定更加響亮和聲名遠揚了。不知道他在高三時能不能帶領總北第三次奪冠,如果能如愿的話,小野田無疑將成為高中自行車歷史上的不可逾越的傳奇。
小野田對卷島是真愛。之前卷島去了英國之后小野田跟丟了魂兒似的,后來得知在英國的卷島收到了他的信后他又突然涌現無限力量,滿狀態復活了,這關系都挺有點基情了。卷島對小野田也真好,為了能給他加buff從英國回來了。卷島加的buff果然效果拔群,再加上小野田之前的hime hime的那首歌加的buff,令小野田最終超水平發揮。感覺小野田如果沒buff的話,未必贏得了真波,但一旦buff上頭,激發出潛力,就成了能超越真波認知的存在。我感覺真波對自己特別自信,總顯得一副游刃有余的樣子,我其實并不怎么喜歡這個角色,看到最后小野田贏了他令我很是開心。
不管此作再怎么試圖正面描寫手島,我對手島也實在喜歡不起來。他各方面太平庸,身體素質上沒啥長處,在智謀上也沒什么突出的,而他的話卻非常多,老是說個沒完。他的存在對于總北奪冠起到的價值極其有限。我對另一個庸才青八木的印象都比他好,至少青八木還有個膨脹身體的絕技,也算是個有用的獨有的skill。雖然手島非常努力,但光靠努力是完全彌補不了天賦的差距,畢竟對手也都在玩命努力。在我來看他這樣的庸才就把騎行當做愛好就好了,沒必要去參與競技自行車賽,搞得自己很辛苦。
我一直都很不喜歡葦木場,居然第9話拿一整話時間講述他和手島小學和初中時候的故事。我不喜歡葦木場一方面是因為其形象極其不符合我的審美,另一方面是他的那種娘娘腔、gay里gay氣的感覺令我相當不適。沒想到葦木場和手島曾經那么基情。在第9話的回憶中,二人鬧矛盾,后來葦木場懂得手島的良苦用心后傷心地在大雨里一個人騎車,渾身淋濕,感覺就像情侶吵架并失去對方,之后一方悲痛欲絕的情景。手島屬于老好人的類型,特別為別人著想,在這一話里把他的這種老好人的形象表現得更充分了。
鳴子最后還挺會減重,不僅扔了水壺,居然還扔了車座。燃盡的鳴子直接就躺地上了,擔心他扔的水壺和車座會給后面的人帶來危險。
第17話今泉和御堂筋拼得夠兇的,居然還一起滑著過彎、跳起來跨越障礙物。沒想到今泉長進不小,能在御堂筋面前不落下風。不過到后來今泉莫名其妙突然沒油了,挺邪門兒。本來他領先后面的人很多,至少也能得個季軍,結果后來騎得慢慢悠悠,季軍都失去了,這顯得太莫名其妙。
御堂筋依然是綜合能力的王者,簡直跟開掛一樣,一個人就能干翻一批人。給人感覺他就像妖魔鬼怪,而且身影巨大,就像巨人一樣給人極強的壓迫感,飛速騎過時就像鋼鐵列車轟隆隆地疾馳駛過一樣。這家伙真是要用駭人來形容。
雖然御堂筋覺得友情、隊友關系沒有什么用處,是可以隨意拋棄的,但看到最后鳴子拼命追趕御堂筋的時候,我倒也發覺隊友情誼還是挺有用的,關鍵是能加buff,并不是多余的累贅。
第18話的御堂筋太奇怪了,之前還騎得賊快,仿佛一切都在他的盤算之內,而過了水坑后突然就歇菜了,真邪乎,這劇情也太唐突了。似乎是御堂筋太缺愛了,特定條件下心病突然發作了吧。
真波看起來總是很輕松,總是帶著自信的微笑,顯得是個神奇的人。沒想到第22話中,真波因為之前箱學在高中聯賽輸給了總北,使他陷入黑暗,心中誕生的巨大的壓力令他過度勉強自己訓練,顯得非常辛苦。女班長對他真是真愛,老主動繞著他轉,耐心聽他傾訴。不知道真波是否哪怕有一丁點已經喜歡上了女班長。感覺女班長對她的愛意和好意如果得不到回報的話挺可惜的。
TechnoRoid: 超越意志(overmind)
總評:93
總之,我先說一句話,此作是一部很不起眼的準寶藏番。此作表面看似是泥巴,實際上耐心挖下去后會發現藏有金子和寶石。真正有品味的人才可能發覺此作的優秀之處。
此作世界中的仿生人是指外貌和人類幾乎難以分辨的人形的機器人。此作講述與眾不同的具有內心的4個仿生人和他們偶遇的小男孩芝浦繪空共同經歷的故事。在故事中他們一起經歷了各種各樣的事,使這四個仿生人的內心得到不斷的成長并產生了人類的感情,同時他們與繪空的關系也逐漸加深。期間這四個仿生人作為偶像組合KNoCC通過演藝不斷挑戰巴比倫塔的闖關活動,在這個過程中陷害機器人的反機器人組織的幕后黑手也被逐漸揭開。
此作剛一開始,畫面上出現了上松范康的名字,我就知道此作大抵是音樂題材的作品,果不其然,音樂和表演在此作里占了挺大的戲份。
此作給我的第一印象不好、不上檔次,畫面顯得比較樸素,劇情也沒能在短時間內令我感覺到有什么吸引力,看第一話時我的整體觀感還是負面明顯大于正面,沒打算追。只是因為第一話里大叔被機器人推下熔爐令我好奇,再加上氖恩的外貌特別美、繪空和四個仿生人之間的情感描寫得還不錯,使我姑且又看了第一話,然后發現此作或許還真的有追下去的價值。當時我回想起《Extreme hearts》那部動畫,也是一開始感覺作品其貌不揚,沒追的興趣,但越到后來越喜歡,極其慶幸沒棄掉,于是乎我盼望此作也能像《Extreme hearts》那樣是一部寶藏番,就湊合看了下去。結果,還真是發現越到后面此作的劇情越吸引我,角色也越令我喜歡,當初沒匆忙棄掉真是太好了。我感覺能追下來此作的人應該很少,肯定此作非常不人氣。看完此作后我衷心感謝有字幕組愿意把這么冷門的作品做下來。
這部作品是一部內涵積極、有愛的作品。此作展現了人類與仿生人的相互理解、相互溝通、消除隔閡的過程,同時也描寫了仿生人逐漸理解人類的各種感情、心靈成長的過程。原本理應沒有自我人格的仿生人開始逐漸有了自己的心事和情感,而一個人非常孤獨的繪空也終于有了陪伴他的家人。作品里對人物間的羈絆描寫得很好,看完整部作品能令我深刻體會到KNoCC的四個仿生人和繪空對彼此是多么的重要、對彼此多么重視和關愛。此作有的劇情還挺感人的,比如第5話鈷博特陪伴大齡腎衰竭的貓走完生命最后旅程的故事看得我有點唏噓不已。總的來說,此作的很多局部并不完美,制作方面還有很多明顯可以改進的地方,但作品的創作思想、整體故事的構思,都相當不錯。如果經過精雕細琢,此作上93.7分都不成問題。
此作前、中期整體比較慢熱,最最精彩的就是最后幾話,能把此作追到這里的話你就贏了!
第10話真是超展開、神回!令我相當吃驚,甚至震撼。這劇情絕了,伏筆全都抖出來了。原來繪空的父親芝浦博士當年在搞機器人開發,他深愛上了自己的學生愛麗莎貝,結果愛麗莎貝卻和別人結婚了,生下孩子繪空后不久因車禍去世了。痛苦中的芝浦決心要將繪空養育成為最優秀的人,于是找了在四個方面分別具有杰出長處的年輕人陪伴繪空成長,結果這四人有一次因為事故半死不活。芝浦博士于是強行闖入醫院把受重傷的四個人的記憶下載下來,從而能將他們復制為仿生人繼續陪伴繪空,但這也令四人因為頭腦超負荷而死亡。此作的四個仿生人正是以這四個人類為原形創造的,并且由于植入了芝浦博士開發的kokoro程序而擁有了心靈。但還沒來得及上傳四個真人的記憶,在這之前瘋狂搞AI、拿自己做測試體并長期嗑藥芝浦博士就已經掛了,導致四個仿生人需要從0開始構建自己的內心。
芝浦博士也真是夠慘的,一個搞技術的天才大齡光棍,終于有人(愛麗莎貝)溫暖他、關心他,令他心動不已,結果她卻被其他男人占有,最后還死亡了,芝浦博士真夠可憐的。他試圖將愛麗莎貝的人格復制到自己創造的有一定靈魂的AI愛麗莎身上,結果也沒能成功。要是愛麗莎貝沒死,而愛麗莎最后能變得有和愛麗莎貝相同的頭腦,從而陪伴芝浦博士一生,給他以心靈的慰藉,那就好了。芝浦博士也真是夠狠的,為了下載那四個人的記憶,居然做出了等同于殺人行為,真令我沒想到。總之,命運對芝浦博士真是殘忍。
第11話16:30開始,繪空和KNoCC四人在法庭上配著杰出的BGM的情感流露的那一段表現力絕贊,把我感動了,令我深切感受到溫暖。這是關于繪空和四個仿生人關系描寫的高潮,充分表現了他們的羈絆是多么深厚,也顯得無情地將四個仿生人格式化真是太殘忍和無法容忍。看到這里,我深感,哪怕是仿生人,只要你為他創造了自主意識,或者他誕生了自我意識,就應當像人一樣尊重他們,要么從一開始就斬斷其任何誕生自我意識的可能性。
最后一話KNoCC的舞臺演出非常好,給此作大為加分,是OP的重新編曲和填詞。盡管這一段在3D模型、畫面效果方面還有改進的余地,但這個演出真是充分傳遞出了愛和溫度,表達出的情感非常飽滿,看得我眼中盈溢淚水,真配得上是令KNoCC戰勝強敵獲勝的超精彩演出,這段表演也足以讓那些對仿生人持偏見的觀眾體會到他們也確實具有和人類一樣溫熱的內心。
我感覺KNoCC組合中的鈷博特、kei、Chrom的人設中規中矩,四人中最大亮點就是那個紅色長發的仿生人氖恩,其人設真是絕了,是神來之筆,神來之筆,神來之筆(很重要所以說三遍)!!!起初從形象看我本以為TA是妹子,居然TA剛一說話是秀吉那種boku感的少年聲音,令我詫異。不過這沒有令我有絲毫遺憾,因為本身我就很喜歡偽娘(限二次元)。氖恩的外貌設定真是超級驚艷,皮膚白皙,相貌格外精致秀氣,身材纖細干練,眼神顯得冰冷銳利,同時還有點輕微的神秘感。他露出的潔白的鎖骨和腰肢真是秀美誘人。每次畫面里有他出場時我就盯著他看,看完此作后發現我總共已給他截了100張圖。第6話專門講氖恩在大學的墻壁上作畫的事,這一話看得我挺滿足,從頭到尾都是氖恩。沒想到不僅氖恩本身形象絕美,還很有藝術細胞。kayto給氖恩配音配得也很不錯,是恰到好處的中性,而且稍微偏冷,跟氖恩的人設和氣質絕搭。
作為KNoCC的競爭對手的Kite這角色也不錯。他一開始對KNoCC的這四個仿生人非常厭惡,但其實內心蠻善良的,追求公平競爭,后來還幫助了KNoCC,并且竭盡所能想辦法治好自己重病的妹妹(不知道最終妹妹活下來沒有)。Kite的外形設定挺好而且挺有個性的,真的是很帥氣,而且眼睫毛上一抹藍色更是增添了亮點,他的形象在我印象里和其它作品里的角色沒有近似撞車的。
此作的OP很好。我第一次聽時還沒什么觸動,但后來越聽越覺得這歌在高潮處的激情感很不錯(尤其是畫面中出現此作幾個主角的臉通過碎片效果過渡切換的那一段)。看完整部作品后再聽,更感覺整部歌曲都相當棒。這OP真是耐聽、耐看,令我越來越喜歡。
極主夫道 第二季
總評:92.9
之前追過第一季,蠻有趣的,講一個前黑社會男子龍后來成為家庭主夫后的日常。這個第二季只有5話,風格和第一季沒什么差別,還是每一話由N個小故事組成,很有樂子。對這一季倒沒什么可特別評價的,想說的在第一季時我都評論過了。
冰屬性男子與無表情女子
總評:92.9
此作中男主冰室和女主冬月在路上偶然相遇,短暫的邂逅令二人有點一見鐘情。而后新入職的他倆發現彼此竟然是同事,相當有緣分。此作主要就是講述他倆之間的感情發展,以及和職場同事之間的故事。此作整體來說是一部風格清新的慢節奏的作品,觀感舒服,有的劇情挺有樂趣,有的劇情挺甜、挺暖的。此作人物畫風都不錯,挺符合我的審美。總之是一部不錯的適合休閑觀賞的戀愛番。
冰室和冬月都是挺害羞、挺含蓄的人,二人都在心里喜歡對方。在此作里他倆的感情一點點慢熱挺有趣的,但進展實在太慢了,經過了12話,在此作的世界里經歷了一年,仍然還停留在關系親密的同事、彼此重視的人的關系上。雖然已經歷了不少高能的橋段,但還是沒有明確表白喜歡對方的心意,真是有點急人。
冬月這個女主的設定很不錯,是個外冷內溫的大美人。表面上她是顯得冷淡的人,感情也表現得缺乏波瀾,也沒什么表情,但她的內在卻溫柔和感性,而且很善良,對體質特殊的冰室相當體貼關照。冬月還很喜歡貓咪,這個設定進一步表現出她內心溫柔可愛的一面。冬月長得真是相當漂亮,挺有超凡脫俗的氣質的,此作里出現她的特寫時我都很認真地欣賞。特別是冬月的45度的顏值巨高,特別精致秀氣,令我經常忍不住截圖。冬月這樣的美人當個普通公司的職員真是有點浪費了。一般的人幾乎見不到冬月的笑容,而唯獨冰室能在一些關鍵時刻見得到冬月對他露出的微笑,難得一見的冬月的笑容真是很迷人。
男主冰室這角色設定也不錯,相貌挺俊秀的,為人也好。他是冰女的后裔,緊張時候會凍腳,情緒和狀態異常時會在四周瘋狂吹頭皮屑(誤),還會彈出小雪人,這些有趣的小設定很有意思。冰室這個姓真是和他的設定相當吻合。冰室從外在形象上看是個有點偏冷、性格平淡的男子,但其實里外反差很大,他的內心戲經常很豐富,尤其是對冬月格外心動,剛和她接觸不久就對她暗戀到難以自拔了,對她心心念念,甚至表現得癡情到難以壓抑喜歡的情感。
感覺冰室和冬月這一對真是蠻般配的,外在是帥哥+美女的天作之合,內在又相互體貼關照,對彼此的好感度很高,為對方所吸引。這倆人在一起肯定能過得很幸福美滿。
冰室受熱、發燒后會變成小孩子,這個設定蠻有趣的,小孩子模樣的他挺可愛的,特別稚嫩,無疑肯定會引起阿姨們的興奮。冬月和這樣的冰室在一起就像大姐姐溫柔地照顧小孩子,甚至有點像母女,也挺有愛的。最后一話居然冰室推倒了冬月,這是此作目前最高能的一幕了,可惜推倒冬月的是兒童體的冰室,要是此時冰室是大人狀態,真可能要出事了。
主角的同事狐森是個狐娘,整個角色很有趣,我對她的印象非常好,其性格又俏皮又萌,狐耳很可愛,是個很討人喜歡的角色。狐森也挺漂亮的,她的一些特寫也能令我覺得挺賞心悅目的。她和冷美人冬月的性格有鮮明的反差,其存在很能活躍氣氛。此作里論絕對顏值,冬月最高,而論可愛,狐森第一。每當冰室在旁邊刮暴風雪的時候狐森總會表現得挺興奮,甚至高興地發出小尖叫,每次看到這一幕我都覺得狐森真是超萌超可愛。狐森還喜歡挑逗別人,特別是對男二號冴島,這稍微有點長瀞挑逗學長的味兒。這一對是此作的副CP,可惜進展也很慢,但無疑最后會走到一起,本身冴島在心里已經坦誠對狐森的喜歡了,而狐森對冴島也毫無防備,向他完全敞開大門,只要冴島告白肯定會欣然接受(頂多屆時狐森在表面上嬌羞一下,來個欲拒還迎)。內山夕實給狐森配音配得很好,聲線很輕盈歡脫。其實要是冬月和狐森有百合發展的話我會高興,此作里有一幕倆人之間還橘了一下(第10話打雷把狐森嚇到,冬月安撫狐森時表現得像可靠的男友似的)。
主角的公司里的課長是個佛祖形象,這個角色設計得很有趣,其形象、性格、聲音都極其慈祥溫和,真是活佛在世。他不僅對員工特別關照,甚至還給冬月和冰室做助攻,第6話給了冰室貓咪樂園的雙人券。有這樣的課長真是很難得的事。
音無這個角色本身人物特點不算鮮明,但佐倉給她的配音卻令我印象挺深,聲音明亮有力,是佐倉很典型的個性聲線,給這角色加分了。
第8話出現的冰室的妹妹顏值挺高,居然突然抱向冬月并且說“喜歡你”。冰室突然驚了,趕緊拉開他妹,還說“這家伙,不會是真的看上冬月小姐了吧”,冰室妹妹真是瞬間成了冰室的情敵!這段劇情有意思。要是冬月撇開冰室和他妹妹真交往上就有趣了,也是我很希望看到的。
此作里人物的瞳孔總是輕微晃動,雖然是想表現出心里的波瀾,但有時我覺得做得有點違和,太夸張了。
關于我在無意間被隔壁的天使變成廢柴這件事
總評:92.8
這是一部蠻不錯的青春戀愛番。男主藤宮周是個性格陰沉、自理能力比較差的獨自生活的宅男,有一天在雨中邂逅了在班級里被稱為天使的大美女同學椎名真晝,對在雨中憂郁的她遞去了傘。沒想到椎名居然就獨自住在藤宮的公寓的隔壁。在第一話,椎名就對藤宮主動白給去照顧他,給他送飯、幫他打掃等等,還分文不取,藤宮從此過上了有超級美少女陪伴的夢幻般的幸福生活。此作主要就是講在此之后二人之間不斷增進了解、感情不斷升溫的故事。整部作品觀感不錯,很多劇情很有趣,很多劇情又很甜,而且椎名特別賞心悅目。
此作的一個亮點是男主女主之間的獨特相處方式。很多戀愛番,諸如《不要欺負我,長瀞同學》、《夫婦以上,戀人未滿。》、《小智是女孩啦!》,男主女主之間的關系都很特別,不像一般的情侶。在此作里,藤宮和椎名始終保持著一起生活但不交往的神奇關系,椎名以照顧生活能力差的藤宮為由經常在他那里膩歪,藤宮也欣然接受椎名的好意,并逐漸對她心動起來。隨著劇情的進展,他倆的關系一點點拉進,起初椎名進入和她還不熟的藤宮的房間里給他做飯從常識來說就已經很離譜了,后來還互相以名來稱呼,互相喂食,二人互送情人節禮物,相互打情罵俏...關系越來越親密,好感度越來越提升。椎名經常還在藤宮家里等著,藤宮一回家就上去招呼,別說像戀人了,就跟已婚的妻子一樣。但都到曖昧這份上了,戀人之間做過的事除了肉體接觸外都做了,二人在名義上卻始終堅稱對方只是異性朋友。說不是情人誰都不信,估計連他們自己都不信。
后期椎名的一些話語和動作還顯得是在勾引撩撥藤宮,比如“我喜歡被你觸碰”、“只要你考進前十名,我就答應你的任何要求”、“...應該只會感嘆一下你也是男生,然后成全你”。真是不僅對藤宮毫無戒備,甚至都暗示愿意上壘。椎名還把藤宮吃得死死的,還說什么“我會毫不客氣地把你寵成廢柴的”,說得好像藤宮一輩子都要注定和她在一起了。藤宮也被她撩得逐漸有了非分之想,居然第10話的時候都夢見和她有肌膚之親了!醒來后撩開被子發現自己已經糟糕了(肯定是下面已經黏糊一片了),這真是超真實。話說,藤宮的spring夢真夠爽的,在床上穿著內衣的椎名真是太大了,得感謝藤宮的腦補給觀眾們奉獻了那么大的福利。
雖然此作有不少很科幻的劇情展開,但是他倆關系緩緩升溫的過程描寫得著實不錯。特別值得一提的是第7話,藤宮撫慰因為母親的冷言冷語再次受傷的椎名,這段劇情令二人的關系又有了重要的升華和進展,椎名也把一直以來隱藏的心事第一次向他人袒露了出來。原來她從小就沒受到過父愛和母愛,始終孤獨一人,逐漸學會了在其他人面前掩飾自己的真心,始終努力扮成完美的天使模樣,竟然以為若別人看到她真正的一面就會討厭她。她真是完全不知道自己有多么可愛,她的真實的一面其實才更為迷人。藤宮走進了她的世界對她來說蠻幸運的,終于遇到了一個人真心重視自己、完全接納自己的一切,而且也是自己可以完全依賴、信賴甚至是撒嬌的對象,藤宮這樣的人真是她最需要的人。看到這一話也令我深感二人在各方面都極為般配。
此作主要以藤宮的視角來描寫,觀眾能看到藤宮早已經愛上椎名,對她心動不已,早就想交往了。但椎名對于戀愛的態度始終是個謎,明明她和藤宮做著熱戀中的情侶的事,但她始終不肯公開承認在和藤宮交往,因此令人會懷疑她是不是有什么特殊的顧忌,有什么始終沒有袒露的隱藏的想法。藤宮也是因此始終沒決心和勇氣向椎名告白,萬一只是自己一廂情愿,告白后說不定就沒法和椎名像現在這樣毫無隔閡甜蜜地在一起了。好在是最后并沒有雷。在最后一話,居然當著全校師生的面,椎名正式宣告了藤宮是自己重要的人,這戀愛宣言真是很令我吃驚,椎名夠有魄力的。之前他們的關系一直秘而不宣、遮遮掩掩,這下子竟然令整個學校的人都知道了。在此作的最后終于達到了二人感情的高潮,對彼此好感度都憋足了的二人終于彼此確認了心意,說著肉麻的話,發糖真是甜死了,這個結局很完美。
藤宮這個男主人設不錯。氣質文質彬彬,性格隨和、踏實穩重,品行優良,對椎名非常體貼,十分重視她的感受,真是挺理想化的男主,挑不出他有什么毛病,也確實配得上椎名。藤宮的相貌上是極其規范的二次元男主的形象,是個標準小帥,和很多作品的男主都太撞臉了。在此作的世界里他的顏值被設定為很平庸那種,但我感覺他看起來比他那輕浮又土氣的小伙伴赤澤樹帥得多,可赤澤有女朋友,而藤宮卻一直沒人追他,我感覺挺不科學的。藤宮之前沒女友大抵是因為他平時顯得低調、沒有自信,其實他把頭發打理打理后顯得還真是蠻帥的,在此作里還吸引了周圍人的目光,和椎名站在一起有種天作之合的感覺。藤宮沒人氣倒也好,使得椎名這樣的真正的天使有機會和他最終在一起。椎名之所以愿意主動接觸藤宮,起初關鍵因素是藤宮顯得特別老實,不會胡思亂想和自作多情,也沒任何壞心思,使得椎名和他在一起的時候能令人比較安心,沒有和其他人相處的時候的心理負擔,也不用自我偽裝。而椎名最終能喜歡上藤宮,則在于藤宮確實有內在的個人魅力,逐漸被椎名一點點所發現。
椎名的顏值是真高,頭發光潔亮麗,眼睛清澈明亮,鼻子和嘴小巧可愛,臉頰粉嫩,像洋娃娃一樣。本來她的相貌設定就特精致,而且在此作里繪制得還非常精美(雖然此作后期作畫崩壞也不少),特別是笑容很養眼,容易令我看得入迷,也令我在觀賞此作過程中為她截了上百張圖。PS:剛看此作時我總是莫名地覺得椎名的面容和《剃須。然后撿到女高中生。》里的沙優有那么一丁點像。不僅椎名的外在堪稱完美,此作還把椎名的內在描寫得非常美好,有時還顯得很萌,比如害羞時突然跑掉。看此作有時候會令我覺得:這是何等可愛的生物!椎名實在是太有魅力,難怪即便是藤宮這樣的挺性冷淡的家伙,才到了第二話就被她所吸引了。
椎名的想法挺與眾不同,待人處事方式不像這個年齡的女生,顯得微妙地成熟、冷靜,有自己對現實問題的看法。她雖然在班級里表現得像完美天使,對誰都顯得溫和、易于相處,但其實她內在也是有脾氣、有性格的人,她始終給自己的心靈披著面紗去面對別人,唯有藤宮才接觸到了真實的椎名,并走入了她的世界。在最后一話,班級里還有些低賤的男生不服氣,覺得憑什么默默無聞的藤宮成了椎名重要的人,殊不知他們這些輕浮、膚淺、沒靈魂的生物其實根本都沒被椎名看在眼里,類似于隱式溶劑模型所描述的外環境而已。
我真是羨慕藤宮和椎名,才上高中就能一個人獨占那么大的還挺高級的公寓,而且父母都不在身邊礙事,這也太理想化了,完完全全是給他倆的戀愛創造最佳的條件。單身俊男靚女彼此就住在對方隔壁,能不出事么(PS:令我想起《租借女友》里男主女主也正好是這種情況,但他倆都是大學生,公寓還比此作的破多了)。
說來,在此作的一開始,椎名對隔壁的藤宮顯得也太關心、太上心了,還一丁點防備都沒有,從常理來講真是挺不合邏輯,發展得也太快了。從藤宮的視角來講,此作的展開用《天使降臨到了我身邊》那部作品的標題來形容真是100%貼切,比起此作的標題還更合適。
男主媽真是個神奇的角色。第3話她剛一出場時我還以為是他姐,完全看不出像媽。男主媽顏值真高,也難怪男主那么俊秀。男主媽的性格真是夠開朗和俏皮的,話特別多,發現兒子金屋藏嬌后,一個勁兒熱情地給兒子和椎名做助攻,椎名這樣文靜的少女在她面前完全沒有還口之力,辯解以失敗告終,只得老實接受被她當做兒媳婦兒了,很樂。
此作的ED是椎名翻唱的名曲,相當好聽,柔和舒緩,傳遞出浪漫的感覺,結合著畫面聆聽時心里會被美好的情愫所充斥,椎名輕盈可愛的聲線和這首歌真是相當搭,充分展現出了椎名純潔可愛的內心世界,絕對是一首非常出色的ED!
不當哥哥了!
總評:92.75
此作內容挺新穎的,蠻有趣,人物鮮活,有大量令人喜聞樂見的元素,有些地方尺/恥度還相當大。整體制作蠻精良的。
此作里主角緒山真尋本來是個兩年都沒出門的天天在家里玩游戲看動畫(包括大量工口的)的死宅男,有一天由于被天才妹妹下了她制造的藥,醒來后發現自己成了個初中生年紀的loli。此作主要講被迫性轉之后的男主逐漸接受自己變成妹子的事實,辛苦地習慣作為妹子的生活,然后逐漸結交了同齡的姑娘們,和她們快樂地在一起玩鬧,最終樂不思蜀(回歸男性)了。
此作中男主真是充分體驗了各種當妹子的感受,去澡堂洗澡弄頭發都得半天。居然還體驗了姨媽,這在性轉的動畫里幾乎都沒有提及過,這性轉得可真夠徹底的。
隨著劇情的發展,男主一點點變得愈發有妹子的感覺了,居然還會自己編辮子、弄丸子頭,有女孩子的坐姿,被風吹起裙子時還驚慌,離男性真是越來越遠了。后來他甚至已經變得比很多妹子還可愛了,居然都主動涂了帶顏色的口紅而且還臭美起來。到最后男主已經完全沒有任何男性特征了,似乎完全都忘了自己曾經是男的了,也失去了曾經作為男性時擁有的各種“趣味”,如今就是一個里里外外都可愛和萌噠噠的嬌小幼女,他的妹妹也完全成了他的姐姐般的存在。
如今的男主已經完全回不了頭了,和女孩子們在一起豐富多彩的生活令已經令他不想變回男性了。第12話在浴場里,男主發現自己下面重新出現軟軟的東西的時候沒有絲毫欣喜感,最后還主動喝了妹妹給他的變性藥水,令他保持少女身(這部分場面搞得夠煽情的,好像做好了心理準備要服毒自殺似的)。男主妹妹的哥哥改造計劃不僅大成功,而且還出乎意料的成功,令他都主動想一直成為妹子了。
男主性轉后興趣、性取向、性格都發生了極大的變化,甚至居然會對有男孩子氣的妹子穗月椛害羞,而對美女和福利卻沒有絲毫沖動和念頭。如果男主決定一直作為妹子活下去,不知道他以后會和什么樣的人交往,和妹子交往成了偽百合,和男性交往成了bl,怎么想都很奇怪。
變成妹子后的男主的角色設計和刻畫真心很不錯,他在此作里顯得頗為天真可愛,個子小小的,經常顯得挺萌,一丁點20歲陰郁宅男的影子都沒有。他的CV高野麻里佳配得是真好,聲線清亮可愛,還帶著恰到好處的稚氣感。
男主曾是個腦子里有大量糟糕知識的lsp,還癡迷于工口游戲,果不其然,這樣的人在性轉后肯定立馬想著自我享受一番,這點在此作里表現得很真實。不過他卻還挺純潔,居然洗澡時看自己的果體都那么謹慎。不過他性轉后是完全未發育的loli體,沒什么身材,如果他有他玩的那些游戲里令他興奮的角色的身材,說不定看到鏡子里的自己時就把持不住了(不過也許不會,畢竟已經沒了雄性激素的刺激)。
男主兩年沒出家門,看樣子居然也不用上學,不可思議。也不知道他的父母都干什么去了,哪來的生活費能令他一直家里蹲。
此作對兄妹之間的描寫不少,二人之間像姐妹一樣的玩鬧劇情挺有趣的。妹妹對性轉的哥哥像對妹妹一樣的關懷描寫得也不錯,有一些劇情令我覺得挺溫馨的。不知道妹妹搞哥哥的性轉計劃的初衷是什么,是出于惡作劇心理,還是研究目的,還是什么特殊的興趣,在此作里沒明確交代。妹妹看起來身材平平,不過在澡堂時展現的妹妹的size也不小呢。她做的那個性轉藥水真是神奇,如果能售賣的話,絕對超級受歡迎、迅速賣爆。
男主的中學同學楓真是要用爆r來形容,第3話試泳裝的一幕真是太驚人了!完完全全就是行走的ero、下作的r量。楓的3D歐派的彈性流體感在此作中表現得超絕,妙不可言,而且模型的質感也特別棒,還把bra的勒肉感表現得很逼真精妙。
沒想到美夜居然是百合少女,居然在書店里買百合書,看到這一幕我感到挺驚喜的。目前她只是作為百合的觀測者,希望最終她也能親身陷入百合。美夜發育驚人,13歲的女生居然如此之大,甚至都不遜色于楓。她和楓站在一起時會令飛機場們感到巨大的壓力。二次元真就是可以隨心所欲地設定,可以忽視基本生理發育規律。
此作是三渲二,模型和渲染質量真是相當出色,線條、表情鮮活靈動自然,甚至令人意識不到其實是3D的。此作色彩設計也不錯,整體感覺挺明亮舒暢。
此作的ED的動作幀數極多,相當靈動,有種快放的感覺,這設計有點意思。
怕痛的我,把防御力點滿就對了。 第二季
總評:92.6
我挺喜歡此作的第一季,是當年的一匹黑馬,情節和設定有創意,內容挺有趣,女一號梅普露和女二號莎莉的人設都令我喜歡,而且我特別喜歡看她倆作為百合搭檔時顯得特別美滿、般配的感覺。此作能出新一季很難得。
這一季前一半不錯,后一半令我覺得有意思的劇情不多,大部分都是在打怪,內容有點平淡了。最后的BOSS設定真是挺丑的。雖然此作有不少登場的角色設定得也都不錯,但我還是希望此作里能再多一些梅普露和莎莉二人單獨在一起的描寫。
梅普露繼續變得越來越強,bug的技能越來越多,運營策劃對她的存在很頭疼,老也沒法弄出能難倒她的敵人。梅普露在這一季里還獲得了胳膊能變成一堆觸手的技能,這個很不錯,感覺變成那種形態后有一點《東京食屎鬼》里的角色的感覺。
莎莉的形象和氣質還是那么瀟灑帥氣,棒極了,而且還是總想著梅普露。梅普露和莎莉的配合真不錯,行云流水,互相照應得特別好。第4話還能看到莎莉對梅普露的公主抱,挺美好的。
沒想到那么帥氣的莎莉居然特別怕恐怖和鬼怪,第6層是恐怖元素,害得她都不敢游玩,必須得有梅普露在身邊保護。甚至就連家里沒人時,莎莉都得和梅普露打電話來消除恐懼感。這段劇情令莎莉挺有反差萌的,給這倆人的百合關系進一步添色了。
蜜伊挺有意思的,平時表現得很強勢,但實則是個軟妹子,還不想被別人知道。佐藤利奈給她配軟妹子形象的聲音真是巨好聽,巨軟。從可愛切換成強勢狀態的時候的反差也顯得很有意思。赤發紅瞳的她放火焰系魔法的時候挺有夏娜的感覺的。
無意間變成狗,被喜歡的女生撿回家。(生而為狗 我很幸福)
總評:92.5
這是半長超福利番,創意滿載,各種高能,很有趣味,在半長番里此作絕對是精品。此作的劇情構思真是很不錯,各種出其不意和意想不到,腦洞極大,特別搞笑,槽點滿滿。此作對角色的塑造也挺好,也把犬男主的心理活動描寫得很好。
此作的故事里男主是個高中生,有一天被學校里的一個科學怪才妹子當成了實驗品,變成了濕身的狗了,在寒冷中被他喜歡的同班的女生犬飼加戀抱到家里養了。之后主要就是講述犬男主被加戀各種寵愛和玩弄,見識到了各種無上的美好福利,同時也加深了對犬飼的認識和對她的喜歡,后來彼此建立了不可分割的關系。在劇情中,犬男主想盡各種方法尋找變回人類的線索,最后終于再次見到了那個怪才妹子,不過因為自己的尊嚴以及對加戀的真心的重視,沒有接受她提出的變回人類的方法。說不定男主真就要當一輩子狗,同時也享受一輩子福利了。我感覺男主為了能讓女主開心、不傷害她,哪怕一直當她的狗也心甘情愿。
此作的作畫雖然說不上多出色,但很多地方做得著實挺精良、挺認真的,場面觀賞性頗強,比如第一話犬飼洗澡的過程的觀賞性就很不錯,她還反復自我摩挲,場面賞心悅目。
一些動畫里,犬在對小動物沒任何戒心的妹子那里往往能體會到美好的觸感、觀賞到絕妙的光景,這時候就會引發很多彈幕說“人不如狗”。此作把一只狗能享受的福利做了最大程度的夸大。犬男主真是令人超羨慕,女主犬飼對他沒有絲毫防備,甚至做出許多不可思議的超羞恥的事情。看胖次這種事都已經不值得一提了,在最后一話犬飼居然都給他喂奶了,那場面太不得了了。要是再進一步就真是*番的世界了。
此作大多都是用犬男主的第一人稱,這樣的作品不多見,很多視角很妙,不光裙下風光盡收眼底(要是有no胖次的妹子那就太有意思了,可惜此作沒有),還有身處在妹子的衣服里往上能看到對方的臉部的鏡頭,從下到上的貫通美景一覽無余,真是妙絕。第一人稱時犬男主被犬飼用狗鏈牽著散步的過程挺羞恥的,這視角可能會令一些抖M觀眾有點興奮。
犬飼的人設很不錯,相貌漂亮,還非常豐饒。她平時顯得非常高冷,但意外地一個人面對狗的時候特別放飛自我,果著和狗一起洗澡、隨心所欲地沖著狗犯傻賣蠢,甚至對狗做不可描述的沙雕行為(比如舔鼻子,感覺跟舔...似的)。相應地,轉生成狗的男主在外人眼里真是幸福得不得了。
犬飼有時候會突然對人露出可怕的表情,仿佛有雙重性格似的。她只對狗有興趣,而對人毫無興趣。她對狗的癖好和看法挺變態,甚至表現出怪女人的癖好,比如愛看狗放水。第3話,居然她為了讓犬男主舔她的玉足,故意把小腳趾踢傷,太狠了。這一話居然她還掀起裙子追著男主讓她進入自己的衣服,甚至還坐在地上把裙子撩起來構筑“狗窩”吸引男主鉆進去。做出如此沒有下限的荒唐、嘆為觀止的行為,把觀眾和貓谷都看傻眼了!第4話她還穿上了極其性感養眼的狗娘(但我看起來像貓娘)的服裝試圖扮演動物來和犬男主歡愉,太難以啟齒了,真是想法清奇。這居然被她媽看到了,她立馬一本正經地辯解說是在做俯臥撐,估計她媽也明白其實女兒是在做奇怪的事,就笑而不語了。犬飼的醋意還很大,第3話她看到犬男主在和貓谷密切親熱,還舔她膝蓋,立馬就怒了,然后立馬做出過激行為。她對犬的占有度和親熱度真是絕不肯輸給別人。
犬男主現在的狀態明明是一般男生求之不得的,但他居然還很抗拒欣賞眼前的絕景,面對美好的畫面甚至還試圖閉上眼睛,不可思議,。現實中怎么會有這么正經古板至極的男高中生?不過犬男主在某些時候還挺勇的,完全不顧及自己是個男人,主動做了不少過激的事。雖然他的內心獨白表達的是做這種事是不得已而為之,但他的潛意識里說不定正在興奮地揩油,只是隱藏得很深不讓觀眾知道。
女二號貓谷的人設不錯,她是性格歡脫頭腦簡單的衣著清涼的辣妹,性格挺有趣的,也有點弱勢。她和犬飼從小就認識,關系很近,第2話她和犬飼抱在一起還有點橘。沒想到她很怕狗,第3話犬男主接近的時候她露出的慌張凌亂的樣子挺令人興奮的,這段劇情展現出了貓谷的極度香艷。第3話特別令我討厭的地方是居然出現了蜈蚣(家里怎么會有那么大蜈蚣!?),而且蜈蚣還不打碼,弄得我都沒法好好觀看了。貌似貓谷倒不怎么怕蜈蚣,至少對蜈蚣的害怕還沒有對犬害怕的1/10。
居然犬男主讓貓谷害怕的做法是舔她的密處,真是赤果果地仗著自己是動物一本正經地耍流氓。在此之后男主還多次一本正經地侵犯貓谷,有時顯得做過頭了,貓谷有點可憐。
第5話出現的漂亮妹子月城兔居然喜歡男主,擅自跑到男主家里進行搜索,不可思議。感覺男主也沒什么氣質,居然還蠻受歡迎的,令我想不到。月城兔居然用男主的桌角自娛自樂(你懂的...),這種事情如此直白地出現在此作里真是難以置信!
第7話里犬飼和月城的對決太搞了,倆人各用裙子搭了個狗窩,看犬男主鉆哪個決定誰才是他的主人。后來還相互施展佛山無影腳,真是太有創意了。居然月城還把棒狀骨頭塞進自己的胖次里吸引犬男主,這行為真是令人無語,我擔心她別一個不小心摔個跟頭,然后棒狀骨頭一下子滑進去釀成血案。
總的來說,三個主要女角色里,我最愿意交往的是貓谷。犬飼的里人格有比較狠的一面,比較危險,而月城的頭腦有點奇葩,也不能讓人放心。貓谷外貌出眾,活潑可愛,內在性格和思想也令人安心。
此作的ED畫面有多個,其中一個ED挺有新意的,是現實中的攝像機追拍在學校樓道里跑步的穿短裙露腿的妹子,顯得挺有青春氣息和悅動感的,這監督還蠻有情趣的,呵呵。
D4DJ All Mix
總評:92.45
D4DJ系列的上一季《D4DJ First Mix》是2020年的,是以高中少女樂團組合為題材的動畫,當時我追了,感覺不錯。這一季的劇情是各個隊伍一起參加一個招財貓的企劃,定期舉辦演出,由此帶動當地經濟,故事內容就是她們的練習、演唱和日常。在這一季里D4DJ企劃里多支隊伍各有一部分戲份,而且戲份差不多,也因此第一季的主角Happy around在此作里就不是唯一主角了。
這個企劃里我最喜歡的始終是Happy around,由于這一季為了公平,導致她們的戲份被壓縮得比較有限,因此這一季的打分沒第一季高。不過Merm4id和Peaky P-key我也挺喜歡的。這一季最糟糕的是講Photon Maiden的幾話,劇情著實沒什么看頭,也不是我喜歡的角色,我都是快進看完,給總分拉低了不少。
不得不再夸一下此作的人物模型和渲染。D4DJ的第一季在這方面就非常好,似乎這一季更進一步增強了。此作里3D人物模型做得相當好,基本挑不出瑕疵和不足,而且人物表情特別鮮活靈動,有的時候我都納悶這么好的效果是怎么做出來的,感嘆制作方的技術水準太高了。此作的人物面部是flexible的,而不是rigid的,神情的表現真是一點都不遜于中高質量的手繪作品。
這一季的live制作依舊出色,絢爛紛呈,氣氛渲染得相當好,觀賞性很強,令人很能high起來。要是能親身在現場體驗她們的演出就好了。
Merm4id的演出真是特別養眼,我極其欣賞。妹子穿得極其清涼,展露大片白皙,在舞臺上曼妙的扭腰更是銷魂。特別是瀨戶莉夏的身姿美不勝收。隊伍里那個長發的姑娘松山達利亞的人設很不錯,氣質成熟優雅,顏值很高,事業線也特顯著。日高沙織挺可愛的,歐派真大。第5話她學英文和外國人氣作曲者對話的劇情真是挺有意思的。
第6話很逗,很有創意,居然真秀一覺醒來只能說英語了,太奇怪了。居然大部分妹子英語都白學了,都是普通日常對話而已,就算聽力再差也應該能聽懂1/3才對啊,好多妹子居然一丁點也聽不懂。也不知道為啥真秀是變得只會說英語而不是其它的,要是變得只會說捷克或希臘語的話在同伴里都找不到翻譯了。真秀的CV表現得不錯,英語說得很流暢地道,一點都沒有日式英語的感覺。這一話的末尾部分太邪乎了,Happy around的四人和觀眾都變成僵尸,眼珠子還變得很惡心,造成我極大不適,我頗不喜歡這種強行鬧鬼的劇情。
忍不住要再次夸贊一下鈴玖,她真是特別耀眼和閃耀,性格歡脫,元氣滿滿,閃閃發亮,每次她的出現都能給人帶來歡樂的感覺和暖意。鈴玖的CV西尾夕香對她的性格表現得也是相當好。最后一話她父母給她寄了一個大海螺,吹海螺的場面很有意思。可惜最終的跨年live她沒在臺上吹,要不然跨年倒計時到0的時候猛吹一下大海螺全場得多high啊!
這一季ED剛開始還令我有些吃驚,這不是第一季的ED畫面么,還是DJ明石真秀在那里自嗨,怎么畫面復用了。之后才發現原來是所有樂團的DJ共享畫面。這個ED構思有點意思。
被解雇的暗黑士兵(30多歲)開始了慢生活的第二人生
總評:92
此作畫面做得挺糙的,剛一開始看的話會令人感覺是個粗制濫造的作品,但其實此作的人設和劇情蠻不錯,值得一看。此作整體劇情有趣,最后幾話又挺有溫情,能令人看得挺開心。而且整體的劇情挺連貫,這點不錯。
此作的男主達利艾爾是個人類,但從小被魔族撫養長大,是魔王軍的一員。但魔王軍的頭領從男主養父古蘭巴札換成了他親兒子巴修巴札后,沒有魔法能力的男主就被從魔王軍里解雇,只能自己流浪。他偶然中途遇到了一個年輕可愛的村姑,即此作的女主艾麗卡,在野外救了她后男主就被接納到了村子里。此作主要就是講述30多歲的男主在此之后的人生,經歷了一連串各種各樣不普通的事情。
男主設定得不錯,我還挺欣賞他的。他的三觀很正,品行端正,內心善良,是個老好人,甚至和敵人戰斗時還會擔心對手會不會受重傷。男主的性格討人喜歡,各方面的魅力都挺強,不光瑪麗卡見到他不久后就把他當做夫君了,小弟casida醉酒狀態時甚至還說要和他結婚,他在魔王軍那時候的伙伴利杰特在第4話重新見到他時居然還臉紅,男主簡直把男的都掰彎了。另外,礦場里面的黑臉小可愛們也十分擁戴男主,男主在此作里真是人見人愛,很有人望。
男主是堅定的和平主義者,盡力消除不必要的戰斗。再加上他是魔族養大的人類,對人類和魔族都有感情,這使得他更是十分希望消除雙方長久以來的紛爭。最后他也確實很大程度做到了這一點,成功地讓自己的血緣的父親,即上一屆勇者,和自己的養父,即魔族的天王,彼此消除了仇恨,甚至成為了友人。看到他倆握手言和時我還有點小感動,真是可喜可賀。最后兩位父親甚至還組成了人魔聯合戰線成為了戰友,真是化干戈為玉帛,男主做得真是很好。
杉田智和給男主的配音真是相當不錯,聲線特征和角色形象很搭,給他增色很多,極大地增加了人物的真實和鮮活感,他的很多內心戲有種顯得像自我吐槽的感覺。
女主瑪麗卡令我印象挺深刻的,這角色設定蠻神奇的。她的相貌給我感覺挺可愛的,眼睛很大,有種天然村姑屬性的美感。其性格也好,對男主非常溫柔體貼,是個好妻子,而且還是護夫狂魔,對于那些對自己丈夫有敵意的人都會投過去令其毛骨悚然的殺氣。
瑪麗卡的體格強大到離譜,能一下跳到天上去,看到男主興奮地瞬間迎上去的時候簡直是超音速,那帶球撞人太厲害了,把男主這么強的人都能瞬間撞暈。好在對象是男主,換了身子弱的就要釀成胸殺案了。也不知道瑪麗卡如此強大是怎么回事,明明其父母都很普通,這基因變異也太邪乎了。我高度懷疑瑪麗卡是不是其實是此作里戰斗力最強的。反正至少是她解決了此作里兩個最強者(前代勇者和前代四天王古蘭巴札)之間的戰斗。
有趣的是男主在此作里老是管歐派叫麻薯。瑪麗卡最大亮點之一是其夸張的麻薯,穿的還是低胸,倆麻薯之間的溝壑實在太深了,對視線的吸引力太強了。我本以為應該是瑪麗卡先向男主表白,但瑪麗卡的麻薯誘惑力實在太大,結果沒想到是男主首先向她示意了。男主獲得了如此極品麻薯,肯定超爽。
那個綠色長發的女魔族干部賽維安提斯的著裝真不錯,豐饒的曲線看著很養眼,后來她對男主還產生了好感。其實我挺想看到她勾引男主然后被瑪麗卡捉奸的劇情的。
說來,我感覺此作和《被勇者隊伍開除的馭獸使,邂逅了最強種的貓耳少女》有一個相似之處,都是原來的同伙都是嫌男主沒什么用就把他開除了,在失去他后才意識到他的輔助有多么的重要,而男主也都是單獨闖蕩后逐漸發現自己潛能是多么的厲害,并利用自己的能力幫助他人,結交了與之交心的伙伴,還找到了自己的存在價值。而且這倆男主都是內心寬厚、豁達、善良,比較容易招人喜歡。
恣睢、自負又無能的巴修巴札把男主趕走真是愚蠢至極,完全不知道自己在干多蠢的事,搞砸了魔王軍一切事物。不過在最后他倒是洗白得很徹底,其本性完全不壞,小時候的樣子還顯得相當純真可愛。可惜他就是由于在成長過程中心中誕生了自己過不去的坎,產生了對男主嚴重偏見和嫉妒,導致自己最終走上了歪路。好在他和此作的其它反派一樣,被男主的大度和真誠所感化,最終重新審視了自己,洗心革面重新做人。
艦隊Collection 總有一天,在那片海
總評:91.5
距離上一部艦娘系列的作品已經很遙遠了,突然出來這么個新作令我挺意外的。此作貌似是C2同人社團的艦娘同人作改編的動畫,講述第一游擊部隊第三部隊的艦娘的事,內容就是她們與深海棲艦的作戰以及軍營里的日常。此作只有8話。
此作整體做得有點虎頭蛇尾。剛開始還令我挺滿意,感覺此作不落俗套,經費足,畫面精良,整部作品不浮躁,不主流,挺細膩的,甚至還曾令我覺得此作有點像電影動畫的感覺,覺得此作是一部用心、講究的作品。結果看到后期,真是蠻失望的。早知如此我都不追了。在我來看此作是A級音樂,B+級制作,C-級劇情。此作最爛的就是劇情了。剛開始我還看得津津有味,但到中期感覺故事的節奏很怪,有些地方劇情太慢,戰斗的過程也往往缺乏流暢連貫的感覺。看到后期真是令我覺得劇情索然無味,都不知道在打什么,戰斗進程都令我摸不著頭腦。感覺此作的劇情組織得特別差,邏輯挺迷的,有種外行人做的感覺。最后一話可謂完全爛尾,莫名其妙就結束了,有種“不知道看了個啥”的感覺。明明愛荷華她們都來增援了,怎么結局卻顯得像全軍覆沒一樣,太奇怪了。我覺得此作的監督嚴重失職。此作整體的劇情比較沉重,始終籠罩在大概率面臨失敗、要被擊沉的陰霾中,有的角色還知道自己扮演的是棄子的角色,大多數劇情觀感偏壓抑。
此作最大的優點就是音樂,用杰出來形容毫不過分。此作的配樂的水準和品味很高,非常優美和高雅,聽感相當好,如果有OST的話絕對值得聽聽。感覺這么好的配樂用在此作上太浪費了。
此作的水波制作挺驚艷的,非常細膩,令人看到后不禁心里感嘆:啊!這光!這水!
此作的主角是時雨,我對此角色沒啥興趣。島風明明人氣那么高,要是什么時候能以島風為女主做一部動畫就好了。
時雨的CVタニベユミ的表現令我不怎么滿意。第一話她剛一開口就令我覺得難受,那哪是妹子的聲音啊,明明是那種會自稱boku的少年的聲音,而且還有種老成、老氣感,表現力也不好,和時雨的形象很不相符。感覺>=2線的CV界里隨便抽一個人配大概率都比這強。タニベユミ都不是專職的CV,感覺她配時雨這么關鍵的角色是大人的關系的原因。
第7話,時雨進行身體改造的過程描寫得太敷衍了,沒看到什么喜聞樂見的畫面。感覺改造前后也沒啥變化似的,改造了個寂寞。
第3話山城飛上天和對方BOSS死拼(居然艦艇能飛...),這段邏輯真是挺迷的,明明大和都來了,我不信用巨炮轟不死BOSS,干嘛山城要拼上性命和BOSS一換一。最后山城(顯得是)掛了,我還有點心疼。雖然我對山城算不上多喜歡,但她姐控這一點我還挺中意的。那個BOSS也是個姐控,但她姐似乎是她腦補創造出來的。真是倆姐控的對決。
好在是山城和扶桑最后都沒事,只不過失去了戰斗能力而退役了。這已經算是happy end了,山城沒掛真是奇跡。我感覺山城和扶桑這倆主力艦不怎么中用,沒起到多大價值,反倒有點像活靶子。值得一提的是這姐妹倆的歐派真是不得了,第3話她倆洗澡的那一幕浮起來的脂肪堆顯得真是太夸張了。
頭上戴著小喇叭的雪風真是挺萌的,第6話居然橘子吃太多把引擎吃壞了,這個事件真可愛!不知為啥雪風對時雨挺親近,第5話居然雪風突然就對時雨親上去了,橘得也太突然了。
第6話出現的4個小學生樣子的loli組成的護衛海防艦隊伍很不錯,并排出現的畫面顯得她們萌極了,要是戲份多一些就好了。
榛名在此作里畫得真漂亮,特別養眼,非常清秀柔美,我給她截了不少圖。
此作的提督居然是男的,而且完整露臉了。其長相也太路人了。要是女提督該多好。
此作的OP很特別,是高亢的詠嘆唱法,風格真是夠classic的,很抒情和動情,初次聽的時候那種優美感甚至令我有點隱隱地起雞皮疙瘩,這和主流動畫的OP風格真是大相徑庭。
此作ED的畫面設計怪怪的,上來先是連著放一堆圖高頻率地切換,這種設計真是很少見。ED歌曲還算有可圈可點之處。
呆萌酷男孩
總評:91.4
半長番,是個女性向的比較挺輕松、慢節奏的講述幾個男青年平日經歷的作品。此作有四位男青年,高中生、大學生、專科生、社會人都有,他們的共同特點就是呆萌,平時總是比較冒失,往往犯蠢、犯愣,往往顯得有點笨拙,這些性格特征正是此作所著重刻畫的他們萌點。這些男青年一開始是單獨出現的,前期每一話主要講不同角色,但在后來的劇情中逐漸出現了交集,后來都相互認識了,最后結成了要好的小團體。此作倒是沒有明顯的bl傾向(除了五十嵐X三間貴之外),但個別二人在一起的場面也能令人腦補出腐來。
此作我最感興趣的角色是二見瞬,他老是犯小錯誤,諸如放音樂的時候忘了插耳機線,衣服老穿反,丟三落四,傳球甚至都能傳給對方。但是他的運動能力又很強,長相也小帥,不怎么和女生說話,真是很呆萌酷。他特別特別有意思的一點是自己犯了蠢之后總是故作淡定地掩蓋,逞強表現他做的傻事是他故意而為,這個設定真是相當逗。這樣的男孩應該是很受姐姐們喜歡的那種。
那個紅毛四季蒼真的笑點真是太低了,連自己犯蠢也會自己吭哧吭哧笑起來,真是很奇怪的設定。
公司職員三間貴之的氣質挺不錯,溫文爾雅,性格平靜,按理說應該有女同事追他。不過此作里的四個男主角對女性都從未表現出過任何興趣。他和作家五十嵐之間的故事挺有意思的,互相都知道對方是自己的小學同學,但二人一直都沒說出來,始終保持著工作上的關系。他倆在晚上的公園中的對話,令彼此都知道原來對方還記得自己,還令五十嵐瞬間感到吃驚和驚喜,這倆人挺有愛的,希望他倆能在一起。
此作中明顯有出鏡的女角色也就是咖啡廳的店長了。她的人設不錯,漂亮、可愛、豁達,很關照傻弟弟瞬。
虛構推理 第二季
總評:2-4話93.75,其它話平均89.5
此作的上一季我追了,講上大學的男主櫻川九郎和loli樣子的女主巖永琴子處理怪力亂神的事,有些劇情挺不錯,特別是第一季那個女偶像去世后變成的無頭女幽靈的人設很棒,養眼極了。過了N年出了第二季很難得。
這一季由一系列獨立的故事組成,彼此之間沒啥聯系。第2、3、4話講雪女的劇情真心超好看,可惜雪女的故事就只有這么三話,真令我意猶未盡。要是她作為一整季的女主那就爽歪歪了。第6、7話關于老頭制作的木頭人偶危害城鎮的劇情著實無趣,我都快進兩倍以上看。8、9、10、11關于分老頭遺產、弄清楚其妻子23年前被殺害真相的劇情也令我完全提不起興趣。第5話講男主姐姐和兇宅的事,以及第12話講一社員殺害其妻子的事還算有看頭。第1話講屋子有異響的劇情湊湊合合。總之,在我來看,這一季2、3、4話是絕對要看的精華部分,第5、12話也值得一看,其它的完全可以pass。
先說說第2、3、4話的雪女劇情。
年輕的室井昌幸和伙伴一起去雪山登山時,被嫉恨他的伙伴推下山崖,偶遇了漂游在那里的雪女,二人都覺得對方挺有意思。雪女把他救了之后就分別了。過了10年,成家立業的室井遭遇人生挫折,妻子離異、失去了公司,漫無目的地晃蕩時居然偶遇了假扮人類的吃著蛋筒的雪女。雪女和室井表面上繼續拌嘴,實則二人相性很不錯,再加上雪女貪圖人類的美食,室井愿意給她投食,于是室井就被雪女黏上了。
這雪女的人設我真心特別喜歡!喜歡極了!她的顏值真是相當高,賞心悅目,她那烏黑的長發、白皙的皮膚、清澈的眼眸,令我回想起更衣小夜帶給我的那種感覺。她表面上看起來冰清玉潔,是個冷美人的樣子,實則性格自在、有趣,還很貪吃,品嘗到美味時幸福得臉紅的樣子特別可愛,還有小虎牙和貓嘴的設定進一步給她增添了可愛感。她雖然是妖怪,居然還挺懂事,知道不能老占室井的便宜,要主動獻福利,于是提出用身子作為回報,看到這里我真是大吃一驚,居然這么直球!那福利也太有殺傷力了,魅惑力和誘惑力爆表(是我可扛不住),而且還說只要好好contraception就沒關系,完全就是明著表示愿意和室井搞事。可室井居然繼續一臉淡定,表現得不為所動,不可思議,真懷疑室井在人生挫折后已經徹底拋棄了七情六欲、四大皆空,因而對雪女沒有任何雄性的想法和沖動。雖然室井在離異后,單身生活過得挺郁悶,但那么美好的雪女三天兩頭跑來找他,室井可真幸福!
公主做假定室井有罪的推理真是精彩,我都一度懷疑室井是不是真的是殺害前妻的犯人。公主有理有據的推理把雪女聽得都嚇哆嗦了,如果雪女自己深深信賴和喜歡的室井真的是犯人并且利用自己,對她的心靈的打擊將是毀滅性的。好在果不其然室井是無辜的,但公主排除他是犯人的原因是因為沒有受到雪女的誘惑而OOXX。雖然這理由很無語,但確實也有點說服力。這一話里室井對雪女說當時和他的那位前妻結婚是因為前妻長得像自己在雪山遇難時一見鐘情的雪女,雪女萬萬沒想到自己在看起來平靜、嚴肅、刻板的室井心里一直占據著如此重要的地位,令他如此難以忘懷。這一幕大大加深了二人之間的羈絆,我都有點感動了。室井也真是幸福,自己最愛、最難忘,也是最遙不可及的人,現在就在自己的身邊陪伴著自己。我不禁感嘆雪女和室井之間真是有著難以置信、奇跡般的緣分。雖然他們在一起時間長了是否能融洽相處、是否會產生矛盾,這很難說得準,而且還有壽命差異這個嚴肅的問題,但總之我還是希望他倆能在盡可能長的時間內恩愛地度過。
暗戀室井的飯塚渚很漂亮,直接去追失意的室井多好,居然干出殺害其前妻并且嫁禍給室井的事,真是糊涂,感覺這行為很不可思議,這劇情構思有點牽強。她要是好好表白,至少是先給向室井獻殷勤,比如經常給他去做飯什么的,我認為室井早晚會從了他。
室井真算得上是個好男人,品性和道德很好。前妻曾經試圖毒害他未遂,他卻在離婚時還給她前妻那么好的條件。面對雪女的誘惑時,他還保持著高度的克制和理性。他這樣品行端正、性格老實的人,得不到真愛的話真是太可憐了。此外,他之前還被摯友所害,推下山崖,真是挺慘的。能遇到雪女,以及后來再次相遇,有種人品守恒定律的感覺。
這一季里充分展現出琴子是何等的沒品,琴子已經可以作為“沒品”一詞的配圖了。劇情里琴子的言行無數次令她的風評受害,作者無疑就是要塑造一個表面上看起來像端莊可愛的洋娃娃,名義上是公主,但內在卻極度無節操的角色。這一季里她兩次提醒室井和雪女要注意contraception,還督促他們在床上干活。在第5話里,還N次提到琴子沒品,不光男主她姐這么說,就連房東男和他情人也如是說。后來她還說在眾人面前說什么“做愛做的事”,真是寡廉鮮恥。在這一季最后一話琴子都不忘再次表現自己,對一個陌生人就直接說“因為我今晚要去男友家過夜,突然想要吃點能增強精力的東西,剛好看到那間鰻魚專賣店就走進去了...由于外形和男性的**很像,感覺吃了就能精力百倍呢。今晚的我可是干勁十足!”,這令對方驚嘆“講出如此猥褻的話!”。在最后一幕,琴子居然還對男主說“今晚可不必對我客氣哦,學長”。哎呦,感覺琴子就像生怕別人不知道自己多沒下限一樣。
琴子對男主真是夠主動的,甚至都主動到床上粘他,跟小妖精似的,這跟琴子表面上給人的感覺反差極大。這么有頭腦、特別睿智的少女,誰想得到她會對男主那么專情和執著。而男主到是對她顯得挺冷淡的(但在觀眾看不到的場景里,他說不定表現得很中意琴子)。
第12話很逗。前一半劇情里,有倆社會人在鰻魚飯專賣店里不停地互相叨逼叨,其中一人在不停地虛構推理,完全把琴子的活兒都給搶了,感覺這倆人都神經了,像推理偏執狂一樣。其中戴眼鏡的那個人感到自己推理有誤時,突然低頭道歉,眼鏡瞬間滑落,那一幕樂死了。他倆還不停地一本正經地討論在遠處一個人享用鰻魚飯的琴子的來頭,他倆顯得真是特別滑稽,令人難以理解他們怎么會對一個少女進入鰻魚飯專賣店這事如此較真兒。
這一季的ED是宮野真守唱的,充滿了男性的騷氣感。
Love Live!虹咲學園學園偶像同好會 虹四格動畫
總評:90.9
我很喜歡《Love Live!虹咲學園學園偶像同好會》,這個是記錄虹咲學園日常的泡面番,只有三分鐘。劇情整體給我留下的印象不深。有點可惜的是人物不是手繪而是三渲二,如果手繪的話表情能明顯更靈動些,此作的3D模型顯得有點偏胖,臉部不夠立體。主題曲部分侑和嵐珠以及侑和果林跳舞的小動畫有點橘的感覺。最后一話侑又和雪菜獨處了,聊得很好,雪菜還臉紅了。此時步夢突然出現,還挺慌張的樣子,這令我聯想起曾經步夢以為侑和雪菜出軌的事,不知道這次她是不是又懷疑起侑和雪菜出現了姬情。
之前聽說我很喜歡的優木雪菜的CV楠木燈由于患病而要換人,不過這個泡面番倒還是楠木燈當她的CV,可能這就是她在lovelive官方作品里最后一次登場了吧。
Showtime!2~唱歌的大姐姐也想做
總評:90.45
沒想到此作能出第二季,僧侶番能出第二季的作品很有限。這一季只有8話,繼續講述電視臺節目中的唱歌的大姐姐三奈美和帶一個娃的男主之間的沒羞沒臊的故事。三奈美特別主動地殷勤地向男主獻福利,我始終沒看出男主有什么特別優秀之處值得這么美好的大姐姐這么主動倒貼。男主真是太令人羨慕了。男主的欲望和精力頗為旺盛,基本每一話都要和女主運動,而且花樣還不少,第2話女主試浴衣的時候男主居然就把持不住了,還發生了優衣庫事件。
這一季里狗仔隊偷拍三奈美想爆料緋聞,我覺得三奈美不就是個兒童節目的類似金龜子那樣的主持人么,又不是特別人氣的偶像,怎么狗仔隊對她的私生活那么關切?挺匪夷所思的。好在最終這事被擺平了。狗仔隊真是很令人生厭,跟臭蟲一樣,這些人令我感覺都沒有什么自我尊嚴。
某天早上變成人頭麥克風的我君的人生
總評:90.2
之前有個《180秒能讓你的耳朵幸福起來嗎?》的動畫,講妹子們玩人頭麥,福利不少。這部作品換了個角度,第一話男主轉生成人頭麥,然后享受被妹子們挑弄時的愉悅,這個構思蠻有意思的。人頭麥是杉田智和配音,那種充滿吐槽感的演繹很適合此作。之后,男主每一話又不知怎么轉生成了各種各樣的物體,枕頭、雞、奶嘴、卷尺、托盤什么的,也因此往往能享受到一些福利,比如第2話他變成妹子的自行車坐,能被妹子柔軟私密之處經常坐著,真是幸福。此作里多半內容都是妹子的日常故事,男主能偷聽到少女們的秘話。
第10話格外有趣,男主變成了狗,和《無意間變成狗,被喜歡的女生撿回家。》的設定撞車了!然后男主幻想和妹子一起洗澡,這是那部作品里男主所享受到的極樂世界。沒想到最后妹子說要給他做絕育,把男主驚了,這一幕太樂了。這一話出現的女主她媽真有氣質,真御,真有范兒,跟大明星似的。
此作里小梅畫得很漂亮,黑色雙馬尾,頭發里還摻著紅色,很有個性,也挺令我眼前一亮的。第9話她懷中抱妹子的樣子真開心,真是橘里橘氣的。
藏前顯得酷酷的,有點中性。令我萬萬沒想到,最后一話曝光出她居然喜歡淺草尤莉!之前雖然一些劇情里顯得幾個女生之間有點姬,但沒想到藏前是真心對淺草抱有unambiguously的戀愛感情!可惜最后藏前對淺草的告白居然沒播完就結束了。不知道淺草會如何回應,感覺她應該是個直女吧。最后一話里男主終于以人形現身了,但是是個渾身漆黑的類似穿連體衣的肌肉男,這令我聯想起《夏日重現》里的四手。
]]>
文/Sobereva 2024-Jan-2
這里對我從2023年7月1日開始截止到2023年12月31日觀看的非TV版動畫進行簡單評論。由于這類動畫不好對比和打分,我也就不給出評分了。以往的此系列文章和TV版動畫評論見http://www.shanxitv.org右方的“動畫評論”分類。
黃金拼圖 Thank you!!
類型:劇場版
長度:1h21m
我作為TV版老觀眾對這個劇場版很滿意,內容很有趣,觀感很好。這個劇場版主要講述高三畢業前夕大家一起經歷的故事,包括休學旅游、日常、參加大學考試等劇情。這個劇場版百合濃度很高,有很多高橘的畫面和對話,能看得人挺開心。
令我再次感嘆可憐的人設真出彩,顏值真是相當高,賞心悅目,是此作里我最喜歡的角色。居然可憐留在日本上大學,而忍和愛麗絲一起去英國上大學了,從此可憐和愛麗絲這青梅竹馬終于分開了。
穗乃花真是喜歡可憐,老是想著在修學旅行過程中和可憐單獨拍照,但又不好意思,她還挺漂亮的。可憐的老相好愛麗絲如今和她不在一起,可憐又很友善又很豁達和open,如果穗乃花多主動一些引起可憐的注意力,我感覺穗乃花在大學期間還是挺有希望和她愛慕的可憐發展出親密關系的。
豬熊陽子和小路綾之間繼續橘里橘氣,在這劇場版里綾對陽子表現出各種心動和臉紅。這倆人不僅上了同一所大學,而且之后還租了房子一起同居了,這發展挺不得了,大橘已定!綾是那種看起來學習優秀的孩子,而躁動的陽子的學習成績應該不如她。陽子能和綾考上同一所大學,不知道是不是綾為了能和陽子在一起而遷就她選了好考一些的大學。
忍繼續表現出對愛麗絲的各種癡迷。想不到忍的媽媽當年和愛麗絲的媽媽也有過美好的回憶,可惜后來都和男性結婚了,百合緣分就只能留給她們的女兒繼承了。愛麗絲她媽真是挺漂亮,感覺比愛麗絲更有風采,要是在此作中多出鏡就好了。
看到最后,綾、陽子和可憐去英國看望在那里讀書的忍和愛麗絲的時候,我再次心想,11區的護照就是方便...
總之就是非常可愛 女子高中篇 02話
類型:OVA
長度:23m40s
這個OVA主要講述星空在女子高中當老師時遇到的女學生的事。內容包括司和星空去店里吃拉面、司換上女高中生校服、好幾個女學生來澡堂、星空自己動手在院子里制造水塘。這個OVA很有趣。
那個家境富有的女高中生的設定像極了《愛吃拉面的小泉同學》,走遍各個拉面店并在網上寫評論,感覺作者肯定是借鑒了小泉的人設。
吃拉面的時候星空還挺貼心,看到司點了個巨辣的拉面時,刻意點了個不辣的拉面,免得到時候可愛的妻子被辣到時沒的吃,這令和他們同桌的那個女學生被閃到。
星空的好幾個女學生看上去不漂亮,但到了澡堂里把頭發放下來后一下子顏值增加了許多。這些女學生還蠻有趣的,要是能再在續作中看到就好了。這些女生對星空還挺感興趣的,不過并沒有做出倒貼的行為,也知道他已經有了超級可愛的妻子。但我還是挺想在此作里看到這些女生挑逗星空令他臉紅的畫面的。雖然星空和這些女學生之間什么都沒發生,但還是令司感到吃醋,挺有趣的,她對星空的占有欲還真強。
星空居然憑自己的能力就把水塘造出來了,弄得還有模有樣,就像日本庭院里那種專門建造的水池,真是不簡單,腦力和動手能力真是相當強。
星空和穿了高中制服的司在屋后陰影處除了接吻外都做了什么,令人浮想聯翩,就像男女高中生在學校的角落做不可告人的事一樣。
碧藍航線 女王號令 OVA 01、02
長度:2*24m
我對《碧藍航線》沒有絲毫興趣,一丁點都沒有,興趣是負值。只是偶然看到出了這個OVA就隨便看了看。發現此作簡直就是世界杯,各種壯觀的又大又白。這些人設也太媚宅了。巨r角色的比例實在太高了。
OVA01劇情沒什么營養,內容就是女王伊麗莎白下象棋輸給了英勇,然后英勇當了一天女王,而傻乎乎的伊麗莎白去當了一天下人,算是體驗生活了。劇情沒什么意思,完全就是觀球了。
OVA02劇情也沒啥營養,內容就是要搞陣營聯歡會,伊麗莎白去各個陣營查看準備情況的事。這一話出現了俾斯麥,人設明明就是抄襲艦娘的俾斯麥嘛!
虹咲學園學園偶像同好會 NEXT SKY
類型:OVA
長度:29m40s
之前在TV版最后看到步夢在英國和兩個妹子在一起,令我有點好奇。這個OVA講述其中一個褐皮妹子艾拉來日本體驗偶像文化,大家一起陪她玩的事。感覺這OVA也沒講什么內容。英國的另一個白發的妹子當初令我眼前一亮,可惜在這個OVA里只是打了醬油,沒有什么描寫,感覺她比艾拉的人設更有亮點,希望在以后的作品里她能有一些正經的戲份。可能艾拉以后和她會在英國搞學園偶像活動了。艾拉給我的感覺一般般,個性不怎么鮮明。這OVA里鹽子的戲份不少,之前她一直很正經,甚至有點死板的感覺,結果在這OVA最后終于也決定放飛自我了,是個重要轉變。眾人和艾拉一起逛秋葉原的時候看到了UTX大樓,這一幕令我長嘆,當年UTX的超級學園偶像二賴子再也看不到了,A-Rise組合真的是非常出彩,當年被ll官方浪費掉了太太太太太可惜了!!!這OVA開頭的說唱部分真難聽。居然這OVA有兩個ED。
蠟筆小新 2023年劇場版
這是我于2023年9月初在大阪陪別人看的。其實我一直以來看不上《蠟筆小新》,并且感覺低幼+低俗。但這個劇場版令我感覺挺出乎意料的,做得著實很不錯,歡樂搞笑、驚險刺激、感人催淚的元素全都有,而且這些元素融合得非常理想,毫無生硬和突兀感。整部作品畫面制作和劇情構思都很下功夫,真是一部老少咸宜的用心的佳作。此作的畫面是三渲二,一開始感覺有點奇怪,看過之后感覺此作也只能用這種方式制作,畢竟此作里面大場面相當多,手繪難度太大。而且三渲二效果還挺自然,令觀感并未打什么折扣。
此作主要內容是有兩個隕石撞到了地球上,一個撞到了小新和妹妹身上,另一個撞到了一個性格陰暗、失去理想的男青年身上。那個男青年的人生一直非常灰暗,特別缺愛,作為心靈支柱的他鐘愛的偶像妹子又突然宣布和異性交往了,導致男青年在得到超能力后決定報復社會。小新在此作里利用自己的超能力和他對抗。先是挫敗了他綁架小新所在幼兒園的行動,之后又將變得巨大化的他擊敗。最后男青年失去神志,被超能力和陰暗心理所支配,變成了異形,小新被吸入他肚子里之后接觸到了他過往的記憶和靈魂。這一段內心對話構思得真是相當好,如果沒有這部分,此作就是簡單歡樂過個眼癮的作品,而有了這個部分就給此作內涵和深度拔高了很多,挺催淚的。這個部分很好地展現了外在顯得沙雕的小新的內在品質優良的一面,小新努力和心靈孤單、飽受欺凌的男青年做朋友,友情以及他人的支持也正是那個男青年所最需要的。最終心態非常積極的小新改變了陰暗男青年,并且在小新他爸臭襪子作為武器的情況下戰勝了男青年內心負面情感幻化出的三個反派。最后男青年回歸人形,也在大家的鼓勵下決心好好努力面對人生,可喜可賀。
看了這個劇場版,真是令我再次感到11區的非常多的動畫真是非常用心和良心,哪怕是面向子供的作品也真是做得毫不含糊,反觀某國的低齡動畫那品質真是連低齡兒童都不想看。此作令我對《蠟筆小新》的印象有很大的改觀。結尾看到小新的2024年劇場版的預告,是關于恐龍的,不知道是不是小新要穿越回古代,想來《機器貓》里也有類似的劇情。如果到時候我人在日本的話打算去看。
王者天下3 真人電影(Kingdom 3)
我頗為喜歡《王者天下》的動畫,之前也聽說過有真人電影,但一直沒機會看。2023年9月初我在大阪的TOHO CINEMA意外地看到有這個真人版電影,就果斷看了,票價折合100 RMB。
這個真人電影講的劇情我印象中在動畫版里大部分都有,但是是很多很多年以前的劇情了,應該是王者天下第二季那會兒,講王騎率兵跟趙國交戰的事。王騎讓當時還是百人將的信去偷襲敵人后方,信經歷殊死拼殺后成功斬了敵軍總大將。故事中還回憶了嬴政少時被紫夏帶著逃離趙國時,路途中被追殺的那段故事。
這電影版的前1/3以宮殿里的對話為主,感覺比較沉悶,特別是對于聽不懂日語的人來說。嬴政擺脫追殺,以及后面的戰爭劇情,都做得很不錯,很有看頭。
王者天下原作里的角色普遍都很有個性,也令我這位動畫版老觀眾對此作的選角頗有看法。
王騎演的只能算是及格。那種爆棚的自信,以及骨子里的那種奇妙的幽默和妖嬈感,都演得不錯,但關鍵是欠缺威武感和壓倒性的氣場,他剛走進宮殿時我一瞬間都認不出他來。明明動畫里王騎是個形象高大的武將,結果這真人版里他只是個一般身高的角色,總大將的氣場實在是展現不出來,甚至覺得他是個有點滑稽的角色。
騰的演員基本沒說話,外形并不像動畫版那么顯得像洋人,不過總體氣質還是挺接近騰的,令我比較滿意。
嬴政演得還算可以,但眼神和智慧感和動畫版有明顯差距,80分吧。信演得說不上多出彩,但也能令我滿意,85分。
河了貂居然是橋本環奈演的。之前我覺得她演河了貂大抵只是因為橋本環奈的人氣才被選上,但看了此作之后感覺她演得不錯。橋本環奈長得可愛,而且個子又很小只,演河了貂真是很合適,討人喜歡,換成其她演員也不會明顯更好。可以打93分。可惜河了貂在此電影里主要就是打打醬油,沒什么戲份。
蒙武演得很不錯,長相和動畫版如出一轍,很有野性和霸氣的感覺。美中不足的就是演員個子還不夠高大,要不然就可以接近滿分了。
此作演員選的最糟糕的是羌瘣,完全不及格。完全沒有動畫版的氣質,演員很顯老,明顯不像是靈秀的少女,表情也挺木訥的,一點神采都沒有。不知道當初選角怎么選的,懷疑是不是當初還可以,但過了N年拍第3部的時候其容貌已經發生了很大變化,但又不方便中途換演員。
楊端和的演員還可以,雖然顏值達不到原作中她的level,但整體感覺也還可以了,不令我失望。可惜她在這一部里總共出現了也就10秒鐘,完全是怒領工資(似乎第1或第2部里她有不少細分)
紫夏演的很出色,可以給97分,顏值不錯,特別是演技很好,充分展現出她對嬴政的重視和愛護,最后她為了保護嬴政而被射死的時候相當壯烈。
呂不韋演的很爛,完全沒有動畫版里的氣場,感覺這真人版里他就是個糟老頭子,看不出有任何過人之處。完全不及格。
肆氏一出場就看得出是肆氏,發型很獨特,發際線有幾個尖角。
信的部隊里有好幾個很有個性的小兵,看動畫版的時候我就挺注意到他們,他們在這真人版里被演得活靈活現,這批又不起眼又不可忽視的小角色的選角很不錯。馬桶蓋、大齙牙、大棒槌、老漢等小兵,在此作里全都演得很鮮活。
邪神與廚二病少女 世紀末篇
類型:OVA
長度:23m40s
這是小邪神的OVA。這個OVA的內容繼續無厘頭,又挺有趣的。講述百合鈴成了城保町的獨cai者,然后小邪神到各個地方連哄帶騙呼朋喚友找幫手去找百合鈴對決,美其名曰是推翻獨cai、把百合鈴說得像拉奧似的,實際上就是想找個由頭再次搞掉自己的宿敵。這個OVA的一個巨大槽點是拳四郎居然出現在了此作中,北斗神拳的主題曲的令人印象深刻的伴奏都出現了。小邪神雖然從他那里學了暗殺拳,但底子太弱,再次不知天高地厚地挑戰百合鈴時果然又被虐。更想不到居然百合鈴還從從他面前路過的拳四郎那里學了暗殺拳,這回小邪神被她這招虐得比以往還要慘,腸子紛飛,還像烤魚一樣被木桿貫穿身體叉在公共場所示眾,好慘。小邪神真是從不長記性,如果有下一季肯定還是要沒完沒了作死。也不知道到底以什么方式才能徹底搞死小邪神,有種把她剁碎了包餃子被眾人吃了都還能復活的感覺。這OVA登場角色真夠多的,幾乎所有TV版的角色都出現了,CV成本肯定很高。
文/Sobereva@北京科音 2023-Dec-30
0 前言
耦合簇方法可以通過線性響應(linear response, LR)計算激發態。在常見范疇內,精度和耗時關系是LR-CC3>LR-CCSDR(3)≈EOM-CCSD(T)>LR-CCSD=EOM-CCSD>=CC2。LR-CC3如今被普遍視為是激發態計算的金標準(多參考特征很強以及雙電子激發特征占主導的情況除外),激發能能精確到零點零幾eV。但LR-CC3結合像樣基組只能算得動個位數原子,對更大體系則可以用明顯更便宜的LR-CCSDR(3),精度也已經非常好了。
Dalton程序在耦合簇方面功能豐富,可以做LR-CC3、LR-CCSDR(3)、LR-CCSD、LR-CC2激發能計算,對于LR-CC2/CCSD/CC3還可以得到包括振子強度在內的諸多躍遷屬性。Dalton的輸入文件對于初學者來說比較復雜抽象,本文用盡可能簡單易懂的語言完整介紹一下用免費的Dalton做這些激發態耦合簇計算的方法,使用有C2v對稱性的非常典型的分子甲醛為例。讀者如果對Dalton不熟悉,務必先閱讀我之前寫的《量子化學程序Dalton的編譯方法和運行方式簡介》(http://www.shanxitv.org/463),我假定讀者已經認真看過此文。
值得一提的是,雖然Dalton做LR-CC3計算如今在文獻中很常見,但Dalton在這方面不是最快的,而且代碼還沒有并行化。如果追求更好的效率,可以用專門做耦合簇計算的e^T程序(https://www.etprogram.org),它的LR-CC3是所有程序中明顯最快的。另外,LR-CCSD激發能和振子強度沒任何必要非得用Dalton計算,常用的Gaussian和ORCA的EOM-CCSD也都做得很好,結果和Dalton的LR-CCSD是等同的。Dalton做耦合簇的激發能計算不支持考慮溶劑模型,而G16的EOM-CCSD則可以。還值得一提的是,Dalton的衍生程序LSDalton還額外支持RI-CC2,可以比Dalton的LR-CC2更高效率地計算較大體系,這不屬于本文的范疇。
本文涉及的輸入輸出文件都可以在http://www.shanxitv.org/attach/693/file.zip里獲得。計算用的是2023-Dec-22通過git下載的Dalton最新的開發版。
本文下面的例子首先要重復知名的TBE1激發能測試集(J. Chem. Phys., 128, 134110 (2008))里的LR-CC3/def-TZVP算的甲醛的1-1A2、1-1B1、2-1A1三個單重態的垂直激發能。如文章表1所示,結果分別為3.95、9.18、10.45 eV。此文用的幾何結構是MP2/6-31G*級別優化的,對應的結構文件是file文件包里的H2CO.xyz,本文的計算也將基于這個結構。
1 確定不可約表示順序
為了指定各個不可約表示的激發態各算幾個態,需要先知道C2v點群的各個不可約表示在Dalton程序中的順序。最簡單的方法是做個DFT單點計算,看一開始輸出的不可約表示的順序。為此,我們用Multiwfn(http://www.shanxitv.org/multiwfn)創建這個任務的輸入文件。啟動Multiwfn,然后依次輸入
H2CO.xyz
100 //其它功能(Part 1)
2 //導出各種文件
19 //Dalton
DFT.dal //在當前目錄下產生DFT.dal,對應B3LYP/6-31G*計算設置
H2CO.mol //在當前目錄下產生Dalton格式的體系定義文件H2CO.mol
由于當前計算要考慮對稱性,因此把H2CO.mol里的Nosymmetry刪掉。然后基于DFT.dal和H2CO.mol做計算,從輸出文件DFT_H2CO.out里可以看到在SCF開始之前就輸出了當前自動判斷的點群,以及相應的各個不可約表示的順序
@ Full point group is: C(2v)
@ Represented as: C2v
@ * The irrep name for each symmetry: 1: A1 2: B1 3: B2 4: A2
2 做LR-CC3激發能計算
寫一個文本文件LRCC3.dal,內容如下(這是最簡單寫法,默認設置就已經適合的選項就沒再做額外設置)
**DALTON
.RUN WAVE FUNCTIONS
**WAVE FUNCTION
.CC
*CC INP
.CC3
*CCEXCI
.NCCEXCI
2 1 0 1
**END OF DALTON
對當前體系默認是做閉殼層HF計算得到參考態波函數。**DALTON控制任務類型,.RUN WAVE FUNCTIONS要求算單點并得到波函數。**WAVE FUNCTION設置波函數計算類型,.CC要求做耦合簇計算。*CC INP設置做什么形式耦合簇計算,.CC3要求做CC3計算。*CCEXCI要求做耦合簇的激發能計算,.NCCEXCI下面按照不可約表示的順序指定各個不可約表示的能量最低激發態各計算幾個。當前為了重復TBE1測試集的數據,要求A1算兩個(由此得到1-1A1和2-1A1),B1算1個(得到1-1B1),B2不算,A2算1個(得到1-1A2)。
然后把前述的H2CO.mol復制為H2CO_TZVP.mol,手動把里面的6-31G*替換為Turbomole-TZVP,這是Dalton內置的def-TZVP基組的寫法,對應Dalton目錄下的basis子目錄中的Turbomol-TZVP這個文件的名字。
然后基于LRCC3.dal和H2CO_TZVP.mol做計算,得到LRCC3_H2CO_TZVP.out。本節的文件都已經提供在了前述的file文件包里。打開輸出文件,從里面可以看到如下激發能信息,2-1A1為10.44842 eV,1-1B1為9.18343 eV,1-1A2為3.94711 eV,可見和TBE1原文里的10.45、9.18、3.95 eV完全一致。表中||T1||是單激發貢獻,和TBE1原文里給出的也是一致的。
+=============================================================================+
| sym. | Exci. | CC3 Excitation energies | ||T1|| |
|(spin, | +------------------------------------------------------------+
| spat) | | Hartree | eV. | cm-1 | % |
+=============================================================================+
| ^1A1 | 1 | 0.3502215 | 9.53001 | 76864.734 | 91.00 |
| ^1A1 | 2 | 0.3839723 | 10.44842 | 84272.170 | 91.32 |
+-----------------------------------------------------------------------------+
| ^1B1 | 1 | 0.3374848 | 9.18343 | 74069.360 | 90.93 |
+-----------------------------------------------------------------------------+
| ^1A2 | 1 | 0.1450537 | 3.94711 | 31835.610 | 91.16 |
+=============================================================================+
如果想要計算三重態激發態,就在.NCCEXCI下面的第二行定義。比如下面的寫法,代表除了計算如上那些單重態激發態以外,還計算1-3A2和1-3A1三重態激發態
.NCCEXCI
2 1 0 1
1 0 0 1
如果不需要計算單重態激發態,就寫
.NCCEXCI
0 0 0 0
1 0 0 1
筆者試了幾個Dalton版本,包括2016、2018、2022開發版,算出來的三重態激發能都是錯的,和TBE1表2里的1-3A2和1-3A1三重態激發能明顯不符,而且||T1||嚴重偏低,我認為是bug。如果不利用對稱性,即.mol文件里帶著Nosymmetry,并且.NCCEXCI下面直接指定計算的激發態的總態數,則問題輕得多,可以看到||T1||都顯著高于90%。
值得一提的是LR-CC3是對激發態一個一個獨立計算的,故算的態數越多耗時明顯越高。
如果只需要計算CC3基態能量,去掉*CCEXCI及下面的部分即可,相對于LR-CC3算激發態部分的耗時來說可以忽略不計。
3 做LR-CC3激發能+振子強度的計算
如果對上面算的激發態還要計算振子強度,就創建LRCC3mom.dal文件,寫入以下內容,然后進行計算。其中**INTEGRAL指定要算基函數之間哪些類型的積分,.DIPLEN要求算長度形式的偶極積分,這是因為算振子強度要算躍遷偶極矩,會用到這類積分。*CCOPA模塊用來計算耦合簇的基態到激發態的躍遷屬性,對當前用的Dalton版本支持CCS、CC2、CCSD、CC3,其中CC3僅限單重態激發態。.DIPOLE要求計算長度形式的躍遷偶極矩及振子強度。
**DALTON
.RUN WAVE FUNCTIONS
**INTEGRAL
.DIPLEN
**WAVE FUNCTION
.CC
*CC INP
.CC3
*CCEXCI
.NCCEXCI
2 1 0 1
*CCOPA
.DIPOLE
**END OF DALTON
從輸出文件(file文件包里的LRCC3mom_H2CO_TZVP.out)可以看到各個激發態的躍遷偶極矩和振子強度,比如2-1A1的如下所示,振子強度為0.34867845。這個值很接近TBE1原文表VII里這個態的LR-CC2和LR-CCSD振子強度(這篇文章里沒給CC3的振子強度),分別為0.368和0.374。
Transition from ground state to:
number, multiplicity, symmetry : 2 ^1A1
frequency : 0.3839722612 a.u. 10.44842 e.V. 84272.2 cm^-1
+-----------+-----------------+-----------------+---------------------+
| operator | left moment | right moment | transition strength |
+-----------+-----------------+-----------------+---------------------+
| XDIPLEN | 0.00000000 | 0.00000000 | 0.00000000 |
| YDIPLEN | 0.00000000 | 0.00000000 | 0.00000000 |
| ZDIPLEN | -0.83921304 | -1.62309631 | 1.36212358 |
+-----------+-----------------+-----------------+---------------------+
oscillator strength (length gauge) : 0.34867845
上面表格里的ZDIPLEN(偶極矩的Z分量算符)的transition strength值1.36212358對應的是躍遷偶極矩Z分量的平方。
如果你只需要躍遷偶極矩的特定分量,比如Z分量,可以把.DIPOLE改為
.OPERATOR
ZDIPLEN
這樣耗時會比計算所有分量時稍微低一點。
4 LR-CC2/CCSD/CCSDR(3)激發態計算
LR-CC2、LR-CCSD、LR-CCSDR(3)計算只需要把前面例子里的.CC3分別改成.CC2、.CCSD、.CCR(3)即可。注意LR-CCSDR(3)只能算激發能而無法算包括振子強度在內的躍遷屬性。
這些級別的激發能計算的輸入輸出文件在file文件包里的other目錄下都給了,耗時跟LR-CC3比都不值得一提。整體來說按LR-CC2、LR-CCSD、LR-CCSDR(3)的順序,結果和金標準LR-CC3的越來越接近。比如它們的1-1B1激發能分別為9.348、9.257、9.186 eV,LR-CC3的為9.183 eV。所以LR-CC3激發能很難算得動的時候用LR-CCSDR(3)是很好的平替。
5 凍核設置
Dalton默認是不凍核的。顯然恰當凍核可以節約耗時而幾乎不影響精度。這里演示對H2CO做LR-CC3計算時凍結能量最低兩個分子軌道(對應C和O的1s軌道)的方式。首先得得知它們的不可約表示,從之前B3LYP/6-31G*的輸出文件中可以看到各個占據軌道能量的負值(Koopmans定理對應的電離能,相關知識參考《正確地認識分子的能隙(gap)、HOMO和LUMO》http://www.shanxitv.org/543)。
@ Summary of DFT KT ionization potentials [eV]:
@ Symmetry 1 (A1 ) : 521.511 279.798 28.621 17.429 12.139
@ Symmetry 2 (B1 ) : 10.711
@ Symmetry 3 (B2 ) : 13.454 7.303
@ Symmetry 4 (A2 ) : No IPs in this symmetry
顯然能量最低兩個軌道是A1。
寫一個LR-CC3激發能計算輸入文件LRCC3_FC.dal,內容如下。其中.FROIMP下面第一行指定對各個不可約表示凍結多少個能量最低占據軌道,下面第二行指定對各個不可約表示凍結多少個能量最高空軌道。當前凍結的是能量最低的兩個A1軌道。
**DALTON
.RUN WAVE FUNCTIONS
**WAVE FUNCTION
.CC
*CC INP
.CC3
.FROIMP
2 0 0 0
0 0 0 0
*CCEXCI
.NCCEXCI
2 1 0 1
**END OF DALTON
從file文件包里的此任務的輸出文件LRCC3_FC_H2CO_TZVP.out可看到,現在凍核后耗時是1分49秒,1-1B1激發能是9.18872 eV,之前沒凍核時是3分32秒,1-1B1激發能是9.18343 eV。明顯凍核對激發能的影響可完全忽略不計,而耗時大為降低,因而有重要實際意義。