使用AICD程序研究電子離域性和磁感應電流密度
注:后來發布了AICD 2.0,安裝和使用比此文介紹的老版本方便得多,因此一定要看此文:《使用AICD 2.0繪制磁感應電流圖》(http://www.shanxitv.org/294)
使用AICD程序研究電子離域性和磁感應電流密度
文/Sobereva @北京科音
First release: 2012-Apr-7 Last update: 2015-Jan-26
1 關于磁感應電流密度
在中學就學過,在外加磁場下,閉合導體會產生感應電流阻礙原磁通量變化
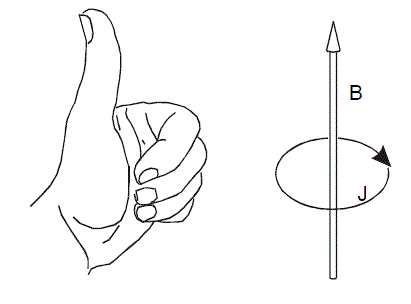
對于分子體系也是一樣,如果體系中電子在整體或者某個部分有很強的離域性,或者說處在這樣區域的電子能夠很自由地運動,比如苯環的pi電子,那么在外加磁場時,會在相應區域會產生一圈明顯的感應環形電流,在相關的原子附近會有較大的電流密度。因此,研究外加磁場下的分子內電流密度分布,對于考察體系的電子離域性,研究芳香性很有幫助。環電流方向與上圖左手規則相同時(稱為diamagnetic或diatropic環電流),電流越大,則芳香性越強;而電流方向越與左手規則相反時(paratropic環電流),電流越大則表現出越強的反芳香性。如果沒有產生明顯凈電流,就是非芳香性。
除了共軛環狀體系,還有人研究了富勒烯在磁場下的誘導電流,被稱為球電流,有興趣者可參閱http://onlinelibrary.wiley.com/doi/10.1002/anie.200462348/abstract
2 關于AICD
感應電流密度是一個矢量場,直接繪制出三維矢量場圖來不便于觀察。通常研究的是一些截面上的感應電流密度二維矢量圖,比如截面可以選取苯環上方1埃的平面用于研究pi電子的感應電流,而直接選取苯環平面的話可以研究sigma電子的感應電流。對于規則的立體體系,比如富勒烯,也可以定義一個球面,比如可以繪制富勒烯球面上1埃的球層上的感應電流密度矢量進行研究。有時候也對感應電流進行定量研究,比如可以積分穿越某個截面的感應電流密度獲得感應電流凈流量。
然而,對于形狀不規則、扭曲的分子,通過選取截面來研究感應電流密度就很不方便了,沒法一目了然地表現出各個位置感應電流密度的大小和方向。而且感應電流密度在電子密度大的地方大,原子核附近電流密度容易掩蓋原子間的電流密度,不便于直接考察電子離域性。另外感應電流密度也與外磁場方向相關,對于復雜體系怎么選取外磁場方向比較好也很難說。
為了提供一個更方便的通過分子磁性質研究離域性、芳香性的途徑,在JPCA,105,3214中作者定義了AICD函數,全稱是Anisotropy of the Induced Current Density,它的定義是
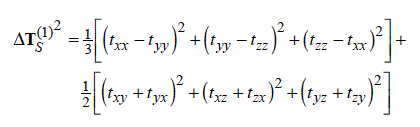
其中t是電流密度張量(3*3矩陣),在空間不同位置是不一樣的。將這個張量與外磁場矢量做點乘,得到的矢量就是感應電流矢量。
AICD本質上反映的是相應位置電子對磁場感應的各項同性的強度。這是一個三維實空間的標量函數,它的大小與電子密度也沒有直接關系,而且不依賴于外場的選取,所以可以方便地通過等值面來研究它的分布以此了解電子離域性。
3 安裝AICD程序
AICD程序是用于繪制AICD等值面和感應電流矢量的程序。它需要讀取Gaussian98/03的NMR計算輸出的電流密度張量信息,然后生成AICD的cube文件和povray渲染器的輸入文件。AICD可以找AICD方法的原作者免費索取(郵件地址見Chem. Rev., 105, 3758上的聯系信息),我也將之傳到了此處/usr/uploads/file/20150609/20150609160036_17152.bz2,這是1.5.7.1版。如果在文章中使用的話還是建議申請一份。
AICD的安裝方法很簡單,將壓縮包解壓,進入其目錄中,運行make all即可,很快就編譯完了,用gnu的編譯器就可以。然后為了方便建個符號鏈接ln -s ./AICD /usr/local/bin/AICD。
使用AICD必須修改Gaussian的源代碼并重新編譯其l1002模塊,以使Gaussian在NMR計算中能輸出電流密度張量信息。這里只考慮Gaussian03版的情況。首先用戶必須自行編譯一遍Gaussian03,對于具體版本號有沒有要求我并不清楚,嚴格按照此文的方法http://www.shanxitv.org/2在Fedora7下編譯G03 C.02 32bit版是肯定兼容AICD的。
在編譯完Gaussian03后,將l1002.F.g03.diff從AICD-1.5.7.1/gaussian_patch目錄中拷到g03目錄下,建議將原先l1002.F和l1002.exe備份一下。
在g03目錄中執行
patch < l1002.F.g03.diff
這樣l1002.F就按l1002.F.g03.diff中的定義進行了修改。
AICD分析是可以分解為各個軌道的貢獻的,具體來說是g03可以正常輸出指定的分子軌道產生的電流密度張量,然而不知為什么在l1002.F.g03.diff中沒有啟用這個功能。如果想啟用這個功能,則應該在patch之后在l1002.F中搜索Call FFRead(IEOF),將它后面的goto 12那行刪掉。然后搜索If(V(IOccMO+I-1).eq.Two) then,在Write(IOut,1010) I這行后面插入以下兩行:
else
Write(IOut,1020) I
AICD程序里大部分注釋是德文,遇見語言障礙時可以使用google翻譯等工具幫助理解。
將之前編譯Gaussian殘留的l1002.a刪掉,原先的l1002.exe改個名作為備份。然后進入csh環境,進入g03目錄,運行source bsd/g03.login,然后執行mg l1002.exe就重新編譯出了l1002.exe。這樣,當Gaussian03中使用NMR=CSGT關鍵詞計算時,在l1002模塊中就會額外輸出電流密度張量、權重和格點位置信息。
4 使用AICD程序的使用
AICD程序并沒有手冊,它的用法可以通過AICD --longhelp命令來查看,但內容比較抽象簡略。我們這里以分析苯為實例進行說明,輸入文件如下。一般情況下,AICD分析使用B3LYP/6-31G*的計算級別就能得到比較有意義的結果。
#P b3lyp/6-31G* NMR=csgt
b3lyp/6-31+g(d) opted
0 1
C 0.00000000 1.39864200 0.00000000
C 1.21125900 0.69932100 0.00000000
C 1.21125900 -0.69932100 0.00000000
C 0.00000000 -1.39864200 0.00000000
C -1.21125900 -0.69932100 0.00000000
C -1.21125900 0.69932100 0.00000000
H 0.00000000 2.48606800 0.00000000
H 2.15299800 1.24303400 0.00000000
H 2.15299800 -1.24303400 0.00000000
H 0.00000000 -2.48606800 0.00000000
H -2.15299800 -1.24303400 0.00000000
H -2.15299800 1.24303400 0.00000000
17 17
20 21
最后的兩行代表要考慮的軌道范圍。17 17代表選擇17號軌道,20 21代表選擇20和21軌道。這三個軌道是苯的三個pi軌道。苯總共有21個雙占據的分子軌道,如果要考慮的是所有sigma軌道,就應該寫
1 16
18 19
如果什么都不寫,就代表考慮所有軌道。
用g03運行此輸入文件,假設輸出文件名叫benzene_AICD.out。從中可以找到這樣的信息
...
Orbital Nr. 15 wird nicht beruecksichtigt.
Orbital Nr. 16 wird nicht beruecksichtigt.
Orbital Nr. 17 wird beruecksichtigt.
Orbital Nr. 18 wird nicht beruecksichtigt.
Orbital Nr. 19 wird nicht beruecksichtigt.
Orbital Nr. 20 wird beruecksichtigt.
Orbital Nr. 21 wird beruecksichtigt.
這表明17、20、21號軌道都考慮了,而其它軌道對電流密度張量的影響都忽略了。在這些內容后面輸出了很長一堆數據,內容包含Gitterpunkte(格點)、Gewichtungsfaktoren(權重因子)、Stromdichtetensor(電流密度張量)。
然后執行AICD -p 80000 -pov -b 0 0 1 -m 4 benzene_AICD.out。
這里-p后面的是格點總數目,越大圖像越精細,cube文件也會越大,分析耗時也越長。-pov代表輸出povray的輸入文件。-b設定的是磁場方向矢量,結果只受矢量方向影響,而不受此矢量的大小影響。-m 4代表輸出的圖像將包含多個視角,如果是-m 2則只包含一個視角。
還有幾個重要的選項也一并說明,其它選項不太重要,請自行摸索。
-l xxx將AICD的等值面設為xxx,默認是0.05。設的越小等值面越大,太小的話會導致等值面被盒子邊緣所截斷而出現窟窿。
-s 使povray渲染的AICD等值面變得平滑,但是AICD要多花很多倍計算時間,體系越大耗時比越大。這并不會對生成的cube文件有任何影響。
-rot a b c 用于調節視角方向,例如90 0 0就是做90度旋轉。這三個量的設定需自行反復嘗試
-plane ox oy oz nx ny nz 通過一個點的坐標(ox oy oz)和面的法矢量(nx ny nz)定義一個平面,povray渲染的AICD等值面將會在這個面上被截斷。
-nc 不輸出AICD的cube文件
-maxarrow length 去除長度超過length的電流密度箭頭。有時候由于數值問題某些箭頭會格外的長。設為2.0往往比較合適。
執行上述命令后AICD會把out文件里的電流密度張量、權重和格點位置都抽取出來到同目錄下.icd二進制文件里,原先的out文件里這些信息就沒了。.icd文件再被后續分析處理。分析速度很快,除非用了-s選項。分析結束后會產生如下文件
benzene_AICD.icd.gz:benzene_AICD.icd的壓縮版本
benzene_AICD.icd80000.gz:這個體系的AICD格點數據的二進制文件
benzene_AICD.icd80000.cub:這個體系的AICD的cube文件。可以通過VMD、Multiwfn、chemcraft、molekel等將之載入觀看AICD等值面。
benzene_AICD.pdb:分子結構文件
以下五個都是povray渲染器輸入文件。文件名和AICD執行參數有關。
benzene_AICD_80000_0.050_0_0_1_Aniso_4.4.Molekuel.inc
benzene_AICD_80000_0.050_0_0_1_Aniso_4.4.Isoober.inc
benzene_AICD_80000_0.050_0_0_1_Aniso_4.4.Rotate.inc
benzene_AICD_80000_0.050_0_0_1_Aniso_4.4.Molekuel.pov
benzene_AICD_80000_0.050_0_0_1_Aniso_4.4.RenderMich.pov
如果還沒裝povray渲染器,可以在這里下載:http://www.povray.org/download/,筆者用的是3.7beta windows版。將這五個povray渲染器輸入文件都拷貝到windows下的某個文件夾中,雙擊后綴是.RenderMich.pov的那個文件,在povray窗口左上角設定好分辨率以及是否開啟抗鋸齒(AA),然后點擊Run按鈕即可渲染出圖像,看到的圖像會同時保存到當前目錄下的png文件中,如下所示
3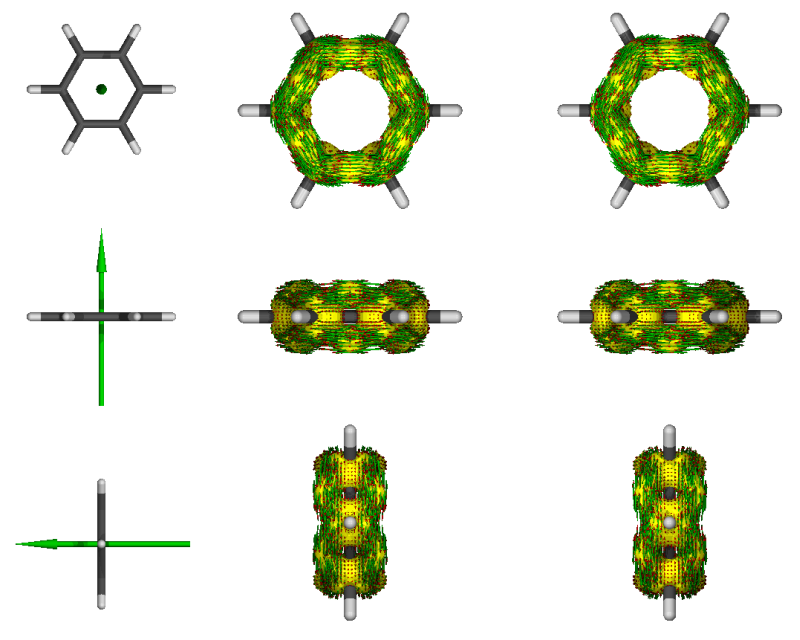
黃色的是AICD等值面,等值面上的箭頭是相應位置感應電流密度矢量,綠色大箭頭是磁場方向。圖太小可能看不清楚,可以在渲染時選擇更高分辨率,也可以在運行AICD命令時將-m 4改為-m 2只顯示其中一個視角。
我們將5個povray輸入文件刪掉,其它文件不動,執行下面的命令就會產生只含單個視角的povray渲染器輸入文件
AICD -p 80000 -pov -b 0 0 1 -m 2 -s benzene_AICD.out
由于已經有了benzene_AICD.icd.gz,程序就不會再試圖從benzene_AICD.out尋找電流密度張量、權重、格點位置并抽取(實際上.out文件中這些信息也已經在上次運行AICD時被截去了)。渲染結果如下
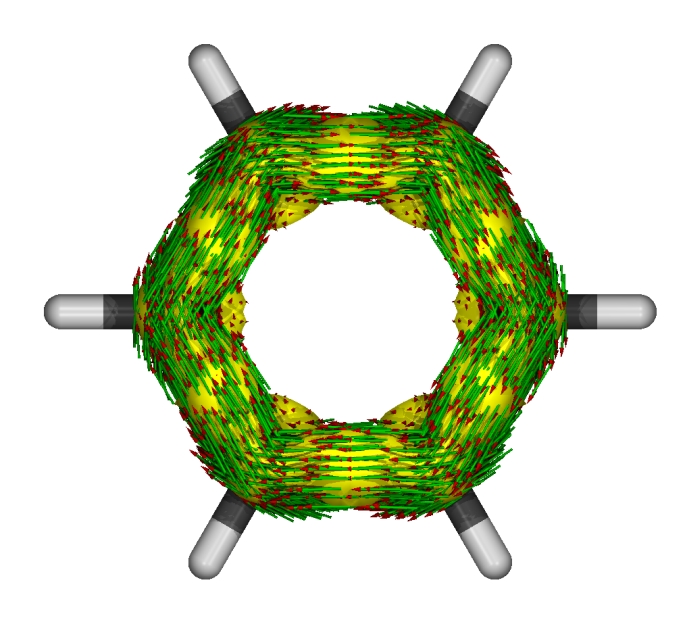
繪制單個視角的圖像時并不會顯示磁場矢量箭頭。但是對照前面多視角的圖可以知道此圖中磁場方向是從屏幕里面指向屏幕外面的。可清楚地看到感應電流方向都是滿足左手規則,展現了苯的pi電子的芳香性。
由于AICD運行時加上了-s選項,所以這次AICD運行耗時長了不少,如果去掉-s選項,會看到渲染出來的圖的等值面不很平滑,而像是多邊形構成的。
在Gaussian輸出文件中將所選軌道設為全部sigma軌道,重新計算,并用AICD分析,繪制出只表現sigma電子的AICD等值面和感應電流密度圖:
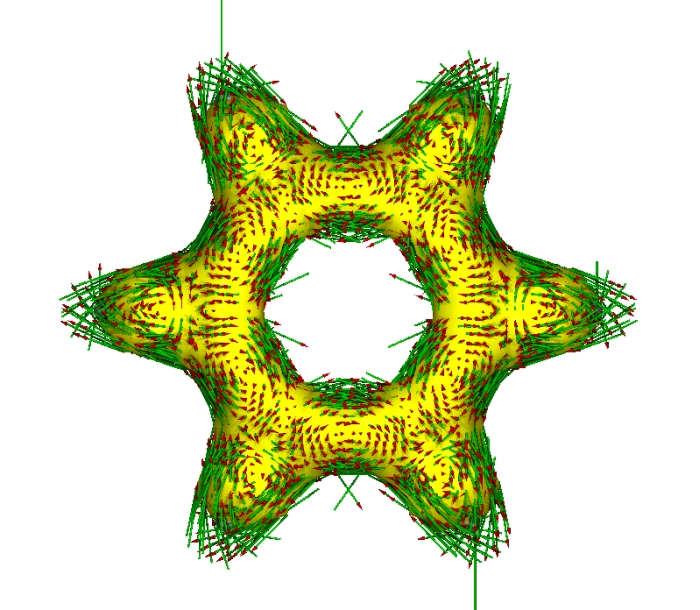
可見,sigma電子并沒有整體離域,而是在局部空間有較大的定域性。在C-C成鍵區域和H原子附近,感應電流密度矢量產生了局部的環流。圖中左上和右下出現了兩個很長的箭頭超出作圖范圍,那是程序bug,不用管它。想去掉的話可以利用-maxarrow參數。
只考慮pi電子、只考慮sigma電子和考慮所有電子時做出來的側視圖如下,依次從上到下
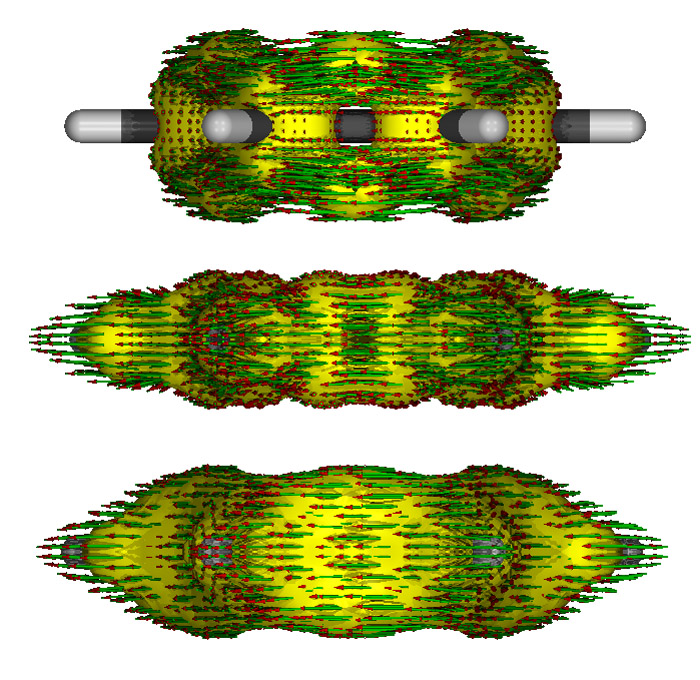
可見,只考慮pi電子時在氫上是沒有AICD等值面的,在C-C sigma鍵的區域也是空著的,這也是理所應當的。只考慮sigma電子時AICD等值面比較扁,sigma鍵區域都被包裹住。考慮所有電子時混合了兩種等值面的特征。
5 其它
AICD有個毛病,就是輸出的圖像可能是當前分子的對應異構體!大家務必注意檢查一下是否是這樣,如果是的話,應自行在圖像編輯軟件里把圖像左右翻轉一下使之對應于原本的分子。
如果用了nosymm關鍵詞的話,AICD程序沒法正確地從Gaussian輸出文件中提取分子坐標,這就需要手動把Gaussian輸出文件里的Input orientation:改為Standard orientation:。
如果想在渲染時讓視野更大一些,可以將.RenderMich.pov文件開頭的location <0, 0, -250>中的-250進行修改,越負視野越大。如果想調節分子朝向,可以在運行AICD時利用-rot選項進行設定,或者直接修改已經生成的.Rotate.inc里面的三個量,和使用-rot是等效的。
在渲染時,默認情況是使用四個點光源對體系進行照明。但如果體系比較長,光源就沒法很好地覆蓋到邊緣。可以自行調整.RenderMich.pov文件開頭的light_source里的<>的數值來改變光源位置。也可以用平行光源,來使整個體系都能被照到,即把原先的light_source用//注釋掉,然后新增一行light_source { < 0, 0, -160 > color rgb < 1.15, 1.15, 1.15 > parallel point_at < 0, 0, 2 >}。rgb里的數值設得越大,光線越強。
使用-m 4時,如果體系略大,在產生的圖像中不同視角的圖像會發生重疊。為避免這個問題,可以修改.RenderMich.pov文件,下面這樣的段落定義的是每個分子結構和磁場示意圖在圖像中的位置和旋轉角度
{ MolUndMag
rotate < 90,0,0 >
translate < -50,0,0 >
}
而諸如下面這樣的則是定義每個AICD子圖在圖像中的位置和旋轉角度
object
{ MolUndIso
rotate < 90,0,0 >
translate < -10,0,0 >
}
如果要研究一批體系,懶得每次都修改的話,可以直接修改模板文件,也就是直接改AICD目錄下的povray目錄里的RenderMich-4.4.pov文件中的相應參數。
如果想只顯示代表電流的箭頭,不顯示等值面的話,在.RenderMich.pov中把以下內容刪掉即可:
object
{ Isooberflaeche
texture
{ pigment { color Yellow }
finish { Plastic }
}
}
本文主要目的是介紹AICD程序的使用方法,限于時間和精力,本文不打算給出AICD的應用例子,建議讀者閱讀AICD綜述性文章Chem. Rev., 105, 3758,這篇文章中作者用AICD方法研究了大量體系。