使用ORCA做從頭算動力學(AIMD)的簡單例子
使用ORCA做從頭算動力學(AIMD)的簡單例子
Sobereva@北京科音
First release: 2020-Dec-13 Last update: 2021-Jul-7
1 前言
基于量子化學方法的動力學一般稱為從頭算動力學(Ab initio molecular dynamics, AIMD),相比于基于一般的經典力場的動力學,其關鍵優勢在于精度高、普適性強、能夠描述化學反應,代價是耗時相差N個數量級。ORCA量子化學程序有不錯的做BOMD形式的從頭算動力學的功能,使用很方便,而且本身ORCA做DFT的效率又高,是做孤立體系AIMD的首選程序之一。雖然有些特性不支持,比如沒法像Gaussian的BOMD那樣直接做準經典動力學,不能根據原子距離等標準判斷什么時候自動結束任務等等,但都不是大問題。對于跑跑普通的AIMD來說,筆者感覺ORCA明顯比Gaussian的ADMP或BOMD更好用(Gaussian的AIMD輸入文件較為抽象,手冊相應部分寫得很爛,而且連個像樣的熱浴都沒有),而且速度也明顯更快。筆者近期發表的文章中對18碳環(cyclo[18]carbon)單體和二聚體做的分子動力學就是用ORCA跑的,分別見Chem. Asian. J. (2020) DOI: 10.1002/asia.202001228和《全面探究18碳環獨特的分子間相互作用與pi-pi堆積特征》(http://www.shanxitv.org/572)中對Carbon, 171, 514 (2021)一文的介紹,可以作為ORCA跑AIMD的范例。
筆者在北京科音(http://www.keinsci.com)的高級量子化學培訓班中會用多達兩百多頁的ppt專門深入詳細講AIMD的模擬,其中也包括ORCA做AIMD的各種細節、大量技巧以及諸多實例。本文只是舉一個簡單的例子,幫助讀者基本了解ORCA如何做AIMD。弄明白這個例子,結合手冊舉一反三,跑其它體系也就沒什么難的了。如果對ORCA一無所知的話,點擊http://www.shanxitv.org右側的“ORCA”分類,筆者之前寫過不少相關文章。ORCA只適合跑孤立體系的從頭算動力學,如果是做周期性體系的從頭算動力學,CP2K是最佳的選擇,在筆者講授的北京科音CP2K第一性原理計算培訓班(http://www.keinsci.com/workshop/KFP_content.html)中有極為全面、系統、詳細的講解并給了大量例子。
本文內容適用于Multiwfn最新版本、VMD 1.9.3、ORCA 4.2.x及以后版本的情況。
2021-Jul-7 針對ORCA 5.0的補充說明:對于2021-Jul-7及以后更新的Multiwfn,進入本文所述的Multiwfn產生輸入文件的界面后,可以通過選項-11選擇適合的ORCA版本,默認為ORCA 5.0及以后版本,而下文內容對應的是4.2.x版的情況。對于>=5.0版,Multiwfn自動設的熱浴是CSVR,比之前版本唯一支持的Berendsen熱浴更好,而且同樣普適。而且在run后面多加了CenterCOM,這是從5.0版本開始支持的消除整體質心運動的選項,而下文里提到的constraint add center這一行就沒有了。
2 實例:[Al(H2O)6]3+與NH3之間的質子轉移
ORCA的MD模塊的開發者網站上有個真空中[Al(H2O)6]3+與NH3之間的質子轉移的動畫,如下所示
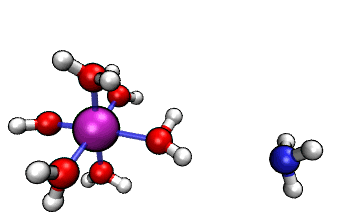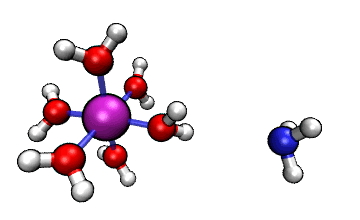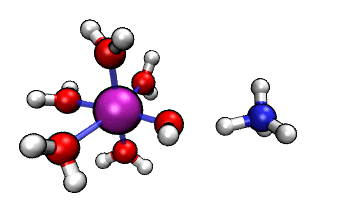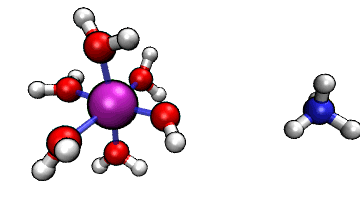
可見NH3向帶正電的[Al(H2O)6]3+逐漸靠近,水上的一個質子轉移到了氨氣分子上,然后由于靜電互斥,NH4+就逐漸遠離[Al(H2O)5(OH)]2+了。這里我們試圖重現這個過程。下面提到的文件都可以在http://www.shanxitv.org/attach/576/file.zip中獲得。
我們首先獲得[Al(H2O)6]3+的基本合理的結構。當然用ORCA優化也可以,這里筆者習慣性地用Gaussian來優化。在GaussView里搭建Al(H2O)6,保存為Al(H2O)6_optfreq.gjf,將關鍵詞改為# B3LYP/6-31G* opt freq,將電荷改為3,然后用Gaussian運行之,就得到了優化后的[Al(H2O)6]3+結構。再用GaussView打開輸出文件Al(H2O)6_optfreq.out,把一個氨氣分子畫在一個水的旁邊,如下所示,然后保存為complex.gjf。
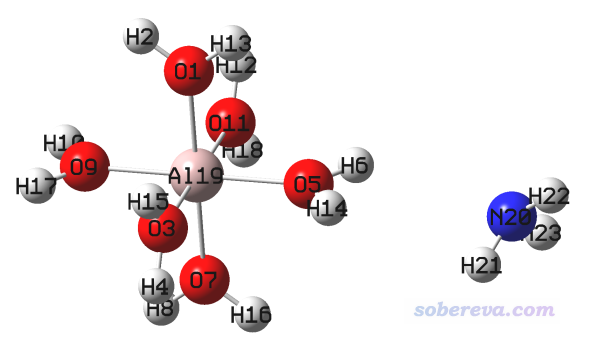
Multiwfn程序(http://www.shanxitv.org/multiwfn)有很便利的生成ORCA常見任務的輸入文件的功能,見《詳談Multiwfn產生ORCA量子化學程序的輸入文件的功能》(http://www.shanxitv.org/490),這里我們用Multiwfn生成ORCA的AIMD任務的標準輸入文件。
啟動Multiwfn,載入complex.gjf,然后輸入
oi // 生成ORCA輸入文件
0 // 選擇任務類型
6 // 分子動力學
1 // 計算級別用B97-3c
在當前目錄下就得到了ORCA的AIMD任務的標準輸入文件complex.inp。
在complex.inp里面將相應幾行改成下面這樣:
%maxcore 10000
%pal nprocs 10 end
timestep 1.0_fs
initvel 298.15_K
* xyz 3 1
現在的complex.inp的完整內容如下。以#為開頭的行代表后面的設置被注釋掉了,不會生效,想啟用可以去掉#。此文件里也有大量Multiwfn自動添加的注釋,只要一看注釋馬上就明白相應的行是什么含義,巨貼心,都省得查手冊了。
! B97-3c noautostart miniprint nopop
%maxcore 10000
%pal nprocs 10 end
%md
#restart ifexists # Continue MD by reading [basename].mdrestart if it exists. In this case "initvel" should be commented
#minimize # Do minimization prior to MD simulation
timestep 1.0_fs # This stepsize is safe at several hundreds of Kelvin
initvel 298.15_K no_overwrite # Assign velocity according to temperature for atoms whose velocities are not available
thermostat berendsen 298.15_K timecon 30.0_fs # Target temperature and coupling time constant
dump position stride 1 format xyz filename "pos.xyz" # Dump position every "stride" steps
#dump force stride 1 format xyz filename "force.xyz" # Dump force every "stride" steps
#dump velocity stride 1 format xyz filename "vel.xyz" # Dump velocity every "stride" steps
#dump gbw stride 20 filename "wfn" # Dump wavefunction to "wfn[step].gbw" files every "stride" steps
constraint add center 0..22 # Fix center of mass at initial position
run 2000 # Number of MD steps
end
* xyz 3 1
[坐標部分]
*
下面簡單說一下complex.inp里這些設置的含義、為什么這么設。
? B97-3c是一個又便宜又不錯的計算級別,在ORCA里還自動會啟用RIJ加速,速度很快,因此很適合跑AIMD,描述當前體系沒有問題。B97-3c的介紹見《詳談Multiwfn產生ORCA量子化學程序的輸入文件的功能》(http://www.shanxitv.org/490)的相應部分。但B97-3c也不是什么時候都能用,比如18碳環用純泛函描述皆失敗,見筆者在Carbon, 165, 468 (2020) 里的討論,顯然就不能用這個了,筆者跑涉及18碳環的AIMD的時候都用的是ωB97X-D3。
? %pal nprocs 10 end代表用10核。筆者當前任務實際上是在雙路E5-2696v3共36個物理核心的機子上跑的,但卻故意用了10核。這是因為根據筆者以前的測試,發現ORCA做DFT的AIMD的并行效率不理想,尤其是對于小體系,用核數太多甚至反倒速度更慢(大家可以對實際情況短暫跑比如5步實測一下設多少核速度最快)。一般來說就設10核就行了,當機子里有明顯更多核的時候,可以跑多個AIMD任務來充分利用計算資源,但應當對CPU內核進行綁定,否則AIMD計算速度可能顯著降低,見《通過設置CPU內核綁定降低ORCA同時做多任務的耗時》(http://www.shanxitv.org/553)。
? %maxcore 10000代表每個ORCA進程最多用10000MB。其實完全沒必要這么大,普通泛函下的AIMD不怎么耗內存,給1000都絕對夠了。
? restart ifexists這句被注釋掉了。如果你的AIMD想續跑,且當前目錄下有之前跑出來的與當前任務同名的后綴為.mdrestart的文件,可以取消注釋,任務就會延續之前AIMD最后的狀態續跑,新軌跡會在原有軌跡文件后面續寫。
? minimize這句被注釋掉了。如果取消注釋的話,動力學開始前會自動在當前級別下做幾何優化。
? timestep 1.0_fs代表動力學步長用1 fs。對于此例常溫下的模擬,1 fs步長是合適的,改大的話可能造成動力學不穩定,而改小的話跑同樣的時間長度需要更多步數導致需要更多耗時。如果追求絕對穩妥,或者是在明顯更高溫度下模擬,可以用0.5 fs。
? initvel 298.15_K代表根據298.15 K溫度通過Maxwell分布隨機初始化原子速度。no_overwrite代表如果之前已經有初速度信息了(比如可以是續跑時從之前的mdrestart文件里繼承來的),就不再產生新的初速度。
? thermostat berendsen 298.15_K timecon 30.0_fs代表用Berendsen熱浴控溫在298.15 K(此例筆者用的ORCA 4.2.1只能用這個熱浴,據悉從ORCA 5.0開始可以用更好的velocity rescale熱浴),時間常數為30 fs,通常這個時間常數是合適的。
? dump position stride 1 format xyz filename "pos.xyz"代表每一步都往當前目錄下的pos.xyz文件里寫入當前的坐標,因此pos.xyz是多幀xyz格式的軌跡文件。此格式的介紹見《談談記錄化學體系結構的xyz文件》(http://www.shanxitv.org/477)。
? dump force和dump velocity開頭的行都被注釋掉了,這兩行用于把模擬過程中的原子受力和原子速度分別保存到相應xyz文件里。dump gbw開頭的行也被注釋掉了,它可以在MD過程中每隔指定步數保存一次gbw文件,之后可以通過寫腳本調用Multiwfn對它們進行批量分析,從而考察動力學過程中電子結構變化,得到豐富的有化學意義的信息(如動力學中的電荷轉移情況、成鍵變化情況、電子定域性變化等等,在北京科音高級量子化學培訓班里筆者會給出很多這種例子)
? constraint add center 0..22代表將當前整個體系(0號到22號原子)的質心約束在初始位置。之所以這么做,是因為盡管ORCA在產生初速度時已經去掉了整體平移的速度分量,但實際模擬過程中由于數值問題,仍然往往會看到有明顯的整體漂移的現象,因而有礙觀察(在VMD里觀看軌跡時還得做一下align才能消掉)。因此直接把質心位置約束住就沒有這個問題了。
? run 2000代表總共跑2000步,當前步長是1 fs,即最多跑2 ps。當前這個模擬的目的是觀察到質子轉移,跑多長時間合適并不好預估,所以可以一次先跑2000步看看,若不夠到時候再續跑。值得一提的是,至少對于筆者現在用的ORCA 4.2.1,我發現如果一次跑的步數很多,達到2000步左右之后,之后每一步的耗時會有逐漸的上升趨勢,原因不明。因此我建議每次跑最多不宜超過3000步,如果需要跑更長的話,最好分多段來跑。
現在用ORCA運行complex.inp,模擬過程中可以看到每一步的實時情況:
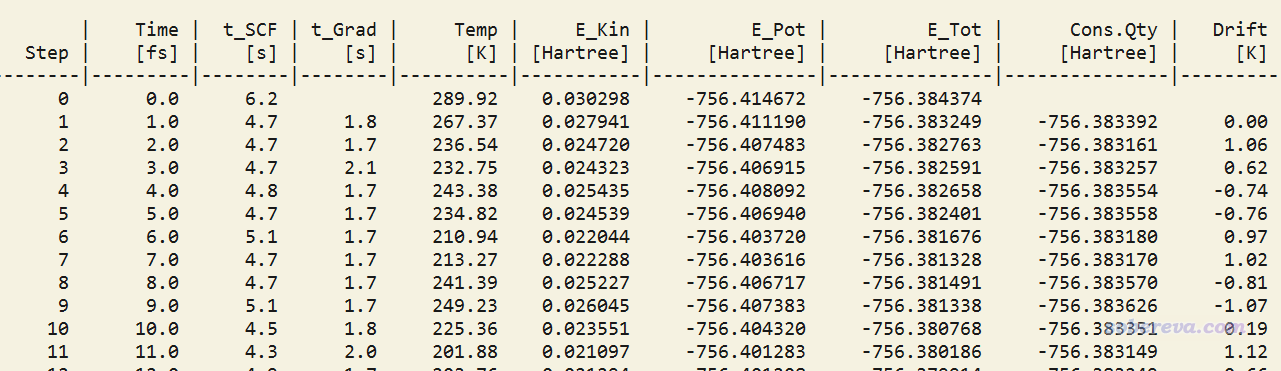
Step是當前的步數,Time是當前的時間,t_SCF和t_Grad分別是算這一步的SCF過程和計算受力的耗時,二者加和就是這一步的總耗時。可見每一步耗時約6~7秒,乘以要跑的步數就可以估計跑完整個任務的耗時。后面還可以看到每一步的溫度、動能、勢能等信息。由于當前體系原子數很少,溫度相對于熱浴的參考溫度波動大是很正常的事。而且由于用了熱浴,所以總能量E_tot也有明顯波動。
VMD是觀看動力學軌跡最好的程序,可以在http://www.ks.uiuc.edu/Research/vmd/免費下載。建議在模擬過程中,隔一陣子就用VMD把pos.xyz載入進去,看看當前的動態行為如何、跑成什么結構了。跑到289步的時候,筆者看了一眼pos.xyz,發現質子不僅已經完全轉移,而且NH4+都已經跑走一定距離了,所以就沒必要繼續跑了,故把ORCA直接殺掉了。
模擬過程中ORCA還產生其它一些文件。complex.md.log是ORCA的MD模塊輸出的信息,相當于整個輸出文件中的子集。complex.mdrestart是用來續跑的文件,每一步都會往里面寫入當前步的時間、坐標、速度、受力等信息。complex-md-ener.csv是把每一步的時間、耗時、溫度、能量等信息以csv格式保存的文件。還有其它一些零碎的文件,通常不是一般用戶關心的,這里就不說了,都可以放心刪掉,留著也沒用。
現在我們來看模擬軌跡。建議大家根據《VMD初始化文件(vmd.rc)我的推薦設置》(http://www.shanxitv.org/545)里說的修改VMD的初始化文件,從而添加自定義命令bt,這樣在播放軌跡的時候對每一幀會自動重新判斷成鍵關系。
啟動VMD,載入pos.xyz(在本文文件包里已提供,共290幀)。然后在文本窗口輸入bt,按回車,使得每幀都更新連接關系。然后再輸入bw,按回車使得背景變為白色。選Graphics - Representation,將Drawing Method改為CPK。然后點擊VMD界面右下角的三角播放動畫,會看到如下結果。如果想在VMD圖形窗口中顯示出幀號或時間,看《使VMD播放軌跡時同步顯示幀號》(http://www.shanxitv.org/13)。
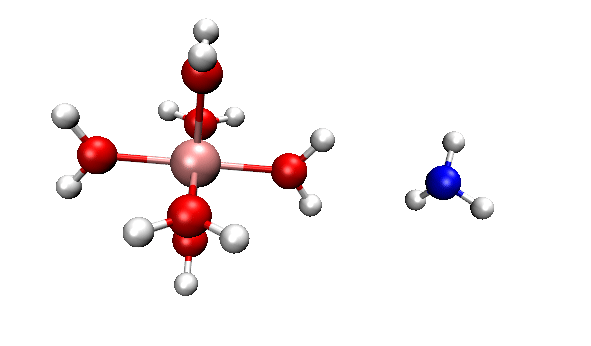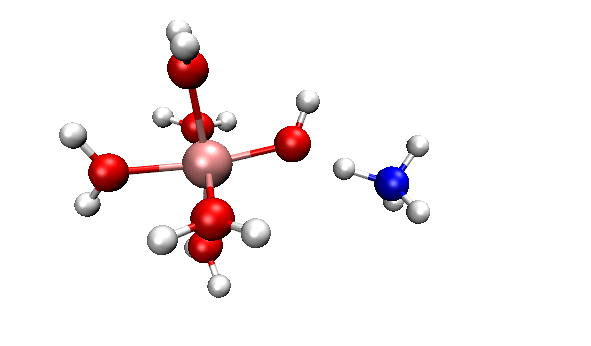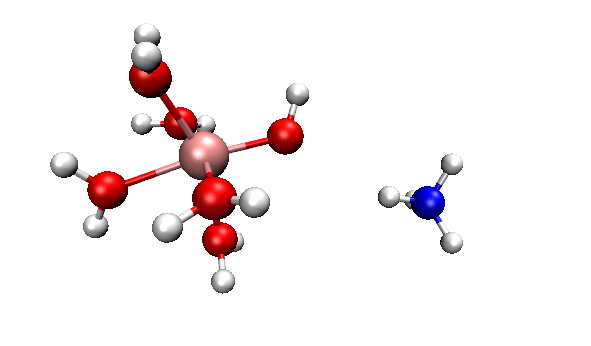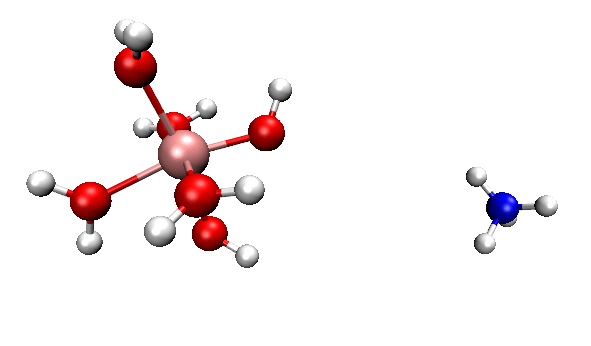
可見,我們跑出來的動力學現象和本文一開始的那個官方動畫幾乎完全一致。都是兩個反應物先接近,然后形成復合狀態時質子在二者之間震蕩,最后NH4+跑掉。
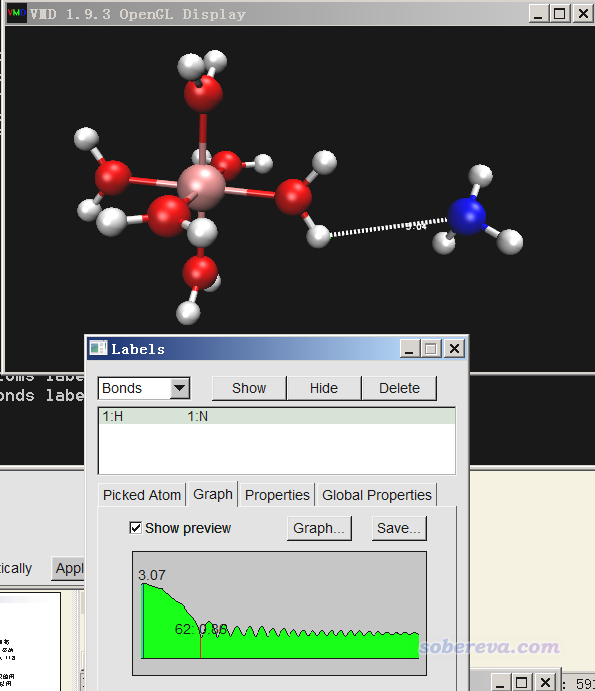
如果想把質子轉移情況通過距離隨時間的變化曲線方式呈獻給讀者,可以在點擊VMD的三維圖形窗口后,按鍵盤上的2鍵(之后按r可以恢復為默認的旋轉模式),然后點擊兩個原子正中心,二者之間就會增加Bond label(默認是以白色字顯示距離,在黑背景下才看得清楚),這里筆者把N和轉移過去的質子之間增加了Bond label。然后進入Graphics - Labels,切換到Bonds,選Graph,點擊Show preview復選框,然后點擊1:H 1:N這項,就會看到距離變化已經顯示在預覽窗口了,如下所示,其中紅色和藍色豎條標記的分別是最小距離和最大距離位置和數值。
如果點擊Graph按鈕,就會把曲線顯示在大窗口中,如下所示。可見,在大約60幀,也即60 fs左右,N-H鍵就算是基本形成了,之后N-H鍵不斷振動。
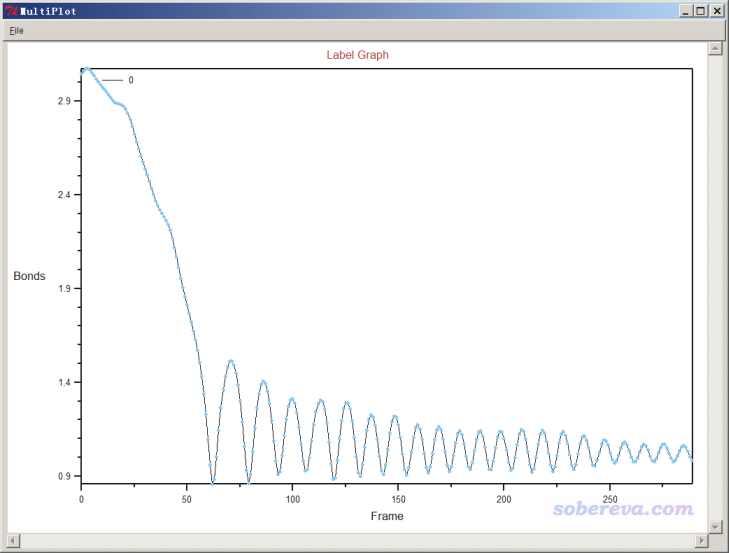
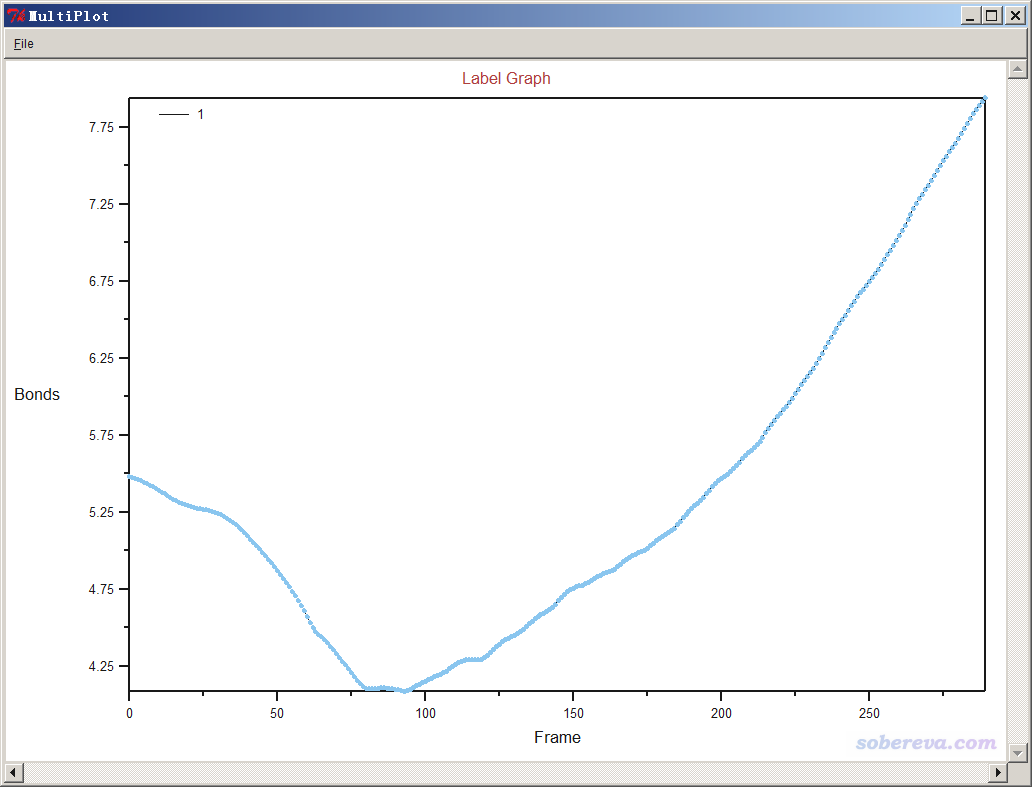
以類似方式,我們可以標記Al與N的距離,隨時間變化如下所示。可見Al與N先接近,質子轉移完畢后,二者就逐漸遠離了。
在Labels窗口里還可以點擊save,把距離變化數據保存到文本文件里,之后可以導入Origin等程序里繪制曲線。
用VMD還可以測量角度、二面角的變化,分別是在圖形窗口里按3然后點三個原子、按4然后點四個原子進行標記,之后在Graphics - Labels里觀看。
3 總結&其它
通過上面的例子,可以看到ORCA做AIMD是相當容易的,只要把Multiwfn支持的含有結構信息的文件(如pdb/gjf/xyz/mol/mol2/fch等等,見Multiwfn手冊2.5節)載入Multiwfn,敲幾下鍵盤產生標準AIMD任務的輸入文件,然后根據實際情況稍微改幾個設定就可以跑了。
以幾十核的一般雙路服務器的運算能力,ORCA里用B97-3c跑幾十原子有機體系的幾十ps的動力學不是特別困難的事。不過,能跑的時間尺度仍遠遠比不上xtb跑半經驗層面DFT的GFN-xTB方法的動力學,xtb跑動力學的粗淺介紹和例子看此文的相應部分:《使用Molclus結合xtb做的動力學模擬對瑞德西韋(Remdesivir)做構象搜索》(http://bbs.keinsci.com/thread-16255-1-1.html)。因此,拿ORCA跑DFT的動力學之前,先拿xtb初步跑跑,找找感覺,大體摸索出自己期望的現象能出現的條件(如溫度、初始結構、反應物相對位置和碰撞方式等),然后再用DFT跑通常是比較好的做法,免得做昂貴的DFT的MD試來試去把時間都耽誤了。
本文只涉及了VMD一丁點皮毛,VMD對于做動力學的人是必須玩得非常溜的。筆者在北京科音分子動力學與GROMACS培訓班(http://www.keinsci.com/workshop/KGMX_content.html)里對VMD有非常深入全面的介紹,包括tcl腳本的編寫。利用VMD的tcl腳本可以對軌跡做千變萬化的分析,有些分析諸如質心距離變化、平面間夾角變化、某幾何變量分布統計、不同結構出現比率等,是必須靠寫腳本才能實現的。
光是分析分析動力學過程的能量、結構變化是很膚淺的。利用Multiwfn,可以對ORCA跑的動力學的全過程的電子結構做深入透徹的分析,從而考察化學鍵、弱相互作用、電荷分布等等在動力學過程中的變化,由此能夠從提供深入的視角,使研究文章信息更豐富、明顯更上檔次。非常建議詳細閱讀《詳談Multiwfn的命令行方式運行和批量運行的方法》(http://www.shanxitv.org/612),里面第4節專門講了怎么做這樣的分析,你會發現特別容易也特別有價值。
如果Multiwfn創建ORCA做動力學輸入文件的功能對你的實際研究產生了幫助,希望在寫文章的時候提及諸如The input file of ab-initio molecular dynamics was prepared with the help of Multiwfn program并引用Multiwfn原文,這是對Multiwfn此功能開發最好的支持。