談談pi-pi相互作用
談談pi-pi相互作用
On the pi-pi interaction
文/Sobereva@北京科音
First release: 2025-Feb-18 Last update: 2025-Feb-19
0 前言
pi-pi相互作用(pi-pi interaction)是化學體系中很常見而且很重要的一種弱相互作用,本文全面談談這種相互作用的各方面特征,以令讀者對其有充分的認識、能在自己的研究中正確分析討論。我在互聯網上廣泛答疑時也時不時看到有關于pi-pi作用的非常錯誤的理解,比如有人被一些文章誤導,竟以為pi-pi作用來自于軌道相互作用或靜電相互作用,而且這種說法還廣泛以訛傳訛,筆者希望通過此文以正視聽。此外,氫鍵、范德華作用等概念在結構化學書里普遍都有,但pi-pi作用鮮有提及,我也推薦相關書籍作者和結構化學教師參考此文的內容將pi-pi作用納入書籍和教學。本文中大量涉及非常流行的波函數分析程序Multiwfn,相關信息見《Multiwfn FAQ》(http://www.shanxitv.org/452)。
我有不少研究文章都和pi-pi作用有密切聯系,是pi-pi作用研究的典型范例,非常推薦讀者閱讀并歡迎引用:
? Theoretical Insight into Complexation Between Cyclocarbons and C60 Fullerene, Chem. Eur. J., 30, e202402227 (2024)。在《全面揭示各種碳環與富勒烯之間獨特的pi-pi相互作用!》(http://www.shanxitv.org/727)中有詳細介紹,專門研究了不同尺寸的碳環和富勒烯之間的pi-pi作用
? Intermolecular interaction characteristics of the all-carboatomic ring, cyclo[18]carbon: Focusing on molecular adsorption and stacking, Carbon, 171, 514-523 (2021)。在《全面探究18碳環獨特的分子間相互作用與pi-pi堆積特征》(http://www.shanxitv.org/572)中有詳細介紹,其中涉及兩個18碳環之間的pi-pi作用
? Molecular assembly with a figure-of-eight nanohoop as a strategy for the collection and stabilization of cyclo[18]carbon, Phys. Chem. Chem. Phys., 25, 16707 (2023)。在《8字形雙環分子對18碳環的獨特吸附行為的量子化學、波函數分析與分子動力學研究》(http://www.shanxitv.org/674)中有詳細介紹,其中涉及18碳環與OPP雙環分子的pi-pi作用
? Comment on “18 and 12 – Member carbon rings (cyclo[n]carbons) – A density functional study”, Mat. Sci. Eng.: B, 273, 115425 (2021)。在《18碳環(cyclo[18]carbon)與石墨烯的相互作用:基于簇模型的研究一例》(http://www.shanxitv.org/615)中有詳細介紹,其中涉及18碳環與石墨烯的pi-pi作用
1 pi-pi作用的基本特征
pi-pi作用的定義和說法比較亂,這里給出我認為最嚴格準確的定義:“pi-pi作用是相距較近的兩個片段上彼此朝向相對的pi電子之間的獨特的色散吸引作用”。這里做幾個注釋,結合后文的實例讀者會了解得更充分:
(1)pi-pi作用可以是分子間的也可以是分子內的,滿足以上定義即可
(2)諸如苯分子里面的pi電子之間的作用不叫pi-pi作用,那屬于共享電子作用
(3)兩套pi電子的分布必須近乎彼此相對才可能算pi-pi作用。而諸如兩套pi電子近乎肩并肩挨著就不能算pi-pi作用,像是不能說環丁二烯里面兩套近乎定域的pi電子之間是分子內pi-pi作用
(4)“相距較近”一般是在4.0-4.5埃以內。也不是說更遠距離就完全沒有pi-pi作用了,只不過由于色散作用隨作用距離r呈-1/r^6快速衰減行為,因此距離稍微一遠pi-pi作用就非常弱了,就不太值得一提了。但相互作用的片段間距離也不能太近,若顯著小于相接觸的原子的范德華半徑之和,則顯著的位阻互斥作用會遠大于pi-pi吸引作用,使得pi-pi作用也相對不值得一提
(5)pi-pi作用最常出現在碳原子間,因為碳最容易帶顯著的pi電子。碳的Bondi和CSD范德華半徑分別為1.70和1.77埃。如果沒有特殊的因素影響相互作用距離的話,在平衡結構(勢能面極小點結構)下,C-C間的pi-pi作用出現在3.4-3.6埃左右是最常見的
(6)pi-pi作用屬于弱相互作用和非共價相互作用范疇。雖然名義上算作弱相互作用,但實際強度可大可小,直接取決于作用面積,詳見后文
(7)有一個詞叫pi-pi堆積(pi-pi stacking),這個詞往往和pi-pi作用混用。但在我來看,這個詞更適合用來形容由于pi-pi作用的吸引效果使得相互作用的兩個部分發生緊密結合從而產生的彼此堆積的結構特征
下圖就是一個再典型不過的存在顯著pi-pi作用的體系,是暈苯的二聚體。高精度理論計算的極小點結構下平面間距大約在3.45埃
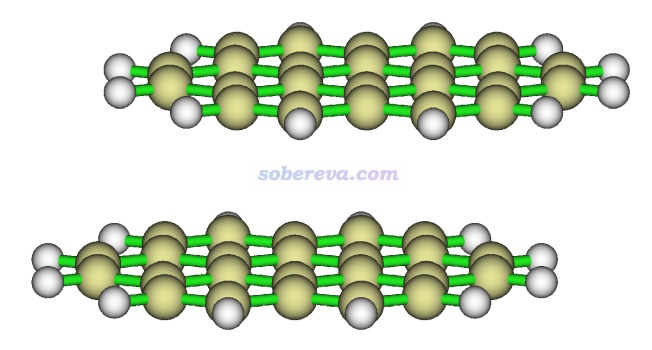
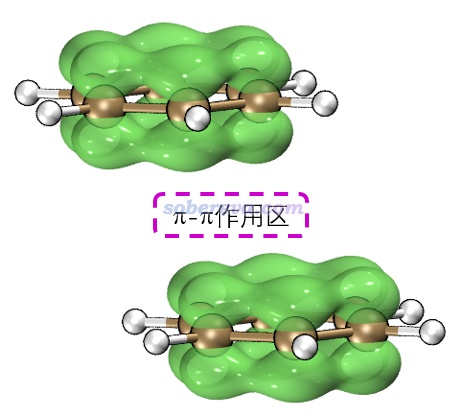
苯二聚體有不同構型,彼此垂直的T型二聚體是能量最低構型,還有一種能量與之非常接近的局部極小點構型是平行位錯構型,屬于pi-pi堆積結構。按照《在Multiwfn中單獨考察pi電子結構特征》(http://www.shanxitv.org/432)的做法繪制出的幾何結構結合pi電子等值面圖如下所示,兩個分子的pi電子的分布非常清楚直觀,可以看到彼此相對。注意并非必須兩個分子的所有pi電子都彼此對著才算pi-pi作用,像下圖這樣哪怕只要一部分對著也同樣算pi-pi作用。而如果完全沒有對著、完全錯開了,那就不算pi-pi作用了。更多的展現pi電子分布的圖見筆者的Theor. Chem. Acc., 139, 25 (2020)一文。
2 pi-pi作用的物理本質
我在《談談“計算時是否需要加DFT-D3色散校正?” 》(http://www.shanxitv.org/413)提到過,原子間相互作用從物理本質上可以基本分為靜電、色散、交換、Pauli互斥、軌道相互作用;我在《談談“計算時是否需要加DFT-D3色散校正?”》(http://www.shanxitv.org/413)里也說過物理本質和相互作用表象之間的關系。pi-pi作用類似于氫鍵、鹵鍵等,是屬于表象層面的,用來描述一種具有特定特征的相互作用,其內在本質就是前面說的來自于相距較近的pi電子之間的色散作用,這一點在Grimme的一篇經典的討論pi-pi作用本質的文章Angew. Chem. Int. Ed., 47, 3430 (2008)中通過嚴格的理論計算和分析對比已經論證得十分嚴格充分,而且也早已是量子化學領域內行人的共識。
有的文章試圖從軌道相互作用解釋pi-pi堆積出現的本質,這是極其有誤導性的,卻還廣泛以訛傳訛。2007年的時候wiki上的詞條說pi-pi作用來自于pi共軛體系間p軌道的重疊,前述Angew文章的腳注中專門對此進行了批判,指出沒有任何量子化學計算能支持這種說法。這也不難理解,本來兩個分子的pi軌道之間重疊程度極低(可以用《分子間軌道重疊的圖形顯示和計算》http://www.shanxitv.org/163介紹的方法考察),遠低于形成共價鍵時原子軌道間的重疊程度,因此有效重疊不足。而且兩個分子的pi占據軌道之間混合會產生全占據的成鍵和反鍵軌道,二者效果充分抵消了,對成鍵沒貢獻;而若一個分子的pi占據軌道和另一個分子的pi非占據軌道混合,意味著要出現分子間pi->pi*電子轉移,這通常又沒有明顯能使體系能量變得更低的內在驅動力(不像常見的較近距離的n->pi*超共軛往往可以明顯降低體系能量)。此外,按照《Multiwfn支持的分析化學鍵的方法一覽》(http://www.shanxitv.org/471)說的,用Multiwfn對pi-pi堆積的兩個分子間去計算衡量兩個部分之間有效共享電子對數的Mayer鍵級、模糊鍵級或離域化指數(DI),會發現數值很接近0,比如《全面揭示各種碳環與富勒烯之間獨特的pi-pi相互作用!》(http://www.shanxitv.org/727)介紹的文章里對18碳環與富勒烯之間算的Mayer鍵級僅為可忽略不計的0.04,也充分體現出pi-pi作用中的軌道相互作用成份基本可忽略不計。所以無論從哪個角度,pi-pi堆積的出現都不是軌道相互作用驅動的,頂多是在色散作用驅動下,pi-pi堆積結構出現后順帶出現了點可忽略的軌道相互作用而已。所以pi-pi作用在主流學術界被歸為非共價相互作用范疇,這是完全正確的。然而有一些論文試圖從軌道相互作用分析pi-pi作用的特征,還擺出一堆軌道圖,比如Phys. Chem. Chem. Phys., 15, 9397 (2013)就是典型文章,切勿把這類文章太當回事,很多都是瞎討論、尬討論。
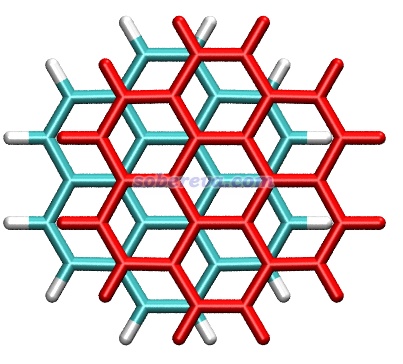
pi-pi堆積的體系有一個普遍特點是在極小點結構下,pi-pi作用區域的原子間通常不是正好對著,而是相互錯位。下圖是暈苯二聚體的俯視圖,其中一個暈苯用紅色顯示,可以很清楚看到錯位特征,即一層的碳對著另一層的六元環中心。一層層堆疊的石墨中每一層之間也同樣是這樣錯位的。出現自發錯位有不同的解釋并且存在一定爭議,有人認為是因為錯位的結構下比嚴格面對面的結構下的原子間的Pauli互斥更低,例如Chem. Sci., 11, 6758 (2020)持這種觀點。也有人認為是因為錯位的結構下的靜電相互作用能更負(靜電吸引作用更強),在Phys. Chem. Chem. Phys., 24, 8979 (2022)中作者通過能量分解論證了這一點并反駁了Chem. Sci.那篇文章。靜電作用引起位錯的原因從邏輯上容易理解:形成pi-pi堆積的兩部分都有豐富的pi電子,面對面構型下pi電子間靜電互斥會較大,錯開后互斥自然就沒那么強了。雖然筆者更同意靜電效應是位錯出現的主導,但我也不否認降低Pauli互斥也可能是產生位錯的另一個驅動力,盡管相對次要。
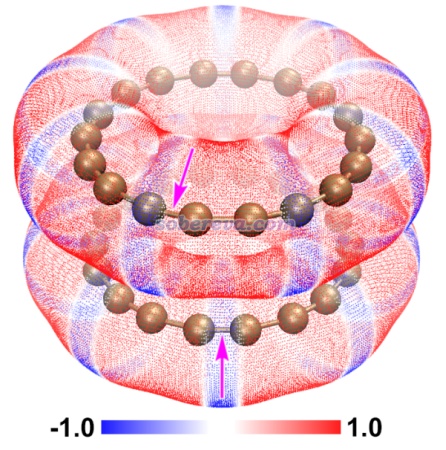
順帶一提,不止是pi-pi堆積二聚體,還有很多其它體系都是色散吸引作用驅動分子間結合,而靜電作用進一步影響幾何結構。《靜電效應主導了氫氣、氮氣二聚體的構型》(http://www.shanxitv.org/209)介紹的筆者的研究就是很好的例子,H2、N2的各種二聚體的相互作用能通過能量分解分析可以發現都是色散作用為主,但不同構型下的靜電作用的差異則直接影響不同構型的穩定性乃至存在與否,并且筆者發現通過靜電勢互補原理可以給出很直觀的解釋。這種靜電勢互補的分析方法也在《全面探究18碳環獨特的分子間相互作用與pi-pi堆積特征》(http://www.shanxitv.org/572)介紹的筆者的論文中也用來解釋為什么18碳環的pi-pi堆積二聚體是錯位的。平面環狀結構的18碳環具有獨特的平面內和平面外兩套pi電子,此文中確認了18碳環能夠形成分子間彼此平行的pi-pi堆積二聚體,下圖是按照《使用Multiwfn+VMD快速地繪制靜電勢著色的分子范德華表面圖和分子間穿透圖》(http://www.shanxitv.org/443)繪制的二聚體極小點結構下的單體的靜電勢著色的分子范德華表面的疊加圖,可以看到兩個碳環表面靜電勢為正和為負的區域在錯位的結構下是以互補的方式重疊的,很清晰地解釋了錯位的產生原因,即這種構型下的靜電吸引作用比一個個碳原子彼此精確對著的時候更強。
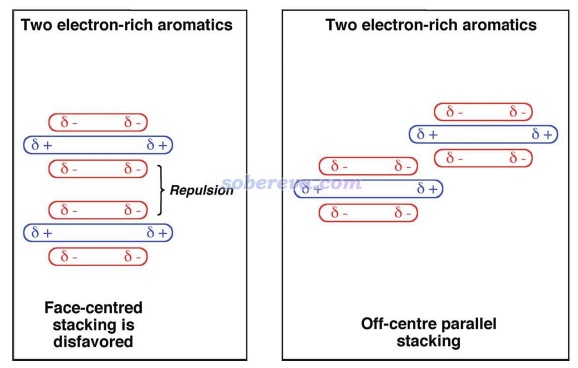
要注意的是,有些文章作者極度夸大了靜電作用對于pi-pi堆積形成的作用,不僅認為平行位錯構型的出現是因為此時靜電吸引作用最有利,還鼓吹pi-pi堆積結構的形成的本質就是靜電吸引作用,這是大錯特錯!最典型的這種文章就是Chem. Sci.,3 , 2191 (2012),這篇文章的發表嚴重誤導了很多讀者。此文雖然是在前述的Grimme的pi-pi作用的研究之后寫的,而且文中還引了那篇文章,但給我的感覺是此文的作者完全沒好好看那篇文章,似乎也完全不懂什么叫電子相關作用,而依然基于古老且過于簡單(忽略了pi電子結構和穿透效應)的Hunter和Sanders的四極矩觀點盲目、主觀、武斷、信誓旦旦地解釋pi-pi堆積的位錯結構形成的本質,而完全沒有可信、嚴格的理論和計算數據作為依據,各種自說自話,并且還錯誤地解釋一些文獻里的理論計算數據,甚至還不承認pi-pi作用的概念。作者還執拗地認為非得是存在精確面對面堆積才能說明pi-pi作用的存在,并因為實際pi-pi堆積體系的極小點結構不是面對面堆積就被他用來否認存在pi-pi作用。此文文中給出了下圖,令一些初學者不明覺厲,誤以為不僅清晰解釋了為什么平行位錯結構比精確面對面結構更有利,還誤以為這種二聚體的形成主要靠的就是靜電吸引作用,完全沒分清楚pi-pi堆積二聚體的形成中色散作用和靜電作用的主次關系。還有不少其它文章如J. Chem. Soc., Dalton Trans., 2000, 3885也是類似地對pi-pi作用存在嚴重誤解、給出誤導性的示意圖,讀者看到這種文章時切勿當回事!
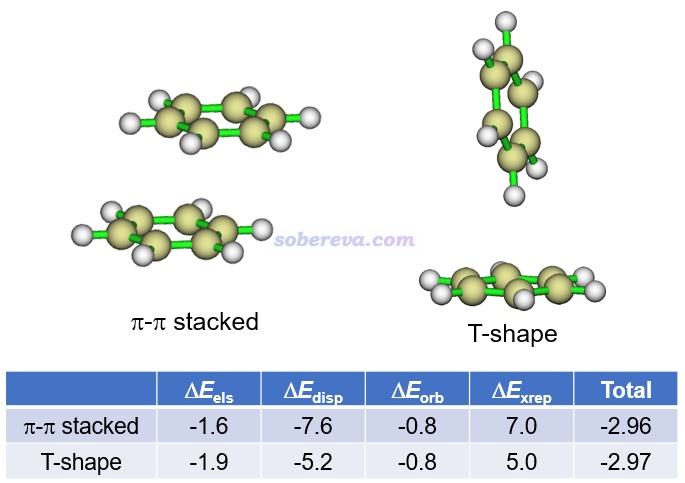
能量分解分析在pi-pi作用的研究中應用得非常普遍,對于認識其本質非常有幫助。這類方法將總的相互作用能分解成不同物理成份。例如將《使用sobEDA和sobEDAw方法做非常準確、快速、方便、普適的能量分解分析》(http://www.shanxitv.org/685)介紹的筆者提出的流行的sobEDAw能量分解方法用于苯的兩種二聚體構型可得到如下結果(計算級別:B3LYP-D3(BJ)/6-311+G(2d,p)并考慮counterpoise,單位為kcal/mol),可見色散項ΔE_disp遠比靜電作用項ΔE_els更負,軌道相互作用項ΔE_orb更是微乎其微,故色散作用對苯的分子間結合起到主導作用。特別是在平行位錯的結構下的ΔE_disp比T構型下的明顯負得多(同時ΔE_els的大小還變小),充分體現出平行位錯結構下才有的pi-pi作用的本質是色散作用,這和上文對pi-pi作用本質的討論相一致。平行位錯結構下,色散作用對總吸引作用項的貢獻百分比是:-7.6/(-7.6-0.8-1.6)*100=76%。
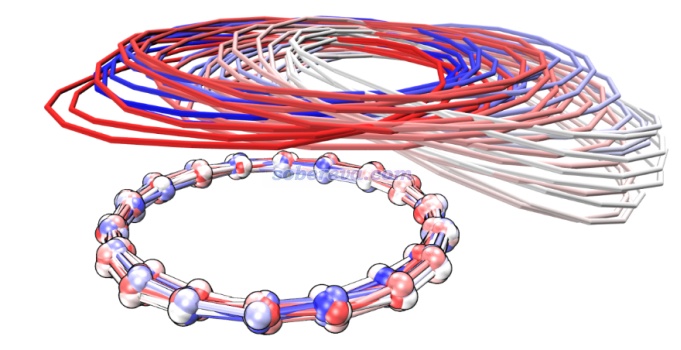
順帶一提,pi-pi堆積的結構在現實環境中一般很容易發生分子間相對滑移(除非有額外的位阻效應等阻礙),這是由于滑移導致能量變化很小。Carbon, 171, 514-523 (2021)文中做18碳環二聚體的勢能面掃描、Phys. Chem. Chem. Phys., 24, 8979 (2022)中做的苯分子平行二聚體的勢能面掃描都體現了這一點。并且由于滑移方向的勢能面很緩,哪怕室溫下也非常容易出現滑移。在Carbon, 171, 514-523 (2021)文中我給出了18碳環二聚體在100K下的4 ps的從頭算動力學的軌跡疊加圖(消除了下方的18碳環的整體運動以著重表現相對運動),如下所示,分子位置根據模擬時間按照藍-白-紅變化,可見在模擬過程中的確出現了顯著的滑移
3 pi-pi作用的強度
pi-pi堆積的極小點結構下的每一對近距離接觸的原子的pi-pi作用都是非常弱的,就是普通范德華作用的程度,但是當涉及pi-pi作用的原子很多時,就可以達到很高的強度,甚至能達到近乎化學鍵的程度。最強的pi-pi作用就是從微觀來看無限延展的石墨中的層間pi-pi作用了。下面看一些實例,以令讀者更好地理解pi-pi作用的強度特征。
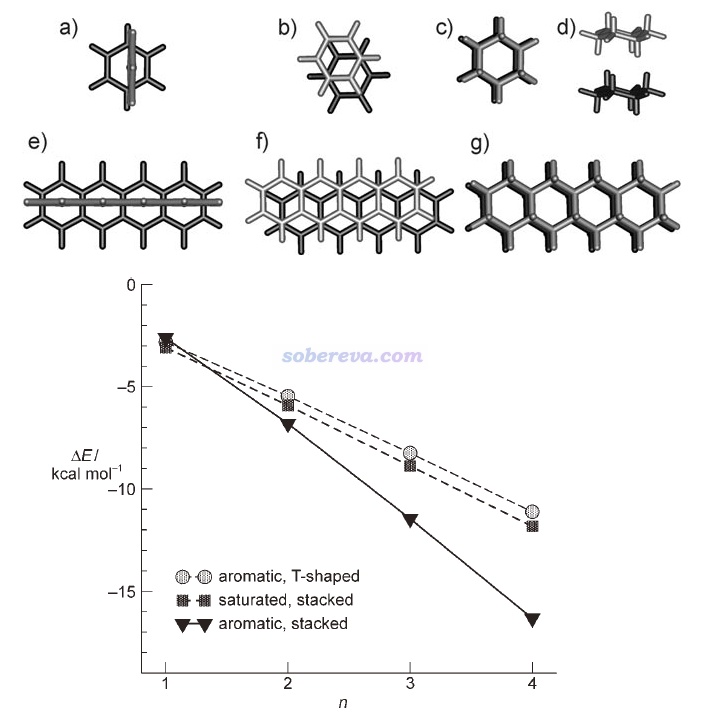
Angew. Chem. Int. Ed., 47, 3430 (2008)文中計算了T型苯二聚體(下圖a)、平行位錯苯二聚體(下圖b)、環己烷二聚體(下圖c和d是兩個視角)的相互作用能,并且還將它們的環數n從1逐漸加到4(分別對應下圖e、f、g的結構),以考察了三種作用類型的相互作用能隨環數的變化,分別對應下圖的aromatic T-shaped、aromatic stacked和saturated stacked三條曲線。由圖可見,當pi-pi作用涉及的原子很少時,即苯二聚體,由于pi-pi作用還不夠強,因此平行位錯二聚體的能量比靜電吸引作用更強的苯T型二聚體略微更高,相互作用強度比起無pi-pi作用特征的環己烷二聚體沒優勢。但隨著環數增加,pi-pi作用強度不斷增大,使得具有pi-pi堆積結構的二聚體的相互作用變得顯著強于不具備這種特征的二聚體。四并苯的二聚體的相互作用能已達到-16 kcal/mol(67 kJ/mol),這已經是水二聚體這種普通強度氫鍵作用能的3倍左右了。
可見,前面提到的Chem. Sci.,3 , 2191 (2012)那篇文章拿兩個苯及其衍生物在液體和晶體中普遍缺少平行位錯構型的出現,以及平行位錯的苯二聚體不是能量最低構型,以此鼓吹pi-pi作用不是一種客觀存在的作用,這明顯是極其狹隘的。
對本文第1節的兩個暈苯的二聚體,在DLPNO-CCSD(T)級別下算出來的相互作用能達到-20 kcal/mol,也是相當強了。
前面給出的18碳環二聚體的相互作用能,在Carbon, 171, 514-523 (2021)中我用ωB97X-V/def2-QZVPP結合counterpoise校正的計算結果為-9.2 kcal/mol(-38.5 kJ/mol),達到平行位錯的苯二聚體的相互作用能的大約三倍,無疑也是非常顯著的pi-pi作用了。
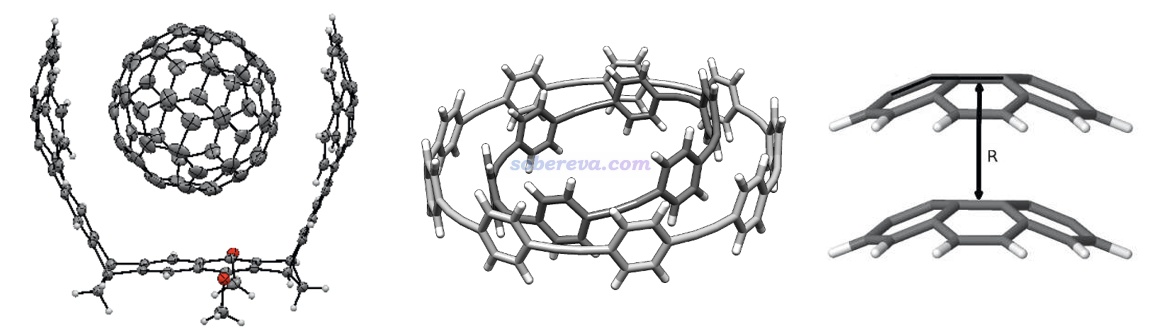
不是只有平面區域之間才可能有pi-pi作用,例如下面三個體系的pi-pi作用區域都是有顯著曲率的。下圖左邊的結構是Org. Lett., 17, 5292 (2015)中合成出的主-客體復合物晶體的一部分,由于客體分子與富勒烯之間的pi-pi作用面積頗大,B97D/QZVP級別計算的相互作用能達到了-50 kcal/mol,已經是弱化學鍵的強度了。下圖右側是碗烯的pi-pi堆積二聚體,相互作用的研究見J. Phys. Chem. A, 116, 11920 (2012)。
4 圖形化展現pi-pi作用區域
《使用Multiwfn做IGMH分析非常清晰直觀地展現化學體系中的相互作用》(http://www.shanxitv.org/621)以及《一篇最全面介紹各種弱相互作用可視化分析方法的文章已發表!》(http://www.shanxitv.org/667)中的綜述詳細介紹的筆者提出的IGMH方法用于圖形化展現片段間相互作用效果極佳,已被廣為使用。IGMH用于展現自定義片段間的pi-pi作用、判斷pi-pi作用是否存在尤為好用,因此在此給出一些簡單例子予以體現。其方法的原理、細節、計算操作見上面的文章。如果你不知道pi-pi作用可能出現在哪,不方便定義片段,則應當用《使用IRI方法圖形化考察化學體系中的化學鍵和弱相互作用》(http://www.shanxitv.org/598)介紹的筆者的IRI方法,可以把體系中所有相互作用區域全都展現出來,也包括化學鍵作用區域。
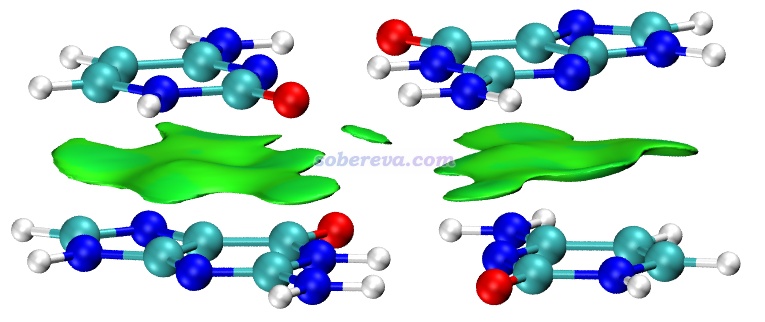
不是只有純碳的pi區域之間才可能有pi-pi作用,pi-pi作用也可以涉及到其它原子。眾所周知,DNA結構中相鄰層的堿基與堿基之間是平行堆疊的,再加上堿基區域有大量pi電子,所以這也屬于典型的pi-pi作用。下圖是從DNA中截取的堆疊的GCGC堿基對,兩層分子各定義為一個做IGMH分析的片段,兩層之間的綠色的IGMH方法定義的delta_g_inter函數的0.004 a.u.等值面非常清晰、優雅地展現出了pi-pi作用的主體區域。圖中還有個面積較小的等值面出現在O...O之間,雖然C=O鍵的O有pi電子,但這倆O的pi電子不是彼此對著的,所以這就只能算是普通的范德華作用區域了。
下圖是《8字形雙環分子對18碳環的獨特吸附行為的量子化學、波函數分析與分子動力學研究》(http://www.shanxitv.org/674)介紹的筆者的論文中的一張圖,研究的是OPP雙環分子結合兩個18碳環后的復合物。圖中綠色等值面直觀地展現出在什么區域18碳環與OPP主體分子間有顯著的pi-pi作用。可見pi-pi作用主要出現在OPP和18碳環近距離接觸的體系的兩端,而OPP靠中間區域離18碳環較遠因此缺乏pi-pi作用。順帶一提,18碳環是極少有的同時具有平面內(in-plane)和平面外(out-of-plane)兩套全局離域的pi電子的體系,當前體系中18碳環主要憑借的是平行于環方向分布的pi電子與OPP大環上的垂直于苯環的pi電子產生pi-pi作用,這是極為新穎、獨特的pi-pi作用形式。如果讀者對碳環類體系感興趣,非常建議進入http://www.shanxitv.org/carbon_ring.html觀看筆者發表的所有碳環相關的理論研究和對應的深入淺出的評述文章。
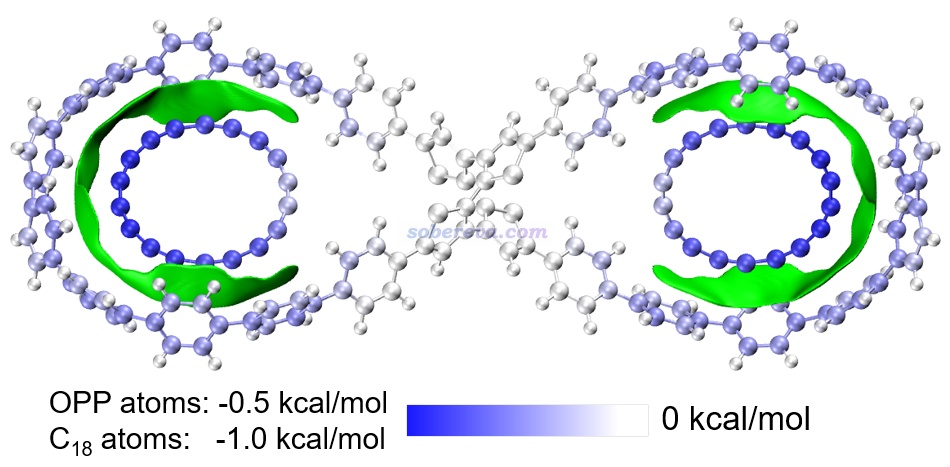
上圖中還對利用Multiwfn的基于力場的能量分解(EDA-FF)功能計算了各個原子對分子間相互作用的貢獻并對原子進行了著色,展現出了更豐富的信息。這種分析見《使用Multiwfn做基于分子力場的能量分解分析》(http://www.shanxitv.org/442)。
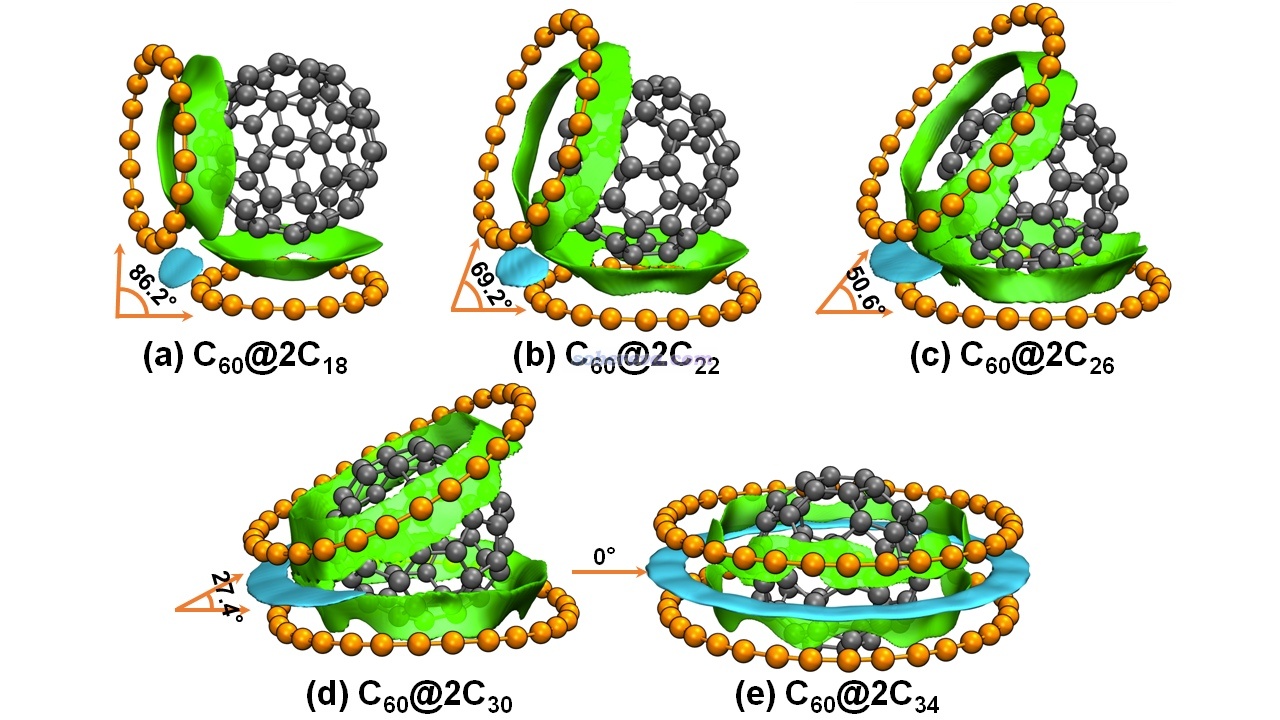
在《全面揭示各種碳環與富勒烯之間獨特的pi-pi相互作用!》(http://www.shanxitv.org/727)介紹的文章中,筆者全面研究了碳環與富勒烯的相互作用,其中給出了下圖,展示了不同尺寸的兩個碳環與富勒烯形成的三聚體。并且為了直觀區分富勒烯-碳環和碳環-碳環兩種pi-pi作用,分別將對應的等值面用綠色和青色著色。可見此圖極好地將體系中兩種pi-pi作用出現的主要位置展現了出來,比起在文中只是給出個幾何結構圖信息豐富得多。
標準的IGMH圖是通過sign(λ2)ρ函數對等值面著色的,ρ是電子密度。按照IGMH方法的標準的色彩刻度著色的話,ρ接近0的區域的等值面都是綠色。由于pi-pi作用區域、普通范德華作用、極弱的氫鍵(如C-H作為氫鍵給體時)等情況的作用區域的電子密度都很低(0.01 a.u.以內),因此相應的標準方式著色的IGMH等值面都會是綠色或很接近綠色。因此解釋IGMH圖的等值面時必須結合不同特征的弱相互作用的基本定義,不能單純根據顏色去亂解釋。比如網上有人問我“下面這個圖里紅圈部分是pi-pi作用嗎?”,這實在是匪夷所思的問題。跨越那個等值面和右下方苯環的pi電子區域直接作用的是一個氫原子,氫原子又沒pi電子,怎么可能是pi-pi作用!?而且右下方的苯環也明顯不可能和左上方的苯環有pi-pi作用,一方面二者離得老遠,另一方面二者的pi電子區域也根本不相互對著,顯然無論如何也不可能算是pi-pi作用。
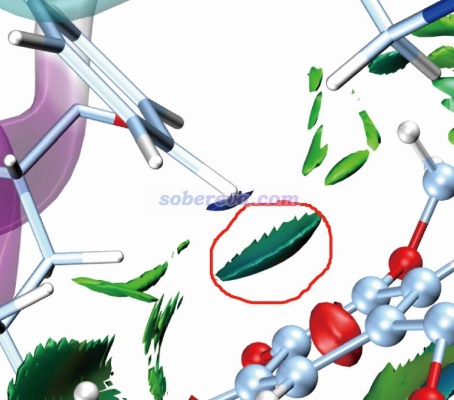
那么上圖中紅圈里的等值面算什么作用?明顯這是pi-氫鍵,C-H作為氫鍵給體,其中氫帶一定正電荷(這一點用H的原子電荷就能體現,原子電荷的相關知識見《一篇深入淺出、完整全面介紹原子電荷的綜述文章已發表!》http://www.shanxitv.org/714這篇綜述),右下角的苯環上的pi電子令苯環上方顯負電性而作為氫鍵受體(用靜電勢可直接體現,參見http://bbs.keinsci.com/thread-219-1-1.html里匯總的靜電勢相關資料和我的相關博文)。
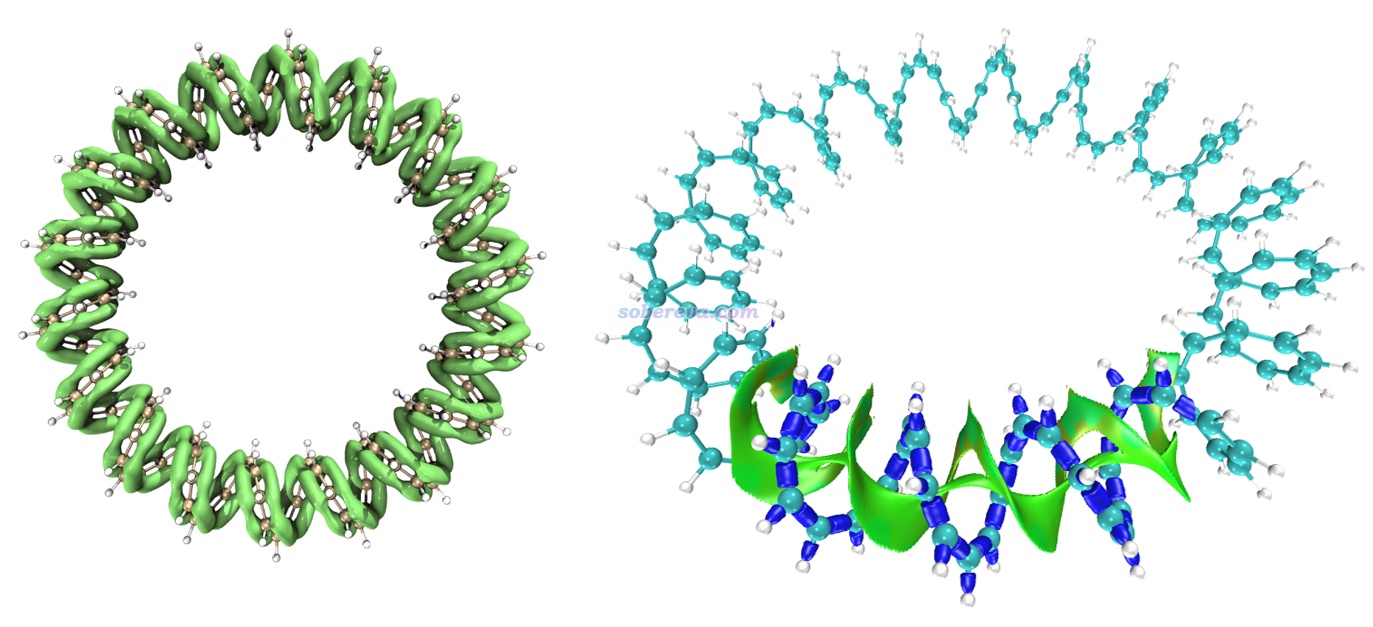
下面再舉一個IRI圖展現pi-pi作用區域的例子。Multiwfn的原文之一J. Chem. Phys., 161, 082503 (2024)里給出了144-輪烯的LOL-pi函數的等值面圖,如下圖左側所示,此圖清晰直觀地展現出了這個全局大共軛體系的pi電子的主要離域路徑。根據此圖展現的pi電子的分布情況,憑直覺就知道這個體系里肯定存在pi-pi作用。下圖右側是對這個體系的其中一個局部區域繪制的IRI等值面圖,藍色的等值面展現出了化學鍵作用區域,綠色的等值面清楚直觀地展現出了pi-pi作用區域。可見這個體系非常有趣,pi-pi作用區域綿延不斷貫通整個體系!也正是有分子內pi-pi作用的存在,此體系才能形成螺旋狀結構,要不然就散了(如同用不能描述色散作用的理論方法優化DNA結構時結構會散掉)。
5 衡量pi-pi作用強度的方法
這一節說一下如何衡量pi-pi作用強度。最簡單的方法就是計算pi-pi堆積二聚體的相互作用能,作用能越負說明作用越強。但如果要討論二聚體的熱力學穩定性,則需要計算結合自由能,溶劑環境中還得考慮溶劑模型。相關知識參見《談談分子間結合能的構成以及分解分析思想》(http://www.shanxitv.org/733)。復合物AB在特定結構下,A和B的相互作用能的最常規的計算方法就是用E(AB)-E(A)-E(B)方式手動計算,做sobEDAw能量分解(http://www.shanxitv.org/685)時也會順帶給你相互作用能。
以上述方法算相互作用能的一個問題是,如果兩個分子之間不僅僅有pi-pi作用,還有其它作用(如氫鍵),那么得到的只是總相互作用能。如果想只得到pi-pi作用能,有幾個辦法可以用:
(1)用《使用Multiwfn做基于分子力場的能量分解分析》(http://www.shanxitv.org/442)介紹的EDA-FF能量分解方法,將兩個分子的pi作用區域定義為兩個片段,讓Multiwfn給出基于分子力場的這兩個部分的相互作用能,取其中的范德華作用能(即色散作用和交換-互斥作用之和)。如果你只需要色散部分,還有另一種做法,見《使用Multiwfn圖形化展現原子對色散能的貢獻以及色散密度》(http://www.shanxitv.org/705)。這兩種做法還都可以給出具體原子產生的貢獻,并可以對原子進行著色以便通過圖像直觀展現和考察
(2)如果分子間同時有pi-pi作用和氫鍵,且其它作用可忽略不計,可以按《透徹認識氫鍵本質、簡單可靠地估計氫鍵強度:一篇2019年JCC上的重要研究文章介紹》(http://www.shanxitv.org/513)介紹的方法使用Multiwfn做拓撲分析估計出氫鍵作用能,從總相互作用能中扣掉之作為pi-pi作用能
(3)對體系進行恰當改造,基本保留每個分子參與pi-pi作用的部分,然后將總相互作用能近似當成pi-pi作用能。
如果是分子內的pi-pi作用能的估計,還可以參考《計算分子內氫鍵鍵能的幾種方法》(http://www.shanxitv.org/522)里的說明舉一反三處理。
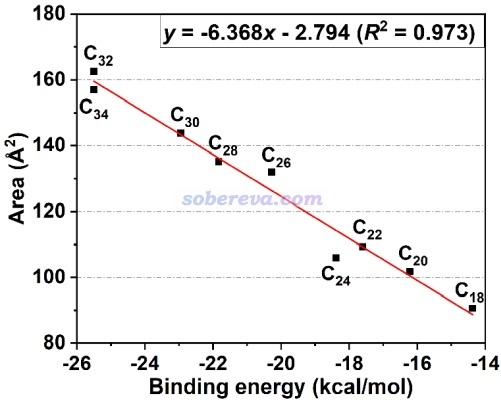
在特定條件下,pi-pi作用強度和IGMH的等值面面積有密切的正相關性。《全面揭示各種碳環與富勒烯之間獨特的pi-pi相互作用!》(http://www.shanxitv.org/727)介紹的文章中給出了下圖,是不同原子數的碳環與富勒烯之間的相互作用能和描述pi-pi作用的IGMH等值面面積的關系,可見有挺理想的線性關系。通過這種關系,可以直接根據pi-pi作用區域的等值面面積估計對應的pi-pi相互作用能。這種面積的計算方法見《計算IGMH等值面的面積和體積的方法》(http://www.shanxitv.org/738)。
Chem. Commun., 48, 9239 (2012)提出的LOLIPOP方法值得一提。基于單個分子的波函數文件,就可以用Multiwfn非常容易地計算體系中的各個六元環的LOLIPOP指數,此值越小說明這個環發生pi-pi堆積的能力越強,原文對不同體系以苯分子作為探針分子進行了測試證實了這一點。Multiwfn手冊3.100.14節有LOLIPOP的詳細介紹,4.100.14節有計算實例。
6 不同理論方法描述pi-pi作用的能力
色散作用的更深層本質是電子的庫侖相關作用,因此做量子化學或第一性原理計算時,若想描述好pi-pi作用,用的方法必須能描述好庫侖相關,顯然高精度后HF方法都沒問題,比如CCSD(T)在計算pi-pi作用上可以算是金標準。至于低級別的后HF方法MP2則傾向于明顯高估pi-pi作用,絕對不要用。
對于特別常用的DFT,描述pi-pi作用的能力基本等同于描述普通色散作用的能力,如果泛函原本描述色散作用爛(如PBE、PBE0)或者完全不能描述(如B3LYP),就必須帶色散校正,參考《談談“計算時是否需要加DFT-D3色散校正?” 》(http://www.shanxitv.org/413)、《DFT-D色散校正的使用》(http://www.shanxitv.org/210)、《談談量子化學研究中什么時候用B3LYP泛函優化幾何結構是適當的》(http://www.shanxitv.org/557)。本來就帶色散校正的wB97M-V、wB97X-D3等直接就能很好描述pi-pi作用。M06-2X描述pi-pi作用雖然定性正確但不算太好,加了零阻尼DFT-D3色散校正后對pi-pi作用的描述有明顯改進,但還是不如B3LYP-D3(BJ),對比測試見考慮了很多pi-pi作用體系的L7測試集的原文(J. Chem. Theory Comput., 9, 3364 (2013))。雙雜化泛函由于帶有MP2項,所以都有描述pi-pi作用的能力,但一般在考慮了色散校正后才會變得足夠好,如revDSD-PBEP86-D3(BJ)。
實際上DFT-D那種形式的色散校正在原理上對于描述pi-pi作用并非很理想,因為pi電子不是繞著原子核球對稱分布的,而DFT-D校正能的公式依賴的只是原子間距離(這里不考慮三體校正項),沒體現出pi電子在原子核周圍的具體分布特征。不過這倒也不是明顯問題,常用的DFT-D3、DFT-D4色散校正對于描述pi-pi作用從實際效果上來看并沒有什么問題。
在半經驗方法層面,專門考慮了對色散作用描述的GFN2-xTB、PM6-D3H4X'、PM6-ML在表現pi-pi作用方面優秀,見J. Chem. Theory Comput., 21, 678 (2025)里3圖基于L7測試集的測試,PM6-D3和PM7只能算是定性正確。至于沒專門考慮對色散作用描述的諸如PM6、AM1等方法則完全失敗。
主流的分子力場,如GAFF、AMBER、CHARMM、OPLS-AA、MMFF94等,對pi-pi作用的描述雖然跟像樣的量子化學方法比算不上出色,但至少也算定性正確,因為它們都有描述色散作用的能力。J. Chem. Inf. Model., 49, 944 (2009)的測試專門體現了這點(不過這篇文章也有不少漏洞和槽點)。
7 疏水作用與pi-pi作用的關系
眾所周知,疏水效應使得水環境下非極性物質傾向于發生聚集,本質是溶劑的熵效應。兩個石墨烯片段在水中會自發堆積在一起,這算疏水作用還是pi-pi作用?實際上二者都有,對于堆積結構的形成起到協同作用。疏水效應更為普遍,無論兩個溶質的接觸區域是否有pi電子,只要溶質是基本無極性的,在水中都有疏水作用促使它們發生結合。而對于pi電子區域暴露的兩個溶質,疏水效應則在pi-pi作用的基礎上進一步促進了它們的pi-pi堆積結構的出現。如《全面揭示各種碳環與富勒烯之間獨特的pi-pi相互作用!》(http://www.shanxitv.org/727)介紹的文章的理論計算所示,在水環境下碳環與富勒烯之間的結合自由能的大小顯著大于在真空下,充分體現了這一點。
順帶一提,前述的Chem. Sci.,3 , 2191 (2012)一文居然誤以為溶劑環境下pi共軛體系間出現堆積結構僅僅是因為溶劑效應,并似乎試圖靠這個否認真空環境下也存在pi-pi相互作用,甚至說the terms "pi-stacking" or "pi–pi interactions" do not describe any physically meaningful interaction,實在是難以理喻!!!PS:這樣的文章能通過Chem. Sci.的審稿真是離譜。
8 總結&判斷pi-pi作用的標準
本文系統地對pi-pi作用的各個方面進行了介紹,包括其基本特征、物理本質、強度范圍、圖形化展現方法、考察強度的方法、理論計算方法的精度、疏水作用與它的關系。通過本文,讀者應該已經對pi-pi作用有了較全面的了解,能夠進行正確的分析討論,并且能認識到哪些文章或書籍里的說法是有誤導性的。我也推薦讀者接下來閱讀本文一開始提到的筆者的一系列和pi-pi作用有關的研究文章和對應的介紹博文。
最后再總結一下pi-pi作用的常規判斷標準,便于讀者能確切判斷哪些作用算是pi-pi作用。通過下面第1、2條就可以進行粗略判斷,3、4、5可以作為進一步檢驗。一般意義的pi-pi作用應當能同時滿足所有條件
(1)相互作用的兩部分都有pi電子且其分布彼此相互對著。如果拿不準有沒有pi電子分布、分布朝向如何,可以按照《在Multiwfn中單獨考察pi電子結構特征》(http://www.shanxitv.org/432)介紹的方法直接把pi電子密度等值面圖畫出來,一目了然
(2)相互作用的pi電子之間離得不太遠。比如明顯超過5埃的直接就可以忽略了
(3)用Multiwfn做IGMH分析(如果得不到波函數文件或體系太大難以算得動的話,可以改用極便宜且只依賴于原子坐標的mIGM),在等值面數值調到諸如0.003 a.u.這樣較小的值時,在預期出現pi-pi作用的區域能明顯看到等值面
(4)兩部分之間的Mayer鍵級或模糊鍵級或離域化指數非常小(遠小于0.1),故而軌道相互作用可基本忽略。注:如《Multiwfn支持的分析化學鍵的方法一覽》(http://www.shanxitv.org/471)所述,Multiwfn在主功能9里計算鍵級之前可以用選項-1定義兩個片段,之后做鍵級計算時會給出兩個部分之間的總鍵級,即片段間每一對原子的鍵級的總和
(5)使用sobEDAw等能量分解方法分析,色散項應當占所有吸引項總和的大部分,軌道相互作用項應當占比微小