使用sobEDA和sobEDAw方法做非常準確、快速、方便、普適的能量分解分析
使用sobEDA和sobEDAw方法做非常準確、快速、方便、普適的能量分解分析
文/Sobereva@北京科音
First release: 2023-Aug-31 Last update: 2023-Sep-24
1 前言
計算化學領域的能量分解分析(energy decomposition analysis, EDA)方法很多,思想各有不同,幾種常見的在《Multiwfn支持的弱相互作用的分析方法概覽》(http://www.shanxitv.org/252)中的能量分解部分有簡要介紹,在量子化學波函數分析與Multiwfn程序培訓班(http://www.keinsci.com/workshop/WFN_content.html)里有系統的介紹。雖然也有基于分子力場的能量分解,比如《使用Multiwfn做基于分子力場的能量分解分析》(http://www.shanxitv.org/442)里介紹的EDA-FF,但絕大多數能量分解方法是基于量子化學的,并且其中大多數的目的是將體系中用戶自定義的兩個或多個片段間的相互作用能進行分解,得到不同物理成份的貢獻,從而能夠深入了解片段間相互作用的主要本質(靜電、色散、交換-互斥、軌道相互作用等),以及橫向對比不同情況下相互作用的內在差異,例如《透徹認識氫鍵本質、簡單可靠地估計氫鍵強度:一篇2019年JCC上的重要研究文章介紹》(http://www.shanxitv.org/513)介紹的研究氫鍵的文章中使用了流行的SAPT能量分解方法充分考察不同體系中氫鍵的本質和差異并總結出了規律、《靜電效應主導了氫氣、氮氣二聚體的構型》(http://www.shanxitv.org/209)介紹的文章里用DFT-SAPT考察了不同構型的氫氣/氮氣二聚體相互作用的主導因素。
下文說的能量分解一律指基于片段和量子化學的能量分解方法。這類方法之前已經有很多,但在筆者看來各有各的缺點或局限性,尚沒有一個完全同時滿足耗時低、使用簡單方便、普適性強、精度好、物理意義清晰的能量分解方法。特別是對于最流行的Gaussian量子化學程序的用戶,目前尚沒有一個不需要額外代價就能做能量分解的方法。為了解決這個難題,筆者基于如今非常流行的色散校正的DFT理論提出了名為sobEDA的能量分解方法,同時還提出了一個它專用于考察弱相互作用的變體sobEDAw。在筆者來看,這兩個方法完全掃清了廣大量子化學研究者做能量分解的一切障礙。此方法目前已經正式發表在JPCA上,非常建議讀者仔細閱讀:
Tian Lu, Qinxue Chen, Simple, Efficient, and Universal Energy Decomposition Analysis Method Based on Dispersion-Corrected Density Functional Theory, J. Phys. Chem. A, 127, 7023 (2023) https://doi.org/10.1021/acs.jpca.3c04374
同時,此文還給出了筆者開發的名為sobEDA.sh的Bash shell腳本,可以很容易地將非常流行的量子化學程序Gaussian和波函數分析程序Multiwfn(http://www.shanxitv.org/multiwfn)相結合做sobEDA、sobEDAw能量分解。
在下文第2節,筆者將對sobEDA和sobEDAw的特點以盡量簡短的語言進行介紹,使得讀者能夠有最低限度的知識正確理解和使用這兩種方法,完整、全面的介紹和討論請仔細閱讀上述原文。第3節將會對sobEDA.sh腳本的基本用法做簡要介紹。第4節將會給出一系列具體例子充分展示如何用sobEDA.sh對各種類型體系和情況做sobEDA和sobEDAw能量分解。最后在第5節將對本文進行總結。
2 sobEDA和sobEDAw簡介
2.1 sobEDA/sobEDAw的主要特點
首先讓大家快速了解一下sobEDA/sobEDAw方法及實現它們的sobEDA.sh腳本的關鍵優點,從而知道為什么sobEDA和sobEDAw極其值得推薦!
? 門檻低&方便:只要是裝有Gaussian(G16 A.03及之后)和Multiwfn程序的Linux機子就直接能跑sobEDA.sh腳本。此門檻很低,因為Gaussian是用戶規模最大、使用得最普遍的量子化學程序,雖然收費但不算貴,安裝也很簡單。Multiwfn是用戶最多的波函數分析程序,完全免費,在Linux機子上的安裝也不費事。
sobEDA.sh的輸入文件的創建又容易又簡單。用戶僅僅需要懂得Gaussian算DFT單點能的輸入文件怎么寫就能順利使用,而這屬于幾乎所有量子化學研究者的最基本常識。
? 快:在像樣的雙路服務器下,算含有幾百個原子的體系都完全沒問題,耗時僅比DFT算整個體系貴一倍多,也即DFT計算單點能能算得動什么體系,就能對什么體系做sobEDA/sobEDAw能量分解。而且計算過程中對內存和硬盤的消耗微乎其微。
? 普適:兩個和多個片段、閉殼層和開殼層體系、主族和含過渡金屬體系、平衡結構和非平衡結構、化學鍵作用和弱相互作用,全都適用!
? 合理:各種量子力學效應都充分考慮,結果準確、物理意義清楚,并且與之前廣為接受的很多主流能量分解方法的結果高度吻合。
由于sobEDA、sobEDAw沒有明顯缺點,又有以上眾多優點,因此筆者十分建議把sobEDA、sobEDAw作為做能量分解的默認的方法。此外,這兩種方法的原理和實現很簡單,任何其它量子化學程序也都可以非常容易地將之實現在其中。
已有人在體驗過sobEDA后評論道(http://bbs.keinsci.com/thread-39400-1-1.html):在修回的文章里想添加EDA部分,本來是想用SAPT做的,結果計算速度慢只算的動SAPT0不說,有些結構還死活不收斂,弄得我心力憔悴。在Sobevera老師的推薦下在截至日期的前幾天轉用了他新開發的sobEDA,試用下來的效果只能用一句話形容,“令人震驚的好”!
2.2 sobEDA方法簡介
首先要明確一點,sobEDA及下一節介紹的其變體sobEDAw是對于特定幾何結構下片段間相互作用能進行分解,因此不考慮單體的變形能(即片段從其孤立狀態極小點結構變成它在復合物中的結構過程中的能量升高量)、不考慮熱力學效應(比如結合過程中焓或者自由能熱校正量的變化)、不考慮溶劑效應(即能量分解是對真空環境而言的)。如果要明確考慮這些效應,可以自己手動計算并作為單獨的能量項予以討論,也令能量分解給出的信息更為豐富,其過程也一點都不復雜,具體公式在前述的JPCA文章的2.1節末尾給明了。
sobEDA和sobEDAw都是基于考慮了色散校正的DFT方法定義的。如果不了解色散校正的話,看《DFT-D色散校正的使用》(http://www.shanxitv.org/210)和《談談“計算時是否需要加DFT-D3色散校正?” 》(http://www.shanxitv.org/413)。sobEDA和sobEDAw目前只考慮了普通泛函,精度已經足夠滿足實際研究需要,更昂貴的雙雜化泛函目前不納入考慮。
下面來看一下sobEDA的思想、各個能量項是怎么來的。
化學體系的實際狀態的波函數可以視為是由各片段的波函數經歷三個步驟構成的,如下所示,這里為了表示簡單只考慮了A、B兩個片段。ΨA+ΨB對應的是片段之間沒有任何相互作用的狀態,ΨAΨB對應的是兩個片段間已經出現了相互作用,但電子結構沒有發生變化,因此整體的占據軌道是這兩個片段占據軌道的并集,這稱為準分子狀態(promolecule state)。Ψ0AB是進一步考慮這倆片段的電子之間的Pauli互斥令占據軌道變形(滿足正交關系)之后的波函數,也叫做凍結態(frozen state)。最后的ΨAB是真實狀態波函數,也相當于常規DFT計算在SCF收斂后產生的波函數。
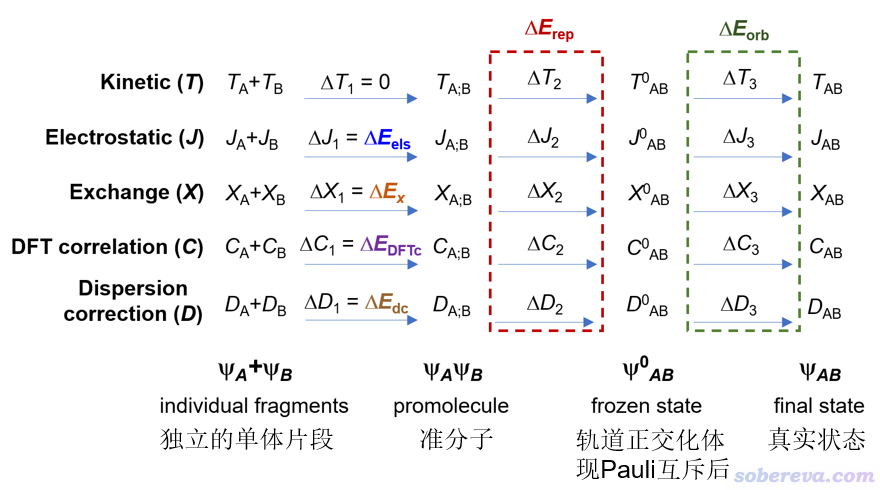
如上圖可見,體系狀態的每個變化過程都可能伴隨著動能(T)、經典靜電作用能(J)、交換能(X)、DFT相關能(C)、色散校正能(D)的變化。上圖中彩色字對應的是sobEDA方法分解出來的相互作用中的各個物理成份,顯然它們的加和精確等于色散校正的DFT方法以常規的超分子方式(即整體能量減各個片段能量)算的相互作用能ΔE_int,它可以寫為
ΔE_int = ΔE_els + ΔE_x + ΔE_DFTc + ΔE_dc + ΔE_rep + ΔE_orb
sobEDA給出的以上相互作用成份的物理含義如下
ΔE_els:片段間經典靜電作用能,els代表electrostatic。可正可負
ΔE_x:交換作用能,x代表exchange。數值為負,由DFT交換泛函貢獻,對雜化泛函顯然其中還牽扯到一部分HF交換能。這體現交換相關效應對片段間相互作用能的貢獻
ΔE_DFTc:DFT相關能。DFTc代表DFT correlation。一般數值為負,由DFT相關泛函貢獻。體現庫侖相關效應對片段間相互作用能的貢獻
ΔE_rep:Pauli互斥能,rep代表repulsion。數值為正。體現不同片段的電子間為了滿足Pauli互斥原理造成的能量升高
ΔE_orb:軌道相互作用能,orb代表orbital。數值為負。來自于片段內和片段間占據軌道與非占據軌道間混合導致的能量變化,物理上體現片段內電子分布被極化、片段間電子轉移對能量的總影響,共價作用也由此項體現
ΔE_dc:色散校正能。dc代表dispersion correction。數值為負。物理含義取決于具體情況,見下文
為了簡化討論,可以將ΔE_x與ΔE_rep相加作為交換-互斥項ΔE_xrep,這樣的項在很多能量分解方法里都有。如果不需要單獨討論色散作用的話,ΔE_DFTc和ΔE_dc還可以相加作為庫侖相關項ΔE_c,因為色散校正在本質上來說主要是對DFT相關泛函描述不好的長程庫侖相關部分進行彌補。
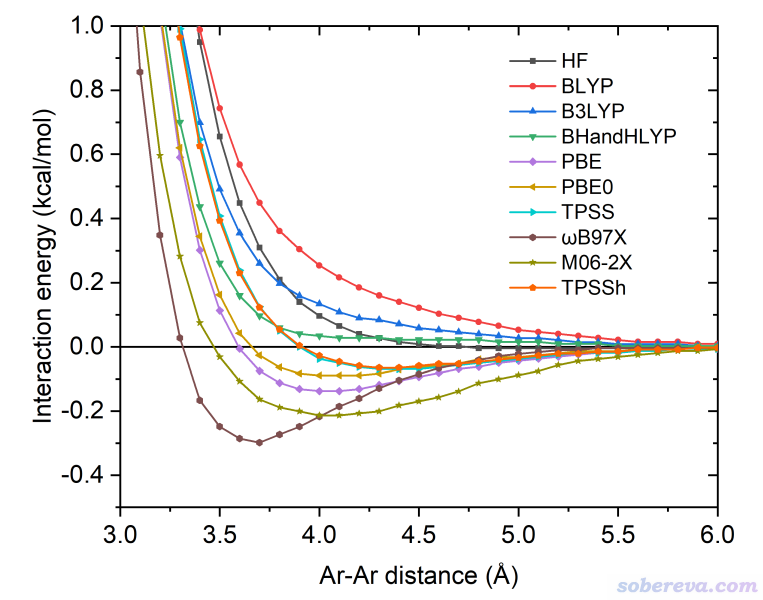
在目前的sobEDA、sobEDAw實現中,色散校正項ΔE_dc是由極為流行的DFT-D3(BJ)色散校正方法來計算的,原理上也可以用其它色散校正方法,如DFT-D4(http://www.shanxitv.org/464)、VV10等。特別要注意的是,僅當你用的泛函是完全無法描述色散作用的泛函,ΔE_dc才能解釋為色散作用能。什么泛函是完全無法描述色散作用的?在sobEDA原文里用一些方法對Ar二聚體做了能量隨距離的掃描,如下所示。眾所周知Ar和Ar之間是最典型的范德華作用,色散作用就是其中起到吸引作用的部分。可見BLYP、B3LYP、BHandHLYP在勢能曲線上都不存在極小點,即它們完全無法描述色散作用,因此sobEDA結合BLYP-D3(BJ)、B3LYP-D3(BJ)、BHandHLYP-D3(BJ)使用時,ΔE_dc才有明確的物理意義、可以被解釋為色散作用能。
值得一提的是,sobEDA/sobEDAw給出的軌道相互作用能ΔE_orb還可以用Multiwfn進一步通過ETS-NOCV方法進行分解,能得到不同類型軌道相互作用的能量貢獻以及對應的電子密度變化,對于探究片段間軌道相互作用本質極其有用!詳見《使用Multiwfn通過ETS-NOCV方法深入分析片段間的軌道相互作用》(http://www.shanxitv.org/609)。
sobEDA的計算耗時相當低,具體實現的細節和流程在前述的JPCA原文2.1節里說了,總共耗時僅僅是DFT計算整個體系的單點能的兩倍左右。所以但凡DFT算單點沒壓力,sobEDA方法做能量分解也必定輕輕松松。對如今較好的雙路服務器來說,基于Gaussian的DFT功能來實現sobEDA,對于好幾百原子都不費勁。
2.3 sobEDAw方法簡介
在說sobEDAw之前,先專門說一下色散作用項。一般意義上的色散作用指的是隨作用距離r具有-1/r^6衰減行為的長程庫侖相關作用。但是當分子間不遠不近,比如在分子復合物的極小點結構下,分子間庫侖相關作用既有中程的也有長程的。《使用PSI4做對稱匹配微擾理論(SAPT)能量分解計算》(http://www.shanxitv.org/526)中介紹的SAPT方法在對分子間弱相互作用的能量分解方面用得很普遍。SAPT(及其變體DFT-SAPT、SAPT(DFT)等)的色散作用項ΔE_disp包含了分子間全部庫侖相關作用,也就是說,當分子間距離不很遠時,SAPT的ΔE_disp項不僅包括了一般意義的色散作用,實際上還包含了中程庫侖相關作用。因此還有人特意管ΔE_disp叫做dispersion-like(而非dispersion)項。ΔE_disp由此也在一定程度上高估了一般意義的色散作用強度。盡管SAPT的色散項不那么“純粹”,SAPT在分子間弱相互作用的能量分解方面特別流行,而且很多文章都用SAPT給出的ΔE_disp和靜電作用項ΔE_els的比值來對相互作用體系進行分類,說誰是靜電主導、誰是色散主導、誰是混合型作用,比如非常知名的S66弱相互作用測試集的原文里就是這樣做的。前面說了,sobEDA結合比如B3LYP-D3(BJ)時的ΔE_dc對應的是“純”色散部分,因此在分子團簇極小點結構附近的ΔE_dc的大小勢必明顯小于ΔE_disp,也因此沒法把sobEDA的結果和SAPT的直接對比。顯然這不是因為sobEDA不合理,而是在于SAPT的色散項定義的特殊性所致。
考慮到以上問題,為了在sobEDA框架內能得到與SAPT相對比的結果,特別是令色散/靜電作用的比例能與SAPT的基本吻合、消除由內在定義差異帶來的明顯分歧,我提出了sobEDA的變體,名為sobEDAw,其后綴w特意強調此方法和SAPT一樣是專門用于對弱(weak)相互作用能進行分解的。sobEDAw的能量分解方式為
ΔE_int = ΔE_els + ΔE_xrep + ΔE_orb + ΔE_disp
其中ΔE_int、ΔE_els、ΔE_orb都和sobEDA完全一樣,但sobEDAw將交換-互斥作用項ΔE_xrep和色散作用項ΔE_disp定義為
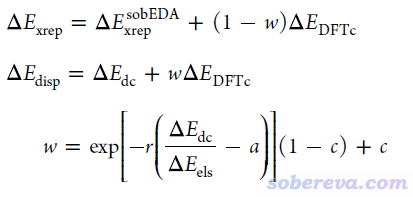
可見,sobEDAw引入了一個介于[0,1]的系數w,用來將sobEDA的ΔE_dc和一部分ΔE_DFTc組合成sobEDAw的ΔE_disp,而剩下的ΔE_DFTc和sobEDA的ΔE_xrep組成了sobEDAw的ΔE_xrep。w的計算除了依賴于sobEDA的ΔE_dc和ΔE_els外,還依賴于針對每種泛函/基組的組合而擬合的一套參數c、a、r。擬合的目標是令sobEDAw的ΔE_disp/ΔE_els比例與SAPT的盡可能一致,實際當中是對S66測試集與其文中的DFT-SAPT的數據盡可能吻合來擬合的。從sobEDA原文的圖2(d)可見這個目的充分達到了,因此sobEDAw可以很好地代替昂貴的SAPT。
由上可見,sobEDAw是對sobEDA的進一步延伸,當sobEDA數據算出來后,通過非常簡單的公式一瞬間就能進一步得到sobEDAw的結果。做sobEDAw分析也因此必然順帶產生sobEDA的數據。
大家都知道計算弱相互作用需要對基組重疊誤差(BSSE)問題做恰當的考慮,相關介紹見《談談BSSE校正與Gaussian對它的處理》(http://www.shanxitv.org/46)。以counterpoise方式校正BSSE問題,絕大多數情況可以在基組層面令弱相互作用能的計算精度進行提升。在sobEDA或sobEDAw能量分解計算中,如果計算每個單體都使用整個體系里所有原子的基函數,則給出的ΔE_int及其各個能量成份就都相當于是counterpoise方法校正后的。
下面的表格是前述JPCA文中的表1。表中體現了非常常用的泛函B3LYP-D3(BJ)結合不同基組對S66測試集計算的相互作用能與極高精度CCSD(T)/CBS參考值的誤差(kcal/mol),MAE是平均絕對誤差。w.CB代表with complex basis functions,也即計算單體時使用整個復合物的所有基函數,理解成在sobEDAw分析時考慮了counterpoise校正也可以。
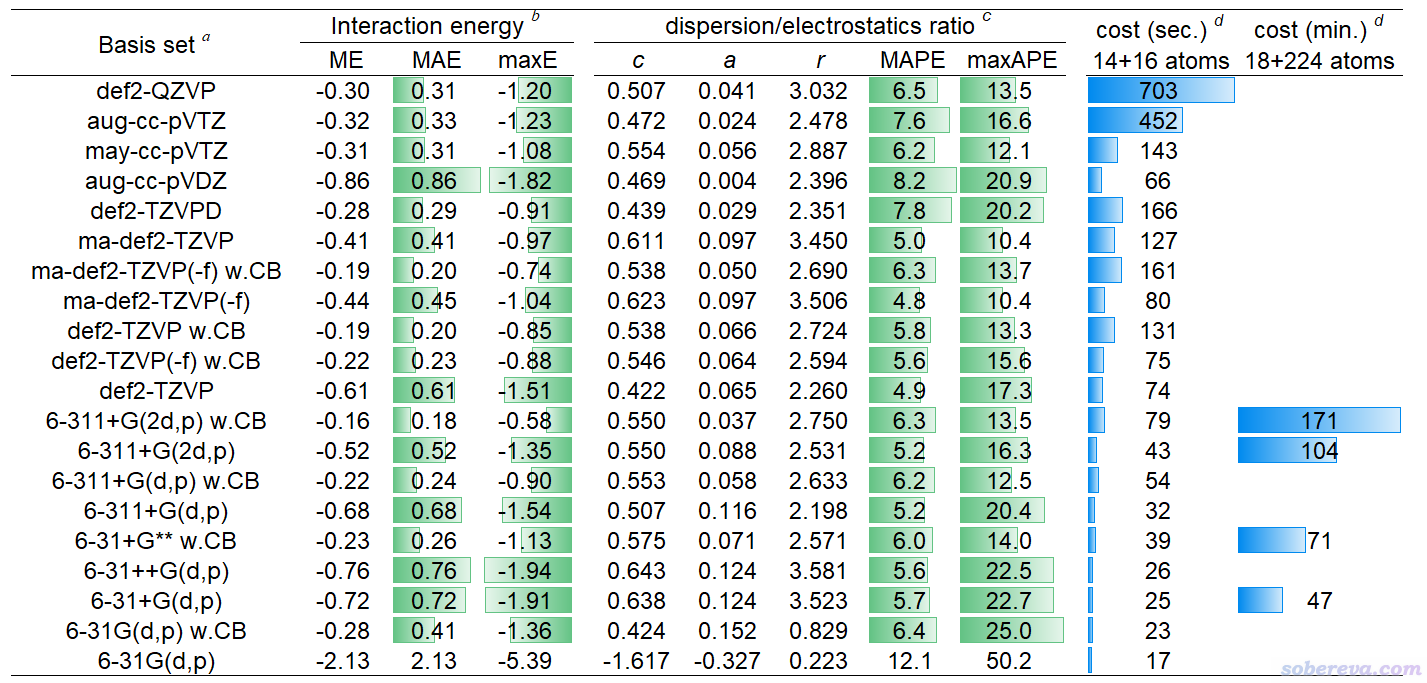
從以上表格可見6-31+G** w.CB是圖便宜首選,精度已經不錯,MAE才區區0.26 kcal/mol。要求更好精度且可以接受更高耗時的話推薦用6-311+G(2d,p) w.CB。def2-TZVP w.CB也值得推薦,性價比雖然不及6-311+G(2d,p) w.CB,但好處是支持的元素非常多,前六個周期都有定義。嫌def2-TZVP w.CB太貴的話也可以用更便宜的def2-TZVP(-f) w.CB,這是去掉了主族元素的f極化函數的版本,精度基本沒打折扣。平時就用這幾個基組做sobEDAw就足夠了,上表里的其它基組都沒什么值得額外特別推薦的。
上表里也給出了對S66測試集用sobEDAw計算的ΔE_disp/ΔE_els比例與DFT-SAPT的平均絕對誤差,以及對這些基組擬合的c、a、r參數。可見B3LYP-D3(BJ)結合上面推薦的幾個基組時的平均絕對百分比誤差(MAPE)才區區6%左右,表現都很理想,和昂貴的DFT-SAPT數據的吻合度相當高。
以上表格也體現了耗時情況。最后一列的18+224原子的體系是《8字形雙環分子對18碳環的獨特吸附行為的量子化學、波函數分析與分子動力學研究》(http://www.shanxitv.org/674)里介紹的主-客體復合物體系。在《淘寶上購買的雙路EPYC 7R32 96核服務器的使用感受和雜談》(http://www.shanxitv.org/653)介紹的96核機子上,筆者用B3LYP-D3(BJ)/6-31+G** w.CB對這么大的體系做sobEDAw能量分解僅僅花了71分鐘,可見耗時很低。只要有耐心,這樣的條件對500原子體系做能量分解都是可行的。如果是對14+16原子的體系,總共才39秒就完了事。
考慮到B3LYP-D3(BJ)并不總適合所有情況,因此筆者在JPCA文中表2里還給出了其它幾種很常見、適合不同情況的泛函結合前述幾種基組的c、a、r參數,以及誤差統計,可見都表現良好。
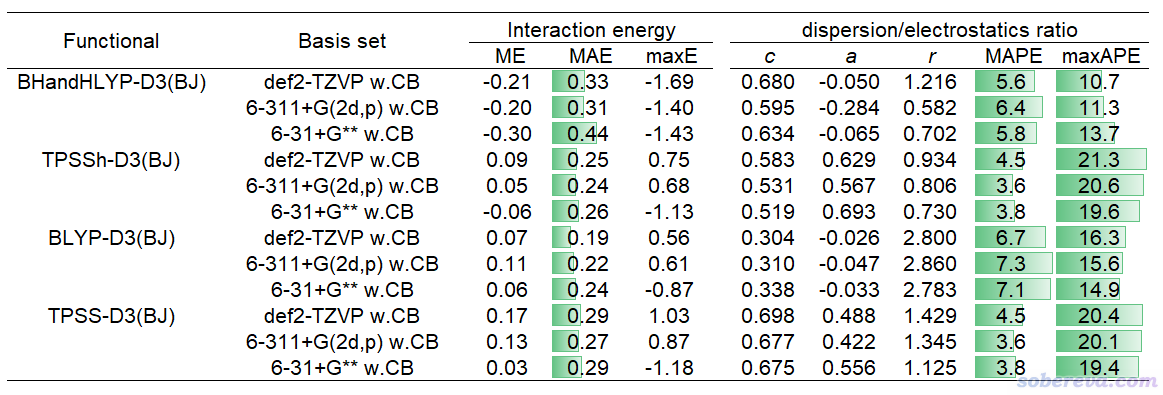
從上一節中的Ar-Ar勢能面掃描曲線看到,PBE、PBE0對色散作用的描述較為明顯,而M06-2X對色散作用的描述更是很充分,它們從原理上就不能結合sobEDAw,筆者也發現無法對它們擬合出合理的參數。因此以上表格沒有對這些也很常見的泛函進行考慮。表格里的那些的泛函已經足夠用了。
2.4 sobEDA和sobEDAw搭配泛函和基組的建議
鑒于之前有一些用戶對于sobEDA和sobEDAw分析搭配的泛函和基組有點糊涂,也有些人缺乏量子化學研究經驗、不太懂泛函和基組的選擇,這一節專門強調一下這個問題。
首先要明確一點,sobEDA和sobEDAw都可以用于研究弱相互作用,后者的額外好處是其色散項與流行的SAPT給出的可以相對比。對于化學鍵程度的強相互作用,或者片段間既有強相互作用也有弱相互作用,則只適合sobEDA,而sobEDAw和SAPT都不適合這類情況。
(1) 做sobEDAw用的泛函和基組
sobEDAw計算用的級別(泛函+基組)必須是已經有了現成的c、a、r參數的才行。在上面提到的JPCA原文的表1、2中,已經給了B3LYP-D3(BJ)結合大量基組的參數,以及其它幾種知名泛函TPSS-D3(BJ)、TPSSh-D3(BJ)、BLYP-D3(BJ)、BHandHLYP-D3(BJ)結合3種值得使用的基組的參數,因此它們都可以結合sobEDAw使用。如果你非要用其它級別的,則需要自行擬合參數。擬合用的程序、數據集和做法見http://www.shanxitv.org/soft/sobEDAw_fit.zip。一般完全沒必要自己擬合。
先說泛函方面。非常流行的B3LYP-D3(BJ)適合作為sobEDAw一般情況下默認用的泛函。但如《談談量子化學研究中什么時候用B3LYP泛函優化幾何結構是適當的》(http://www.shanxitv.org/557)所說的,B3LYP-D3(BJ)算pi大共軛體系往往不好,算過渡金屬配合物也很一般。對于pi大共軛體系建議改用離域性誤差較小的BHandHLYP-D3(BJ),對過渡金屬配合物建議用通常表現更好的TPSSh-D3(BJ)。其它情況可以隨機應變,例如如《簡談量子化學計算中DFT泛函的選擇》(http://www.shanxitv.org/272)所示,TPSS和TPSSh算Cu、Ag、Au團簇較好,因此算這種團簇對小分子的物理吸附做sobEDAw能量分解可以用TPSS-D3(BJ)和TPSSh-D3(BJ)。
再說基組方面。要求準定量準確的話,搭配6-31+G(d,p) w.CB即可,要求更高精度用6-311+G(2d,p) w.CB。若有的元素是這倆沒定義的,改用def2-TZVP w.CB或更便宜的def2-TZVP(-f) w.CB。def2-TZVP(-f) w.CB的耗時和6-311+G(2d,p) w.CB差不太多。
(2) 做sobEDA用的泛函和基組
原理上sobEDA可以結合任意泛函(雙雜化除外)和任意基組使用,因為sobEDA自身不依賴任何經驗參數。實際用的級別只要是計算當前體系片段間相互作用能較好、耗時你能接受的即可。從實際角度來說,在泛函方面,一般用B3LYP-D3(BJ)做sobEDA就可以,如果是計算過渡金屬與配體間相互作用,用TPSSh-D3(BJ)或PBE0-D3(BJ)通常更好。基組方面,對主族元素用6-311G(d,p),過渡金屬用SDD贗勢結合標配基組就不錯。若想要更好結果,或者對6-311G(d,p)沒定義的元素,可以用def2-TZVP。更多情況的泛函和基組的推薦在本文限于篇幅沒法展開介紹,請參閱這些文章里關于泛函和基組的討論:《簡談量子化學計算中DFT泛函的選擇》(http://www.shanxitv.org/272)、《談談量子化學中基組的選擇》(http://www.shanxitv.org/336)、《談談贗勢基組的選用》(http://www.shanxitv.org/373)。
sobEDAw是沒法用混合基組的,因為不同基組牽扯的擬合參數不同。而sobEDA可以混合基組。
sobEDA不是必須用DFT-D3(BJ),用其它任何色散校正都可以,也包括DFT-D3(0)。JPCA原文中sobEDAw擬合參數時只考慮了DFT-D3(BJ)的情況因此只能用這個。
無論是sobEDA還是sobEDAw,都不能結合ωB97XD、CAM-B3LYP等范圍分離泛函。不清楚哪些是范圍分離泛函的話看《不同DFT泛函的HF成份一覽》(http://www.shanxitv.org/282)。這不是sobEDA/sobEDAw理論的原因,而在于Gaussian程序(至少截止到目前撰文時最新的G16 C.02為止)的Link 608模塊對范圍分離泛函的情況沒法給出總能量當中的各種成分,也因此下面的sobEDA.sh腳本無法提取需要的信息。如果以后Gaussian改進了這點,sobEDA/sobEDAw也就沒這個限制了。
2.5 sobEDA和sobEDAw的準確性和可靠度
在sobEDA原文的第3節里通過8個涉及截然不同情況的例子(中性和離子、閉殼層和開殼層、平衡構型和非平衡構型、二聚體和三聚體、單重鍵和多重鍵、有機/無機分子和過渡金屬配合物,等等)。充分證明了sobEDA和sobEDAw的準確、可靠、普適和極高的實際應用價值。本文限于篇幅就不再復述一遍了,請讀者務必閱讀原文以對sobEDA和sobEDAw的實際應用有充分的認識,相信這些例子能令很多讀者受到不少啟發。
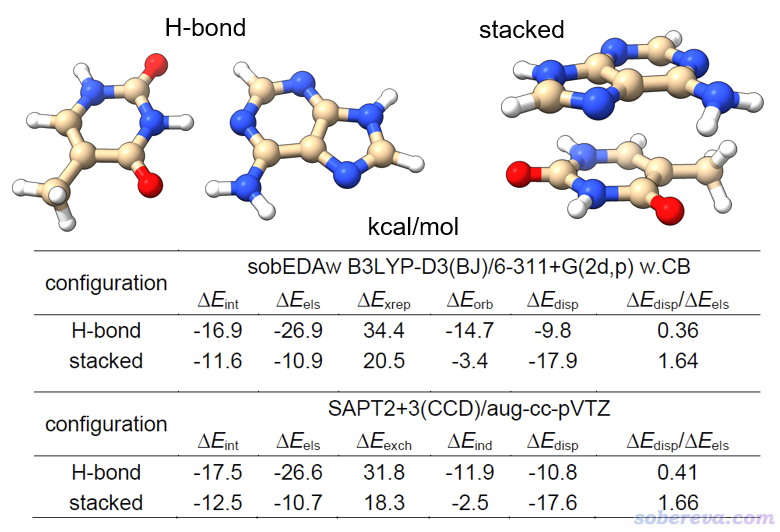
這里就僅僅展示原文里的一個例子,腺嘌呤-胸腺嘧啶氫鍵和堆積型二聚體。它有氫鍵和pi-pi堆積兩種構型,作用強度和作用本質都不同。下圖是sobEDAw和高階的SAPT的結果的對比,其中SAPT2+3(CCD)/aug-cc-pVTZ雖然精確但極為昂貴。可見盡管二者在原理上差異極大,但不僅兩個方法算的總相互作用能差異很小,而且各項都能相符得很好,ΔE_disp/ΔE_els吻合度相當高,因此便宜的sobEDAw完全可以代替很昂貴的高階SAPT。雖然SAPT里最低階的SAPT0相對便宜,但耗時還是顯著高于sobEDAw結合合適的計算級別,同時精度還差不少。此外,做SAPT的最主流程序PSI4的SCF收斂性比Gaussian有較大差距,改用基于Gaussian+Multiwfn做的sobEDAw就沒這個煩惱了。
3 sobEDA.sh腳本的使用
sobEDA/sobEDAw的原理非常簡單,只要基于DFT的程序的開發者有意向,非常容易地就能在程序里實現。sobEDA.sh是專門結合Gaussian和Multiwfn實現sobEDA/sobEDAw分析的Bash shell腳本,在這一節以盡量簡短的語言介紹一下基本使用方法。sobEDA.sh腳本文件可以在http://www.shanxitv.org/soft/sobEDA_tutorial.zip下載,并且里面的pdf文檔是此腳本最完整、詳細的使用說明,建議讀者有時間時完整看一遍。
3.1 使用要求
sobEDA.sh腳本的運行環境必須滿足以下幾點:
(1)必須是Linux環境。不一定非得是實體Linux系統,也可以是比如Windows下用VMware虛擬機創建的Linux客戶機、Windows下的WSL環境等
(2)機子里已正常安裝Gaussian,比如可以通過g16命令正常調用Gaussian 16。必須是G16 A.03及以后版本。不會裝Gaussian的話見《Gaussian的安裝方法及運行時的相關問題》(http://www.shanxitv.org/439)
(3)機子里已正常安裝Multiwfn,比如直接輸入Multiwfn命令就可以正常啟動。Multiwfn可以在http://www.shanxitv.org/multiwfn免費下載,必須是2023-Jun-25以后更新的版本。安裝方法見Multiwfn手冊2.1.2節。圖安裝省事也可以裝noGUI版,此時不需要額外裝任何圖形庫,但此版本的Multiwfn不具備任何和作圖有關的功能和圖形界面。
(4)dos2unix命令必須可以正常使用。機子里沒裝的話就裝上,比如Redhat/Rocky Linux/CentOS系統里用yum install dos2unix即可迅速安裝上。
3.2 sobEDA.sh的運行方法
(1)把sobEDA.sh放在任意目錄,并用chmod +x [文件路徑]命令給它加上可執行權限
(2)根據實際情況恰當修改sobEDA.sh里面的設置
(3)將整個體系的結構保存為system.xyz并放在當前目錄下
(4)在當前目錄下創建fragment.txt,在里面定義各片段的原子序號、凈電荷、自旋多重度
(5)在當前目錄下創建template.gjf,作為sobEDA.sh產生Gaussian輸入文件的模板文件
(6)輸入sobEDA.sh腳本的路徑啟動之
sobEDA.sh啟動后會自動調用Gaussian和Multiwfn進行計算、提取和處理數據,最后把結果輸出在屏幕上。運行中途產生的Gaussian的gjf文件、out文件、chk/fch文件都會自動保留,用戶可以自行查看。用Multiwfn載入fch文件后還可以考察各個任務產生的波函數的狀態,比如根據《使用Multiwfn觀看分子軌道》(http://www.shanxitv.org/269)的做法觀看里面的軌道,根據《談談自旋密度、自旋布居以及在Multiwfn中的繪制和計算》(http://www.shanxitv.org/353)的做法計算自旋密度和自旋布居。
下面說一下具體細節。
3.3 sobEDA.sh的設置
用文本編輯器打開sobEDA.sh腳本,就可以在開頭部分看到各種設置,里面有非常清楚的注釋,這里只說幾個重點:
? gau和mwfn變量分別對應調用Gaussian和Multiwfn用的命令,應根據實際情況設置。比如你的機子上的noGUI版Multiwfn是用Multiwfn_noGUI命令啟動的,顯然設為mwfn="Multiwfn_noGUI"
? sobEDAw=0代表做sobEDA分析,=1代表在sobEDA分析后還輸出sobEDAw的結果
? iCP=0代表不用counterpoise校正,=1代表用(對應基組帶w.CB后綴的情況)。分解弱相互作用的時候強烈建議用iCP=1以改進結果,分解化學鍵作用時則不建議iCP=1,不僅浪費很多時間,還可能令結果更差,見《計算化學鍵鍵能時考慮BSSE不僅是多余的甚至是有害的》(http://www.shanxitv.org/381)。
用sobEDAw的時候必須在sobEDA.sh里設置c、a、r參數,分別對應parm_c、parm_a、parm_r變量。為了方便用戶,sobEDA.sh腳本里已經自帶了一批常用級別的參數(和JPCA原文里表1、2一致),默認都是被注釋掉的狀態(行首帶#)因此不生效。例如腳本里可以看到
# B3LYP-D3(BJ)/6-31+G(d,p) with iCP=1
#parm_c=0.575;parm_a=0.071;parm_r=2.571
# B3LYP-D3(BJ)/6-311+G(2d,p) with iCP=1:
#parm_c=0.550;parm_a=0.037;parm_r=2.750
# BHandHLYP-D3(BJ)/6-31+G(d,p) with iCP=1:
#parm_c=0.634;parm_a=-0.065;parm_r=0.702
...略
你用哪種級別就把哪種情況的參數取消注釋,即去掉開頭的#。比如要用B3LYP-D3(BJ)/6-311+G(2d,p) w.CB,就把以上部分改為
# B3LYP-D3(BJ)/6-31+G(d,p) with iCP=1
#parm_c=0.575;parm_a=0.071;parm_r=2.571
# B3LYP-D3(BJ)/6-311+G(2d,p) with iCP=1:
parm_c=0.550;parm_a=0.037;parm_r=2.750
# BHandHLYP-D3(BJ)/6-31+G(d,p) with iCP=1:
#parm_c=0.634;parm_a=-0.065;parm_r=0.702
...略
顯然,其它級別的參數也都可以自行定義在里面。對于sobEDAw=0的情況,就完全不用管這些sobEDAw參數設沒設、怎么設的了。
3.4 system.xyz文件
system.xyz文件包含當前整個體系的坐標。不了解xyz格式的話看《談談記錄化學體系結構的xyz文件》(http://www.shanxitv.org/477)。產生這個文件極為簡單,比如Multiwfn載入pdb、mol、mol2等主流坐標格式的文件(支持什么其它的看http://www.shanxitv.org/379),或載入Gaussian/ORCA優化任務的輸出文件(會讀取最后一幀結構),然后在主界面輸入xyz,再輸入system.xyz,在當前目錄下就有了這個文件了。
3.5 fragment.txt文件
此文件里定義能量分解有幾個片段、各對應哪些原子。比如下面的例子代表sobEDA/sobEDAw分析是對3個片段間相互作用能做分解,第1個片段的凈電荷為0、自旋多重度為1,原子序號為1-3,7,10-12。第2個片段的凈電荷為-1、自旋多重度為1,原子序號為4-6。第3個片段的凈電荷為+1、自旋多重度為1,原子序號為8,9。注意所有片段中的原子的并集必須對應整個體系。
3
0 1
1-3,7,10-12
-1 1
4-6
1 1
8,9
自旋多重度寫為負值時表示片段帶的是beta單電子。比如下面的例子,是兩個片段,片段1包含1-4號原子,是三重態,單電子為alpha自旋。片段2包含5-8號原子,也是三重態,但單電子為beta自旋。
2
0 3
1-4
0 -3
5-8
3.6 template.gjf文件
這是Gaussian輸入文件的模板文件,里面記錄了計算過程要用的關鍵詞。[geometry]部分會被腳本自動替換成實際要算的坐標。此文件里凈電荷和自旋多重度設的是對整個體系計算時用的(算各個片段時用的是fragment.txt里定義的)。#P和nosymm關鍵詞是必須有的。ExtraLinks=L608關鍵詞結合此文件末尾的數字用來讓Gaussian輸出當前總能量中的各個成份,它們是要被sobEDA.sh腳本自動提取和處理的。在此基礎上,若遇到SCF不收斂,可以自行加入幫助SCF收斂的關鍵詞,比如scf(novaracc,noincfock)之類的,參考《解決SCF不收斂問題的方法》(http://www.shanxitv.org/61),但有的關鍵詞不兼容,比如guess=AM1等。
#P b3lyp/6-311+G(2d,p) em=gd3bj ExtraLinks=L608 nosymm
<---空行
title line
<---空行
0 1
[geometry]
<---空行
-5 5
<---空行
<---空行
上面例子末尾的-5對應當前用的泛函在Gaussian中的內部IOp參數,必須根據實際情況設置,需要查閱Gaussian IOps手冊的IOp(3/74)部分,見http://gaussian.com/overlay3/#iop_(3/74)。一些比較常見的泛函的這個值列在這里了:-73 (MN15), -55 (M06-2X), -54 (M06), -53 (M06L), -35 (TPSSh), -13 (PBE0), -5 (B3LYP), -6 (B3PW91), -3 (BHandHLYP), 402 (BLYP), 1009 (PBE), 2523 (TPSS)。上面例子末尾第二個數字(5)不用動它,這是要求Link 608模塊也用Gaussian 16做SCF過程默認用的ultrafine積分格點。
要注意一些泛函的DFT-D3(BJ)參數在Gaussian里沒內置。比如TPSSh的D3(BJ)參數起碼到Gaussian 16 C.02仍然不是內置的。這種情況需要按照《DFT-D色散校正的使用》(http://www.shanxitv.org/210)里的做法自行定義。
為了用戶方便,在http://www.shanxitv.org/soft/sobEDA_tutorial.zip其中的templates目錄下已經提供了幾種常見的泛函的模板文件,比如template_BHandHLYP-D3(BJ).gjf就是對應BHandHLYP-D3(BJ)的情況,把它改名為template.gjf,并且修改里面的基組、電荷、自旋多重度為實際情況,就可以用了。
雖然sobEDA/sobEDAw是基于色散校正的DFT定義的,但也可以不加色散校正,即去掉em=gd3bj,此時輸出的色散校正項將為0。
3.7 常見問題
這一節回答幾個使用sobEDA.sh的常見問題。
(1)怎么讓sobEDA.sh計算時并行?
《Gaussian的安裝方法及運行時的相關問題》(http://www.shanxitv.org/439)里說了,Default.Rou里設置比如-P- 96就可以默認用96核并行,-M-可以設置用的內存上限。也可以直接在template.gjf里用%nproc和%mem里設置并行核數和內存上限。
(2)為什么sobEDA.sh最后給出的總相互作用能和我自己用整體能量減去各個片段能量得到的不符?
原理上是必然相符的,不相符有以下常見可能:用的幾何結構不一樣、用的基組不一樣、用的泛函不一樣、影響能量的某些設置不一致(比如溶劑模型、積分格點精度)、counterpoise校正考慮得不一樣、凈電荷和自旋多重度設置有差異、你自己做的或者sobEDA.sh自動做的某些單點計算恰好收斂到了不穩定波函數。仔細對比自己的Gaussian輸入輸出文件和sobEDA.sh產生的Gaussian輸入輸出文件必定能找到導致差異的原因。
(3)sobEDA和sobEDAw能帶溶劑模型么?
本文2.2節一開始說了,sobEDA/sobEDAw是對真空下相互作用能做的分解,也顯然不能直接在template.gjf寫上scrf關鍵詞帶溶劑模型。想考慮溶劑模型要按JPCA原文2.1節末尾說的,自己計算各個體系在真空和溶劑模型下的單點能,通過簡單運算得到溶劑效應對相互作用能的貢獻。
(4)sobEDA.sh運行中途出錯怎么辦?
首先要知道,sobEDA.sh按順序計算這些狀態:各個孤立片段(fragment[序號].gjf)、準分子態(promol.gjf)、凍結態(frozen.gjf)、以凍結態波函數為初猜的最終態(final.gjf)。只有fragment[序號].gjf和final.gjf在計算過程中需要做SCF直到收斂。腳本還利用Multiwfn產生準分子態的波函數、創建輸入文件等工作。
sobEDA.sh計算過程中在干什么、做到哪一步了,在輸出信息中顯示得非常清楚,從中可以很容易了解到是在哪個環節上出了錯。如果是調用Gaussian運行時出錯,應仔細看當前目錄下sobEDA.sh產生的相應任務的Gaussian的out文件末尾,結合Gaussian基本使用常識搞清楚原因。如果從提示中看到是Multiwfn執行出錯,>99%的可能性是Multiwfn沒有按手冊2.1.2節寫的正確安裝,導致無法正常啟動,或者由于可調用內存上限太少而無法執行。
絕對不能用太老版本的Multiwfn和Gaussian,sobEDA.sh需要用什么版本在本文3.1節明確說了。
sobEDA.sh中途涉及用Gaussian自帶的formchk和unfchk工具進行chk和fch之間的轉換。如果要算幾百原子的大體系,有可能由于自帶的這倆程序可以調用的內存上限不夠而無法轉換而造成任務失敗。解決方法是在用戶主目錄下的.bashrc里加上比如export GAUSS_MEMDEF=20GB然后重新進入終端,以使得這些Gaussian輔助程序能用的內存上限有20GB,這就足夠大了。
如果死活弄不清楚為什么出錯,又確信自己的輸入文件肯定沒毛病、Multiwfn自帶的例子都能正常跑完,可以在計算化學公社論壇http://bbs.keinsci.com的“量子化學”版發帖求助(選擇[綜合交流]分類),上傳所有給sobEDA.sh用的輸入文件。
順帶一提,sobEDA.sh腳本注釋得特別清楚,有點shell腳本常識就能很容易理解里面的內容,沒常識的話看《詳談Multiwfn的命令行方式運行和批量運行的方法》(http://www.shanxitv.org/612)里面關于shell腳本的相關介紹。sobEDA.sh做的幾個主要計算在sobEDA的JPCA原文7025頁左上角部分都有描述。用戶也可以結合實際需要對sobEDA.sh腳本進行適當的修改和調試。
(5)用Gaussian自帶的基組時sobEDA.sh能正常運行,但是使用自定義基組、混合基組,或者引用外部基組文件時sobEDA.sh中途出錯怎么回事?
顯然沒定義對。一定記得先仔細看《詳解Gaussian中混合基組、自定義基組和贗勢基組的輸入》(http://www.shanxitv.org/60)了解怎么正確使用gen和genecp、格式是什么樣。sobEDA官方教程里“Cu+與H2O間的相互作用”都是用genecp的具體例子。如果是靠@來引用外部基組/贗勢文件,記得外部基組/贗勢文件末尾不要有空行,否則當此文件被展開后,相當于基組/贗勢部分末尾出現了多余的空行,導致Gaussian無法讀取到最末尾的比如-5 5這樣的信息。
4 實例
sobEDA/sobEDAw官方教程http://www.shanxitv.org/soft/sobEDA_tutorial.zip里面有非常豐富而且講解得特別清楚易懂的例子,充分涵蓋了各種情況下的能量分解,全都弄懂了就可以把sobEDA.sh用得游刃有余。例子包括:
? 對H2O...NH3相互作用在B3LYP-D3(BJ)/6-311+G(2d,p) w.CB下做sobEDA能量分解
? 對OC-BH3相互作用在B3LYP-D3(BJ)/6-311G*下做sobEDA能量分解
? 對Cu+與H2O間的相互作用在TPSSh-D3(BJ)/SDD&6-311G*下做sobEDA能量分解
? 對CH3-NH2相互作用在B3LYP-D3(BJ)/def2-TZVP下做sobEDA能量分解
? 對水四聚體的四個分子間相互作用在BHandHLYP-D3(BJ)/6-31+G(d,p) w.CP下做sobEDAw能量分解
? 利用腳本對CH3-CN相互作用在不同間距下做sobEDA能量分解,并且繪制成“相互作用成分 vs 片段間距”曲線圖
? 利用腳本對苯...水相互作用在不同間距下做sobEDAw能量分解并且繪制成曲線圖
? 獲得孤立狀態→準分子態、準分子態→凍結態、凍結態→最終態這三個過程中完整的能量成分變化
此外,教程文件包里還提供了sobEDA/sobEDAw原文里的諸多例子用的原文件,包括:
? 對18碳環...三重態氧氣間的相互作用在BHandHLYP-D3(BJ)/6-311+G(2d,p) w.CB級別下做sobEDAw能量分解
? 對(CO)5Cr=CH2配位鍵相互作用在TPSSh-D3(BJ)/6-311G*&SDD級別下做sobEDA能量分解
? 對C6F6...Cl-離子相互作用在BHandHLYP-D3(BJ)/6-311+G(2d,p)級別下做sobEDA能量分解
? 對HC-CH乙炔中的三重鍵在B3LYP-D3(BJ)/def2-TZVP級別下做sobEDA能量分解
在下面,就只把H2O...NH3相互作用的sobEDAw能量分解這個最簡單的例子說一下,其它的請務必看sobEDA/sobEDAw官方教程里的講解(若在這里把所有英文例子翻譯成中文也沒什么意思)。此體系的結構圖如下所示,是經過幾何優化過的
H2O...NH3在B3LYP-D3(BJ)/6-311+G(2d,p) w.CB下做sobEDAw分析能量分解用到的所有文件在官方教程文件包里的HOH-NH3目錄下都提供了。system.xyz就是上圖對應的結構文件,是經過優化過的水與氨氣的二聚體極小點結構。template.gjf和本文3.6節示例的那個是一致的,不再累述。fragment.txt內容如下,就是定義兩個片段,也沒什么可說的
2
0 1
1-3
0 1
4-7
在此目錄下的sobEDA.sh里可見iCP=1、sobEDAw=1,說明要做sobEDAw分析并且考慮counterpoise。并且可以看到parm_c=0.550;parm_a=0.037;parm_r=2.750這一行開頭的#被去掉了,說明這次sobEDAw計算用這套c、a、r參數,對應于當前要用的B3LYP-D3(BJ)/6-311+G(2d,p) w.CB級別的參數。
把HOH-NH3目錄下的sobEDA.sh、system.xyz、fragment.txt、template.gjf都拷到當前目錄下,先運行chmod +x ./sobEDA.sh加上可執行權限,然后運行./sobEDA.sh |tee result.txt即可開始計算,并很快得到結果。結果既顯示在了屏幕上,也同時輸出到了result.txt里,如下所示
(計算過程輸出的細節信息略...)
*************************
***** Final results *****
*************************
Total interaction energy: -7.18 kcal/mol
Physical components of interaction energy derived by sobEDA:
Electrostatic (E_els): -12.15 kcal/mol
Exchange (E_x): -6.40 kcal/mol
Pauli repulsion (E_rep): 20.28 kcal/mol
Exchange-repulsion (E_xrep = E_x + E_rep): 13.88 kcal/mol
Orbital (E_orb): -5.38 kcal/mol
DFT correlation (E_DFTc): -2.70 kcal/mol
Dispersion correction (E_dc): -0.83 kcal/mol
Coulomb correlation (E_c = E_DFTc + E_dc): -3.53 kcal/mol
可見總相互作用能為-7.18 kcal/mol,同時sobEDA方法定義的各個物理成份都給出得相當清楚。為了便于討論,也把ΔE_x與ΔE_rep的加和值ΔE_xrep給出了、把ΔE_DFTc和ΔE_dc的加和值ΔE_c給出了。
然后看到的是sobEDAw的結果,如下所示。由于sobEDA和sobEDAw的相互作用能是相同的,差異僅在于物理成份的劃分上,因此總相互作用能就沒再輸出一遍。由提示可見96.26%的ΔE_DFTc與ΔE_dc相組合得到了sobEDAw的ΔE_disp項,即0.9626*(-2.70)-0.83 = -3.43 kcal/mol。其余的ΔE_DFTc與sobEDA的ΔE_xrep組合得到了sobEDAw的ΔE_xrep項,即(1-0.9626)*(-2.70)+13.88 = 13.78 kcal/mol。sobEDAw給出的ΔE_orb和ΔE_els與前面給出的sobEDA相應項完全一致。
Variant of sobEDA for weak interaction (sobEDAw):
Note: 96.26% DFT correlation is combined with dispersion correction to yield a SAPT-like dispersion term
Electrostatic (E_els): -12.15 kcal/mol
Exchange-repulsion (including scaled DFT correlation): 13.78 kcal/mol
Orbital (E_orb): -5.38 kcal/mol
Dispersion (E_disp): -3.43 kcal/mol
由以上數據可見,HOH...NH3氫鍵作用是靜電相互作用所主導的,ΔE_els占總吸引作用的-12.15/(-12.15-5.38-3.43)*100% = 58%。其次軌道相互作用也有不可忽視的貢獻,占總吸引作用的26%。色散作用對此氫鍵的形成貢獻最小。交換-互斥作用對結合起到了顯著不利因素,抵消掉了總吸引作用的66%,這是普遍現象。
腳本運行完畢后在當前目錄下還可以看到fragment1.gjf/out/fch、fragment2.gjf/out/fch、promol.gjf/out/chk/fch、final.gjf/out/chk這些中間過程產生的文件,現在已經完全沒用了,可以直接刪掉,感興趣的話也可以自己查看以了解腳本都調用Gaussian做了哪些計算。
值得一提的是,《透徹認識氫鍵本質、簡單可靠地估計氫鍵強度:一篇2019年JCC上的重要研究文章介紹》(http://www.shanxitv.org/513)里介紹的筆者的J. Comput. Chem., 40, 2868 (2019)一文中算的42種氫鍵復合物中也包含了HOH...NH3氫鍵復合物,文中在精度很高且非常昂貴的SAPT2+(3)δMP2/aug-cc-pVTZ下得到的相互作用能為-6.47,靜電、交換互斥、誘導(本質同軌道相互作用)、色散的貢獻分別為-11.70、12.63、-4.16、-3.23。當前sobEDAw計算得到的這5個值分別為-7.18、-12.15、13.78、-5.38、-3.43,可見相符程度極好!SAPT2+(3)δMP2給出的色散/靜電比例為0.276,此例sobEDAw給出的為0.282,相符巨好!
5 總結
本文全面、完整介紹了筆者提出的基于很流行的色散校正DFT理論的sobEDA和sobEDAw能量分解方法,以及基于Gaussian和Multiwfn實現這兩種分析的sobEDA.sh腳本。sobEDA/sobEDAw方法分析準確度高、普適性強、耗時低,而sobEDA.sh用起來十分容易,對于量子化學初學者來說也沒有什么門檻,輸出信息頗為易于理解。sobEDA/sobEDAw結合sobEDA.sh可謂掃清了廣大量子化學研究者做能量分解的各種困難,相信在不久的將來就會成為很流行的方法,非常推薦在大家實際研究中廣泛使用sobEDA/sobEDAw并向同行積極推廣!
發表的研究工作中使用sobEDA或sobEDAw方法時請記得引用方法原文J. Phys. Chem. A, 127, 7023 (2023)。由于sobEDA.sh腳本利用了Gaussian和Multiwfn程序,因此也請同時按照Multiwfn官方網站上或程序啟動時的提示正確引用Multiwfn的原文,以及引用Gaussian程序。