使用Multiwfn通過ETS-NOCV方法深入分析片段間的軌道相互作用
使用Multiwfn通過ETS-NOCV方法深入分析片段間的軌道相互作用
文/Sobereva@北京科音 2021-Jul-31
0 前言
強大、免費的波函數分析程序Multiwfn(主頁:http://www.shanxitv.org/multiwfn)支持極為豐富的化學鍵分析方法,見《Multiwfn支持的分析化學鍵的方法一覽》(http://www.shanxitv.org/471)。其中已支持不少從軌道相互作用層面上考察特定片段間的相互作用的方法,比如
? CDA方法可以從片段軌道混合成復合物軌道的角度對片段間電荷轉移情況予以深入分析,見《使用Multiwfn做電荷分解分析(CDA)、繪制軌道相互作用圖》(http://www.shanxitv.org/166)。
? 原始的NAdO分析方法經筆者擴展后,可以將特定兩個片段間的總離域化指數(本質類似于片段間的總Mayer鍵級)轉化為一批軌道的形式,從而能圖形化展現和直觀分析,見《使用鍵級密度(BOD)和自然適應性軌道(NAdO)圖形化研究化學鍵》(http://www.shanxitv.org/535)。
? 兩個片段間的以自然原子軌道(NAO)為基的總Wiberg鍵級可以在Multiwfn中分解為兩個片段間每一對原子軌道殼層之間的相互作用的貢獻,從而便于探究對片段間結合起主要貢獻的殼層,見Multiwfn手冊3.11.8節的介紹和4.9.4節的例子。
從2021-Jul-28更新的版本開始(絕對不要用更老的版本!),Multiwfn正式支持另一種十分流行的分析片段間軌道相互作用的方法,稱為ETS-NOCV (Extended Transition State - Natural Orbitals for Chemical Valence),可譯為“擴展過渡態-化學價的自然軌道”。這個方法由Ziegler等人于J. Chem. Theory Comput., 5, 962 (2009)提出,如今已被廣泛應用于不同類型體系的研究。它最常用來考察片段間的化學鍵作用,但也有不少文章拿它來考察氫鍵、鹵鍵等弱相互作用(雖然軌道相互作用并非弱相互作用的關鍵構成,但通過ETS-NOCV對它分析無疑可以提供更多的考察弱相互作用的視角)。
下面第1節筆者先簡要介紹ETS-NOCV的原理,然后在第2節說一下Multiwfn的ETS-NOCV分析模塊的功能和特性,之后在第3節給出豐富的ETS-NOCV分析例子,最后在第4節對本文進行總結。本文比較長,但只要仔細看過一遍后,就會發現使用Multiwfn做ETS-NOCV分析特別簡單而且極具實際價值,也沒有什么難理解的地方。
1 ETS-NOCV原理簡述
在Multiwfn手冊3.26.1節有ETS-NOCV分析方法的非常詳細、全面、透徹的說明,強烈建議大家仔細閱讀。由于在本文寫公式不太方便,筆者也不想重復一遍手冊里已詳細交代的信息,因此在這里只是用淺顯的語言簡單描述一下ETS-NOCV大概是怎么回事,至少足矣讓讀者能理解后文的分析實例。
首先應了解到,兩個片段A、B從分離得無窮遠的孤立的狀態逐漸接近變成復合物AB(或者成鍵形成新分子,后文不做區分),在此過程中能量的變化可以寫為以下關系(注意這絕不是唯一的總能量變化的劃分方式,只是來自ETS-NOCV方法原文的思想)
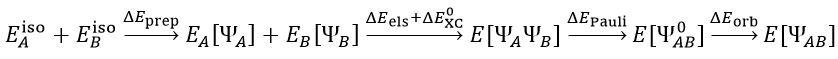
其中E_iso(A)、E_iso(B)分別代表A和B在孤立狀態下的能量。A和B在結合時首先經歷preparation階段,令其幾何結構從它們的孤立存在下的最穩定狀態變成它們在復合物中所處的狀態,這部分的能量變化是ΔE_prep。上式中E_A[ΨA]和E_B[ΨB]分別代表A和B在復合物結構時其單獨存在時的能量,ΨA和ΨB是此時各自的波函數。ΨAΨB是A和B各自單獨存在時波函數的簡單乘積(此時片段的電子結構尚未受到另一方的影響),E[ΨAΨB]是這個波函數對應的復合物的能量,它與E_A[ΨA]+E_B[ΨB]之間的能量差可視為來自A和B之間的靜電作用(ΔE_els)和交換-相關能的變化(ΔE_0_XC)。A和B形成復合物后,由于二者間有Pauli互斥,為了滿足Pauli不相容原理,A和B的占據軌道都會發生變形,從而令彼此之間滿足正交關系(原本的軌道在片段內正交但在片段間不正交),此時這倆片段的所有的占據軌道構成的復合物的波函數是Ψ_0_AB,也可以說這是A和B波函數的反對稱乘積,它對應的復合物能量E[Ψ_0_AB]與原本A和B波函數簡單組合(相乘)對應的復合物能量E[ΨAΨB]之差是Pauli互斥能ΔE_Pauli。Ψ_0_AB也被稱作凍結態(frozen state),這是片段間尚未發生軌道相互作用但已考慮Pauli互斥的假想態。由于A、B片段間存在軌道相互作用,會造成電荷轉移(一方的占據軌道與另一方的空軌道混合)和電子極化(片段的占據軌道與它自己的空軌道混合),這使得復合物的波函數由Ψ_0_AB進一步變化為最終狀態ΨAB,這個過程的能量變化是軌道相互作用能ΔE_orb(orbital interaction energy)。這里的ΨAB就相當于對復合物做單點計算得到的波函數。其實上面這一系列過程的劃分有明顯的人為任意性,這里就不深究和多說了。
之所以說上面這些,是為了讓大家知道ETS-NOCV的分析對象是什么。簡單來說,ETS-NOCV就是把上面的ΔE_orb這一項拆分成一批NOCV pair的貢獻,即每個NOCV pair對于片段間結合都有相應的能量貢獻。而且每個NOCV pair對軌道相互作用造成的電子密度差還有各自的貢獻,并且可以將NOCV pair的密度作圖以了解它對應的軌道作用方式。因此,ETS-NOCV分析能給出各種形式的軌道相互作用的能量和對應的密度變化,這無疑對于考察片段間作用本質極有幫助。
在ETS-NOCV提出之前實際上是先有的NOCV (Natural Orbitals for Chemical Valence)方法,然后作者將其在1977年提出的ETS (Extended Transition State)能量分析方法相結合才進一步提出的ETS-NOCV。我們先看NOCV是怎么回事。
密度差大家都熟悉,利用整體與各個片段間的密度之差可以展現片段結合前后電子密度的變化,在《使用Multiwfn作電子密度差圖》(http://www.shanxitv.org/113)有相關例子。密度矩陣是體系的電子波函數的一種矩陣形式的變相描述,軌道相互作用對密度矩陣的影響可以寫為實際復合物的密度矩陣與凍結態的密度矩陣之差ΔP_orb = P[ΨAB] - P[Ψ_0_AB]。將ΔP_orb做對角化就得到了一批本征矢和本征值,每個本征矢對應于一個NOCV軌道的展開系數,每個NOCV軌道有相應的本征值,所有的本征值加和恰為0。
由于軌道相互作用造成的密度變化是下式的Δρ_orb,這叫做軌道變形密度(orbital deformation density)。φ(i)和v(i)分別代表第i個NOCV軌道波函數和它的本征值,N是所有NOCV軌道的數目。
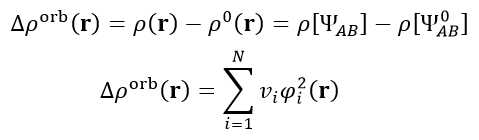
由上式可見,NOCV理論可以把軌道相互作用造成的密度變化分解為各個NOCV軌道的貢獻,貢獻的權重就是NOCV軌道的本征值。通常在研究片段間相互作用時人們只是簡簡單單地繪制一個整體減片段的密度差圖,此時片段間的各種形式的軌道相互作用和Pauli互斥作用全都一鍋粥地攪合在一起,經常無法從圖像上準確、完整地洞悉作用本質,往往得靠主觀臆測。而如果使用NOCV方法對軌道變形密度進行分解分析,則不同方式的軌道相互作用就能被區分得很清楚,明顯便于深入討論。計算復合物時有多少個基函數,就會得到多少個NOCV軌道,雖然其數目很多,但一般只有很少量NOCV軌道的本征值較大、對變形密度影響顯著并因此值得被討論,而其余那些本征值很小的NOCV軌道都可以在討論時直接忽略掉。
由于ΔP_orb矩陣的特性,其本征矢NOCV軌道的一個特點是成對出現的,即如果有一個NOCV軌道本征值是0.3,則必定會有另一個NOCV軌道本征值是-0.3。因此,我們可以把所有N個NOCV軌道匹配成N/2個NOCV pair,并得到每個NOCV pair對軌道變形密度的貢獻,如下所示。-i代表與i配對的NOCV軌道序號,v(-i)=-v(i)。
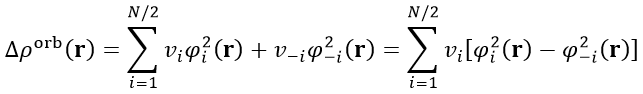
基于NOCV pair來討論不僅明顯更為方便,物理意義也清楚,因為配對的兩個NOCV軌道可以認為來自于同一種軌道相互作用。
最后再來看ETS-NOCV是怎么回事。ETS理論證明了軌道相互作用能可以以如下方式計算
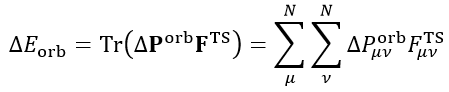
其中F_TS是擴展過渡態的Fock矩陣(對于DFT計算來說是Kohn-Sham矩陣,后文不作區分)。注意擴展過渡態和平時我們說的過渡態完全是兩碼事,擴展過渡態是復合物幾何結構下的一個虛構的電子結構狀態,對應的是前述的實際復合物波函數ΨAB與凍結態波函數Ψ_0_AB之間的中點,它可以用這兩個波函數的平均的密度矩陣來構造Fock矩陣得到。
ETS-NOCV將ETS和NOCV相結合,就得到了如下關系,即每個NOCV軌道都對軌道相互作用能有相應的貢獻。例如i軌道的貢獻為ΔE_orb(i),可以寫為這個軌道的本征值乘上以NOCV軌道為基的F_TS矩陣的(i,i)對角元(以NOCV為基的F_TS上面寫了個波折號以注明),實際上這個對角元也等于相應的NOCV軌道的F_TS算符的期望值,可以理解為NOCV軌道的能量。
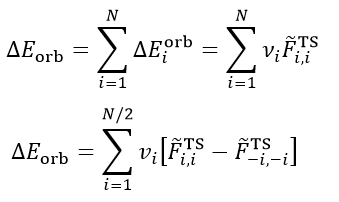
如上面第二個式子可見,也可以根據NOCV軌道的配對關系,獲得各個NOCV pair對軌道相互作用能的貢獻。通常只有很少幾個NOCV pair對軌道相互作用能的貢獻相對較大,這些是我們討論的重點,其余的都可以忽略。注意NOCV pair的本征值的大小和它對應的軌道相互作用能貢獻的大小并沒有必然的相關性,只能說如果本征值非常小,那么由于上式中的v非常小,則必然它對軌道相互作用能的貢獻也很小。
2 Multiwfn中的ETS-NOCV分析
Multiwfn中的ETS-NOCV分析功能的詳細說明見手冊3.26.2節,這里只是做簡要介紹。
2.1 ETS-NOCV分析模塊的特征
Multiwfn的ETS-NOCV分析功能是主功能23,具有以下功能:
? 計算NOCV軌道波函數、本征值、能量
? 觀看NOCV軌道的等值面圖、將其波函數格點數據導出為cub文件
? 計算NOCV pair對軌道相互作用能的貢獻
? 觀看NOCV pair的等值面圖、將其密度格點數據導出為cub文件
? 計算NOCV pair和NOCV軌道的成份(使用SCPA方法)
? 可視化準分子狀態(promolecular)、frozen state狀態、實際狀態的整體的軌道波函數
? 可視化Pauli變形密度(Δρ_Pauli)、軌道變形密度(Δρ_orb)以及總變形密度(Δρ)
雖然Multiwfn的ETS-NOCV功能很多,但由于筆者花了極大心思令其使用盡可能簡單方便,所以你會發現用起來相當容易、順手。
前面介紹ETS-NOCV理論的時候只用了兩個片段為例,但Multiwfn的ETS-NOCV功能為了靈活考慮,可以支持無數多個片段,分析的是所有片段之間總的相互作用。
有一點需要注意:Multiwfn的ETS-NOCV功能無法產生前述的對應擴展過渡態的Fock矩陣(F_TS)來計算NOCV能量,作為近似,Multiwfn使用整體的實際波函數對應的Fock矩陣來計算NOCV能量,也因此此時所有NOCV軌道能量之和與軌道相互作用能不是恰好相同的。不過這個近似對于實際分析來說沒明顯問題,不至于誤判NOCV軌道或pair起到的作用。尤其是對于軌道相互作用通常不起關鍵貢獻的弱相互作用體系,Multiwfn的這個近似造成的影響更是可忽略不計。順帶一提,如果你想要得到準確的軌道相互作用能ΔE_orb,可以用Multiwfn結合Gaussian做簡單能量分解,看Multiwfn手冊4.100.8節。
2.2 ETS-NOCV分析的輸入文件
用戶需要提供整體和每個片段的波函數文件,可以用fch、molden、mwfn等含有基函數信息的文件作為輸入文件,怎么產生見《詳談Multiwfn支持的輸入文件類型、產生方法以及相互轉換》(http://www.shanxitv.org/379)。必須是HF或者DFT(雙雜化泛函除外)波函數,必須是限制性閉殼層或者非限制性開殼層,不支持限制性開殼層(RO)。整體和片段的波函數文件在計算時必須用相同計算級別,而且片段里的原子坐標必須和整體的一致。整體中每個片段里的原子出現順序必須是連續的,而且載入片段波函數文件時要按照片段在整體中出現的順序載入,否則無法通過ETS-NOCV分析功能的自動檢測。
(讓片段內原子序號連續的小技巧:在GaussView里把兩個片段之間的所有鍵都斷開,然后進入Atom list editor,選Edit - Reorder - All atoms (except the first) by bonding,這樣每個片段里的原子序號就都是連著的了,之后保存文件做計算即可)。
做ETS-NOCV分析沒必要用太高質量的波函數。對于Gaussian用戶,通常產生要用的波函數文件的流程是:
(1)創建整體的結構,并保證每個片段里的原子序號是連著的
(2)用中等級別如B3LYP-D3(BJ)/6-311G*優化整體,并通過%chk保留chk文件,之后把chk用formchk轉成fch文件
(3)從優化后的整體的笛卡爾坐標中直接取出各個片段的笛卡爾坐標分別保存成gjf文件,使用和整體相同的級別進行單點計算,必須加上nosymm關鍵詞避免坐標被自動平移/旋轉造成和整體坐標不對應(見《談談Gaussian中的對稱性與nosymm關鍵詞的使用》http://www.shanxitv.org/297)。之后把得到的各個片段的chk轉成fch文件
如果你在計算整體時通過gen或genecp使用了混合基組,而且你對Gaussian的默認規則不是特別熟悉的話,建議在計算片段時始終都加上5d關鍵詞強制要求用球諧型基函數,否則可能出現比如算整體用了球諧型基函數而計算某片段時用了笛卡爾型基函數的情況,造成所有片段的基函數數目加和與整體不對應,此時沒法做ETS-NOCV分析(相關知識見《談談5d、6d型d殼層基函數與它們在Gaussian中的標識》http://www.shanxitv.org/51)。
顯然,絕對不能對片段進行幾何優化,否則片段的坐標就和整體不一致了。片段的坐標一定要從優化后的整體中直接提取。
如果整體或某片段是開殼層的,則ETS-NOCV分析將會按照開殼層形式來做,此時alpha和beta自旋的情況會分別進行計算和輸出。
對于ORCA用戶結合Multiwfn做ETS-NOCV分析,產生輸入文件的流程和上述大體相同,用orca_2mkl把計算產生后的gbw文件轉換成molden文件當Multiwfn的輸入文件即可(作用和Gaussian的fch相同)。ORCA里用不著類似nosymm的關鍵詞,因為ORCA不會自動調整體系朝向;也不需要類似5d的關鍵詞,因為ORCA總是用球諧型基函數。
NWChem、GAMESS-US、Molpro、Dalton等其它量化程序的用戶也都可以結合Multiwfn做ETS-NOCV分析,只要按《詳談Multiwfn支持的輸入文件類型、產生方法以及相互轉換》(http://www.shanxitv.org/379)所述產生含有基函數信息的文件即可,過程大同小異,這里不再累述。
3 實例
下面給出豐富的實例介紹怎么用Multiwfn做ETS-NOCV分析。只有第一個例子介紹得盡可能詳細,并把ETS-NOCV分析模塊里各種功能都涉及一遍,而之后的例子就寫得相對簡略了,且基本只是通過ETS-NOCV pair來進行討論。請讀者務必在實際研究中舉一反三。作為演示目的,計算級別用的比較便宜,大多都是B3LYP/6-31G*,大多用的是用戶最多的量子化學程序Gaussian產生的fch文件作為輸入文件。
3.1 簡單的閉殼層相互作用例子:OC→BH3
這個例子我們把COBH3分子分為CO和BH3兩個片段考察它們之間的軌道相互作用。這個作用主要是CO的碳與BH3的硼形成的配位鍵,CO和BH3都是閉殼層的,所以屬于閉殼層相互作用。COBH3、CO和BH3的Gaussian輸入文件在Multiwfn自帶的examples\ETS-NOCV\COBH3\目錄下都提供了。COBH3.gjf是優化整體的任務,CO.gjf和BH3.gjf都是單點任務,里面的坐標是筆者之前從COBH3.gjf任務產生的優化后的結構中提取的。這三個任務計算后得到的fch文件也都提供了。
3.1.1 ETS-NOCV數據的定量分析
啟動Multiwfn,然后輸入
examples\ETS-NOCV\COBH3\COBH3.fch
23 // ETS-NOCV分析
2 // 兩個片段
examples\ETS-NOCV\COBH3\CO.fch // 片段1的波函數文件
examples\ETS-NOCV\COBH3\BH3.fch // 片段2的波函數文件
由于COBH3.gjf里先出現的是CO然后是BH3,所以載入片段文件的時候也必須按這個順序載入。然后馬上就會看到下面的NOCV信息,之后進入到后處理菜單(NOCV信息可以隨時在后處理菜單選0 Print NOCV information重新輸出到屏幕上,或者選-4 Export NOCV information to a plain text file導出到一個文本文件里)
--------------- Pair and NOCV orbital information --------------
There are totally 26 NOCV pairs and 51 NOCV orbitals
NOCV orbitals whose eigenvalues are smaller than 1.0E-03 are not shown
Note: Energies of NOCV orbitals have not been evaluated, so they are all zero
Pair Energy | Orbital Eigenvalue Energy | Orbital Eigenvalue Energy
1 0.00 1 0.56514 0.00 51 -0.56514 0.00
2 0.00 2 0.40205 0.00 50 -0.40205 0.00
3 0.00 3 0.40204 0.00 49 -0.40204 0.00
4 0.00 4 0.13007 0.00 48 -0.13007 0.00
5 0.00 5 0.03704 0.00 47 -0.03704 0.00
6 0.00 6 0.03703 0.00 46 -0.03703 0.00
7 0.00 7 0.03419 0.00 45 -0.03419 0.00
8 0.00 8 0.00834 0.00 44 -0.00834 0.00
9 0.00 9 0.00273 0.00 43 -0.00273 0.00
Sum of NOCV eigenvalues: 0.00000
如上面的提示所示,總共有51個NOCV軌道,其中軌道本征值小于0.001的沒有被輸出。從以上信息可以看到NOCV pair和NOCV軌道的序號是怎么對應的,比如NOCV pair 1對應1號和51號NOCV軌道,這倆軌道本征值一個是0.565一個是-0.565,正好相反。也可以看到,基本上只有前三個NOCV pair的本征值相對較大,而且它的相對來說就可以忽略了(這里說的NOCV pair的本征值是指它對應的NOCV軌道中本征值為正的那個值,后同)。NOCV pair的本征值越大,說明這種NOCV pair所描述的軌道作用方式對密度差的影響也越大。某NOCV pair的本征值大是它對片段間結合有較大能量貢獻的基本前提。
由以上輸出可見,目前所有NOCV軌道的能量都為0,這是因為軌道能量還沒被計算。想要計算能量需要提供Fock矩陣,有兩種方式:(1)提供一個含有Fock矩陣的文件,詳見Multiwfn手冊附錄7了解哪那些文件可以用,比如Gaussian通過自帶的NBO模塊導出的.47文件就可以用 (2)直接讓Multiwfn通過整體的波函數文件(即COBH3.fch)里的軌道能量和軌道展開系數反解出Fock矩陣。用(2)比較方便,不需要再提供額外的文件,因此這里我們用這個做法。遂在后處理菜單中選擇選項-2 Generate Fock/KS matrix and evaluate NOCV orbital energies,然后Fock矩陣就生成了,并且NOCV軌道和pair的能量也算出來了,NOCV信息再次被輸出到屏幕上,如下所示
Pair Energy | Orbital Eigenvalue Energy | Orbital Eigenvalue Energy
1 -77.88 1 0.56514 -120.88 51 -0.56514 16.93
2 -15.83 2 0.40205 -139.60 50 -0.40205 -100.22
3 -15.84 3 0.40204 -139.62 49 -0.40204 -100.23
4 -5.53 4 0.13007 -44.60 48 -0.13007 -2.06
5 -0.41 5 0.03704 -18.25 47 -0.03704 -7.19
6 -0.41 6 0.03703 -18.19 46 -0.03703 -7.13
7 -0.65 7 0.03419 21.33 45 -0.03419 40.48
8 -0.06 8 0.00834 -308.53 44 -0.00834 -301.42
9 -0.02 9 0.00273 -1661.86 43 -0.00273 -1655.69
Sum of NOCV eigenvalues: 0.00000
Sum of pair energies: -116.63 kcal/mol
可見上面的輸出中有每個NOCV軌道的能量和pair的能量,單位是kcal/mol。NOCV pair的能量就是它對應的兩個NOCV軌道的能量與本征值相乘的加和,比如1號NOCV pair的能量-77.88 kcal/mol就是通過0.56514*(-120.88)-0.56514*16.93算出來的。如上所示,所有的NOCV pair能量加和是-116.63 kcal/mol,這是CO和BH3間總軌道相互作用能的近似值。
你可能注意到,9號NOCV pair對應的9和43號NOCV軌道的能量特別負,但是本征值卻非常小,導致NOCV pair的能量大小也微乎其微,這是何故?實際上,這倆軌道分布區域特別接近原子核,有內核軌道的特征,所以能量特別負;但由于片段間軌道相互作用主要涉及的是價層電子,因此這倆NOCV軌道的參與程度極低,所以本征值非常小。
順帶一提,如果你在Gaussian計算時用了彌散函數,此時Gaussian有可能會去除一些線性依賴基函數,這種情況下讓Multiwfn直接生成Fock矩陣會不成功。所以,如果沒必絕對必要帶彌散函數的一律話不要帶。如果必須帶彌散函數,比如是陰離子體系,那么若遇到Fock矩陣無法被Multiwfn直接生成的情況,你需要選擇-1 Load Fock/KS matrix and evaluate NOCV orbital energies并且輸入一個含有Fock矩陣的文件的路徑,支持哪些文件看手冊附錄7。
3.1.2 可視化NOCV pair
前面我們看到NOCV pair 1的能量-77.88 kcal/mol占總軌道相互作用能-116.63 kcal/mol的大半部分,因此通過考察它的密度圖形我們就能知道CO和BH3的軌道相互作用的主要來源是什么。NOCV pair 2和3的能量是簡并的,都是-15.8 kcal/mol,它們對片段間結合的貢獻也不算很小,因此也值得對它們作圖考察一下什么樣的軌道相互作用起到了次級貢獻。
在后處理菜單里選2 Show isosurface of NOCV pair density就可以進入觀看NOCV pair密度的等值面圖的界面。因為只有先計算了密度的格點數據之后才能觀看其等值面,所以程序會先讓你設置格點。格點設置界面里有豐富的選項控制格點數和格點分布,缺乏相關常識的話看《Multiwfn FAQ》(http://www.shanxitv.org/452)里的Q39。由于當前體系很小,我們就選擇2 Medium quality grid,如提示所示這會計算大約512000個點。為了方便用戶找感興趣的NOCV pair,故屏幕上又把NOCV信息顯示了一遍。現在想看第幾個NOCV pair就輸入幾,我們先輸入1看一下1號NOCV pair的密度,然后圖形界面就蹦出來了。默認的等值面數值是0.005 a.u.,可以在窗口右側的文本框里改,當前的等值面圖如下所示,箭頭是我手動標的
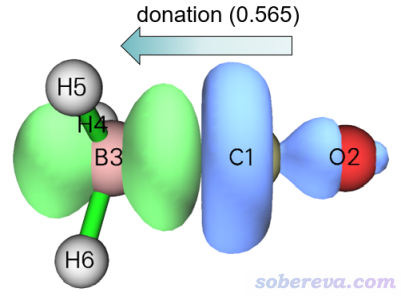
上圖中藍色和綠色分別對應負值和正值部分,NOCV pair對應的軌道相互作用導致電子從藍色區域往綠色區域轉移。上圖明顯體現的是電子從CO往BH3上整體轉移,更確切來說,是往BH3的朝著CO的空p軌道轉移,因為綠色等值面有明顯的兩個瓣,顯然對應的是p軌道的特征。NOCV pair的本征值0.565近似對應了這種作用方式的電子轉移量(不是精確對應,因為NOCV軌道1和51的分布還是存在少量重疊的,見下一節的圖,故二者的密度在構成pair的密度時有所相互抵消)。NOCV pair 1的密度所展現的作用特征明顯是CO與BH3形成配位鍵作用,因此可以說CO向BH3的配位作用對這倆分子的結合貢獻是-77.88 kcal/mol(即NOCV pair 1的能量)。
再來看看描述CO與BH3的次要作用的NOCV pair 2是什么情況。點擊當前圖形窗口右上角的RETURN按鈕返回,然后輸入2,NOCV pair 2的密度的等值面圖就顯示出來了。將等值面數值適當調小到0.002 a.u.便于觀察其主體特征,此時的等值面圖如下所示
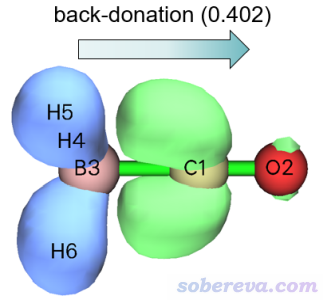
明顯,NOCV pair 2描述的是B-H的σ軌道的電子向CO的反π空軌道轉移,我們可以稱之為反饋(back-donation)作用。NOCV pair 2的能量是-15.8 kcal/mol,是個不大不小的值,體現出反饋作用比NOCV pair 1描述的配位作用弱得多,但也不可忽視。這個例子充分展現出ETS-NOCV的獨特價值,如果僅僅是按照常規方式畫密度差圖的話,是難以認識到有這種反饋作用的。
感興趣的讀者可以再看一下NOCV pair 3的密度等值面,會發現特征和NOCV pair 2是相似的,也是σ(B-H)→π*的反饋作用。
Multiwfn還允許一次性觀看一批NOCV pair密度總和的等值面。比如我們想看看所有其它NOCV pair(4~26號)的總密度是什么樣的,于是我們就輸入4-26。然后在文本窗口中可看到
Sum of energies of selected pairs: -7.09 kcal/mol
這是所有選擇的NOCV pair的能量總和,可見只是個很小的值,所以其它形式的軌道相互作用都對片段間結合的貢獻微乎其微。在圖形窗口中可看到下圖
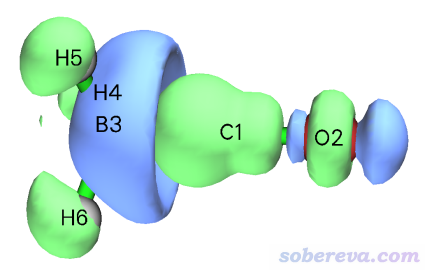
這個等值面的形狀比較復雜,不太容易說清楚對應的是什么作用。但多多少少還是能看出來體現了一些BH3到CO的反饋作用,并且片段內有電子密度的極化(電子密度的變形),因為倆片段區域內電子密度都是有增有減。
之后我們輸入q返回到后處理菜單。由于我們之前已經設置過一次格點了,所以之后再進入任何涉及設置格點的選項就不會讓你再次重新設置了。如果想改格點設置的話,在后處理菜單選-5 Set grid for calculation of various densities。
NOCV pair的密度也可以讓Multiwfn導出為cub文件,即選擇7 Export cube file of NOCV pair density。之后可以把cub文件在VMD等可視化程序里顯示成等值面,屆時可以更靈活地調節圖像效果和視角。很推薦用《在VMD里將cube文件瞬間繪制成效果極佳的等值面圖的方法》(http://www.shanxitv.org/483)里面的腳本繪制,圖像效果又非常好又特別方便。
通常的ETS-NOCV分析像以上這么考察一下NOCV pair的密度圖、能量、本征值就夠了。但為了通過本例讓讀者了解更多相關細節、更好地理解和運用ETS-NOCV分析,下面再多介紹一些。
3.1.3 可視化NOCV軌道
在后處理菜單選1 Show isosurface of NOCV orbitals就可以進入觀看NOCV軌道的圖形界面,里面的選項和用法與Multiwfn的主功能0基本一致,在《使用Multiwfn觀看分子軌道》(http://www.shanxitv.org/269)里有全面介紹。與此同時,文本窗口再次輸出了NOCV信息便于你尋找考察的軌道。圖形界面右下角的列表里點擊相應序號就可以顯示出相應NOCV軌道等值面圖,也可以直接在文本框里輸入軌道序號然后按回車。前面我們知道,NOCV pair 1對應的軌道是1號和51號,這里我們就繪制它們的波函數圖形,見下圖下方兩個子圖,等值面數值由默認值改為了更大的0.15以便觀察。同時,為了讓大家明白到底NOCV pair 1的密度是怎么來的,下圖上方把它的0.01 a.u.等值面圖附上了,并把計算公式明確給出了。
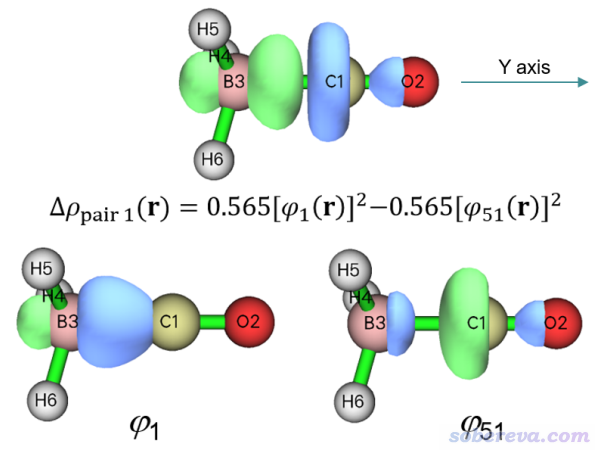
可見,本征值為正的1號NOCV軌道主要是在BH3那一邊,有點類似于接受電子的軌道的意味,而本征值為負的51號NOCV軌道主要在CO那一邊,有點類似于貢獻電子的軌道的意味。
3.1.4 考察NOCV軌道和pair的成份
為了更深入且定量了解NOCV軌道和pair的本質,可以在后處理菜單里選14 Calculate composition of NOCV orbitals and pairs,然后輸入要考察的NOCV pair的序號,此時它以及與之對應的NOCV軌道的成份都會被輸出。成份用的是SCPA方法算的,見《談談軌道成份的計算方法》(http://www.shanxitv.org/131)和《分子軌道成分的計算》(http://sioc-journal.cn/Jwk_hxxb/CN/abstract/abstract340458.shtml)里的相關介紹。進入選項14后輸入1,則NOCV pair 1以及它對應的NOCV軌道1和51的成份就馬上輸出了,如下所示。為了避免輸出的信息量過大,默認只有對NOCV pair貢獻超過0.5%的基函數和殼層被輸出,這個閾值可通過settings.ini里的compthres進行修改(改完后需要重啟Multiwfn)。
Contribution of each basis function to NOCV pair/orbitals:
Basis Type Atom Shell Orb. 1 Orb. 51 Pair 1
1 S 1(C ) 1 0.09 % 1.13 % -0.58 %
2 S 1(C ) 2 1.05 % 4.09 % -1.72 %
4 Y 1(C ) 3 1.81 % 5.53 % -2.10 %
6 S 1(C ) 4 15.77 % 49.68 % -19.17 %
8 Y 1(C ) 5 5.07 % 5.06 % 0.00 %
19 Y 2(O ) 9 0.01 % 1.60 % -0.90 %
23 Y 2(O ) 11 0.02 % 0.68 % -0.37 %
32 S 3(B ) 14 9.89 % 9.27 % 0.35 %
34 Y 3(B ) 15 42.83 % 10.50 % 18.27 %
36 S 3(B ) 16 11.57 % 9.02 % 1.44 %
38 Y 3(B ) 17 4.37 % 1.23 % 1.77 %
47 S 4(H ) 20 1.99 % 0.09 % 1.07 %
49 S 5(H ) 22 1.97 % 0.09 % 1.06 %
51 S 6(H ) 24 1.97 % 0.09 % 1.06 %
Contribution of each basis function shell to NOCV pair/orbitals:
Shell Type Atom Orb. 1 Orb. 51 Pair 1
1 S 1(C ) 0.09 % 1.13 % -0.58 %
2 S 1(C ) 1.05 % 4.09 % -1.72 %
3 P 1(C ) 1.81 % 5.53 % -2.10 %
4 S 1(C ) 15.77 % 49.68 % -19.17 %
5 P 1(C ) 5.07 % 5.06 % 0.00 %
9 P 2(O ) 0.01 % 1.60 % -0.90 %
11 P 2(O ) 0.02 % 0.68 % -0.37 %
14 S 3(B ) 9.89 % 9.27 % 0.35 %
15 P 3(B ) 42.83 % 10.50 % 18.27 %
16 S 3(B ) 11.57 % 9.02 % 1.44 %
17 P 3(B ) 4.37 % 1.23 % 1.77 %
18 D 3(B ) 0.55 % 0.09 % 0.26 %
20 S 4(H ) 1.99 % 0.09 % 1.07 %
22 S 5(H ) 1.97 % 0.09 % 1.06 %
24 S 6(H ) 1.97 % 0.09 % 1.06 %
Contribution of various types of shells to NOCV pair/orbitals:
Type Orb. 1 Orb. 51 Pair 1
s: 45.14 % 74.98 % -16.87 %
p: 54.10 % 24.59 % 16.67 %
d: 0.76 % 0.42 % 0.19 %
f: 0.00 % 0.00 % 0.00 %
g: 0.00 % 0.00 % 0.00 %
h: 0.00 % 0.00 % 0.00 %
Contribution of each atom to NOCV pair/orbitals:
Atom Orb. 1 Orb. 51 Pair 1
1(C ): 23.99 % 65.82 % -23.64 %
2(O ): 0.28 % 2.94 % -1.50 %
3(B ): 69.40 % 30.30 % 22.10 %
4(H ): 2.12 % 0.32 % 1.02 %
5(H ): 2.11 % 0.31 % 1.01 %
6(H ): 2.11 % 0.31 % 1.01 %
上面依次給出了各個基函數、各個殼層、各個角動量、各個原子對NOCV pair和軌道的貢獻,內容非常容易理解。某一項對NOCV pair 1的貢獻相當于它對1號和51號NOCV軌道貢獻值乘上各自本征值的加和。上面給出的成份值和我們之前看到的等值面圖所展現的情況一致,比如NOCV pair 1在給電子的C上面的成份為明顯的負值(-23.64%),而在接受電子的B上的成份為明顯的正值(22.10%)。NOCV pair的所有成份的加和恰為0%,而各NOCV軌道的所有成分加和恰為100%。
根據基組的定義,我們可以知道哪些基函數和哪些原子軌道相對應,從而討論哪些軌道對NOCV pair或軌道有什么樣的貢獻。如果你不明白對應關系怎么判斷,或者有時從定義上不好判斷,參看《利用布居分析判斷基函數與原子軌道的對應關系》(http://www.shanxitv.org/418)。從當前用的6-31G*基組的構成以及上面給出的各個基函數的貢獻可以看出,2號和6號S型基函數一起對應的是C的2s原子軌道,它對NOCV pair 1的貢獻是-1.72%-19.17% = -20.89%,占了負值的絕大部分,因此C的2s原子軌道就是NOCV pair 1密度為負部分的主要來源,至少可以說CO與BH3的配位作用導致電子很大一部分從這個原子軌道上轉移走了。34和38號基函數一起對應的是C的2py軌道(Y軸方向在上一節的圖里標注了,即CO軸線方向),它倆對NOCV pair 1的貢獻是18.27%+1.77% = 20.04%,是正值部分的主要來源,因此可以說CO和BH3的配位作用使得B的2py軌道獲得了很多電子,這也正對應NOCV pair 1密度等值面圖里在B上有綠色的順著Y軸伸展的兩個瓣的等值面。
注意SCPA方法不能在有彌散函數的情況下使用(如果必須帶彌散函數而且只關心的是NOCV里原子的成份,可以從后處理菜單返回Multiwfn的主菜單,此時內存里記錄的軌道就是NOCV軌道,可以照常用主功能8里的能兼容彌散函數的軌道成份分析方法算軌道中的各個原子的成份,比如Hirshfeld、Becke方法)。
3.1.5 觀看Pauli變形密度、軌道變形密度和總變形密度
Pauli變形密度(Δρ_Pauli)體現的是由于片段間的Pauli互斥導致的密度變化,即片段間占據軌道正交化之后的電子密度減去兩個片段孤立狀態下密度的簡單疊加。軌道變形密度(Δρ_orb)體現的是由于片段間的軌道相互作用導致的密度差(即實際電子密度減去凍結態的電子密度)。總變形密度Δρ是Δρ_Pauli和Δρ_orb的加和,也即是通常我們用整體密度減去各個片段密度得到的密度差。可以通過后處理菜單中的3 Show isosurface of Pauli deformation density、4 Show isosurface of orbital deformation density、5 Show isosurface of total deformation density這三個選項分別將這三種變形密度的等值面直接繪制出來,如下所示
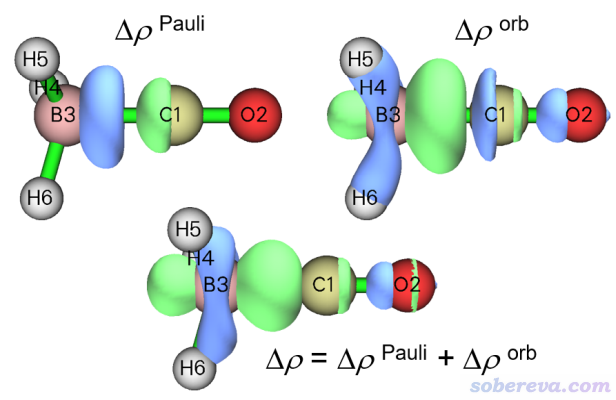
從Δρ_Pauli的圖上可以看出Pauli互斥使得電子在BH3和CO之間的區域有顯著下降,有一大塊藍色等值面。相反,Δρ_orb圖中在BH3和CO之間有一大塊綠色區域,體現出軌道相互作用使成鍵區域電子密度顯著增加,這也是形成共價鍵時的典型情況。從總變形密度上看,明顯是軌道相互作用令成鍵區域密度增加的程度高于Pauli互斥導致成鍵區域密度降低的程度,因此總變形密度圖中B和C之間明顯為正。
值得一提的是,Δρ_orb等同于所有NOCV pair密度的總和,也因此如果你在2 Show isosurface of NOCV pair density里輸入1-26選擇所有NOCV pairs的話,看到的圖和上面Δρ_orb的圖是完全一樣的。Δρ_orb體現了各種軌道相互作用的總效果,即配位作用、反饋作用、極化等都隱藏在了里面。若你不做NOCV分析的話,是無法將它們區分開來獨立、清晰地討論的,而只能根據等值面圖來試圖臆測里面存在哪些作用,非常含糊和主觀。不過NOCV也不是唯一的分解分析方法,本文一開始提到的CDA分析、NAdO分析也都可以幫助我們從軌道角度洞察片段間軌道相互作用的本質,只不過視角不同。
3.1.6 觀看準分子軌道、凍結態軌道和實際的復合物軌道
“準分子軌道”就是指由單體的軌道簡單組合出的復合物軌道,其實就是把各個單體的各個軌道用復合物的基函數展開后簡單地合并到一起(此時某片段的軌道在其它片段的基函數上展開系數為0),因此準分子軌道的圖形直接等同于原本單體軌道的。前面提到,片段間的Pauli互斥會造成片段的占據軌道變形,從而令兩個片段的占據軌道間滿足正交關系。具體來說,Multiwfn是按照ETS-NOCV原文,令兩個片段占據軌道彼此間做Lowdin正交化來構造考慮Pauli互斥之后的軌道的,此時的軌道也即前述的凍結態波函數的軌道。凍結態中的非占據軌道和準分子的非占據的那些是相同的。“實際的復合物軌道”是指復合物波函數文件里的分子軌道。
如果你想觀看上述三類軌道,可以在后處理菜單分別選11 Visualize promolecular orbitals、12 Visualize frozen state orbitals、13 Visualize actual complex orbitals。選其一后,相應波函數里的占據軌道的信息會輸出在屏幕上:
Orb: 1 Ene(au/eV): 0.000000 0.0000 Occ: 2.000000 Type:A+B
Orb: 2 Ene(au/eV): 0.000000 0.0000 Occ: 2.000000 Type:A+B
Orb: 3 Ene(au/eV): 0.000000 0.0000 Occ: 2.000000 Type:A+B
Orb: 4 Ene(au/eV): 0.000000 0.0000 Occ: 2.000000 Type:A+B
Orb: 5 Ene(au/eV): 0.000000 0.0000 Occ: 2.000000 Type:A+B
Orb: 6 Ene(au/eV): 0.000000 0.0000 Occ: 2.000000 Type:A+B
Orb: 7 Ene(au/eV): 0.000000 0.0000 Occ: 2.000000 Type:A+B
Orb: 31 Ene(au/eV): 0.000000 0.0000 Occ: 2.000000 Type:A+B
Orb: 32 Ene(au/eV): 0.000000 0.0000 Occ: 2.000000 Type:A+B
Orb: 33 Ene(au/eV): 0.000000 0.0000 Occ: 2.000000 Type:A+B
Orb: 34 Ene(au/eV): 0.000000 0.0000 Occ: 2.000000 Type:A+B
從序號的連續性上可以看出,1-7號是CO的占據軌道,8-30號是CO的非占據軌道,31-34號是BH3的占據軌道,剩余的是BH3的非占據軌道。為了讓大家清晰地理解Pauli互斥會令占據軌道出現何種變形,下面把準分子和凍結態的7號軌道的圖形繪制了出來,等值面數值設為了很小的0.02以便能看出差異
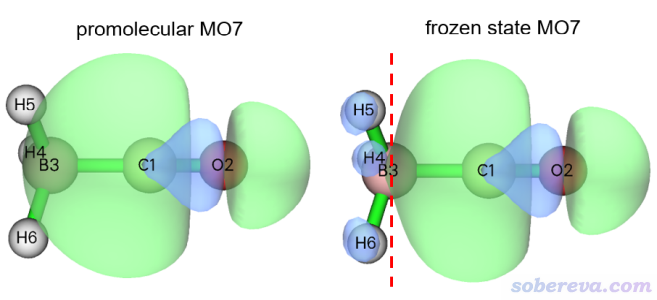
可見,CO原本的這個軌道(左圖)在BH3部分是沒有波節的,但為了滿足Pauli互斥,在凍結態的時候它在BH3的地方出現了節面,上圖中用紅色虛線標注了,這是為了這個軌道能與BH3的所有占據軌道都滿足正交關系而產生的。這種形式的軌道變形增加了它在BH3區域波函數的平均曲率,導致軌道動能上升,也因而軌道能量上升。也正是由于這種效應,Pauli互斥導致凍結態的復合物能量比準分子態的更高,它們的能量差就是常說的Pauli互斥能(前述的ΔE_Pauli)。
3.2 簡單的開殼層相互作用例子:乙烷
此例用ETS-NOCV考察乙烷中的兩個甲基自由基CH3之間的軌道相互作用。這個是開殼層相互作用,兩個片段都拿出一個單電子用于形成C-C鍵。產生此例用的波函數文件的步驟和前例一樣,gjf文件和fch文件都在examples\ETS-NOCV\ethane\目錄里提供了。顯然,CH3片段的計算必須設二重態。
啟動Multiwfn之后輸入
examples\ETS-NOCV\ethane\ethane.fch // 整體的波函數文件
23 // ETS-NOCV分析
2 // 兩個片段
examples\ETS-NOCV\ethane\CH3_1.fch // 第一個CH3片段
examples\ETS-NOCV\ethane\CH3_2.fch // 第二個CH3片段
n // 不翻轉第一個CH3片段的自旋
y // 翻轉第二個CH3片段的自旋
可見此例和前面閉殼層的例子不同的是,程序會問你是否翻轉片段的自旋。自旋翻轉就是讓片段里alpha和beta電子的信息相互交換。必須恰當設置翻轉,以使得各個片段的alpha電子數的加和等于整體的alpha電子數,對beta電子也是一樣。像此例,兩個CH3在Gaussian計算時都是5個alpha電子、4個beta電子,如果都不設翻轉的話,加起來就是10個alpha電子、8個beta電子,這和乙烷里9個alpha電子和9個beta電子不符了。所以兩個片段中必須翻轉其中一個的自旋,翻哪個都一樣。而且,也只有翻轉其中一個的自旋,才對應于一邊提供一個alpha單電子、一邊提供一個beta單電子的情況,這樣才能形成一個典型的共價鍵,要是兩邊都給的是alpha單電子明顯不可能成鍵。
接下來,在后處理菜單選選項-2產生Fock矩陣并計算NOCV能量,然后看到下面的輸出
--------------- Pair and NOCV orbital information --------------
There are totally 42 NOCV pairs and 84 NOCV orbitals
NOCV orbitals with absolute eigenvalues smaller than 1.0E-03 are not shown
Note: All energies are given in kcal/mol
----- Alpha NOCV orbitals -----
Pair Energy | Orbital Eigenvalue Energy | Orbital Eigenvalue Energy
1 -93.24 1 0.44471 -117.62 42 -0.44471 92.06
2 -1.20 2 0.04959 -28.00 41 -0.04959 -3.76
3 -1.20 3 0.04959 -28.00 40 -0.04959 -3.76
4 -0.85 4 0.04192 8.65 39 -0.04192 28.92
5 -0.85 5 0.04192 8.65 38 -0.04192 28.92
6 -1.09 6 0.03679 -19.92 37 -0.03679 9.79
7 -0.48 7 0.02349 -4.18 36 -0.02349 16.23
8 -0.01 8 0.00125 -2808.79 35 -0.00125 -2799.86
----- Beta NOCV orbitals -----
Pair Energy | Orbital Eigenvalue Energy | Orbital Eigenvalue Energy
22 -93.24 43 0.44471 -117.62 84 -0.44471 92.06
23 -1.20 44 0.04959 -28.00 83 -0.04959 -3.76
24 -1.20 45 0.04959 -28.00 82 -0.04959 -3.76
25 -0.85 46 0.04192 8.65 81 -0.04192 28.92
26 -0.85 47 0.04192 8.65 80 -0.04192 28.92
27 -1.09 48 0.03679 -19.92 79 -0.03679 9.79
28 -0.48 49 0.02349 -4.18 78 -0.02349 16.23
29 -0.01 50 0.00125 -2808.79 77 -0.00125 -2799.86
Sum of NOCV eigenvalues: Alpha= 0.00000 Beta= 0.00000
Sum of pair energies: Alpha= -98.93 Beta= -98.93 Total= -197.86
由于當前是以開殼層方式做的ETS-NOCV分析,所以alpha和beta自旋的情況分別輸出了,beta的序號排在alpha之后。由于兩個CH3是對稱的,所以alpha和beta部分的輸出看起來一樣。alpha和beta軌道的總相互作用能都是將近-100 kcal/mol,加起來總共大約-200 kcal/mol。看起來這遠大于C-C鍵的鍵強,但別忘了Pauli互斥會極大地產生抵消作用。
為了考察兩個CH3之間的軌道相互作用特征,我們對alpha和beta中能量最大的NOCV pair的密度繪制等值面圖。輸入
2 // 顯示NOCV pair密度的等值面
2 // 中等質量格點
1 // NOCV pair 1。從能量上看它描述了絕大部分alpha軌道相互作用,其它alpha的NOCV pair的貢獻和它相比可忽略不計
類似地,也繪制NOCV pair 22密度的等值面,由上面的輸出可見它是對beta軌道相互作用起絕對主導的。此外,也繪制NOCV pair 1和22密度之和的等值面,即輸入1,22。這些等值面圖一起在下面展示了,圖上標的q是轉移的電子量,也即NOCV pair的本征值
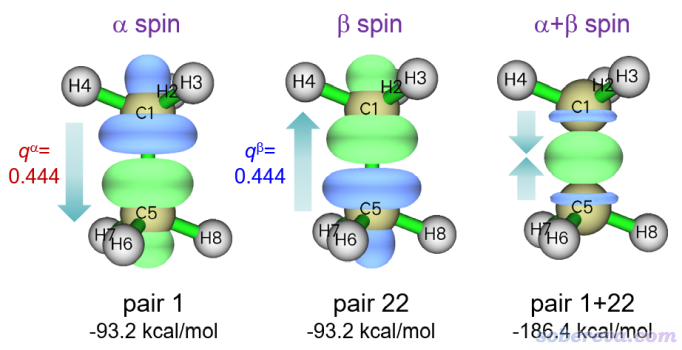
此體系上方的CH3是帶alpha單電子的,下方的帶beta單電子。由上圖可清晰地看到,上方的CH3將一部分alpha單電子轉移到了下面的CH3的alpha空軌道上,下方的CH3將一部分beta單電子轉移到了上方的CH3的beta空軌道上。二者密度相加對應的總密度變化是C-C鍵區域的密度顯著增加。而在甲烷的兩端,alpha和beta電子密度的變化在求和時則大幅抵消了。
再來看看其它NOCV pair對應的作用。NOCV pair 2和3是簡并的,對alpha軌道相互作用貢獻都只有-1.20 kcal/mol,二者的0.0015 a.u.的密度等值面如下
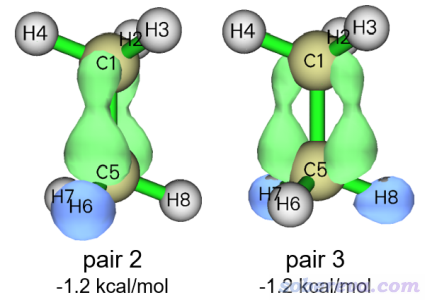
從圖上可見在H的部分電子密度降低了,而在垂直于C-C鍵的兩側電子密度增加了,因此我們可以估計這種作用是C-H的sigma電子向C-C的未占據的pi軌道轉移所致的。這一點通過觀看NOCV pair對應的NOCV軌道可以了解得更清楚。但這種軌道相互作用產生影響基本可以完全忽略不計,因為NOCV pair 2和3對結合能的貢獻微乎其微。
兩個CH3之間沒有什么顯著的sigma鍵以外的軌道相互作用,這點也可以通過計算兩個CH3之間的Mayer鍵級認識到。啟動Multiwfn后載入ethane.fch,之后輸入
9 // 鍵級分析
-1 // 定義兩個片段
1-4 // 第一個CH3
5-8 // 第二個CH3
1 // 計算Mayer鍵級
從屏幕上可見兩個片段間的總Mayer鍵級是0.996,幾乎正好1.0,說明基本就是個標準的單重鍵作用。而之前考察的COBH3中,CO與BH3的Mayer鍵級則是1.378,顯著大于1,明顯在OC→BH3配位作用以外還有其它重要的軌道相互作用,正對應于ETS-NOCV分析所揭示的反饋作用。
3.3 多重鍵的相互作用例子:乙烯
這回以考察乙烯中兩個CH2之間的雙鍵作用為例展現一下ETS-NOCV分析多重鍵作用。注意在計算CH2的時候要設為三重態,而且在載入時讓其中一個CH2的自旋翻轉,即一個是↑↑CH2一個是↓↓CH2,這樣兩個CH2間才能靠兩對電子來形成雙鍵。順帶一提,本身CH2在孤立狀態下的基態也是三重態。本例涉及的Gaussian輸入文件和計算產生的fch文件在examples\ETS-NOCV\ethene\里都提供了。
啟動Multiwfn之后輸入
examples\ETS-NOCV\ethene\ethene.fch // 整體的波函數文件
23 // ETS-NOCV分析
2 // 兩個片段
examples\ETS-NOCV\ethene\CH2_1.fch // 第一個CH3片段
examples\ETS-NOCV\ethene\CH2_2.fch // 第二個CH3片段
n // 不翻轉第一個CH2片段的自旋
y // 翻轉第二個CH2片段的自旋
-2 // 產生Fock矩陣并計算NOCV能量
現在看到下面的輸出
--------------- Pair and NOCV orbital information --------------
There are totally 38 NOCV pairs and 76 NOCV orbitals
NOCV orbitals with absolute eigenvalues smaller than 1.0E-03 are not shown
Note: All energies are given in kcal/mol
----- Alpha NOCV orbitals -----
Pair Energy | Orbital Eigenvalue Energy | Orbital Eigenvalue Energy
1 -63.11 1 0.57168 -125.97 38 -0.57168 -15.57
2 -99.78 2 0.38673 -101.82 37 -0.38673 156.21
3 -2.28 3 0.07016 -66.15 36 -0.07016 -33.70
4 -1.64 4 0.05624 13.27 35 -0.05624 42.50
5 -1.73 5 0.04700 -26.85 34 -0.04700 9.99
6 -0.48 6 0.02675 -62.39 33 -0.02675 -44.32
7 -0.01 7 0.00136 -2733.67 32 -0.00136 -2723.71
----- Beta NOCV orbitals -----
Pair Energy | Orbital Eigenvalue Energy | Orbital Eigenvalue Energy
20 -63.11 39 0.57168 -125.97 76 -0.57168 -15.57
21 -99.78 40 0.38673 -101.82 75 -0.38673 156.21
22 -2.28 41 0.07016 -66.15 74 -0.07016 -33.70
23 -1.64 42 0.05624 13.27 73 -0.05624 42.50
24 -1.73 43 0.04700 -26.85 72 -0.04700 9.99
25 -0.48 44 0.02675 -62.39 71 -0.02675 -44.32
26 -0.01 45 0.00136 -2733.67 70 -0.00136 -2723.71
Sum of NOCV eigenvalues: Alpha= 0.00000 Beta= 0.00000
Sum of pair energies: Alpha= -169.05 Beta= -169.05 Total= -338.09
可見,對于alpha和beta自旋,都只有兩個NOCV pair起關鍵貢獻,其它的基本可忽略。使用選項2把對應alpha和beta主要軌道相互作用的共4個NOCV pair(1、2、20、21)的密度等值面都分別畫出來,并且也把對應sigma作用和對應pi作用的兩種自旋的NOCV pair密度加和后繪制成等值面。它們的0.01 a.u.等值面圖如下所示
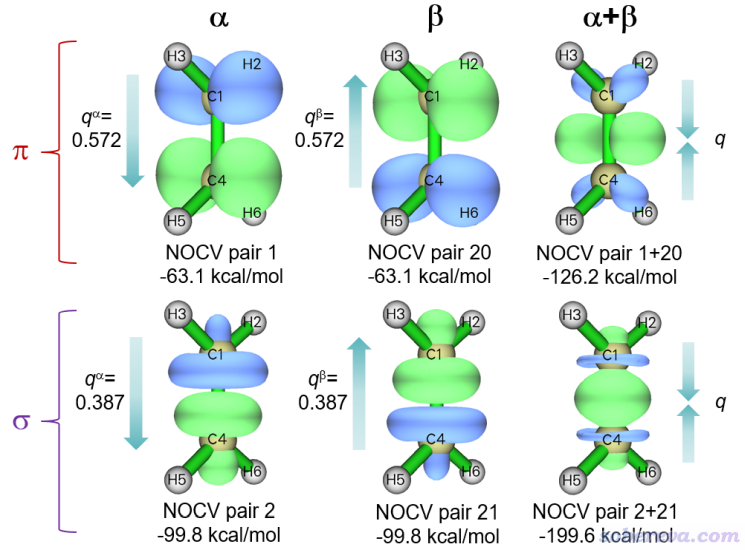
可明顯看出,NOCV pair 1和2分別對應alpha電子的sigma和pi作用,NOCV pair 20和21分別對應beta的sigma和pi作用。CH2之間的sigma作用特征無論從密度上還是能量上都和乙烷的CH3之間的sigma作用情況很相似。CH2之間的pi作用使得電子密度在C-C鍵的兩側都有顯著增加,體現出pi鍵的共價作用特征。從能量上可以看出每個自旋的pi作用對結合的貢獻(-63.1 kcal/mol)明顯比sigma作用(-99.8 kcal/mol)更弱,這是普遍現象。乙烯的sigma總作用-199.6 kcal/mol比前例乙烷的-189.4 kcal/mol略大,這體現出乙烯的pi作用與sigma作用之間有一定協同性。
3.4 弱相互相互作用的例子:A-T堿基對
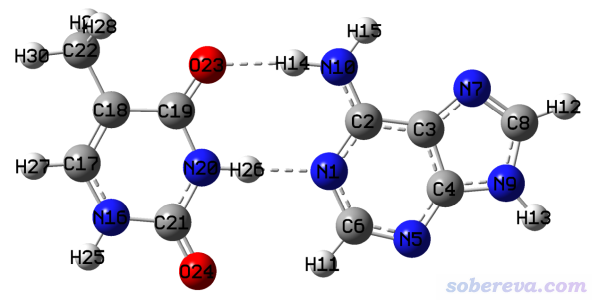
此例我們用ETS-NOCV來研究一次弱相互作用,被考察對象是腺嘌呤(A)與胸腺嘧啶(T)形成的堿基對(AT)之間氫鍵作用,結構如下所示,虛線標注了形成氫鍵的地方。這個結構是文獻里優化好的,所以本例就不再優化了。
雖然如《透徹認識氫鍵本質、簡單可靠地估計氫鍵強度:一篇2019年JCC上的重要研究文章介紹》(http://www.shanxitv.org/513)里所述,像這種普通強度的氫鍵是靜電作用所主導的,軌道相互作用只起到相當次要的因素,但考察一下軌道相互作用終究還是可以提供研究問題更多的視角,是有益的。
這里筆者使用流行的ORCA程序的5.0.1版在ωB97M-V/def2-TZVP級別下對AT、A、T分別做單點計算,當然用Gaussian也可以。如果對ORCA缺乏了解,參看http://www.shanxitv.org右側的ORCA分類里的諸多文章。相應的ORCA輸入文件是examples\ETS-NOCV\AT\目錄下的AT.inp、A.inp、T.inp,對它們計算后就得到了同名的gbw文件,按照《詳談Multiwfn支持的輸入文件類型、產生方法以及相互轉換》(http://www.shanxitv.org/379)所述,通過orca_2mkl將它們轉化為.molden文件,這些文件可以在http://www.shanxitv.org/multiwfn/extrafiles/A-T_base_pair_molden.zip直接下載。
啟動Multiwfn之后輸入
AT.molden // 整體的波函數文件
23 // ETS-NOCV分析
2 // 兩個片段
A.molden // 腺嘌呤(A)的波函數文件
T.molden // 胸腺嘧啶(T)的波函數文件
-2 // 產生Fock矩陣并計算NOCV能量
從屏幕上顯示的NOCV信息可見,當前一共有多達40個NOCV pair的本征值都大于默認的輸出閾值而被輸出,由于信息量太大考查起來稍微不便。因此我們輸入以下命令
-3 // 修改NOCV本征值的閾值
0.02 // 此值明顯大于默認值
0 // 重新顯示NOCV信息
此時輸出的NOCV pair數就少多了,如下所示
--------------- Pair and NOCV orbital information --------------
There are totally 328 NOCV pairs and 655 NOCV orbitals
NOCV orbitals with absolute eigenvalues smaller than 2.0E-02 are not shown
Note: All energies are given in kcal/mol
Pair Energy | Orbital Eigenvalue Energy | Orbital Eigenvalue Energy
1 -11.20 1 0.20598 -15.91 655 -0.20598 38.45
2 -4.05 2 0.11820 -18.61 654 -0.11820 15.66
3 -0.79 3 0.07198 -91.72 653 -0.07198 -80.71
4 -0.79 4 0.07066 -77.74 652 -0.07066 -66.59
5 -0.59 5 0.04312 -29.08 651 -0.04312 -15.45
6 -0.34 6 0.03551 -40.19 650 -0.03551 -30.49
7 -0.27 7 0.02859 -24.18 649 -0.02859 -14.72
8 -0.15 8 0.02619 -56.34 648 -0.02619 -50.77
9 -0.15 9 0.02032 -16.97 647 -0.02032 -9.45
Sum of NOCV eigenvalues: -0.00000
Sum of pair energies: -18.33 kcal/mol
可見只有1、2號NOCV pair對應顯著的軌道相互作用,其它的都可以忽略不計。所有NOCV pair的能量之和只有-18.33 kcal/mol,與前面的例子相比可見軌道相互作用在弱相互作用中的強度遠不及在化學鍵作用中的強度。
現在用選項2分別繪制NOCV pair 1、2的密度等值面圖,結果如下所示。繪制時必須用很小的等值面數值0.002 a.u.,否則沒法明顯看到等值面,這也是因為弱相互作用中的軌道相互作用引起的電子密度變化程度相當小。
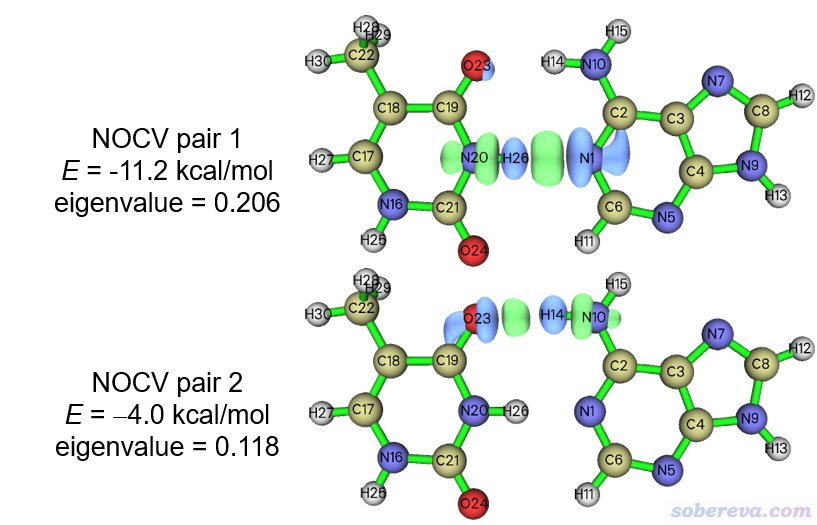
可見NOCV pair 1和2分別對應N20-H26...N1和O23...H14-N10氫鍵的軌道相互作用,在氫和氫鍵受體原子之間有一塊密度增加區域,這體現出即便是靜電主導的氫鍵作用,也是伴隨著些許共價作用成分的。單從軌道相互作用能角度來看,N20-H26...N1比O23...H14-N10更強。讀者感興趣的話,可以用Multiwfn按照《透徹認識氫鍵本質、簡單可靠地估計氫鍵強度:一篇2019年JCC上的重要研究文章介紹》(http://www.shanxitv.org/513)里基于atoms-in-molecules (AIM)理論提出的方法估算一下這兩個氫鍵各自的氫鍵鍵能是多少,做法在Multiwfn手冊4.2.1節有示例。
3.5 過渡金屬配合物一例:(CO)5CrCH2
此例演示一下ETS-NOCV分析過渡金屬配合物。例子是(CO)5CrCH2,我們要研究(CO)5Cr和CH2片段間的相互作用。
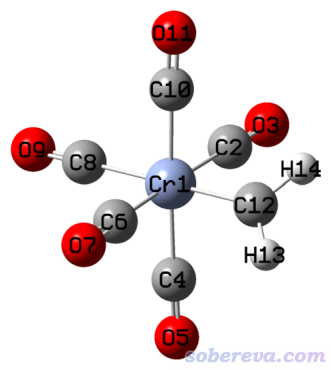
如《簡談量子化學計算中DFT泛函的選擇》(http://www.shanxitv.org/272)所述,TPSSh是計算過渡金屬配合物不錯的選擇,故此例我們用這個泛函。Cr用SDD贗勢結合標配的基組,配體用6-311G*基組。相應的Gaussian輸入文件以及計算后得到的fch文件在examples\ETS-NOCV\(CO)5CrCH2\目錄下都提供了。
注意ETS-NOCV的分析結果直接受到參考態(由計算片段時設定的電荷和自旋多重度決定)選取的影響,這有一定任意性。一般來說,計算片段時應當讓片段的電子結構與它在整體中盡量相似,怎么設置需要具體問題具體分析。在(CO)5CrCH2中,可以視為單重態的CH2將它的孤對電子用于與Cr的空軌道形成配位鍵,所以CH2在計算時應當用單重態,而非用它在孤立狀態時的基態(三重態)。順帶一提,由于CH2以這種方式對待,前面提到的preparation energy里此時既包含了CH2在結合時結構變化造成的能量改變,也包含了CH2從三重態基態變成單重態的能量改變。由于(CO)5CrCH2整體是單重態,CH2在此例也當成單重態,顯然(CO)5Cr只能作為單重態對待。
啟動Multiwfn然后輸入
examples\ETS-NOCV\(CO)5CrCH2\(CO)5CrCH2.fch
23 // ETS-NOCV分析
2 // 兩個片段
examples\ETS-NOCV\(CO)5CrCH2\(CO)5Cr.fch // 片段1
examples\ETS-NOCV\(CO)5CrCH2\CH2.fch // 片段2
-2 // 產生Fock矩陣并計算NOCV能量
此時輸出的NOCV信息為
Pair Energy | Orbital Eigenvalue Energy | Orbital Eigenvalue Energy
1 -42.57 1 0.94516 -139.72 247 -0.94516 -94.68
2 -72.70 2 0.71995 -114.71 246 -0.71995 -13.74
3 -3.49 3 0.16177 -28.10 245 -0.16177 -6.51
4 -1.45 4 0.08316 -60.03 244 -0.08316 -42.63
5 -0.58 5 0.05439 -52.30 243 -0.05439 -41.65
6 -0.77 6 0.04558 54.27 242 -0.04558 71.17
...略
可見只有前兩個NOCV pair對片段間的結合有顯著貢獻,其它的可以忽略。現在用選項2繪制它們的密度等值面圖,0.01 a.u.的等值面如下所示。注意由于這個體系不算很小,所以讓你設置格點的時候最好選High quality grid而非前面用的Medium quality grid,這樣等值面才不會有棱有角。
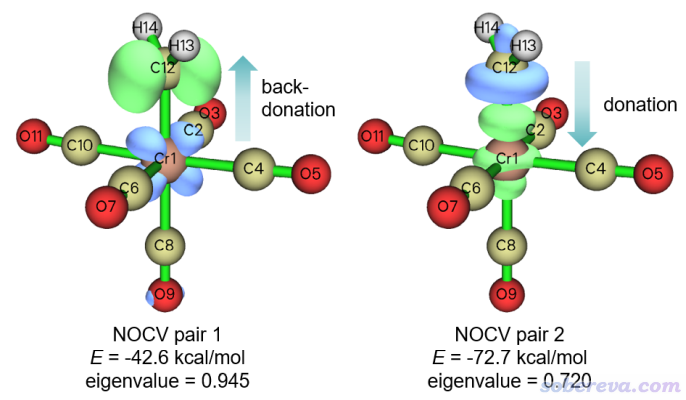
哪怕不用Multiwfn對NOCV pair做成分而分析,而是只看上面的等值面圖,從形狀和顏色上也可以判斷出NOCV pair 2對應的是配位作用,CH2的孤對電子往Cr的dz^2空軌道上轉移,這對應形成了sigma鍵。而NOCV pair 1則明顯體現Cr的某個占據的d軌道的電子往CH2的垂直于片段平面的空軌道上轉移,這對應形成了pi鍵。可見ETS-NOCV再次清晰地展現出兩個片段之間的相互作用本質,光是繪制一個普通的密度差圖是沒法分析這么透徹的。從能量上也可以看到sigma作用能-72.7 kcal/mol比pi作用能-42.6 kcal/mol強得多,這是典型現象,前面的乙烯的例子也體現了對同一個鍵sigma作用會比pi作用更強這一點。也注意對應pi作用的NOCV pair 1的本征值0.945比對應sigma作用的NOCV pair 2的本征值0.720更大,說明pi作用造成的電子密度變化程度更大。這又一次體現出NOCV pair的能量和本征值并沒有正相關性,造成電子密度變化大的作用未必造成的能量變化就大。
3.6 多于兩個片段的相互作用一例:乙炔三聚化成苯的過渡態
前面都是兩個片段考察極小點結構的軌道相互作用。最后一個例子,演示一下ETS-NOCV分析用于過渡態結構,而且是三個片段之間的總的軌道相互作用,從而體現ETS-NOCV分析的靈活性和普適性。
本例的分析對象是乙炔三聚化成苯分子的過渡態結構,這個反應示意圖如下所示。筆者之前有個博文曾以這個反應為例考察反應過程的電子結構變化,建議看看:《通過鍵級曲線和ELF/LOL/RDG等值面動畫研究化學反應過程》(http://www.shanxitv.org/200)。
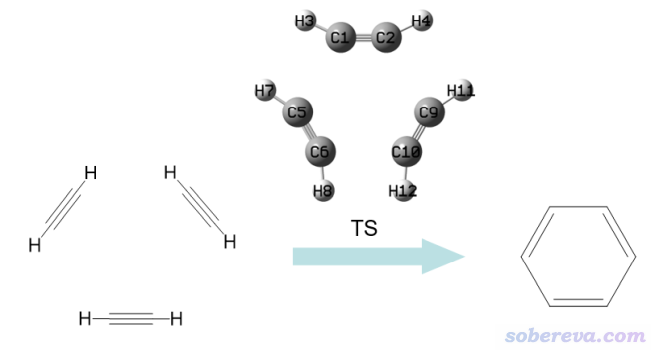
examples\ETS-NOCV\C2H2_trimerization_TS\目錄下有四個單點任務的gjf文件,TS.gjf里的結構是我以前用B3LYP/6-31G*優化好的過渡態結構,1.gjf、2.gjf、3.gjf分別對應過渡態結構下的三個變形了的乙炔。運行這些文件得到相應的fch文件。
啟動Multiwfn,然后輸入
23 // ETS-NOCV分析
3 // 三個片段
examples\ETS-NOCV\C2H2_trimerization_TS\1.fch // 片段1
examples\ETS-NOCV\C2H2_trimerization_TS\2.fch // 片段2
examples\ETS-NOCV\C2H2_trimerization_TS\3.fch // 片段3
-2 // 計算NOCV能量
然后可以看到以下輸出
There are totally 51 NOCV pairs and 102 NOCV orbitals
NOCV orbitals with absolute eigenvalues smaller than 1.0E-03 are not shown
Note: All energies are given in kcal/mol
Pair Energy | Orbital Eigenvalue Energy | Orbital Eigenvalue Energy
1 -25.50 1 0.57545 -95.07 102 -0.57545 -50.76
2 -25.50 2 0.57545 -95.07 101 -0.57545 -50.76
3 -0.93 3 0.09515 -66.85 100 -0.09515 -57.12
4 -0.93 4 0.09515 -66.85 99 -0.09515 -57.12
5 -1.31 5 0.07688 -114.89 98 -0.07688 -97.89
6 -1.50 6 0.07168 62.87 97 -0.07168 83.83
7 -0.33 7 0.03245 3.82 96 -0.03245 14.08
8 -0.33 8 0.03245 3.82 95 -0.03245 14.07
9 -0.13 9 0.02029 -0.40 94 -0.02029 6.16
可見NOCV pair 1和2是簡并的,出現這樣的情況是因為過渡態結構具有D3h點群對稱性。現在用選項2繪制他們各自的密度等值面以及二者加和的等值面,0.005 a.u.等值面數值下的圖像如下所示
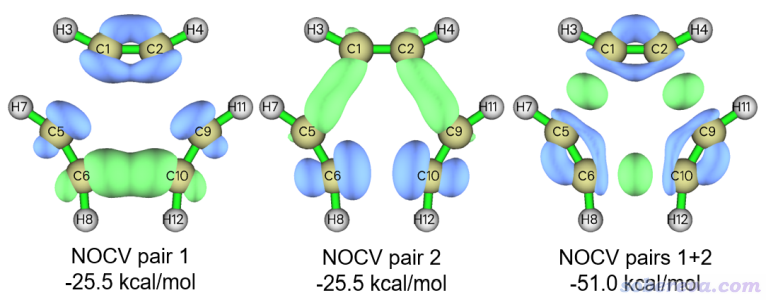
從NOCV pair 1和2各自的密度等值面上看不出明確的化學意義,但是二者的加和對應的軌道相互作用意義很明確,可見在三個即將形成的C-C鍵之間都有一塊綠色等值面,因此體現的是相鄰乙炔間的C-C鍵的sigma作用。這兩個NOCV pair的能量總和-51.0 kcal/mol已經不小了,這說明在過渡態結構下三個已變形且靠近的乙炔之間的sigma軌道相互作用已經很明顯了。此例也反映出,當一些NOCV pair存在簡并性,而且單獨看的時候不好指認作用特征時,應當將它們加和之后再考察。還值得注意的是,上圖中藍色等值面在平面內pi作用區域,這體現出在電子凝聚于C-C間形成新的sigma鍵的時候是以乙炔原有pi鍵上的電子減少為代價的。
眾所周知,苯環上是有六中心pi作用的。在三個乙炔三聚成苯的過渡態結構下,是否變形的乙炔之間也已經形成了pi作用?上面可見sigma作用此時已經一定程度形成了。這個問題也可以通過ETS-NOCV分析來回答,于是我們依次觀看其它的NOCV pair的密度圖,找找看是否有對應pi作用的。可以發現NOCV pair 3、4和pi作用有直接關系,因為它們在體系平面上有個節面,見下圖。但光看其中任何一個NOCV pair也是說不清楚對應的意義,所以下圖右邊也給出了這兩個NOCV pair密度加和的等值面。由于作用特別弱,所以必須把等值面數值設到特別小0.0002 a.u.的時候才能清楚觀察其特征。
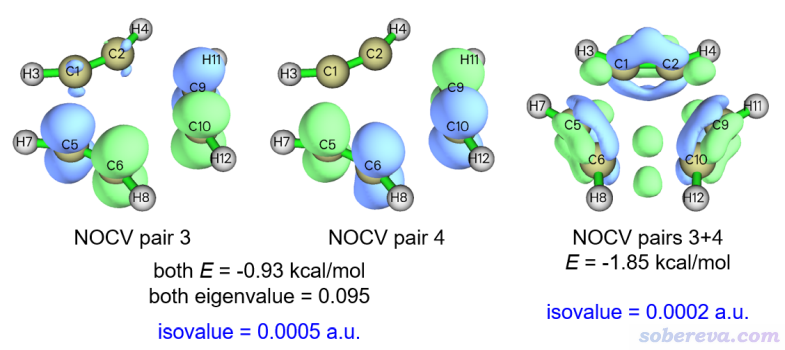
如NOCV pair 3+4的等值面所示,在相鄰乙炔間即將形成的每個C-C鍵的上、下方都有一塊綠色等值面,對應電子在此聚集,體現出確實在過渡態結構時也有一定的pi作用。然而其對應的軌道作用能僅有-1.85 kcal/mol,可以說微乎其微,所以過渡態結構下的pi作用的實際效果可以完全忽略不計。這點通過鍵級也能體現,按照《在Multiwfn中單獨考察pi電子結構特征》(http://www.shanxitv.org/432)里的做法去計算一下C1-C5之間的pi電子的Mayer鍵級,數值僅為0.01,這進一步說明確實pi作用此時還沒有明顯形成(而對于苯的結構來計算pi Mayer鍵級,數值則有將近0.5)。
4 總結
ETS-NOCV已被越來越多的研究者用于深入分析片段間的軌道相互作用的內在特征,對于討論化學鍵的本質有不可替代的重要價值,對于理解弱相互作用體系中的單體間的軌道相互作用亦有很大幫助。ETS-NOCV分析與Multiwfn另支持的CDA (Charge decomposition analysis)分析和NAdO (natural adaptive orbital)分析有明顯的互補性,同時運用可以得到更全面的視角。
本文對ETS-NOCV的原理進行了簡要的講解,介紹了Multiwfn中ETS-NOCV模塊的特征,并且提供了大量的分析例子,涉及到各種類型的作用和體系。把這些例子全都做一遍,認真領會分析思想,就可以輕松地運用ETS-NOCV考察各類情況下的軌道相互作用。由這些例子可見,Multiwfn的ETS-NOCV模塊使用極盡簡單、方便和靈活,已將ETS-NOCV分析的門檻降到了最低,預期以后Multiwfn做ETS-NOCV分析將成為理論化學研究者們從軌道角度討論作用本質的最常用的手段之一。