使用Multiwfn做電荷分解分析(CDA)、繪制軌道相互作用圖
使用Multiwfn做電荷分解分析(CDA)、繪制軌道相互作用圖
文/Sobereva @北京科音
First release: 2012-Nov-7 Last update: 2021-May-28
1 前言
電荷分解分析(Charge decomposition analysis, CDA)是基于片段軌道的概念提出的一種將分子片段之間電荷轉移分解為軌道的貢獻,從而能更深入了解電荷轉移本質的方法。這個方法很有意義,如今已被廣泛使用。在Multiwfn (http://www.shanxitv.org/multiwfn)從2.6版開始加入了這個功能,并且能夠直接繪制出軌道相互作用圖。本文將詳細介紹這個方法的原理,并通過一些實例介紹一下在Multiwfn中的操作。
2 原理
2.1 片段軌道
在介紹CDA之前先說一下片段軌道(Fragment orbital)的含義。比如對于一個復合物AB,片段A的片段軌道就是指A部分在保持與復合物狀態相同的幾何結構下,單獨做量化計算得到的軌道(本文所謂“軌道”指分子軌道或后HF方法產生的自然軌道)。A的各個片段軌道是正交歸一的,B的各個片段軌道也是正交歸一的,但A的片段軌道和B的片段軌道之間往往不是正交的,而是有重疊的。復合物AB的軌道可以視為是由A的片段軌道和B的片段軌道相互混合而構成的。這在結構化學教科書里就有很多例子,尤其是討論配合物中金屬與配體成鍵問題時,經常會有圖片描述金屬的d軌道與配體的軌道混合構成配合物的軌道,實際上此時金屬和它的原子軌道就相當于A和A的片段軌道,配合物和它的分子軌道就相當于B和B的片段軌道。
在量子化學計算中,通常復合物的軌道是以基組所定義的原子中心的基函數來描述的,有多少個基函數就會產生多少個軌道。若計算AB、A、B時所用的基組都一致,且假設計算AB時有NC個基函數,其中NA個在片段A上,另外NB個在B上,則計算復合物時就得到了NC個復合物的軌道,對這兩個片段進行單獨計算時就會產生NA個A的片段軌道和NB個B的片段軌道。此時可以令這NA+NB個片段軌道作為新的基函數代替原先的NC個原子中心基函數來展開這NC個復合物的軌道,這實際上就是一個基函數的變換。由于基函數數目不變,前后都是NC個,所以這個變換并沒有使信息損失。經過這樣的變換,就得到了片段軌道在復合物軌道中的展開系數,也得到了片段軌道間的重疊矩陣。利用這樣的信息,我們也可以像討論原子在分子軌道中的成分一樣討論片段軌道在復合物軌道中的成份,這對于了解片段軌道是以何種方式組合成復合物軌道是非常有用的。
2.2 CDA及其廣義化
CDA方法由Dapprich和Frenking在J. Phys. Chem., 99, 9352 (1995)當中被提出,它直接利用了片段軌道的概念對A、B間的電子轉移進行了分解。它定義了三個量:
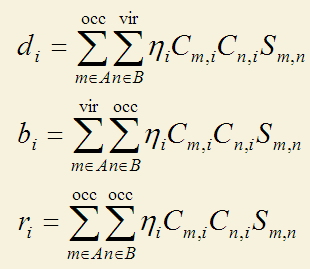
i是復合物軌道標號,m和n分別是A和B片段軌道的標號。其中occ和vir代表加和是對于占據軌道和非占據軌道(虛軌道)。η是復合物軌道占據數。C的m,i矩陣元代表片段軌道m在復合物軌道i中的系數。S是片段軌道間的重疊矩陣。
上式中d代表donation,其d_i表現的是經由復合物軌道i,A的電子向B轉移的量,這是由A的占據軌道與B的虛軌道相混合而導致的。b代表back donation,也就是由B向A轉移的電子數。為什么這兩項是這么定義的可以這么來理解:m和n片段軌道在i復合物軌道中的重疊布居數可以視為是此軌道中m和n共享的電子數。對于d_i,由于m和n分別是占據軌道和虛軌道,所以共享的電子是完全來自于m的。按照Mulliken的均分處理,重疊布居應當有一半劃分給m,另一半劃分給n,也就是等于說m轉移電子給了n,轉移的數目就是重疊布居的一半。在d_i和b_i的加和號當中的項就正是m和n的重疊布居的一半,m是占據的還是n是占據的決定了轉移的方向,所以d_i和b_i能夠代表電子的donation和back-donation的量。
r_i體現了占據的片段軌道在i中的相互作用。正值時表明重疊布居為正,說明在i中占據片段軌道的電子向兩個片段重疊區域聚集(至少不是互斥),此時i也體現了成鍵軌道的特征。如果r_i為負,i中表明由于占據片段軌道間的電子互斥,使得重疊區域電子減少,而向非重疊區域轉移。所有復合物軌道的r項之和幾乎一定是負值,因為滿占據的軌道間總是以互斥效應為主導。由于互斥后電子密度分布發生了變化(極化),故r也被稱為互斥極化項。
上述CDA定義有局限性。首先它只能用在閉殼層體系。其次,盡管復合物軌道可以使用自然軌道(其占據數為非整數),然而片段軌道卻沒法用自然軌道,因為式中沒有將片段軌道的占據數考慮進去,而只是用占據和非占據來描述。為了解決這兩個局限性,寡人提出了廣義化的CDA的定義,如下所示,這個定義就是Multiwfn中所用的定義,詳見筆者寫的《廣義化的電荷分解分析(GCDA)方法》(物理化學進展,4,111-124 (2015) http://www.hanspub.org/journal/PaperInformation.aspx?paperID=16370),使用Multiwfn做CDA分析時除了引用Multiwfn程序原文外也應同時引用此文章,引用的時候寫J. Adv. Phys. Chem., 4, 111-124 (2015), http://dx.doi.org/10.12677/JAPC.2015。
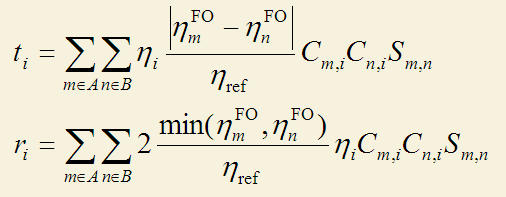
在這個廣義化的CDA定義中,對于開殼層體系,alpha和beta電子是單獨處理的。比如計算alpha部分時,i,m,n就只循環alpha軌道。這個廣義化的形式將片段軌道的占據數直接考慮了進去,故片段軌道也可以是后HF方法得到的自然軌道了。式中η_ref是參考占據數,對于閉殼層軌道是2.0,對自旋軌道是1.0。d_i項的計算方法是在計算t_i時,只對m片段軌道占據數大于n片段軌道占據數時進行加和,計算b_i時就是只對m占據數小于n時進行加和。之所以把m和n的占據數的差值引入進去并不難理解:如果m和n的占據數都一樣,顯然二者間誰也不會向另一方轉移電子,而二者占據數差值越大,則轉移程度越大。當其中一個是完全占據而另一個是完全沒有占據時,結果就會自動恢復為原始CDA的定義,也就是說廣義化的CDA計算的d和b在原始CDA能用的時候結果是與原始定義一致的。廣義化的CDA定義中r_i項里面有個取最小值的函數min(),它的含義是兩個片段軌道間的互斥作用由二者都有的電子數來決定。之所以引入了因子2,是因為這樣的話r_i里的每一項就直接對應于m和n在i中的重疊布居了(實際上這也是由i貢獻的m和n之間的所謂的Mulliken鍵級),而非像原始定義中那樣是重疊布居的一半,這樣物理意義更明確。對于原先CDA適用的情況,即片段軌道占據數為整數時,廣義化的CDA得到的r_i是原先CDA定義的r_i的二倍。
值得注意的是,在CDA原文中雖然公式是對的,但是文中實例中的d、b、r數值不對,都比公式定義的大了一倍。原作者自己開發的CDA程序和AOMix程序給出的d、b、r的結果也都比原公式的大了一倍。所以如果要想令Multiwfn得到的d、b項與它們的作對比,應該先乘以2。而Multiwfn的r項已經是原先定義的二倍,所以不用再乘2了。QMForge也能做CDA分析,但所得d、b、r居然是原先CDA定義的四倍!
2.3 ECDA
貢獻的電子和反饋的電子數的差值,即d-b項,某種意義上可以視為是從片段A向B凈轉移的電子數。但是在J. Am. Chem. Soc., 128, 278當中作者認為這種觀點有問題,因為他們覺得d、b項當中不光體現了電荷轉移部分(CT),還包含了電子極化部分(PL)。所謂的電子極化就是電子密度的變形,可認為是在片段內,此片段的虛軌道和此片段的占據軌道混合而產生的。因此為了只得到CT,就得排除PL部分的影響。于是他們提出了擴展的CDA方法(extended CDA, ECDA),給出了以下關系:
PL(A) + CT(A→B) = A的全部的占據片段軌道在全部虛復合物軌道中的成份之和,乘上Occ
PL(A) + CT(B→A) = A的全部的虛據片段軌道在全部占據復合物軌道中的成份之和,乘上Occ
PL(B) + CT(B→A) = B的全部的占據片段軌道在全部虛復合物軌道中的成份之和,乘上Occ
PL(B) + CT(A→B) = B的全部的虛據片段軌道在全部占據復合物軌道中的成份之和,乘上Occ
對于開殼層時,Occ是1.0。對于閉殼層情況,Occ是2.0。ECDA不能用于后HF計算,因為沒考慮軌道占據數。
將以上四項中的前兩項(或后兩項)算出來后,就可以直接得到電子凈轉移數了:
CT(A→B) - CT(B→A) = [ PL(A) + CT(A→B) ] - [ PL(A) + CT(B→A) ]
即電子極化部分被抵消了。
雖然ECDA名義上是CDA的擴展,也都基于片段軌道的概念,但是二者的思路截然不同,在我來看沒什么可比性。CDA的主要妙處在于可以將電子轉移以及占據軌道的相互作用分解到各個復合物軌道上,然而ECDA的用處只不過是算一個總的轉移量而已。而實際上總的轉移量在我來看也沒必要用ECDA來算,因為有更直接、物理意義更清楚的方法來算,也就是通過原子電荷加和來得到片段在復合物中的凈電荷,再與此片段在孤立狀態下的凈電荷求差值,就直接知道了形成復合物過程中從此片段轉移走了多少電子。盡管ECDA意義不是很大,在Multiwfn里還是支持了ECDA。
不少計算原子在軌道中成份的方法也可以用于計算片段軌道在復合物軌道中的成分,如Mulliken方法、SCPA方法、Stout-Politzer方法,見《談談軌道成份的計算方法》(http://www.shanxitv.org/131)中的討論。雖然SCPA是最省事的,只需將系數求平方,然后再乘上歸一化因子就行了,但是我發現SCPA得到的片段軌道在虛復合物軌道中的成份不甚合理,進而得到的CT(A→B) - CT(B→A)項可能比較離譜,而ECDA原文用的是Mulliken方法計算成份,結果看起來比較合理。在Multiwfn的CDA模塊里,凡是涉及到片段軌道在復合物軌道中的成份,默認都是用Mulliken方法計算的。對于占據的復合物軌道的成份,我發現SCPA方法和Mulliken方法給出的結果相差不大。如果你就是想用SCPA方法來算,將Multiwfn目錄下的settings.ini里的iCDAcomp改為2即可,SCPA的好處是可以保證貢獻值在0~100%之間,而Mulliken方法不能保證這一點。
3 現有的支持CDA的程序
相對于其它能做CDA的程序,Multiwfn的CDA功能無疑是最強大、最靈活、最好用的,免費且開源,如今已經被大量文章所使用。Multiwfn的相關知識見《Multiwfn入門tips》(http://www.shanxitv.org/167)。Multiwfn的CDA模塊支持幾乎所有主流量子化學程序,支持開殼層、后HF的情況,可以定義無數個片段。Multiwfn能直接繪制出軌道相互作用圖,操作非常方便快捷,每步操作在屏幕上有明確提示,因此不必記憶復雜的關鍵詞或命令行。Multiwfn的輸出結果也非常容易讀懂。另外,用戶可以查看各個軌道的成份,計算出的片段軌道間的重疊矩陣、片段軌道在復合物軌道中的系數矩陣都可以導出到文本文件里便于用戶進一步分析和處理。此程序的原理、選項、實例在手冊里都有特別詳細的介紹。
還有其它也能做CDA的程序,但都明顯不如Multiwfn,沒有任何使用的必要,但感興趣的話可以了解一下。
CDA理論的作者開發了CDA程序(http://www.uni-marburg.de/fb15/ag-frenking/cda),是免費的。此程序很簡短,只支持閉殼層。此程序所用的CDA的公式也是原先定義的廣義化,對于片段軌道也能支持自然軌道,但其形式和筆者提出的廣義化的CDA的形式不同。此程序手冊里給出的廣義化的CDA公式意義很不明確,但對于原先CDA定義能用的情況下,此廣義化定義給出的結果和原先定義給出的結果一樣。此程序目前支持Gaussian98/03/09的輸出文件。另外此程序還能輸出各個軌道的結合能,但在手冊中原理沒寫清楚(而且其代碼中的算法和手冊里根本不一樣!),沒有對應的文獻,也沒有實例,十分詭異,筆者不建議用這來路不明的軌道結合能。另外程序還會輸出片段相互作用的化學勢和硬度,前者還稍微有點道理,但后者按照概念密度泛函理論,其定義完全是錯的。CDA程序不支持ECDA,亦不能繪制軌道相互作用圖。
AOMix程序(http://www.sg-chem.net)是收費的,CDA模塊是其主要功能之一。此程序只有Windows版。此程序支持原始形式的CDA,但可以用在開殼層。ECDA在此程序中支持,實際上ECDA就是此程序作者提出的。此程序號稱可以繪制軌道相互作用圖,但實際上沒法直接生成圖像,它會產生一些文本文件,必須將這些文件放到Origin、sigmaplot等作圖工具里繪制折線圖,才能得到軌道相互作用圖。而軌道成份、軌道序號之類都得之后自己標上去,很不方便。AOMix這個程序筆者認為使用很不方便,手冊寫得較亂,還得記憶一大堆關鍵詞,而且程序運行過程詭異,輸出文件過于緊湊而不好閱讀。AOMix的功能實際上只相當于Multiwfn的一個很小的子集而已,還明顯不如Multiwfn強大靈活易用。
QMForge程序(https://qmforge.net)是收費的,有圖形界面,不支持Linux,功能甚少,它的CDA功能是基于cclib庫實現的,只支持原始CDA的定義,因此不能用在開殼層、后HF情況。QMForge不支持ECDA,亦不能繪制軌道相互作用圖。此程序功能特別弱,能做的事其實就只是Multiwfn的一個微不足道的子集而已,居然這還好意思賣錢...
4 Multiwfn的CDA模塊的輸入文件及選項
雖然前面討論的都是只有兩個片段的情況,但CDA方法也可以直接用于研究含多個片段的體系,Multiwfn的CDA功能甚至可以定義無限個片段。做CDA分析需要的文件有以下情況
a) Gaussian用戶
需要提供復合物和各個片段的單點任務的fch文件。
注:也可以用Gaussian的輸出文件,此時對片段計算必須使用pop=full關鍵詞,對復合物必須用pop=full IOp(3/33=1)關鍵詞。因為Multiwfn要從復合物的輸出文件中讀取以原子中心高斯函數為基的重疊矩陣,這是要寫IOp(3/33=1)的原因。由于Multiwfn還要讀取復合物及片段的軌道系數,所以必須寫pop=full。由于用fch/fchk文件方便得多,我不建議用Gaussian輸出文件用于CDA目的。
b) 其它量化程序用戶
只要輸入到Multiwfn的文件能給Multiwfn提供基函數信息即可做CDA分析。哪些文件包含基函數信息、怎么生成,參見《詳談Multiwfn支持的輸入文件類型、產生方法以及相互轉換》(http://www.shanxitv.org/379)。比如Gaussian和Q-Chem用戶可以用fch/fchk文件,ORCA、Molpro、NWChem、Dalton等程序用戶可以用Molden文件,GAMESS-US和Firefly用戶可以直接用單點任務的輸出文件(但后綴需要改為.gms)。
輸入文件里片段的坐標必須和復合物坐標一致,因此片段的坐標只需要從已優化的復合物的坐標中直接提取出來就行了,不要單獨對片段進行優化。對于Gaussian用戶,為了避免Gaussian自動將坐標調整到標準朝向而使得坐標無法相互對應,應當恰當使用nosymm關鍵詞,見《談談Gaussian中的對稱性與nosymm關鍵詞的使用》(http://www.shanxitv.org/297)。如果為了保險,就在所有計算時都寫上nosymm關鍵詞!
坐標必須都用笛卡爾坐標,而且片段的定義順序必須和復合物中的完全一致。比如復合物文件中片段的出現順序是A,B,C,那么就必須將A,B,C分別定義為片段1,2,3,而不能定義為諸如2,1,3。并且片段內原子順序、原子坐標也要完全和復合物中的一致。
計算復合物和片段時必須用嚴格相同的基組。理論方法雖然允許不同,但沒有特殊情況的話應當相同,否則結果沒實際意義。
絕對不要用彌散函數,否則結果可能完全沒意義!這在前述筆者寫的《廣義化的電荷分解分析(GCDA)方法》論文中有專門討論。基組用不著很大,def2-SVP就足矣得到合理結果。
注:如果有什么特殊的原因非得使用彌散函數不可,由于此時可能會出現基函數的線性依賴問題,Gaussian計算時應當帶著IOp(3/32=2)選項。這個選項用于避免Gaussian自動刪除線性依賴的基函數,因為一旦刪除了線性依賴基函數,將導致軌道數目少于基函數數目,然而CDA分析卻必須要求基函數數目和軌道數目相同。
特別需要注意的是,在計算含有很重原子體系時(如計算金屬配合物),往往要對這些重原子用贗勢基組,而其它部分用其它基組,這可能會產生基函數數目不符的問題。比如計算Ni(CO)4,對Ni用lanl2DZ(作為片段1),而對4個CO用6-31G*(作為片段2)。在計算整體時,由于用了自定義方式指定基組(即gen或genecp),此時Gaussian會自動用球諧型基函數來計算。對于lanl2DZ,Gaussian也默認用球諧型基函數。然而,當計算(CO)4時,Gaussian對于6-31G*會自動使用笛卡爾型基函數(對于d或更高角動量殼層其基函數的數目會大于球諧型),這就導致兩個片段的基函數數目之和與整體不符,因此CDA無法進行。解決辦法就是計算(CO)4的時候在route section中寫上5d關鍵詞,這就強制讓Gaussian使用球諧型基函數了。如果你對Gaussian決定基函數類型的機制不很清楚,建議凡是使用混合基組的情況,在計算片段時應總加上5d關鍵詞,這樣最保險。關于笛卡爾和球諧型基函數的更多討論見《談談5d、6d型d殼層基函數與它們在Gaussian中的標識》(http://www.shanxitv.org/51)。
對于Gaussian,如果用后HF方法計算,應當再加上density關鍵詞,并且此時只能用Gaussian輸出文件作為輸入。對于閉殼層,pop=full應當改為pop=NO;對于開殼層,應改為pop=NOAB。這樣才能讓自然軌道信息代替HF軌道信息出現在Gaussian輸出文件中。
如果復合物和各個片段中的一個或多個是開殼層任務,則整個CDA分析(包括ECDA等)會被當做開殼層來處理,即分別輸出alpha和beta的結果。
如果復合物和各個片段中的一個或多個是后HF任務,則ECDA分析將會被跳過,而且也沒法繪制軌道相互作用圖,因為自然軌道沒有明確的能量的定義。
做CDA分析的時候片段的狀態選取并不唯一。比如NaCl,既可以把兩個片段當成Na+和Cl-來計算,也可以當成一個Na原子和一個Cl原子計算。關于怎么設定片段合適,在前述的《廣義化的電荷分解分析(GCDA)方法》中有非常詳細的討論,請仔細閱讀。
5 實例
下面通過兩個實際例子來說明CDA、ECDA及軌道相互作用圖的用處,以及在Multiwfn當中的操作。下面例子所用的Gaussian輸出的fch文件,連同相應的Gaussian輸入文件,在Multiwfn壓縮包里examples\CDA的相應目錄下都已提供了。
5.1 實例1:COBH3
這是一個閉殼層相互作用的例子,在COBH3(復合物)當中,CO(片段1)利用孤對電子與缺電子的BH3(片段2)形成配位鍵,也因此CO的電子向BH3發生了轉移。
此例是在HF/6-31G*下計算的(一般都應當用DFT來算,這里只是個例子,用HF也無所謂),復合物的坐標為
C 0. 0. -0.09962
O 0. 0. -1.20782
B 0. 0. 1.5275
H 1.17227 0. 1.79707
H -0.58614 -1.01522 1.79707
H -0.58614 1.01522 1.79707
片段1的坐標為
C 0. 0. -0.09962
O 0. 0. -1.20782
片段2的坐標為
B 0. 0. 1.5275
H 1.17227 0. 1.79707
H -0.58614 -1.01522 1.79707
H -0.58614 1.01522 1.79707
可見,片段1、2的坐標和復合物中是對應的,而且順序是一致的。對自己的體系做CDA分析時務必要滿足這一點。
啟動Multiwfn,輸入以下內容:
examples\CDA\COBH3\COBH3.fch //復合物的Gaussian輸出文件
16 //進入CDA模塊
2 //定義兩個片段
examples\CDA\COBH3\CO.fch //片段1的Gaussian輸出文件
examples\CDA\COBH3\BH3.fch //片段2的Gaussian輸出文件
之后立刻會輸出以下CDA分析結果
Orb. Occ. d b d - b r
1 2.000000 -0.000004 -0.000000 -0.000004 -0.000001
2 2.000000 0.001119 -0.000023 0.001141 0.000326
3 2.000000 -0.000002 -0.000471 0.000469 0.000313
4 2.000000 -0.013250 -0.000704 -0.012546 -0.005676
5 2.000000 0.041648 -0.003309 0.044957 0.232262
6 2.000000 0.037385 -0.020136 0.057521 0.212422
7 2.000000 -0.000543 0.000647 -0.001190 0.022166
8 2.000000 -0.000543 0.000647 -0.001190 0.022166
9 2.000000 0.171353 0.026952 0.144401 -0.741381
10 2.000000 -0.000569 0.043713 -0.044281 -0.038916
11 2.000000 -0.000569 0.043713 -0.044282 -0.038916
12 0.000000 0.000000 0.000000 0.000000 0.000000
13 0.000000 0.000000 0.000000 0.000000 0.000000
14 0.000000 0.000000 0.000000 0.000000 0.000000
15 0.000000 0.000000 0.000000 0.000000 0.000000
......
-------------------------------------------------------------------
Sum: 22.000000 0.236023 0.091027 0.144996 -0.335233
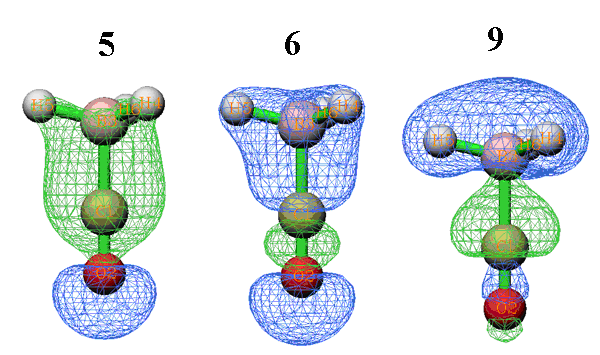
Orb.就是復合物軌道的編號,Occ.是其占據數。從以上數據可見,復合物軌道9的d是最大的,因此表明這個軌道對于CO向BH3的電子轉移有最主要的貢獻。通過形成這個復合物軌道,CO向BH3貢獻了0.171個電子,而BH3向CO反饋了0.0269個電子,故凈轉移可以估算為0.144(即d-b項)。BH3對CO反饋電子主要是來自于復合物軌道10和11,它們是簡并的軌道,b的數值一致,為0.0437。前三個復合物軌道的d、b、r項都接近于0,表明它們對于片段間的相互作用沒什么用處,這是因為它們分別是O、C、B的內核軌道,基本不會參與成鍵。所有的虛軌道的d、b、r項都為0,這是理所應當的,因為它們的占據數都為0。只有對于自然軌道,對應于LUMO及更高階的復合物軌道由于仍有少量電子占據,其d、b、r項才不是精確為0,但由于自然軌道序號是按照占據數由高到低排的,超過LUMO+5的話由于占據數往往已經很低了,d、b、r項已很小,所以Multiwfn中LUMO+5以上的軌道都不會輸出,以避免太冗長(若想全部輸出的話,在接下來出現的菜單里選1)。
以上數據中r(9)非常負,這說明復合物軌道9的形成使得CO和BH3的占據的片段軌道的電子很大程度上從二者的交疊區域移走,從而降低了電子互斥,從下面的它的軌道波函數圖形中也可見在交疊區域有個明顯的節點。而r(5)和r(6)都是不小的正值,因此它們的形成導致占據片段軌道的電子向交疊區域轉移了不少,可以認為體現了片段間成鍵軌道的特征,從下面的等值面圖形中也可看到這兩個復合物軌道都較好地覆蓋了片段間成鍵區域,故有利于成鍵。
在CDA數據后面緊接著輸出了ECDA結果:
Contribution to all occupied complex orbital:
Occupied, virtual orbitals of fragment 1: 680.4194% 8.0593%
Occupied, virtual orbitals of fragment 2: 390.3988% 21.1226%
Contribution to all virtual complex orbital:
Occupied, virtual orbitals of fragment 1: 19.5806% 2291.9407%
Occupied, virtual orbitals of fragment 2: 9.6012% 1678.8774%
PL( 1) + CT( 1-> 2) = 0.3916 PL( 1) + CT( 2-> 1) = 0.1612
PL( 2) + CT( 1-> 2) = 0.4225 PL( 2) + CT( 2-> 1) = 0.1920
The net electrons obtained by frag. 2 = CT( 1-> 2) - CT( 2-> 1) = 0.2304
含義在前面已經介紹過了。一般來說只需要關注最后一行數據,它表明了從片段1向片段2總共凈轉移了多少電子,此例即0.2304。這個數據和CDA的d-b項會有一定出入,因為二者計算方法沒有明顯關聯。ECDA給出的總的凈轉移量更可靠。
值得一提的是,做CDA分析時沒必要非得把供電子的部分定義為片段1。如果將BH3作為片段1,CO作為片段2(當然,復合物坐標中也因此BH3得在CO前面),只不過會使CDA的d和b的數值互換,ECDA的數值變為-0.2304罷了,物理意義沒變。
接下來,會看到一個菜單,各選項含義不言自明。這里我們看看復合物軌道9的具體成份。因此選2,再輸入軌道編號9,就會看到以下內容
Occupation number of orbital 9 of the complex: 2.00000000
Orbital 7 of fragment 1, Occ: 2.00000 Contribution: 25.8560%
Orbital 13 of fragment 1, Occ: 0.00000 Contribution: 1.0798%
Orbital 2 of fragment 2, Occ: 2.00000 Contribution: 57.2921%
Orbital 5 of fragment 2, Occ: 0.00000 Contribution: 14.5640%
只有片段軌道在此復合物軌道中的貢獻值大于等于1%才會被顯示出來(這個值可以用settings.ini中的compthresCDA參數調節)。從數據中可見,復合物軌道9主體是BH3的軌道2,達57.3%。之所以此復合物軌道會導致CO向BH3轉移那么多電子,顯然是因為CO的占據軌道7與BH3的虛軌道5相混合。至于此復合物軌道引起的少量電子的反饋,則主要是緣于BH3的占據軌道2與CO的虛軌道13的很少量混合。至于為何r(9)很負,必然是CO的占據軌道7與BH3的占據軌道2間的互斥作用所致。因此,通過考察軌道成份,軌道的d、b、r項可以分析理解得更加透徹。如果結合軌道圖形來看,則相互作用會更為直觀(軌道圖形可以用Multiwfn的主功能1來看,建議以fch文件作為輸入文件)。例如,下圖左邊是CO的占據軌道7(展現較明顯C的孤對電子特征),右邊是BH3的虛軌道5,從形狀上看二者比較匹配,因此容易混合,并且混合后會導致不少電子轉移。而且,容易想象混合后的形狀會和前面給出的復合物軌道9的圖形較相似,這也解釋了為什么這兩個軌道能夠在復合物軌道9當中占不少成份。
從Multiwfn 3.3.8版開始,還可以直接將某個復合物軌道的d,b,r值分解為片段軌道對兒的貢獻,這使得考察片段軌道之間的相互作用能夠更清晰透徹。這里我們對復合物軌道9進行分解。輸入0返回上一級菜單,選選項6,然后輸入9,然后輸入0.005,這樣對復合物軌道9的d,b,r項任意之一貢獻大于0.005的片段軌道對兒就會被輸出出來。結果如下
FragA Orb(Occ.) FragB Orb(Occ.) d b d - b r
4( 2.0000) 2( 2.0000) 0.000000 0.000000 0.000000 -0.009969
7( 2.0000) 1( 2.0000) 0.000000 0.000000 0.000000 -0.005759
7( 2.0000) 2( 2.0000) 0.000000 0.000000 0.000000 -0.723845
7( 2.0000) 5( 0.0000) 0.176503 0.000000 0.176503 0.000000
7( 2.0000) 8( 0.0000) 0.006221 0.000000 0.006221 0.000000
7( 2.0000) 11( 0.0000) 0.005846 0.000000 0.005846 0.000000
7( 2.0000) 12( 0.0000) -0.023941 0.000000 -0.023941 0.000000
13( 0.0000) 2( 2.0000) 0.000000 0.021958 -0.021958 0.000000
很清晰地可以看到CO的7號軌道與BH3的5號軌道相互作用導致電子向BH3轉移了0.176,這正是9號復合物軌道d項達到0.171的主要因素。
接下來我們繪制軌道相互作用圖,輸入0回到上一級菜單,然后選5進入繪制軌道相互作用圖的菜單中,各個選項的含義不言自明。我們選3,輸入-30,10把相互作用圖能量下限和上限分別設為-30和10eV。然后選1,軌道相互作用圖立刻會顯示在屏幕上:
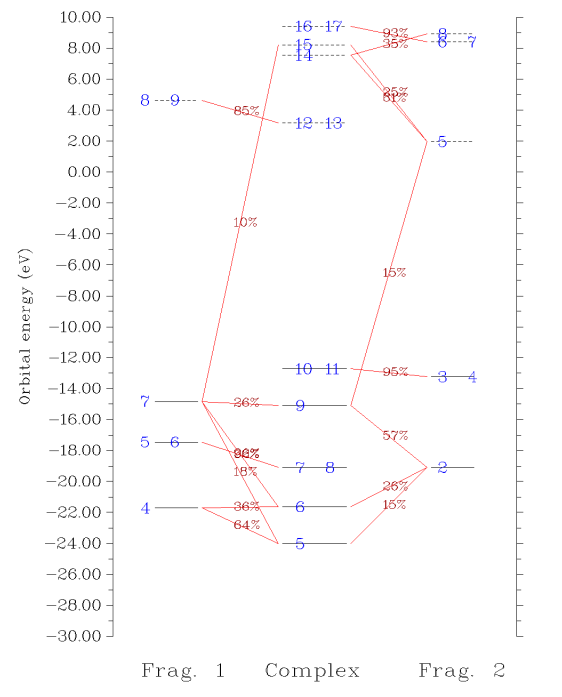
圖中,每個軌道用一個橫杠表示,垂直位置對應其軌道能量。片段1、復合物、片段2的軌道分別在左、中、右部分。占據軌道和虛軌道以實橫杠和虛橫杠分別表示。軌道編號在橫杠上標注了。如果多個軌道編號都標在一個橫杠上,表明它們是簡并軌道。如果片段軌道在復合物軌道中的成份大于指定閾值(默認是10%),則相應的橫杠間會以紅線相連,并在此線上標注出成份具體數值。因此,直接從這樣的圖上,立刻就能看出軌道能級分布,以及各個復合物軌道主要是由哪些片段軌道混合而成的,非常方便。而像諸如復合物的7、8號軌道,只與片段1的5、6號軌道相連,就表明這兩個軌道在形成復合物過程中變化不大,片段2的軌道摻入得不多。
有時候多個文字標簽在圖上發生重疊而看不清楚,可以直接到前述的顯示復合物軌道成份的界面里查看軌道成份;或者,可以嘗試在作圖前通過選項9 Set shifting of composition labels來調節標簽的位置,設的值越接近于1則軌道成份在標注時離片段軌道的橫杠越近,越接近0則離復合物軌道的橫杠越近。默認是0.5,即標注在連線的正中央;也可以選選項7讓成份值不顯示出來,而自行隨后將成份值ps上去。另外還有很多選項用于調節作圖設定,請自行玩弄。
如果你覺得軌道相互作用圖上的連線太亂,不符合自己的要求,可以利用Multiwfn中十分靈活的選項進行修改,見本文文末的說明。
順帶一提,如果你要把軌道相互作用圖發文章的話,應當在繪制界面里選擇保存圖像文件,并且強烈建議選擇pdf格式。因為pdf格式可以無損縮放,并且你會發現文字和線條都特別平滑。可以把打開的pdf文件截圖再保存成其它格式如png、tif,這比起直接讓Multiwfn保存出png、tif圖像格式的線條更光滑。
5.2 實例2:CH3NH2
這個例子是共價(開殼層相互作用)的例子。我們將把CH3NH2當中的CH3視為片段1,NH2視為片段2。
復合物結構已在B3LYP/6-31G**下優化,用于CDA分析的Gaussian輸出文件也是在此級別下產生。片段1、2的計算都在UB3LYP/6-31G**下進行,電荷及自旋多重度都分別是0 2。
啟動Multiwfn,輸入
examples\CDA\CH3NH2\CH3NH2.fch
16
2 //定義兩個片段
examples\CDA\CH3NH2\CH3.fch
examples\CDA\CH3NH2\NH2.fch
n //不翻轉CH3的電子自旋
y //翻轉NH2的電子自旋
CDA用于開殼層時要考慮翻轉電子自旋,這里解釋一下。做CDA分析時各個片段的alpha電子數之和必須等于復合物的alpha電子數,對于beta電子也是這樣。CH3NH2有9個alpha電子和9個beta電子。然而在計算CH3和NH2時,Gaussian都會認為有5個alpha電子和4個beta電子,因此,加和就是10個alpha電子和8個beta電子,這沒法和復合物的情況對上。因此,就必須對其中某一個片段的電子自旋進行翻轉,具體來說,就是令其alpha軌道的占據數、能量、展開系數與beta軌道的進行交換,相應地alpha和beta電子數也交換。此例翻轉的是NH2,這樣處理之后,兩個片段的總alpha和總beta電子數就分別是5+4和4+5了,都和復合物對應上了。若改為翻轉CH3也完全可以,不影響總結果(即總的d、b、r和CT(1->2) - CT(2->1)項),只不過最后得到的alpha和beta的結果會互換。
在屏幕上會看到CDA和ECDA的結果都輸出出來了,并且對于alpha電子和beta電子是分別輸出的。對于此體系的C-N鍵,按照經典理論解釋可以認為是共享一個電子對兒而形成的,CH3相當于是提供其alpha電子的一方,NH2在翻轉了自旋之后,相當于是提供beta電子的一方。所以alpha部分的CDA的d-b項和ECDA分析結果都表明CH3向NH2轉移電子,而beta部分則相反,是NH2向CH3轉移電子。
在CDA部分的末尾會看到CDA的對全部電子的結果,即alpha和beta部分的加和值:
d= 0.044335 b= 0.145181 d - b = -0.100847 r= -0.172318
在ECDA部分的末尾也會看到ECDA對全部電子的結果,也是alpha和beta部分的加和:
CT( 1-> 2) - CT( 2-> 1) for all electrons: 0.1252
由于NH2的電負性明顯強于CH3,所以按道理說電子應該從CH3轉移向NH2。ECDA的結論滿足這一點,并顯示轉移量為0.1252。然而從CDA給出的d-b上來看,轉移方向和預期是相反的。因此,對于開殼層體系,不適合用總的d-b來討論整體電子的轉移方向。但是用每個復合物軌道的d_i、b_i、r_i來討論片段間的作用依然是有意義的。
接下來,如果選選項2進入查看復合物軌道的界面,然后比如輸入6,就會看到第6條alpha復合物軌道中alpha片段軌道的成分,以及第6條beta復合物軌道中的beta片段軌道的成份。輸入0返回上一級菜單。
選5進入繪制軌道相互作用圖的界面。作為例子這次我們繪制beta軌道間的相互作用圖,所以需要先選5來將要做圖的軌道從默認的alpha切換為beta。然后選3,輸入-30,10把相互作用圖能量下限和上限分別設為-30和10eV。然后選1繪制相互作用圖,如下所示
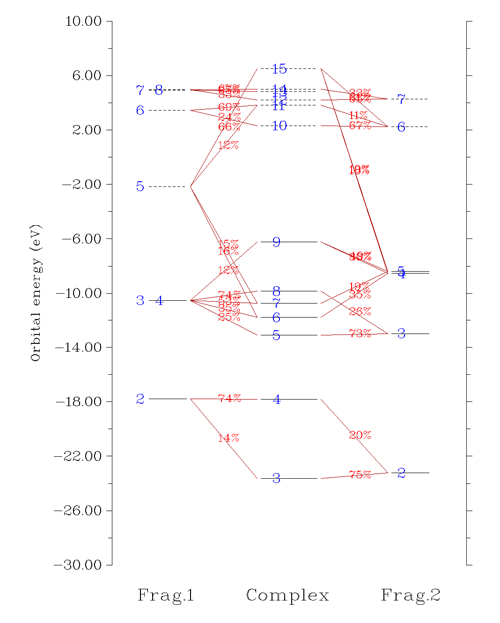
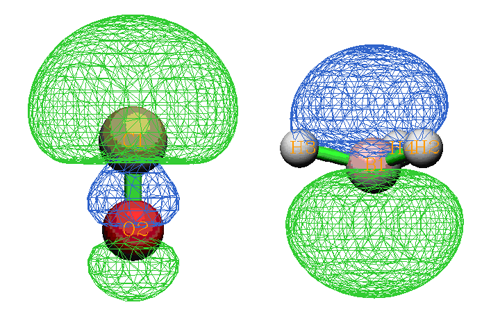
從圖上可以非常清楚看到3、4號復合物beta軌道是由CH3的2號beta軌道和NH2的2號beta軌道混合而成。我們將這部分圖截取出來,并把軌道的圖形附在上面,軌道間如何混合的就變得更直觀了:
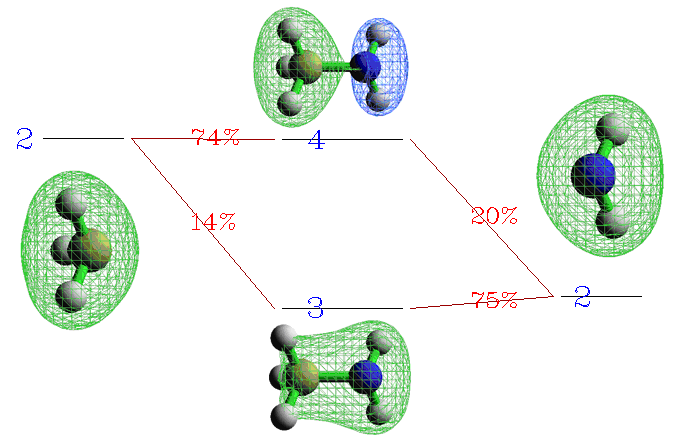
顯然,3號復合物beta軌道是那兩個片段軌道以相位相同方式混合得到,體現了成鍵軌道特征,其中以NH2的片段軌道為主體(占75%的成分)。而4號復合物beta軌道是由NH2的2號beta軌道以相位相反的方式摻入CH3的2號beta軌道而構成,體現了反鍵軌道的特征。
值得一提的是,對此例這樣的開殼層相互作用Multiwfn其實也可以基于限制性開殼層(RO)軌道進行分析,由于alpha和beta軌道特征完全相同,故此時不需要考慮alpha和beta軌道的差異,對alpha和beta自旋繪制的軌道相互作用圖完全相同,因此在討論時往往明顯更容易。見Multiwfn手冊4.16.4節的例子。
5.3 實例3:Pt(NH3)2Cl2
這是個3個片段的例子,Pt2+、(Cl)2 2-以及(NH3)2分別是片段1、2、3。詳細內容懶得貼在這了,大家看Multiwfn手冊4.16.3節就好了,可以把三個片段中每兩個片段間的d,b,r項都得到。
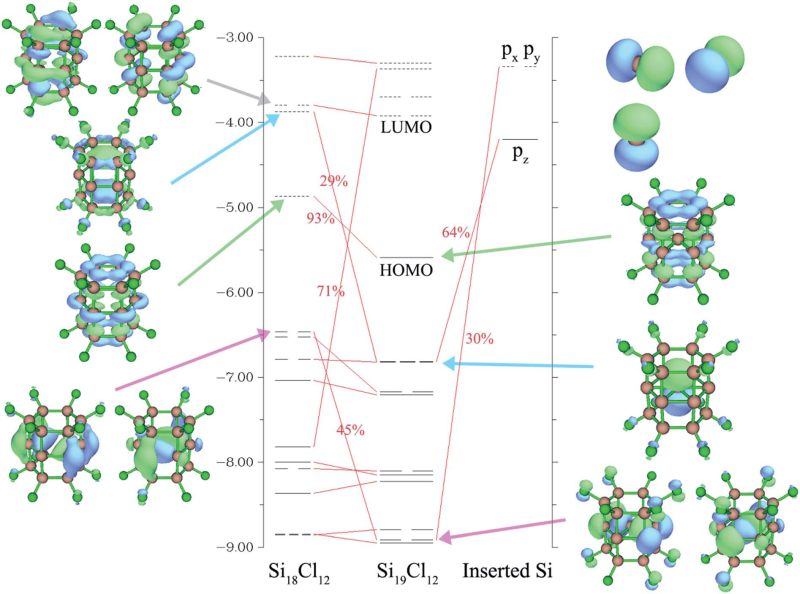
5.4 實例4:Si19Cl12的軌道相互作用圖
筆者在RSC Adv., 5, 78192 (2015)當中考察了Si19Cl12體系中的中心Si原子與Si18Cl12籠的軌道相互作用,繪制了下圖,可見軌道相互作用展現得十分清晰,建議看看此文:/usr/uploads/file/20170105/20170105203001_71053.pdf
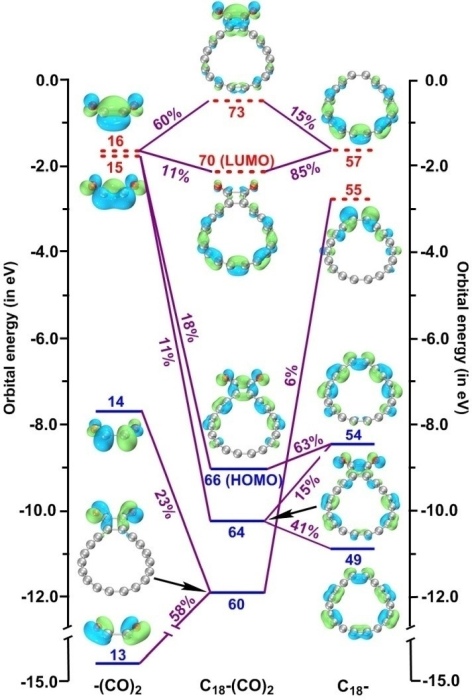
5.5 實例5:C18與(CO)2片段的軌道相互作用圖
筆者在Chem. Eur. J., 28, e202103815 (2022)當中考察了C18-(CO)2體系中的C18與(CO)2之間的軌道相互作用并繪制了下圖,可見軌道相互作用展現得十分清楚。這篇論文在《深入揭示18碳環的重要衍生物C18-(CO)n的電子結構和光學特性》(http://www.shanxitv.org/640)中做了深入淺出的介紹并加入了許多補充信息,包括對下圖的分析。Chem. Eur. J.這篇文章是Multiwfn程序和軌道相互作用圖很好的應用范例,非常建議讀者閱讀和作為范例引用。
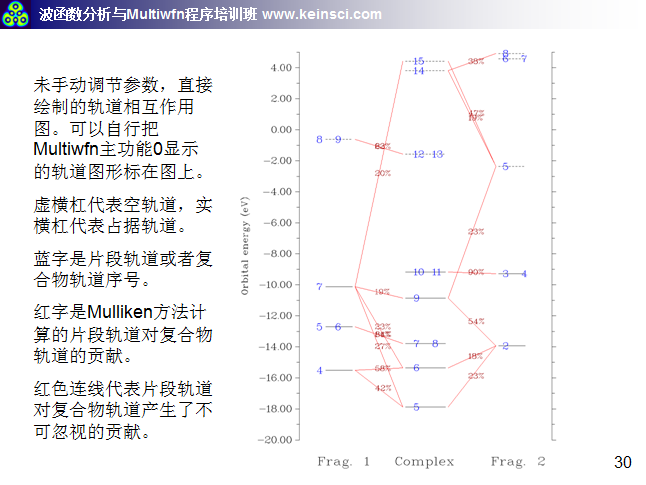
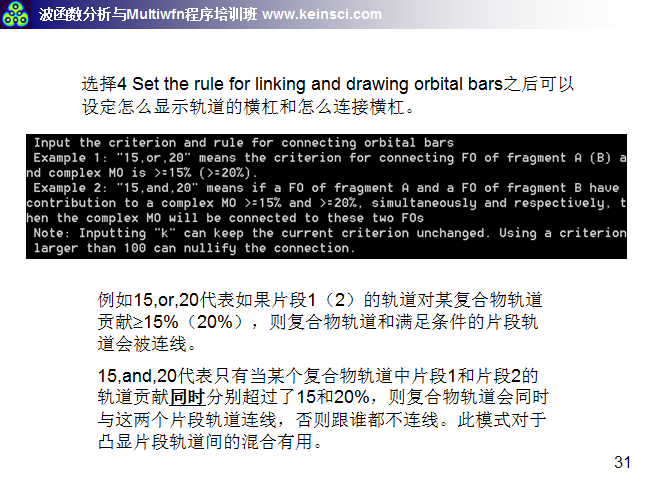
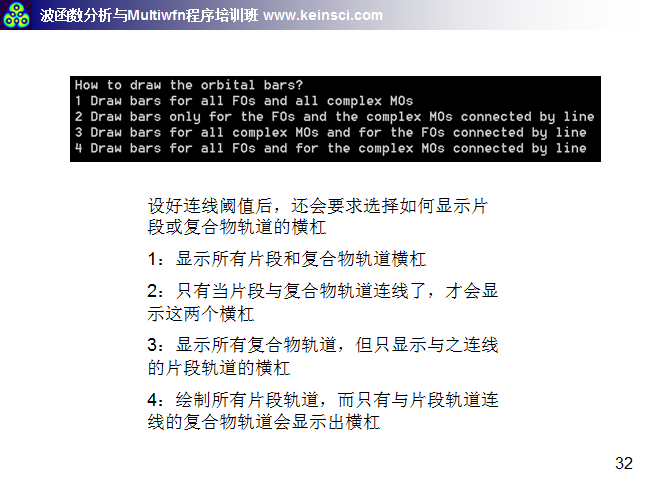
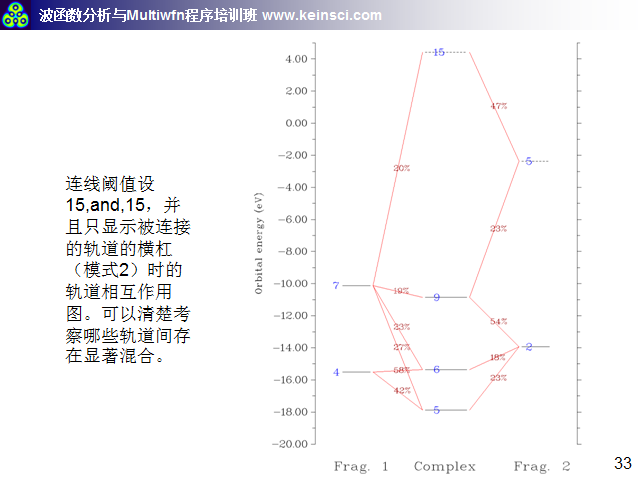
6 關于繪制軌道相互作用圖的連線方式說明
經常有人問,Multiwfn繪制的軌道相互作用圖密密麻麻看不清楚,應該怎么調節。一方面要調節的是軌道相互作用圖的能量范圍,讓能量范圍只對應自己感興趣的那些軌道能量范圍就好。另一方面就是調節連線規則和繪制橫杠的方式,這里把筆者講授的量子化學波函數分析與Multiwfn培訓班(http://www.keinsci.com/workshop/WFN_content.html)里相關的幾頁ppt貼在這里,便于讀者理解。