使用Multiwfn圖形化展現原子對色散能的貢獻以及色散密度
使用Multiwfn圖形化展現原子對色散能的貢獻以及色散密度
Using Multiwfn to graphically exhibit atomic contribution to dispersion energy and dispersion density
文/Sobereva@北京科音 2024-Apr-14
0 前言
色散作用是分子間和分子內弱相互作用中的關鍵組成部分,在《談談“計算時是否需要加DFT-D3色散校正?”》(http://www.shanxitv.org/413)中我做過簡要介紹,在“量子化學波函數分析與Multiwfn程序培訓班”(http://www.keinsci.com/workshop/WFN_content.html)的“弱相互作用的分析”一節里有深入全面介紹。本文將介紹從2024-Apr-13起,Multiwfn程序加入的計算原子對色散能貢獻的功能,是主功能21中的子功能4。這個功能可以非常方便快速地給出每個原子對體系中色散能的貢獻,結合VMD可以直觀地通過原子著色展示,還能計算任意方式定義的兩個片段間的色散作用能。雖然《使用Multiwfn做基于分子力場的能量分解分析》(http://www.shanxitv.org/442)介紹的EDA-FF功能也可以實現這些分析,但原理不同,EDA-FF是基于力場來得到的,而且使用前需要先指認原子類型、寫分子列表文件,用起來明顯沒本文介紹的功能省事。此外,本文介紹的功能還支持周期性體系,這是EDA-FF目前不支持的。本文介紹的功能還可以把原子對色散作用的貢獻轉化為實空間函數,便于通過等值面圖觀看,還能直接給出不同化學環境下原子貢獻的色散能的差值從而得到更豐富信息、提供更多分析討論的素材。相信讀者看過本文的例子后會感到此文介紹的功能頗有實際價值。
Multiwfn可以在官網http://www.shanxitv.org/multiwfn免費下載,必須使用2024-Apr-13及以后發布的版本。不了解Multiwfn者建議參看《Multiwfn入門tips》(http://www.shanxitv.org/167)和《Multiwfn FAQ》(http://www.shanxitv.org/452)。使用本文的功能發表文章時必須在正文引用Multiwfn啟動時提示的程序原文,給別人代算時也必須非常明確告知對方這點。
下面第1節簡要介紹此功能的基本原理,第2節介紹使用方法,之后結合各種例子演示典型的用法。
1 原子對色散能貢獻的計算原理
我在《使用sobEDA和sobEDAw方法做非常準確、快速、方便、普適的能量分解分析》(http://www.shanxitv.org/685)介紹的sobEDA能量分解的原文里專門討論了DFT-D3色散校正能和色散作用的關系,推薦仔細看看。如果不了解DFT-D3者也務必先看看《DFT-D色散校正的使用》(http://www.shanxitv.org/210)和《談談“計算時是否需要加DFT-D3色散校正?”》(http://www.shanxitv.org/413)。對于B3LYP這樣本身對色散作用描述能力為0的泛函,DFT-D3色散校正能可以近似視為是色散能,JCTC, 20, 1923 (2024)將D3校正能與耦合簇方法算的高精度色散作用能進行對比也印證了這一點。在不考慮三體耦合項的情況下,DFT-D3總色散校正能是每一對原子間色散作用能Edisp(i,j)的加和。因此,i原子對體系色散能的貢獻可以計算為Edisp(i)=∑[j≠i]Edisp(i,j)/2。兩個片段間的色散作用能等于二者間的每一對原子間色散作用能的總和。
對周期性體系也可以計算i、j原子間色散作用能Edisp(i,j),是i與當前晶胞和周圍所有鏡像晶胞里的j原子的色散作用能的總和。當然,鏡像晶胞不可能考慮無窮多,用個足夠大的截斷閾值即可。要注意對孤立體系顯然Edisp(i,i)=0,但對周期性體系它一般不為0,因為當前晶胞里i原子和它的周期鏡像之間也有色散作用。周期性體系中原子對色散能的貢獻為Edisp(i)=∑[j≠i]Edisp(i,j)/2+Edisp(i,i)。
JCTC, 20, 1923 (2024)還提出色散密度(dispersion density)的概念,它相當于把每個原子對色散能的貢獻展寬成高斯函數并疊加構成三維實空間函數ρ_disp,從而可以作圖展現,如畫成等值面圖,或者用于給原子球或電子密度等值面著色等。ρ_disp的定義如下式所示。α一般取0.5,ε_A是前述方式計算的A原子對色散能的貢獻,R_A是A原子坐標。
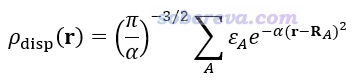
不同化學環境下(比如m和n兩個結構下)共有的某個原子A貢獻的色散能之差記為Δε_A,將上式的ε_A替換為Δε_A得到的就是Δρ_disp函數。如果計算Δρ_disp用的核坐標是m結構下的,那么可以把Δρ_disp用于給m結構下的原子球進行著色,或者畫成等值面附在其結構圖上。
順帶一提,DFT-D3色散校正能本質上是利用原子的C6色散系數進行計算的,推薦看《使用Multiwfn計算原子的C6色散系數》(http://www.shanxitv.org/709)了解更多相關知識。
2 Multiwfn分析原子對色散能貢獻功能的使用
任意含有結構信息的文件,如xyz、pdb、mol2、gjf、fch、mwfn等,都可以作為本文介紹的功能的輸入文件,介紹見《詳談Multiwfn支持的輸入文件類型、產生方法以及相互轉換》(http://www.shanxitv.org/379)。如果是周期性體系,載入的文件還必須包含晶胞信息,如cif、帶有Tv(平移矢量)的gjf文件、POSCAR、gro等,參看《使用Multiwfn非常便利地創建CP2K程序的輸入文件》(http://www.shanxitv.org/587)里的說明。
當前功能必須借助Grimme的dftd3程序,而且必須是我的魔改版,下載地址為http://www.shanxitv.org/soft/dftd3_TLmod.zip,里面有源代碼,dftd3.exe是Windows下的可執行文件,無后綴的dftd3文件是Linux下的可執行文件。必須把Multiwfn的settings.ini文件里的dftd3path參數設為當前機子里dftd3程序的實際路徑,否則Multiwfn沒法調用之,例如Windows下你把dftd3.exe放在了D:\study\dftd3_TLmod\目錄下,就得設成dftd3path= "D:\study\dftd3_TLmod\dftd3.exe"。對于Linux,別忘了還得給dftd3程序用chmod命令加上可執行權限。
啟動Multiwfn,載入輸入文件,進主功能21的子功能4,根據需要選擇恰當的選項、根據屏幕上的提示操作即可。Multiwfn的此功能計算原子間色散作用能是基于B3LYP參數的DFT-D3(BJ)色散校正得到的。
下文所有例子涉及到的輸入輸出文件都可以在http://www.shanxitv.org/attach/705/file.zip中找到。VMD用的是1.9.3版,可以在http://www.ks.uiuc.edu/Research/vmd/免費下載。
3 實例1:考察螺烯中原子貢獻的色散能和色散密度
此例考察一下6-螺烯中的哪些原子對色散作用貢獻最為顯著。啟動Multiwfn,然后輸入
examples\helicene.xyz //螺烯的結構文件
21 //能量分解分析
4 //分析原子對色散作用的貢獻
1 //計算各個原子貢獻的色散作用能
此時看到以下信息。可見當前體系色散能,也即B3LYP參數下的DFT-D3(BJ)色散校正能,為-74.468 kcal/mol,各個原子的貢獻值也給出了,加和等于總值
Total dispersion energy: -74.468 kcal/mol
Atomic contribution to dispersion energy
1(C ) -2.681 kcal/mol
2(C ) -2.072 kcal/mol
3(C ) -2.068 kcal/mol
4(C ) -2.650 kcal/mol
5(C ) -3.432 kcal/mol
...略
40(H ) -0.569 kcal/mol
41(H ) -0.511 kcal/mol
42(H ) -0.614 kcal/mol
現在Multiwfn問你是否導出atomdisp.pqr,輸入y,現在當前目錄下就有了atomdisp.pqr。其中pqr格式中專用于記錄原子電荷的那一列(倒數第三列)現在記錄的是原子對色散能的貢獻。
接下來用VMD對原子著色以直觀展示原子對色散能的貢獻。將atomdisp.pqr載入VMD,Graphics - Representation里把Drawing Method設CPK,Coloring Method設Charge,Trajectory標簽頁里把色彩刻度下限和上限分別設為-5和5。此時看到的圖如下所示,由于VMD默認用的色彩刻度是紅-白-藍,因此此圖中顏色越紅說明原子的色散能越負、與其它原子間的色散作用越強。
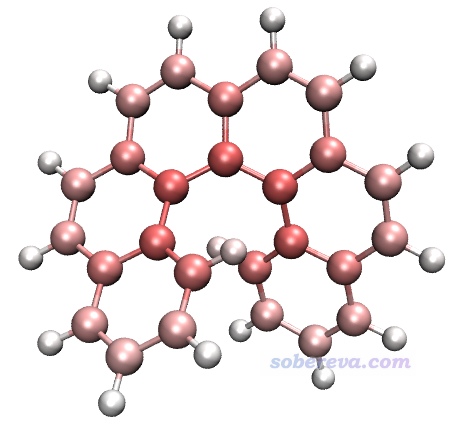
由上圖可見,碳原子對色散能的貢獻遠高于氫的,而且越靠螺烯中央的碳貢獻越大。原因容易理解,因為越靠中央的碳原子能和越多的其它原子產生色散作用。
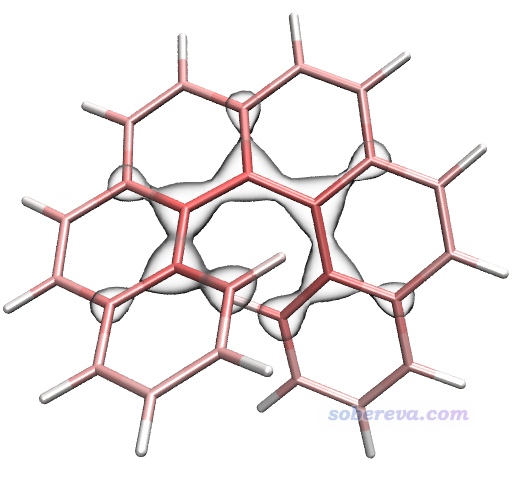
下面計算色散能密度。當前界面里選擇選項2 Calculate dispersion density for current system,然后選Medium quality grid,馬上當前目錄下就有了dispdens.cub,這是色散能密度的cub文件。將之載入VMD,Graphics - Representation界面里把Drawing Method設為Isosurface,然后Draw設為Solid Surface,Show設為Isosurface,Isovalue設為-0.15,Material設BlownGlass(并確保Display - Rendermode已經選了GLSL),再把之前的分子結構顯示方式設為Licorice,Bond Radius用0.1,此時看到下圖,圖中的等值面分布明確體現出螺烯靠內區域比其它區域對色散能有相對更顯著的貢獻。
4 實例2:計算無窮烯中的片段之間的色散作用能
Multiwfn文件包自帶的examples\NICS_scan目錄下的infinitene.pdb是無窮烯的結構文件。如下圖所示,綠色的8個碳原子和藍色的8個碳原子彼此對著,距離又較近,且接近平行,因此可以認為二者之間有pi-pi作用(可以通過《使用IRI方法圖形化考察化學體系中的化學鍵和弱相互作用》http://www.shanxitv.org/598介紹的方法可視化展現)。這里計算一下這兩個片段間的色散作用能,它和pi-pi作用強度有密切關系,因為pi-pi作用的吸引部分的本質就是色散作用。但注意不能把色散作用能直接當做pi-pi作用能,因為還同時有顯著的交換-互斥部分不可忽視。
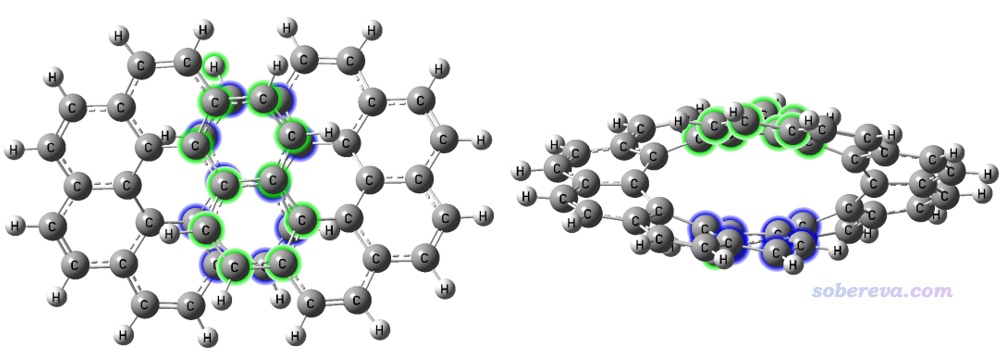
啟動Multiwfn,依次輸入
examples\NICS_scan\infinitene.pdb //無窮烯的結構文件
21 //能量分解分析
4 //分析原子對色散作用的貢獻
6 //計算兩個片段間的色散作用能
1,3,5,35-38,68-69,71 //上圖中第1個片段里的原子序號
25-26,28,30-31,42-44,46,48 //上圖中第2個片段里的原子序號
Multiwfn馬上就輸出了片段間色散作用能:
Dispersion interaction energy between the fragments: -13.000 kcal/mol
Multiwfn還可以直接給出某個片段對總色散作用能的貢獻。比如要得到上圖第一個片段的貢獻,就選擇選項5 Calculate contribution of a fragment to dispersion energy,然后從屏幕上會看到總色散能為-165.089 kcal/mol。之后再輸入1,3,5,35-38,68-69,71,就得到了這個片段的貢獻量:
Dispersion energy contributed by this fragment: -35.550 kcal/mol
這個值等于片段與片段自己,以及自己與其它部分的色散作用能之和。它占總色散能的-35.550/(-165.089)*100%=21.5%。
5 實例3:Actos不同構象下色散作用的差異
之前我在《利用Gaussian或ORCA程序把分子結構拉直的幾種方法》(http://www.shanxitv.org/594)里講了柔性的Actos分子的能量最低構象是彎曲的,但也可以加外力給它拉成直線型構象。彎曲和直線構象的xyz文件已提供在了本文文件包中的Actos目錄下,分別為curly.xyz和linear.xyz。這一節我們考察一下彎曲構象下各個原子對色散能的貢獻相對于直線型時的變化。啟動Multiwfn,然后輸入
curly.xyz //Actos彎曲構象的結構文件
21 //能量分解分析
4 //分析原子對色散作用的貢獻
3 //計算當前與另一個體系的原子對色散作用貢獻的差值
[回車] //感興趣的是當前體系(curly.xyz)中的所有原子
linear.xyz //Actos直線構象的結構文件
[回車] //感興趣的是linear.xyz中的所有原子
現在屏幕上告訴你后載入的linear.xyz的色散能為-57.553 kcal/mol,先載入的curly.xyz色散能為-69.757 kcal/mol,可見彎曲構象下色散作用更強。屏幕上還顯示了各個原子對色散能的貢獻在兩個結構下的差異。
然后輸入y,當前目錄下就產生了diffatomdisp.pqr,其中charge屬性記錄的是curly.xyz的各個原子貢獻的色散能減去linear.xyz的各個原子貢獻的色散能,而此文件里的原子坐標和curly.xyz一致。將此文件用VMD按照實例1的做法作圖,色彩刻度范圍用-1.0到1.0 kcal/mol,得到下面左圖(色彩刻度條是從Graphics - Color - Color Scale里摳出來再用powerpoint手動做的),下面右圖是根據元素名著色以便對照(黃色是硫)。
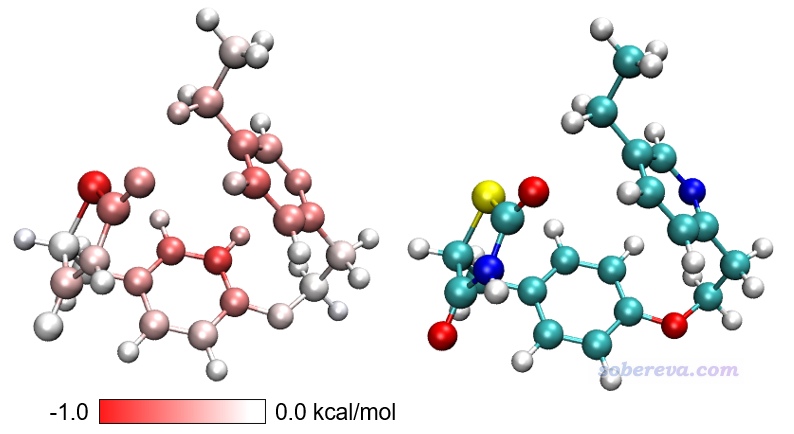
上圖越紅的原子對應從直線構象變成卷曲構象過程中對色散作用貢獻得越多的原子,可見色散作用增強主要出現在構象蜷縮后能夠與其它原子整體距離變得更近的原子上,而主要分布在邊角部分的白色原子的色散作用則沒有顯著改變。
Multiwfn也可以給出色散密度的差值格點數據并作圖。在當前界面里輸入
4 //計算當前與另一個體系的色散密度的差值
[回車] //感興趣的是當前體系(curly.xyz)中的所有原子
linear.xyz //Actos直線構象的結構文件
[回車] //感興趣的是linear.xyz中的所有原子
3 //高質量格點
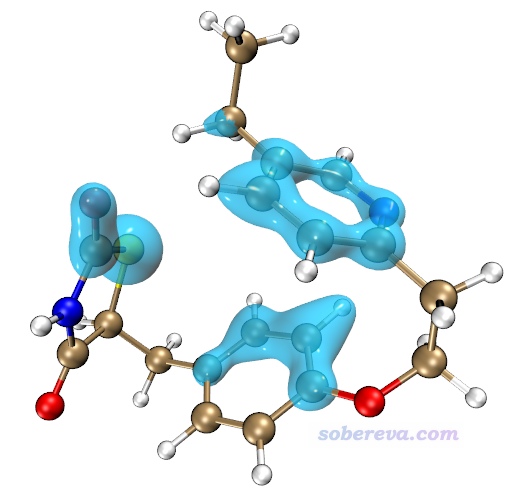
現在當前目錄下就有了dispdensdiff.cub。使用《在VMD里將cube文件瞬間繪制成效果極佳的等值面圖的方法》(http://www.shanxitv.org/483)里的腳本對其繪制±0.025的等值面,得到下圖,藍色對應負值等值面(-0.025,單位kcal/mol/Bohr^3)。可見此圖把卷曲造成色散作用顯著增強的區域展示得很清楚。
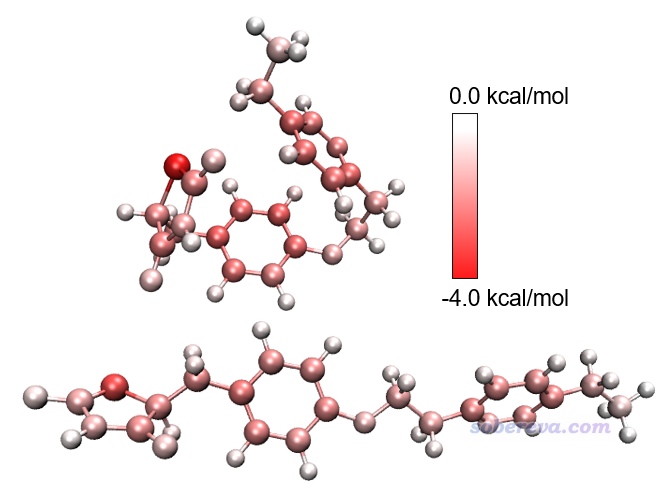
下面是對Actos兩種構象各自分別繪制的原子貢獻的色散能的著色圖。由此圖可見,如果不把兩個構象的數據求差,直接觀看各自顏色的話,并不好分辨各個原子對色散作用的貢獻在兩個構象之間有什么差異,這體現出求差的重要性。值得一提的是,兩個構象下最紅的原子都是硫原子,它比其它元素原子對色散作用的貢獻都更強,這在于它有明顯更大的動態極化率。
6 實例4:沸石吸附甲苯
這一節演示一下對周期性體系做前述分析。我在“北京科音CP2K第一性原理計算培訓班”(http://www.keinsci.com/workshop/KFP_content.html)的結構優化部分講了個例子,是沸石吸附甲苯的,這一節使用這個結構作為例子。由于甲苯的極性很低,因此沸石與甲苯間的吸引作用多半是色散作用貢獻的,因此將色散作用分解為原子的貢獻來考察可以得到有益的信息。
CP2K在PBE-D3(BJ)/DZVP-MOLOPT-SR-GTH級別下對沸石+甲苯做結構優化得到的restart文件是本文文件包里的zeolite-mol目錄下的opt-1.restart,作為Multiwfn的輸入文件就可以從中載入優化后的原子坐標和晶胞信息。只要有了晶胞信息,本文介紹的Multiwfn的功能就會自動在色散作用計算時考慮周期性。當然,用第2節提到的cif等含有晶胞和原子坐標信息的格式作為輸入文件也都是可以的。
我們先對這個體系繪制一下原子對色散能貢獻的著色圖。完全按照第3節的例子操作,只不過把輸入文件換成opt-1.restart,得到的結果如下。色彩刻度用-8到8 kcal/mol范圍。由于當前體系有晶胞信息,所以Multiwfn導出的pqr文件里也有晶胞信息,故載入到VMD后在文本窗口里輸入pbc box命令可以顯示出盒子。為了清楚起見,對應217-231號原子的甲苯部分用大圓球方式顯示,并且用了正交視角(Display - Orthographic)。
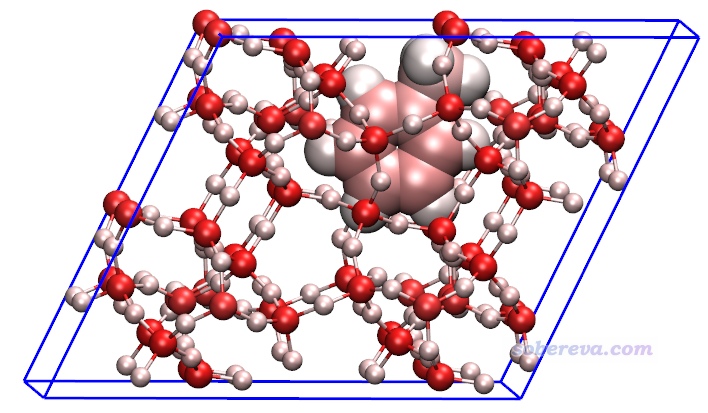
由上圖可見,對色散作用貢獻最大的是硅原子,比氧、碳都大得多,氫的貢獻最小。
上圖并不能直接展現沸石中哪些原子與甲苯的色散作用最強。想考察這個問題,需要令沸石+甲苯體系中的沸石原子(1-216號原子)對色散能的貢獻與獨立的沸石體系中的原子對色散能的貢獻求差,下面就這么做一下。值得注意的是,從上圖中可見,甲苯在當前晶胞的靠邊的位置,若把甲苯挪到晶胞中央,無疑更便于觀看,因此在考察色散作用之前先對結構做一些修改。
啟動Multiwfn,載入前述的opt-1.restart,然后輸入
300 //其它功能(Part 3)
7 //幾何操作。這個功能在《Multiwfn中非常實用的幾何操作和坐標變換功能介紹》(http://www.shanxitv.org/610)中有詳細介紹
24 //對體系進行平移以使得選擇的區域在晶胞中央
217-231 //甲苯部分的原子序號
22 //由于平移后會有原子露出晶胞,用這個選項把原子都卷進盒子以便觀察
-4 //將當前體系導出為cif文件
zeolite-mol.cif //導出的文件名。對應沸石+甲苯
17 //只保留體系的一部分
1-216 //保留沸石部分,因此甲苯部分會被刪掉
-4 //將當前體系導出為cif文件
zeolite.cif //導出的文件名。對應沸石部分
現在當前目錄下就有了zeolite-mol.cif和zeolite.cif,在本文的文件包的zeolite目錄下也提供了。下面開始做色散作用的分析。重啟Multiwfn,然后依次輸入
zeolite-mol.cif //沸石+甲苯的結構文件
21 //能量分解分析
4 //分析原子對色散作用的貢獻
3 //計算當前與另一個體系的原子對色散能貢獻的差值
1-216 //感興趣的原子是當前體系(zeolite-mol.cif)中的沸石部分的原子(前216號)
zeolite.cif //沸石的結構文件
[回車] //感興趣的原子是zeolite.cif中的所有216個原子,也正對應于zeolite-mol.cif的1-216號原子
y //導出diffatomdisp.pqr
將diffatomdisp.pqr載入VMD,用Charge屬性進行著色,色彩刻度范圍用-0.8到0.8,看到下圖。
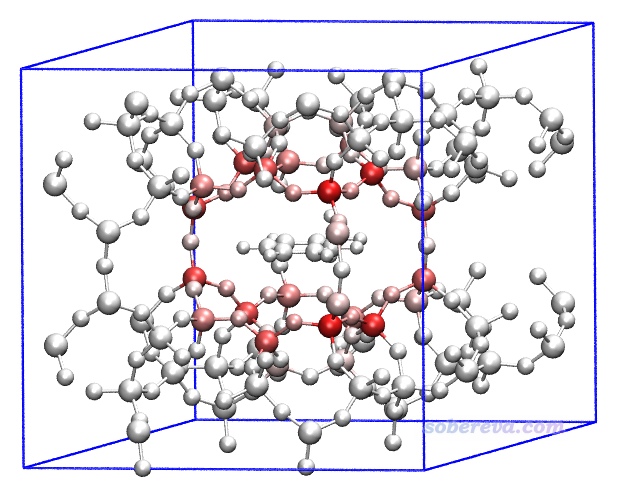
上圖中甲苯是白色,是因為它不在前面我們定義的zeolite-mol.cif的感興趣的原子范圍內,因此它的數據完全為0。距離甲苯較近的沸石原子對色散能的貢獻發生了較大變化,由于zeolite-mol.cif和zeolite.cif中沸石部分的結構是相同的,因此圖中的顏色完全體現了沸石的各個原子與甲苯之間的色散作用能。由圖可見色散作用隨距離衰減得非常快(有1/r^6衰減行為),基本上只有與甲苯最近一層的沸石原子才與它有很顯著的色散作用。值得一提的是,在《使用Multiwfn做IGMH分析非常清晰直觀地展現化學體系中的相互作用》(http://www.shanxitv.org/621)文中也有這個體系,通過IGMH方法直觀地展現了甲苯與沸石之間的相互作用,這是另外一種分析視角。
上圖還可以再改一改變成下面這樣,把與甲苯相互作用顯著的沸石的原子展現得明顯更清楚。具體來說,Graphics - Representation里建立三個Rep:一個Rep顯示甲苯,用Name著色、Licorice方式顯示、BrushedMetal材質;一個Rep顯示整個沸石,選擇語句為serial 1 to 216、Licorice顯示(Bond Radius用0.1)、Charge著色、Transparent材質;一個Rep專門顯示沸石中的原子數值小于-0.2的部分,選擇語句為charge<-0.2、CPK顯示、Charge著色。
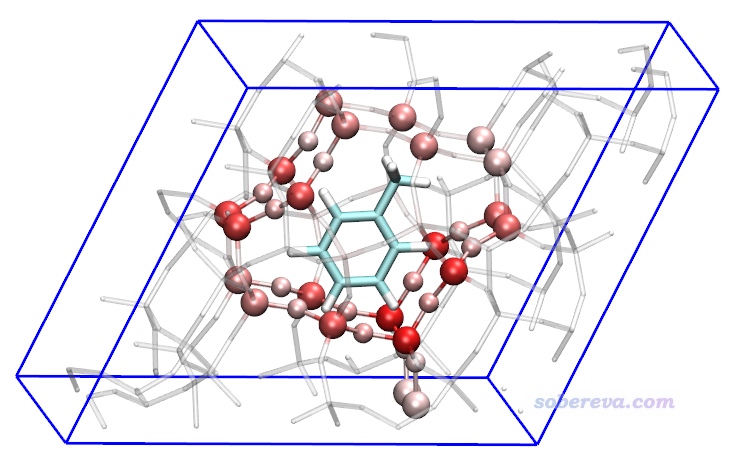
在這一節的最后強調一下,做原子的色散能貢獻或者色散密度的差值分析時,對兩個體系分別選擇的感興趣的原子范圍中的原子數必須相同,而且其中原子順序也必須相同,否則顯然原子的色散能貢獻在求差時就錯亂了。
7 總結
實際化學體系中色散作用幾乎無處不在,本文介紹的從2024-Apr-13更新的Multiwfn中引入的新功能可以非常便利、快速、直觀地展現各個原子對色散作用的貢獻,還能轉化成實空間函數色散密度通過等值面等方式表現三維空間中哪些區域對色散作用貢獻顯著,而且還能對同一個體系的不同結構之間以及兩個相關體系之間給出原子對色散能貢獻的差值,從而更清楚地了解色散效應起到的作用。顯然此功能對于與色散作用關系密切的問題的研究有重要幫助,值得在實際研究中考慮使用。值得一提的是,Multiwfn中的范德華勢分析也與色散作用的研究密切相關,非常推薦閱讀《談談范德華勢以及在Multiwfn中的計算、分析和繪制》(http://www.shanxitv.org/551)和《使用Multiwfn對靜電勢和范德華勢做拓撲分析精確得到極小點位置和數值》(http://www.shanxitv.org/645)。