利用Gaussian或ORCA程序把分子結構拉直的幾種方法
利用Gaussian或ORCA程序把分子結構拉直的幾種方法
文/Sobereva@北京科音
First release: 2021-May-4 Last update: 2021-Jun-15
1 前言
有人問怎么把一個分子從已有的較為卷曲的構象轉化成比較平直的構象。這有一些實際意義,比如通過genmixmem程序構造磷脂雙層膜、用packmol程序構造表面活性劑的囊泡,都需要用戶提供分子比較平直的構象的結構文件。一個非常簡單、直觀、效果又好的做法是使用SAMSON可視化程序里的twist工具,筆者在《生成混合組分的磷脂雙層膜結構文件的工具genmixmem》(http://www.shanxitv.org/245)里還提供了我錄的一段操作視頻。但可惜如今SAMSON程序免費版當中已經沒有twist工具了,需要買收費版才行(雖然收費版也可以申請免費試用)。還有一個辦法是在GaussView等程序里一個一個改二面角,但無疑這非常費時費力,特別是分子可旋轉的鍵很多的時候。
在此文,筆者介紹如何利用Gaussian和ORCA量子化學程序以不同方式實現把分子從卷曲的結構拉直,里面用到的做法對于讀者研究其它一些問題可能也會有啟發。本文使用的例子是《將Confab或Frog2與Molclus聯用對有機體系做構象搜索》(http://bbs.keinsci.com/thread-20063-1-1.html)中的例子,即Actos分子。通過Molclus程序(http://www.keinsci.com/research/molclus.html)搜索出的此體系的能量最低構象如下,我們后文將要把此結構拉直:
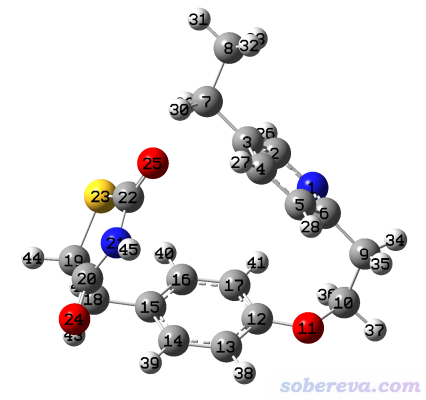
本文涉及的所有文件可以在http://www.shanxitv.org/attach/594/file.rar下載,上面的結構是文件包里的Actos.xyz。Gaussian使用的是G16 B.01版,ORCA是4.2.1版。
2 通過ORCA拉直分子:給兩個原子間加外力
筆者寫過不少與ORCA的安裝和使用有關的文章,參看http://www.shanxitv.org/category/ORCA/,相關常識性問題這里就不再說了。在ORCA中優化時可以給兩個原子在連線方向上加外力驅使它們遠離,很適合用于拉直分子。對于上面圖中展示的Actos分子,顯然我們應當把最末端的兩個重原子,即8和25號之間拉遠。為此,寫一個ORCA輸入文件:
! AM1 opt nopop
%geom
POTENTIALS
{ C 7 24 3.0 }
end
end
* xyz 0 1
[Actos.xyz里的坐標]
*
這里POTENTIALS下面花括號里的C代表constant force。注意原子序號是從0開始算的。3.0是把倆原子間拉遠的力的大小,單位是nN。數值大小不是很重要,一般個位數大小就可以。數值太小的話拉不開,而太大的話會最終把一些化學鍵過度拉長、鍵角被過度拉大(不過鍵伸縮、鍵角彎曲相對于二面角扭轉來說是很剛的,只要外力不是特別大這就不是明顯問題)。這里用的AM1半經驗方法是ORCA里能直接用的幾乎最便宜的方法(雖然ORCA也支持力場計算,但還得搞參數,這里不考慮),注意此方法不支持并行計算。如果嫌AM1太low,也可以用稍微好一點的HF-3c(Hartree-Fock結合額外校正項),對不太大的體系也非常便宜。
用ORCA運行上面的輸入文件,三分鐘就算完了,最終得到的結構如下(即file文件包里ORCA目錄下的Actos.xyz),可見完美地實現了我們的目的,分子被拉得很直
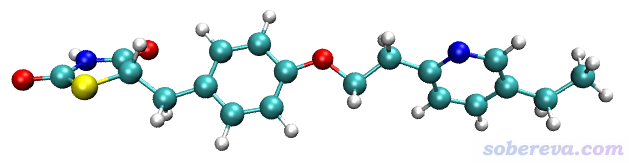
file文件包里的Actos_trj.xyz是優化軌跡,感興趣者可以將之拖到VMD程序(http://www.ks.uiuc.edu/Research/vmd/)里看看優化過程。順帶一提,考察加外力對化學體系的影響專門有人做過大量研究,參見Chem. Rev., 112, 5412 (2012)、Angew. Chem. Int. Ed., 58, 5232 (2019)。
3 通過Gaussian拉直分子
在Gaussian程序中不支持直接給原子加外力,但有其它的方法也可以實現把分子拉直,將在下面幾節介紹。
3.1 方法1:柔性掃描
Gaussian中做柔性掃描在《詳談使用Gaussian做勢能面掃描》(http://www.shanxitv.org/474)里筆者詳細介紹過。顯然,做兩端的兩個原子距離逐漸拉長的柔性掃描最終就可以得到被拉得平直的結構。寫一個這種任務的Gaussian輸入文件,如下所示
%nprocs=1
#T UFF opt(loose,nomicro,modredundant)
Generated by Multiwfn
0 1
[Actos.xyz里的坐標]
8 25 S 50 0.5
這里有幾個需要注意的地方:
(1)為了耗時盡可能低,這里使用了Gaussian支持的最便宜的方法,即分子力場。這里用的是較粗糙但支持元素最廣、用著最省事(不需要指定原子類型)的UFF力場。由于本例只需要得到一個較為平直的構象,對質量什么沒要求,所以UFF就夠了。
(2)柔性掃描每一步相當于一次限制性優化,為了節約時間,用了loose收斂限降低每一步掃描的耗時。
(3)opt里必須寫nomicro。因為分子力場的優化在Gaussian里有專用的代碼,不受modredundant設置的控制。而加了nomicro后,就會用通用的幾何優化代碼,雖然遠沒有分子力場專用的優化代碼快,但此時可以設置柔性掃描。
(4)分子力場計算時,至少對于這個體系,并行計算會令耗時不降反升,所以這里靠%nprocs=1刻意要求不用并行計算。
(5)掃描的步長太小的話把分子拉直需要的步數太多,顯然不行,而步長太大則容易導致中途報錯(此時還沒充分拉直),根據我的經驗步長設0.5埃左右比較合適。由于事先往往不好估計拉直時兩個原子相距多遠,所以可以把掃描步數設得比較大(比如本例設50步),當分子已經被拉得很直后,繼續掃下一步時程序通常會報錯,此時用GaussView載入輸出文件查看掃描軌跡,若最后一幀結構就是想要的平直的結構的話就取最后一幀結構即可。如果步數一開始設小了,導致最后沒有完全拉直也沒關系,拿最后一步的結構繼續做拉長的掃描即可。
(6)當前體系是之前我用DFT優化過的結構,已經很合理了,所以沒有明確靠geom=connectivity指定原子間連接關系,而直接讓Gaussian根據當前結構猜連接關系并用于力場計算。如果初始結構沒有優化過,最好讓GaussView保存出帶有恰當連接關系的輸入文件以免自動判斷的連接關系有誤。
使用Gaussian對上面的輸入文件進行計算,算了1分鐘左右掃描到第31步后報錯,報錯前最后一步結構如下所示,可見是我們想要的平直的結構。
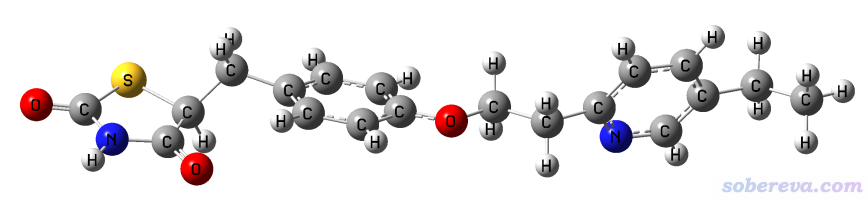
3.2 方法2:利用外電場加外力
Gaussian里可以加外電場,外電場會給帶電粒子施加額外的力,在《一篇文章深入揭示外電場對18碳環的超強調控作用》(http://www.shanxitv.org/570)里介紹的筆者的文章中有很多介紹和討論。因此,可以在UFF力場計算時,把25號原子坐標凍結住,而讓8號原子帶+1.0原子電荷,然后按照下圖所示向X軸正方向加足夠強度的電場,這樣8號原子就會受電場力往X正方向運動,從而實現拉直分子的目的。
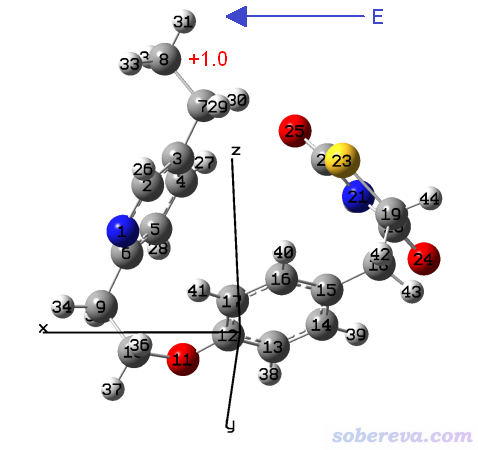
此例輸入文件如下
%nprocs=1
# UFF opt(loose,nomicro,modredundant) field=x-300 nosymm
Generated by Multiwfn
0 1
N 2.96734200 -1.21688000 2.24577900
C 2.32051000 -1.62618600 3.34057500
C 1.63462400 -2.84168400 3.45382100
C 1.64984500 -3.67567100 2.33272400
C 2.32098400 -3.26708600 1.18384000
C 2.96534100 -2.02795600 1.17397000
C 0.96378000 -3.24289700 4.74461900
C--1.0 1.95041300 -3.88295500 5.73536500
C 3.62426800 -1.48867000 -0.07067000
[略]
25 F
此例C--1.0代表對這個8號原子不手動指定原子類型(兩個橫杠之間沒寫內容),原子電荷為+1.0。沒設原子電荷的原子的原子電荷默認為0。Gaussian里電場方向和習俗相反,因此field=x后面是負號。電場強度用300(0.03 a.u.)一般就可以,如果發現最終結果怪異,如有些鍵被拉得太長,結構都扭曲了,應當適當減小電場以減小外力;如果分子拉得還不夠直,可以稍微增大電場來加大外力。nosymm必須寫,要不然結構被自動弄到標準朝向下后外電場相對于分子的方向就和期望不符了,關于nosymm更多信息見《談談Gaussian中的對稱性與nosymm關鍵詞的使用》(http://www.shanxitv.org/297)。nomicro還是要寫,要不然凍結設定不生效,其電場設置也不會對優化起作用。
用Gaussian計算上面的輸入文件,68步后收斂,耗時才十幾秒。最終結構如下,可見是我們想要的
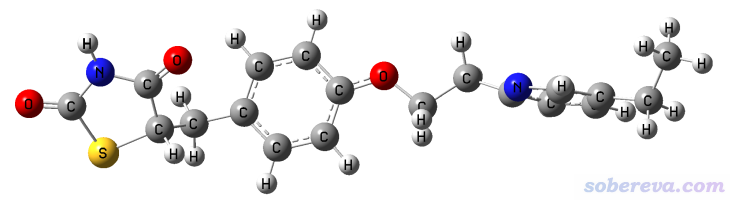
3.3 方法3:令兩端的原子彼此間受到靜電斥力
這個做法超級簡單,而且超級快速(直接用分子力場的專用優化代碼),比前述兩種方法通常更值得推薦。原理很簡單,即給兩端的原子都設數值較大的相同符號的原子電荷,二者通過強烈的靜電斥力自然就會彼此遠離,從而把分子拉直。輸入文件如下,只有8號和25號原子設了原子電荷,都設為了+10.0。設多大合適應當看實際情況,比如如果分子特別長,那么原子電荷應設大一些,要不然靜電斥力不夠強。此例輸入文件如下
%nprocs=1
# UFF opt
Generated by Multiwfn
0 1
[略]
C 0.96378000 -3.24289700 4.74461900
C--10.0 1.95041300 -3.88295500 5.73536500
C 3.62426800 -1.48867000 -0.07067000
[略]
S -2.51218200 -3.05838400 3.26014600
O--10.0 -4.37962400 -3.91727700 -0.05061200
O -1.13156500 -5.32519500 2.86775900
[略]
用Gaussian計算,僅花了一秒鐘就算完了!結果如下所示,非常平直,特別理想!
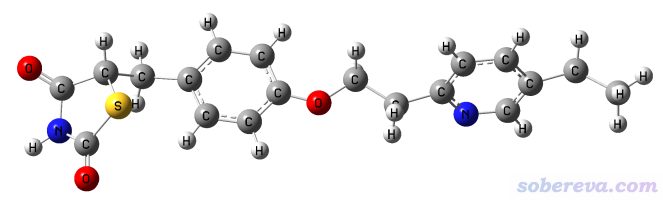
網友還給我發過一個含有200多個原子的彎曲的很長的分子,如下所示
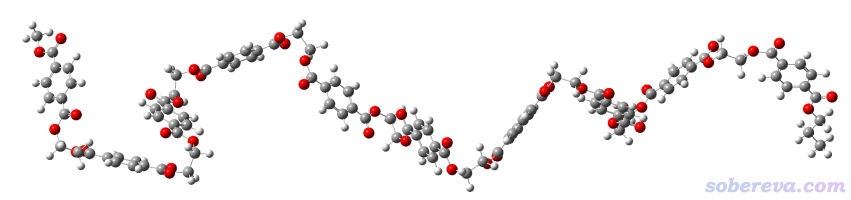
用這一節的做法也很順利地優化成了平直的構象,如下所示。計算僅僅花了5秒鐘。

4 總結
本文介紹了筆者想到的四種把分子從彎曲結構拉成平直的方法,思路各有不同。實際當中會遇上各種特殊情況,比如可能需要同時拉直兩條鏈(如磷脂的兩條尾巴),應當在理解這些方法思想的基礎上根據實際情況恰當選擇、靈活運用。
本文的做法適合一般中、小分子體系。如果是大分子體系,比如蛋白質,比較適合GROMACS等專門的基于分子力場計算的程序。在GROMACS里面可以用pull相關的選項做拉伸動力學(見比如《在Gromacs中模擬金納米線拉伸過程(含視頻)》http://www.shanxitv.org/153),也可以將體系一端的原子定義為凍結組(freezegrps)而另一端定義為受常外力的組(acc-grps,結合accelerate設置)從而拉開。
值得一提的是,本文介紹的雖然是把分子拉直的方法,但實際上也可以將這些方法應用到研究環狀、籠狀、簇狀等化學體系對外力造成的拉伸和壓縮問題的研究上。
補充:使用Avogadro程序拉直分子的做法
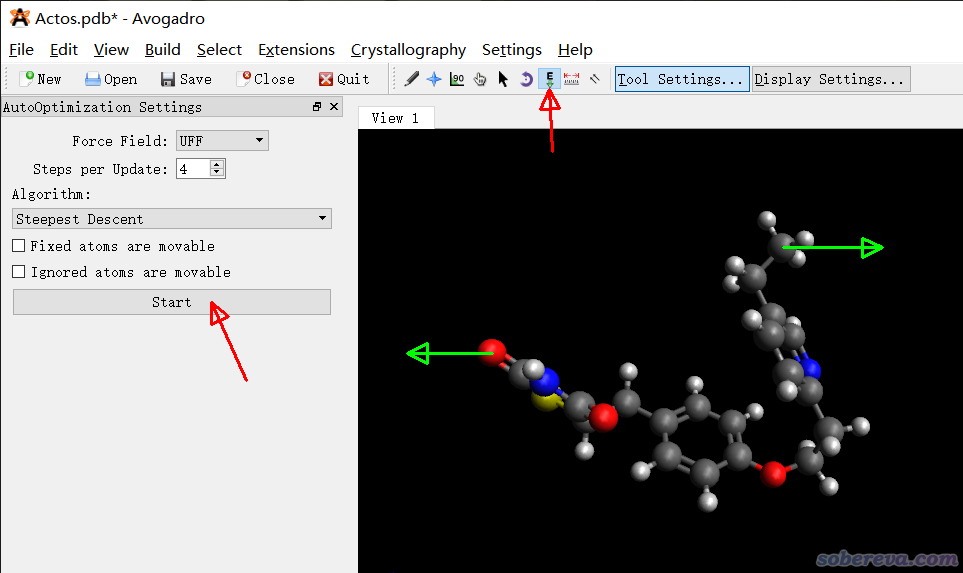
通過Avogadro可視化程序也可以拉直分子,此程序可以在http://avogadro.cc免費下載。在此程序中拉直分子比較直觀,直接按住鼠標拖拽即可,這和SAMSON可視化程序里的twist工具非常類似。啟動Avogadro后,先載入pdb之類的結構文件,點擊下圖上方箭頭所指的按鈕,然后再點擊下圖左側箭頭所指的Start按鈕,之后按住鼠標左鍵拖動特定原子即可,按照下圖的綠色箭頭分別把兩端的原子往兩頭拉就能最終拉直。
Avogadro的這個功能實際上是持續通過指定的力場優化體系幾何結構,因此當你拖動某個原子時,其它原子的位置會自發地弛豫來降低能量,因此鍵長、鍵角、二面角始終能保持比較合理的狀態。但我發現Avogadro的這個功能拉直小分子不錯,而拉直第3節最后那個特別長的聚合物則很難實現,而且特別卡頓。