Using GaussView element coloring scheme in VMD
文/Sobereva@北京科音 2022-Sep-16
GaussView的元素配色總體上來說不錯,而且對所有元素都定義了唯一的顏色,在File - Preference - Colors - Element Colors中可以看到所有元素的顏色,如下所示

VMD是免費、靈活、強大而且超級流行的化學體系可視化工具。在VMD里也可以按照元素著色,見《在VMD程序里對不同元素的原子用不同顏色顯示的方法》(http://www.shanxitv.org/624)。然而,至少對于筆者撰文時的最新的正式版1.9.3版來說,VMD自帶的元素著色定義很少,對絕大多數元素都是統一用的褐色,導致很多情況下沒法區分不同元素,而且顏色也不美觀。這里介紹在VMD里使用GaussView里元素顏色定義的做法。
從http://www.shanxitv.org/attach/652/gview_color.tcl下載gview_color.tcl文件,將之放到VMD目錄下(即VMD啟動后在文本窗口里輸入pwd命令后顯示的目錄),然后在VMD的vmd.rc文件末尾加入一行proc gview {} {source gview_color.tcl}。如果你不了解vmd.rc文件的話,看《VMD初始化文件(vmd.rc)我的推薦設置》(http://www.shanxitv.org/545)。
啟動VMD后,在文本窗口里輸入gview,就可以把默認的元素著色方案替換成和GaussView相同的了。之后載入個含有元素信息的結構文件(比如pdb、xyz等),然后在Graphics - Representation里把Coloring Method設為Element即可看到按元素著色的效果。如果想和GaussView顯示的圖像特征盡可能接近,將Drawing Method設為CPK,Bond Radius設為0.2,Material設為AOShiny。對順鉑體系顯示效果如下,左邊是VMD的,右邊是GaussView的
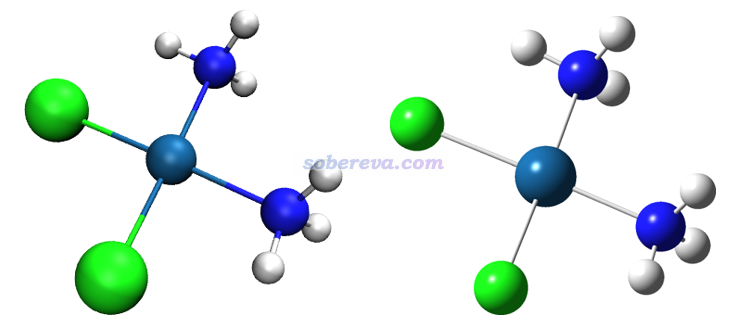
上圖還可以看到在鍵的著色方面有差異,VMD的鍵的兩邊的顏色分別對應兩個原子的,而GaussView都是白色的。如果想讓VMD在這點和GaussView統一,可以把當前的Representation里的Bond Radius設為0使得鍵不顯示,然后再建立一個Representation,用CPK,把Sphere Scale設0,Bond Radius設0.15,Coloring Method選ColorID并指定為白色,之后效果如下所示,可見和GaussView顯示的很接近了
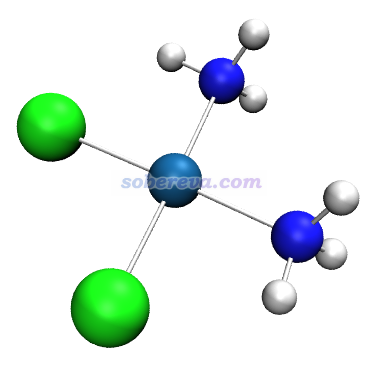
再把原子半徑、光源方向、材質微調一下,就更像GaussView的效果了。不過,GaussView里對多重鍵的顯示效果是VMD里怎么設都模仿不來的。
]]>The way to display atoms of different elements in different colors in VMD program
文/Sobereva@北京科音
First release: 2021-Nov-11 Last update: 2022-Sep-16
1 前言
之前筆者寫了大量的將Multiwfn與VMD相結合的文章,比如《使用IRI方法圖形化考察化學體系中的化學鍵和弱相互作用》(http://www.shanxitv.org/598)、《使用Multiwfn+VMD快速地繪制靜電勢著色的分子范德華表面圖和分子間穿透圖》(http://www.shanxitv.org/443)、《用VMD繪制藝術級軌道等值面圖的方法》(http://www.shanxitv.org/449)等等,并且做法已被大量同行所使用,令VMD被量子化學的研究者們用得也越來普遍,并有越來越多的人在思想家公社QQ群和計算化學公社論壇里問我VMD的使用問題。其中有一個問題被問得越來越頻繁,已經成了半周經問題,也就是怎么對不同元素的原子用不同顏色顯示。為了免得老得重復回答,特此寫一個小文章專門說一下這事。本文的情況是針對VMD 1.9.3版而言的,其它版本的情況可能相同也可能不同。
筆者還有一篇文章介紹了怎么讓VMD對元素的著色和另一個流行的可視化程序GaussView相同,建議閱讀,見《在VMD中使用GaussView的元素著色的方法》(http://www.shanxitv.org/652)。
2 name和element屬性
VMD里每個原子都有各種屬性。name和element在VMD里是每個原子的兩種不同的屬性,name是原子的名字,element是元素周期表里的元素符號(第一個字母大寫,第二個小寫)。在VMD的文本窗口中,輸入[atomselect top all] get name命令可以列出當前體系里所有原子的name,輸入[atomselect top all] get element命令可以列出當前體系里所有原子的element,都是按原子序號順序輸出。
3 不同格式的文件記錄的信息
不同輸入文件提供給VMD的信息是不一樣的,這里對幾種典型的文件來說一下。
? xyz文件:介紹見《談談記錄化學體系結構的xyz文件》(http://www.shanxitv.org/477)。在標準的xyz文件中,第一列是每個原子的元素符號。將xyz文件載入VMD后,name和element屬性值都是元素符號。
注意有些xyz文件不標準,里面記錄的是原子名而非元素符號,比如VMD載入gro文件后直接保存出來的xyz文件里就是原子名。這樣的xyz文件載入VMD后name屬性值是文件里記錄的原子名,element會根據原子名里面的字母部分去猜。比如原子名是HG3的話會被認作汞,因此element被設為Hg。如果猜不出來,比如原子名是CG,則element屬性將為X。注意VMD這么猜元素很容易猜錯,比如alpha碳在氨基酸里的標準原子名是CA,如果xyz文件里記錄的是原子名,VMD就會把它視為是鈣,從而令element屬性值為Ca。
? gro文件:即GROMACS的結構文件。里面對每個原子記錄的是原子名,比如氨基酸里面的碳可以有CA、CB、CG、CG1等等原子名。載入VMD后,name就是這些原子名,而element皆為X。
? mol2文件:同上。
? pdb文件:在標準的pdb文件中,第三列是原子名,最后一列是元素符號。因此,pdb文件載入VMD后可以給VMD分別提供name和element信息,因此element不用根據原子名猜了。但是有些pdb文件不規矩,沒有記錄元素符號的最后一列,這樣的pdb文件載入后element就都為X。
? cub文件:cub文件經常通過VMD程序繪制成等值面圖,見比如《在VMD里將cube文件瞬間繪制成效果極佳的等值面圖的方法》(http://www.shanxitv.org/483)。如《Gaussian型cube文件簡介及讀、寫方法和簡單應用》(http://www.shanxitv.org/125)所述,此文件里沒記錄原子名,但記錄了各個原子的元素在周期表里的序號。此文件載入VMD后,name和element屬性都為元素符號。
還值得一提的是VMD里每個原子還有個type屬性,記錄的是原子類型,可以用[atomselect top all] get type命令查詢。諸如amber的拓撲文件prmtop里記錄了原子類型,mol2格式也記錄了原子類型(但很多程序輸出的mol2文件里原子類型和元素符號相同),因此載入后type屬性就是文件里記錄的原子類型。而xyz、gro、pdb格式里沒記錄原子類型,載入后type屬性值會和name相同。cub格式里也沒記錄原子類型,載入后type、name和element都相同。
4 對原子的著色設置
這里以氯仿為例進行說明,此分子的pdb文件可以在http://www.shanxitv.org/attach/624/CHCl3.pdb下載。
將它載入VMD后,在Graphics - Representation里把Drawing method改為CPK,目前看到的是下圖
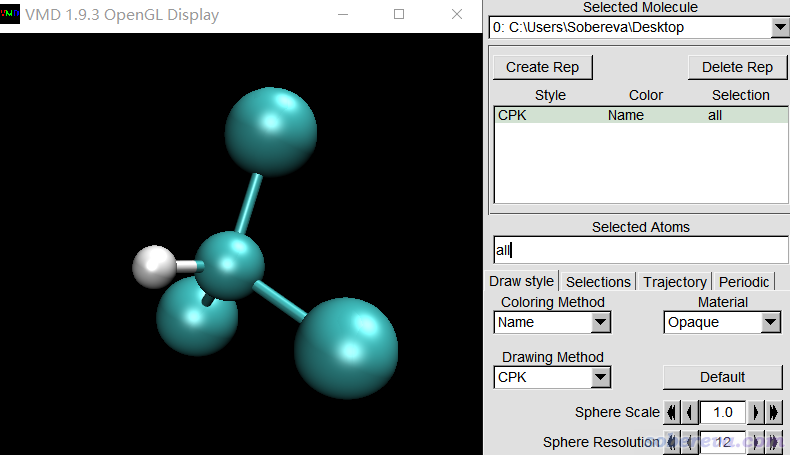
可見,碳和氯的顏色完全一樣,都是青色。為什么沒有區分開?原因很簡單,如上圖所見,Coloring method目前是默認的Name,即根據原子的name屬性決定顏色。我們來看一下當前著色用的顏色定義。進入Graphics - Colors,在Categories里選Name,點擊C,然后看到下圖
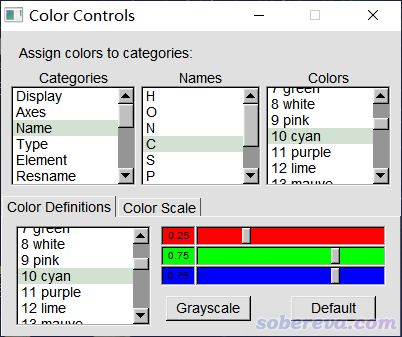
如圖可見,當前根據name進行著色時并不區分C和Cl,只要首字母是C就都按用cyan顏色著色。
如果想按照元素來著色,就在Graphics - Representation里把Coloring method改為Element,此時圖像如下,可見氯和碳的顏色區分開了
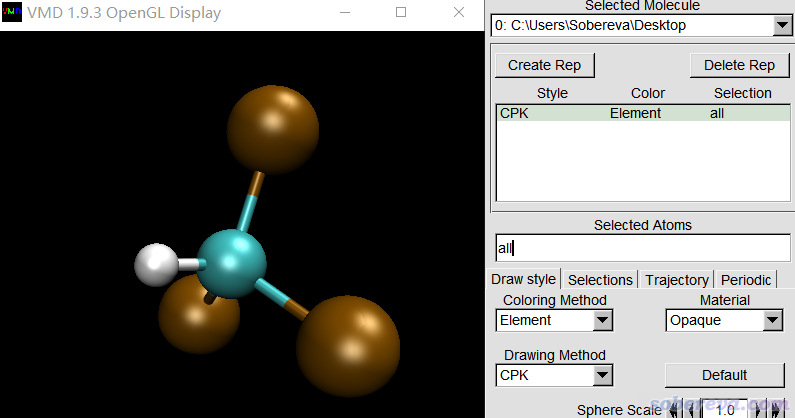
氯用棕色明顯不好看。我們要把它改為常用的綠色,就進入Graphics - Colors,在Categories里選Element,點擊Cl,再選green,此時如下圖所示
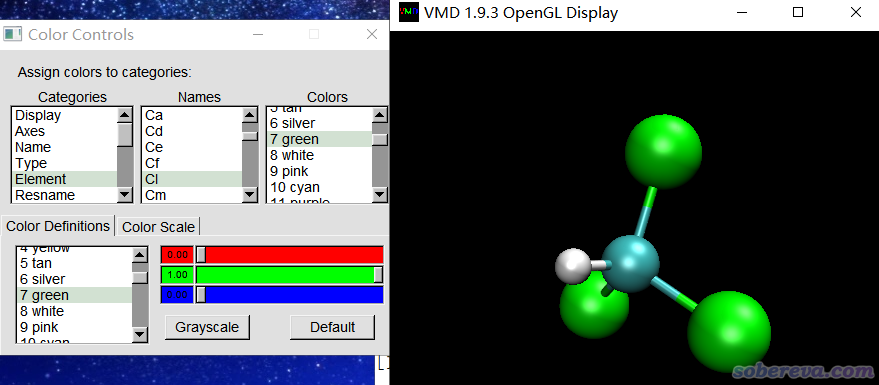
如果想微調green顏色的具體色彩定義,可以在上圖的Color Definitions標簽頁下方修改紅、綠、藍三種顏色分量的大小。
記住,只有載入的是pdb、xyz這樣能直接給VMD提供元素信息的輸入文件,才能像上面這樣按照element屬性來著色。而用比如gro文件的時候,由于不能提供元素信息,就只能按照name來著色了。如果你是GROMACS用戶又想按元素著色,就得把gro轉成比如pdb格式,按照pdb格式規范在合適的列上補上元素信息后再載入;或者用比如gmx editconf -f md.tpr -o md.pdb把tpr文件轉成pdb文件,由于tpr文件里本身有元素信息,所以這樣得到的pdb文件里最后一列直接記錄了元素信息(順帶一提,之后你可以刪除當前僅有的一幀,然后再往這個體系中載入gro文件或軌跡文件,此時載入的只有坐標,而元素信息還是載入pdb時提供給VMD的)。
5 設置默認著色方式
如果你想將element作為默認著色方式,并且Cl元素默認用綠色,免得每次在VMD里還得如上操作一遍,可以在vmd.rc文件末尾加入以下兩行
mol default color Element
color Element Cl green
每次啟動VMD后就會自動執行這兩條命令修改默認設置。如果不了解vmd.rc文件的話,看《VMD初始化文件(vmd.rc)我的推薦設置》(http://www.shanxitv.org/545)。
另外,如果你想默認用CPK方式顯示體系結構,在vmd.rc末尾還可以再加上mol default style CPK。
還值得一提的是VMD有個colordefs.dat文件,記錄了默認的著色用的規則。對于Windows版,此文件在VMD目錄下的scripts\vmd目錄下;對于Linux版,如果安裝到了默認目錄,此文件在/usr/local/lib/vmd/scripts/vmd目錄下。用文本編輯器打開此文件可以看到
Name H white
Name O red
Name N blue
Name C cyan
Name S yellow
Name P tan
Name Z silver
...略
Element C cyan
Element Ca ochre
Element Cd ochre
Element Ce ochre
Element Cf ochre
Element Cl ochre
Element Cm ochre
...略
你可以直接在colordefs.dat里修改按照name著色、按照element著色時默認的色彩,比如可以把Element Cl ochre改為Element Cl green使得程序按照element著色時對Cl元素默認用綠色。這里設置的優先級低于自己在vmd.rc末尾添加諸如color Element Cl green這樣的設置命令。
]]>On the image clarity issues in graphing related to computational chemistry
文/Sobereva@北京科音 2021-Jul-16
0 前言
寡人在網上答疑的時候,總是看到有人問“得到的圖像不清晰怎么辦”、“怎么讓圖像更清晰”這種問題,語義不明,也不知道對方說的“清晰”到底指什么,對方也不說他覺得不清晰是怎么個不清晰法,也不給出個截圖來說明,令人感覺很莫名其妙。筆者忍不住寫個小文,把各種初學者所謂的“不清晰”的可能情況都羅列一下,便于他們自己對照判斷如何解決。此文也有助于一些缺乏相關常識的計算化學工作者了解怎么改進圖像質量。此文內容很多地方用流行的可視化程序VMD(http://www.ks.uiuc.edu/Research/vmd/)來舉例說明,這里是針對VMD 1.9.3版而言的。
1 圖片分辨率太低
關于圖片分辨率的事我在《談談怎么正確認識論文投稿時對圖像分辨率的要求》(http://www.shanxitv.org/511)里專門做過科普。有些人所說的不清晰可能是指圖片的分辨率(當前語境下等于圖片的像素)太低。顯然,讓產生圖片的程序輸出更高分辨率的圖像就完了。比如:
(1)Multiwfn程序(http://www.shanxitv.org/multiwfn)保存一維、二維、三維圖像的分辨率分別通過settings.ini文件里的graph1Dsize、graph2Dsize、graph3Dsize參數來設置。
(2)GaussView通過File - Save Image方式保存圖片的像素由界面上的Enlarge Width and Height by控制。比如如果這里設的是3x,而窗口的大小是400*300,那么保存出的圖像就是1200*900像素。
(3)VMD可以通過File - Render方式保存圖片,如果渲染器選的是Snapshot或者Tachyon (internal, in-memory rendering),那么渲染得到的圖片和OpenGL窗口的大小是相同的。如果想要更高分辨率的圖,要么把窗口拉大再渲染,要么在用第三方渲染器POV-Ray或VMD自帶的獨立的Tachyon渲染器渲染的時候直接指定渲染出的圖片像素。用Tachyon渲染器的時候如何通過命令行指定渲染出的圖片分辨率在《用Multiwfn+VMD做RDG分析時的一些要點和常見問題》(http://www.shanxitv.org/291)里我專門說過,這里不再累述。
有些可視化程序沒有單獨的保存圖片的功能,你只能靠截屏來得到圖片文件。此時你若要想得到高像素的圖像,就應當把窗口最大化,并且把窗口里的體系也放大到盡可能大,然后再截圖。如果還嫌圖片像素不夠高,去找個高分辨率屏幕的機子,在那上面截圖。
2 過度有損壓縮圖像
十分常用的jpg是一種有損壓縮的圖像格式。Photoshop、IrfanView等像樣的圖片編輯或者圖片觀看程序在保存圖像成jpg格式時,都可以選擇jpg文件的質量;設得越高,則圖像質量相對于原圖降低得越少,但圖像文件就越大。顯然,如果保存時圖片質量設得太低,必然得到的圖像會比較模糊,并且還可能伴有色彩失真。
對于科研方面的圖,特別是可能用到文章里的圖,我平時都存成png格式,這是非常好的無損壓縮格式,不會像jpg格式那樣降低圖片質量(代價是尺寸更大),又比bmp那種未經壓縮的無損格式小得多得多,也比常見的tif那種無損壓縮格式尺寸更小。
3 圖像被拉大了
有些人在保存/生成圖片的時候圖像的像素并不是很高,然而他把圖插入到文章里之后,還手動把圖的尺寸給拉大了,這時候圖片能不模糊么?只有ps、eps、wmf等格式儲存的矢量圖才能無損縮放,而png、bmp、tif、jpg等位圖格式只要放大了圖片注定會看起來模糊,這屬于圖像處理的最基本常識。
4 一些程序自動壓縮了圖片
有些人問,怎么圖片原本看起來是清晰的,插入XXX程序之后就不清晰了。這看具體情況。比如powerpoint(筆者這里用的是2016),在“選項”-“高級”的“圖像大小和質量”一欄中,有個選項是“不壓縮文件中的圖像”,這個選項默認是沒有選中的。這種情況下,在保存pptx文件時,對于ppi(和《談談怎么正確認識論文投稿時對圖像分辨率的要求》http://www.shanxitv.org/511里說的dpi本質上相同)過高的圖像,其像素就會自動壓縮到特定的ppi。如果你把一個圖片插入到powerpoint里,并且把圖片尺寸縮小,然后保存pptx文件,此時由于圖片的ppi往往會高于閾值,遂被powerpoint自動壓縮,即當前pptx里的這個圖片的像素已經低于原圖了。之后重新打開這個pptx文件,若再把這張圖拉回原先的大小,就會看到圖片已經模糊了。只有把“不壓縮文件中的圖像”選上才可以避免powerpoint自動壓縮圖像。
還有其它的程序可能也有根據ppi自動壓縮圖片的設置,應仔細去選項菜單里仔細看看,弄不明白的話可以Google搜搜看有無別人遇到過并提供了解決方案。
5 直接截圖,而不是保存圖像
有些可視化程序為了顯示速度比較快,直接在屏幕上顯示的時候用略低的圖像質量,而在保存圖片的時候才自動設高圖像質量。因此,可視化程序凡是有專門的保存圖片的功能的話就不要直接截圖。
對于Multiwfn顯示的三維分子結構圖、等值面圖,特別是使用mesh或者solid face+mesh風格顯示等值面的時候,尤其建議保存成高像素的圖片文件,然后再縮成想要的尺寸,這比起直接在三維窗口里看到的圖像質量好太多了,還順帶起到抗鋸齒效果(見后文)。
6 保存圖像的格式不合適
要分清楚矢量圖和位圖的區別。如果你的圖片沒有平滑的色彩過渡,而是以線條、文字和離散的顏色構成的,那么保存成矢量圖比較好,可以無損縮放,而且線條看起來很平滑。比如在Multiwfn里按照《使用Multiwfn繪制紅外、拉曼、UV-Vis、ECD、VCD和ROA光譜圖》(http://www.shanxitv.org/224)和《使用Multiwfn繪制NMR譜》
(http://www.shanxitv.org/565)繪制光譜圖、按照Multiwfn手冊4.4節的例子繪制等值線圖,我都建議以矢量圖形式保存為pdf格式的文件。如果圖片有連續的色彩過渡,比如Multiwfn、VMD、ChimeraX等程序顯示的等值面圖,以及Multiwfn主功能4繪制的填色圖,則建議用png等位圖格式。
如果違背上面的建議,把線條、文字和離散的顏色構成的圖保存成png等格式,線條看起來可能就不如保存成矢量格式時看著那么平滑。如果把色彩連續變化的圖保存成矢量圖,圖片看起來就會有很難看的色階。
7 被投稿系統降低了圖像質量
在大多數期刊投稿時,在最后確認提交之前,一般投稿系統會生成一個用于預覽的pdf文件。轉換成pdf文件的時候會對圖片自動進行處理,例如把過高分辨率的圖像自動降低分辨率、適當對圖片進行有損壓縮從而避免pdf尺寸太大,這可能導致在預覽pdf文件時發現圖片清晰度變低了,有點糊了。這一般沒辦法避免,也不用太在意,一般也不至于影響審稿。這時候的圖片清晰度和最終出版時候的圖片清晰度沒直接關系。
8 沒用抗鋸齒
顯示3D物體時,如果沒在可視化程序里開啟抗鋸齒,而且顯示器的dpi又不是太高,就會明顯看到3D物體邊緣的線條毛毛糙糙的,可能有的人說的不清晰是指這個。很多可視化程序里都有抗鋸齒(antialiasing)選項,打開之后就能讓邊緣鋸齒感減輕很多,變得平滑不少。有些程序里抗鋸齒有不同級別,越高級別抗鋸齒效果越好,但代價就是對GPU性能要求越高。有些可視化程序里可能沒有抗鋸齒選項,但也沒關系,你可以進入顯卡驅動,往往在里面可以設置強行對某個程序用某種抗鋸齒設定。比如VMD的抗鋸齒選項(Display - Antialiasing)往往沒法直接選,因此你可以在顯卡驅動里直接設,具體怎么操作和你的顯卡的顯示芯片制造商以及驅動版本有關,自行摸索或Google。如果你的顯卡驅動是Windows自帶的,那么是沒有顯卡驅動面板的,應當自行去顯示芯片的廠家官網上下載并裝上。
還有一種人工對圖片實現抗鋸齒的做法是保存很高像素的圖像,然后用Photoshop、IrfanView等程序把圖片縮小(降低像素),此時程序會做重新采樣,可以等效實現抗鋸齒效果。
9 沒有關閉霧化效果
很多程序默認都開啟了霧化效果來體現物體距離屏幕的位置。比如在VMD里,默認是黑背景,并且開著霧化效果(即Display - Depth cueing是打開的)。此時距離屏幕越遠的物體就顯得越黑。但由于VMD對霧化的默認衰減設置不太理想,所以即便是距離屏幕較近的物體看起來也是霧蒙蒙的,在《用Multiwfn+VMD做RDG分析時的一些要點和常見問題》(http://www.shanxitv.org/291)的一開始就有個示意圖。有些初學者由于缺乏常識,就把霧化效果理解成了不清晰。我建議用VMD的人要么關閉Depth cueing,要么在Display - Display Settings里恰當設置Cue Mode,使霧化效果至少不會影響到距離屏幕較近的物體,而只對中、遠距離的物體才生效。
10 繪制等值面圖時格點間距太大
不管什么可視化程序,在繪制某個函數的等值面圖之前都需要獲得這個函數在特定一塊三維空間中均勻分布的各個格點上的函數值,這叫格點數據。格點間距越大,得到的等值面就越粗糙、越有棱有角,格點間距很大時甚至有的地方的等值面可能還不連續,也許有些人說的不清晰就是指這種情況。格點間距如何影響RDG函數的等值面質量在《用Multiwfn+VMD做RDG分析時的一些要點和常見問題》(http://www.shanxitv.org/291)的2.1節做了展現。顯然,等值面效果不好的問題只要把格點間距設小一些即可緩解,在諸如Multiwfn程序里提供了非常豐富靈活的方式設定格點。但無疑減小格點間距的代價是計算的總格點數變多了,耗時增高了。
11 光源設置問題
顯示3D物體時,如果光源設置不當,可能導致有些地方太暗,令有的人覺得不清晰。解決方法就是調節光源設置。比如在VMD 1.9.3里有4個光源,默認開啟前兩個,可以在Display里選擇相應的光源來切換打開或關閉的狀態。另外,在Mouse - Move Light里選擇一個光源后,可以在圖形窗口里拖動鼠標來調節光源位置。而在Multiwfn里,可以在窗口上方的菜單里選擇Other settings - Set lightings,然后選擇哪些光源打開或關閉。
12 顯示風格設得不合理
大多數可視化程序里都可以設置不同的顯示效果,如果設置不當可能會造成看起來不清晰。比如VMD 1.9.3程序默認用的是line方式顯示,而且粗細只有1.0,因此直接顯示出的體系看起來太細,如果是沒有成鍵的單個原子,圖形窗口里會顯示成只有一個像素大小的點,視力不好的同志不僅根本看不清楚,還甚至會以為是VMD顯示有問題,漏掉了原子,筆者在網上答疑時就見過這樣的人。顯然把顯示方式改成諸如CPK、Licorice之類的就能看清楚了。筆者在《VMD初始化文件(vmd.rc)我的推薦設置》(http://www.shanxitv.org/545)中推薦的默認設置中把默認的line風格的線條粗細設大到了2.0也是為了讓默認的顯示效果看著更清楚。
]]>A simple example of performing ab initio dynamics (AIMD) using ORCA
Sobereva@北京科音
First release: 2020-Dec-13 Last update: 2025-May-9
1 前言
基于量子化學方法的動力學一般稱為從頭算動力學(Ab initio molecular dynamics, AIMD),相比于基于一般的經典力場的動力學,其關鍵優勢在于精度高、普適性強、能夠描述化學反應,代價是耗時相差N個數量級。ORCA量子化學程序有不錯的做BOMD形式的從頭算動力學的功能,使用很方便,而且本身ORCA做DFT的效率又高,是做孤立體系AIMD的首選程序之一。雖然有些特性不支持,比如沒法像Gaussian的BOMD那樣直接做準經典動力學,不能根據原子距離等標準判斷什么時候自動結束任務等等,但都不是大問題。對于跑跑普通的AIMD來說,筆者感覺ORCA明顯比Gaussian的ADMP或BOMD更好用(Gaussian的AIMD輸入文件較為抽象,手冊相應部分寫得很爛,而且連個像樣的熱浴都沒有),而且速度也明顯更快。
筆者發表的18碳環(cyclo[18]carbon)的研究論文中,其單體和二聚體做的分子動力學就是用ORCA跑的,分別見《揭示各種新奇的碳環體系的振動特征》(http://www.shanxitv.org/578)對中Chem. Asian J., 16, 56 (2021)和《全面探究18碳環獨特的分子間相互作用與pi-pi堆積特征》(http://www.shanxitv.org/572)中對Carbon, 171, 514 (2021)一文的介紹。在《18碳環等電子體B6N6C6獨特的芳香性:揭示碳原子橋聯硼-氮對電子離域的關鍵影響》(http://www.shanxitv.org/696)中介紹的筆者的Inorg. Chem., 62, 19986 (2023)文章中還用ORCA跑了B6C6N6的高溫動力學以證明其穩定性。在《18個氮原子組成的環狀分子長什么樣?一篇文章全面揭示18氮環的特征!》(http://www.shanxitv.org/696)中介紹的筆者的ChemPhysChem, 25, e202400377 (2024)文章中還用ORCA跑了18氮環的動力學以考察其熱分解行為。這些文章可以作為ORCA跑AIMD的典型范例,很推薦閱讀和引用。
筆者在北京科音高級量子化學培訓班(http://www.keinsci.com/workshop/KAQC_content.html)中會用多達兩百多頁的ppt專門深入詳細講AIMD的模擬,其中也包括ORCA做AIMD的各種細節、大量技巧以及諸多實例,并且此培訓中還會系統講授ORCA程序的使用,因此是使用ORCA專門做AIMD的讀者一定不可錯過的培訓。而本文只是舉一個簡單的例子,幫助讀者快速了解ORCA如何做最簡單的AIMD。注意ORCA只適合跑孤立體系的從頭算動力學,如果是做周期性體系的從頭算動力學,CP2K是最佳的選擇,在筆者講授的北京科音CP2K第一性原理計算培訓班(http://www.keinsci.com/workshop/KFP_content.html)中有極為全面、系統、詳細的講解并給了大量例子。
本文內容適用于Multiwfn最新版本、VMD 1.9.3、ORCA 4.2.x及以后版本的情況。
2021-Jul-7 針對ORCA 5.0的補充說明:對于2021-Jul-7及以后更新的Multiwfn,進入本文所述的Multiwfn產生輸入文件的界面后,可以通過選項-11選擇適合的ORCA版本,默認為ORCA 5.0及以后版本,而下文內容對應的是4.2.x版的情況。對于>=5.0版,Multiwfn自動設的熱浴是CSVR,比之前版本唯一支持的Berendsen熱浴更好,而且同樣普適。而且在run后面多加了CenterCOM,這是從5.0版本開始支持的消除整體質心運動的選項,而下文里提到的constraint add center這一行就沒有了。
2 實例:[Al(H2O)6]3+與NH3之間的質子轉移
ORCA的MD模塊的開發者網站上有個真空中[Al(H2O)6]3+與NH3之間的質子轉移的動畫,如下所示
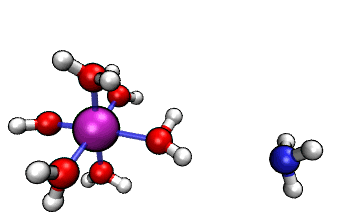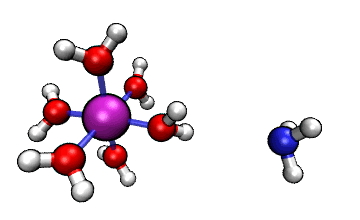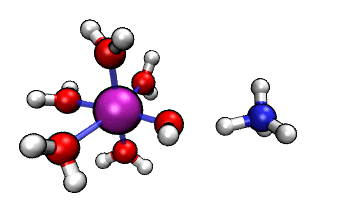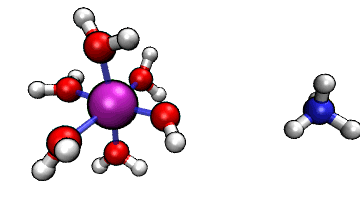
可見NH3向帶正電的[Al(H2O)6]3+逐漸靠近,水上的一個質子轉移到了氨氣分子上,然后由于靜電互斥,NH4+就逐漸遠離[Al(H2O)5(OH)]2+了。這里我們試圖重現這個過程。下面提到的文件都可以在http://www.shanxitv.org/attach/576/file.zip中獲得。
我們首先獲得[Al(H2O)6]3+的基本合理的結構。當然用ORCA優化也可以,這里筆者習慣性地用Gaussian來優化。在GaussView里搭建Al(H2O)6,保存為Al(H2O)6_optfreq.gjf,將關鍵詞改為# B3LYP/6-31G* opt freq,將電荷改為3,然后用Gaussian運行之,就得到了優化后的[Al(H2O)6]3+結構。再用GaussView打開輸出文件Al(H2O)6_optfreq.out,把一個氨氣分子畫在一個水的旁邊,如下所示,然后保存為complex.gjf。
Multiwfn程序(http://www.shanxitv.org/multiwfn)有很便利的生成ORCA常見任務的輸入文件的功能,見《詳談Multiwfn產生ORCA量子化學程序的輸入文件的功能》(http://www.shanxitv.org/490),這里我們用Multiwfn生成ORCA的AIMD任務的標準輸入文件。
啟動Multiwfn,載入complex.gjf,然后輸入
oi // 生成ORCA輸入文件
0 // 選擇任務類型
6 // 分子動力學
1 // 計算級別用B97-3c
在當前目錄下就得到了ORCA的AIMD任務的標準輸入文件complex.inp。
在complex.inp里面將相應幾行改成下面這樣:
%maxcore 10000
%pal nprocs 10 end
timestep 1.0_fs
initvel 298.15_K
* xyz 3 1
現在的complex.inp的完整內容如下。以#為開頭的行代表后面的設置被注釋掉了,不會生效,想啟用可以去掉#。此文件里也有大量Multiwfn自動添加的注釋,只要一看注釋馬上就明白相應的行是什么含義,巨貼心,都省得查手冊了。
! B97-3c noautostart miniprint nopop
%maxcore 10000
%pal nprocs 10 end
%md
#restart ifexists # Continue MD by reading [basename].mdrestart if it exists. In this case "initvel" should be commented
#minimize # Do minimization prior to MD simulation
timestep 1.0_fs # This stepsize is safe at several hundreds of Kelvin
initvel 298.15_K no_overwrite # Assign velocity according to temperature for atoms whose velocities are not available
thermostat berendsen 298.15_K timecon 30.0_fs # Target temperature and coupling time constant
dump position stride 1 format xyz filename "pos.xyz" # Dump position every "stride" steps
#dump force stride 1 format xyz filename "force.xyz" # Dump force every "stride" steps
#dump velocity stride 1 format xyz filename "vel.xyz" # Dump velocity every "stride" steps
#dump gbw stride 20 filename "wfn" # Dump wavefunction to "wfn[step].gbw" files every "stride" steps
constraint add center 0..22 # Fix center of mass at initial position
run 2000 # Number of MD steps
end
* xyz 3 1
[坐標部分]
*
下面簡單說一下complex.inp里這些設置的含義、為什么這么設。
? B97-3c是一個又便宜又不錯的計算級別,在ORCA里還自動會啟用RIJ加速,速度很快,因此很適合跑AIMD,描述當前體系沒有問題。B97-3c的介紹見《詳談Multiwfn產生ORCA量子化學程序的輸入文件的功能》(http://www.shanxitv.org/490)的相應部分。但B97-3c也不是什么時候都能用,比如18碳環用純泛函描述皆失敗,見筆者在Carbon, 165, 468 (2020) 里的討論,顯然就不能用這個了,筆者跑涉及18碳環的AIMD的時候都用的是ωB97X-D3。
? %pal nprocs 10 end代表用10核。筆者當前任務實際上是在雙路E5-2696v3共36個物理核心的機子上跑的,但卻故意用了10核。這是因為根據筆者以前的測試,發現ORCA做DFT的AIMD的并行效率不理想,尤其是對于小體系,用核數太多甚至反倒速度更慢(大家可以對實際情況短暫跑比如5步實測一下設多少核速度最快)。一般來說就設10核就行了,當機子里有明顯更多核的時候,可以跑多個AIMD任務來充分利用計算資源,但應當對CPU內核進行綁定,否則AIMD計算速度可能顯著降低,見《通過設置CPU內核綁定降低ORCA同時做多任務的耗時》(http://www.shanxitv.org/553)。
? %maxcore 10000代表每個ORCA進程最多用10000MB。其實完全沒必要這么大,普通泛函下的AIMD不怎么耗內存,給1000都絕對夠了。
? restart ifexists這句被注釋掉了。如果你的AIMD想續跑,且當前目錄下有之前跑出來的與當前任務同名的后綴為.mdrestart的文件,可以取消注釋,任務就會延續之前AIMD最后的狀態續跑,新軌跡會在原有軌跡文件后面續寫。
? minimize這句被注釋掉了。如果取消注釋的話,動力學開始前會自動在當前級別下做幾何優化。
? timestep 1.0_fs代表動力學步長用1 fs。對于此例常溫下的模擬,1 fs步長是合適的,改大的話可能造成動力學不穩定,而改小的話跑同樣的時間長度需要更多步數導致需要更多耗時。如果追求絕對穩妥,或者是在明顯更高溫度下模擬,可以用0.5 fs。
? initvel 298.15_K代表根據298.15 K溫度通過Maxwell分布隨機初始化原子速度。no_overwrite代表如果之前已經有初速度信息了(比如可以是續跑時從之前的mdrestart文件里繼承來的),就不再產生新的初速度。
? thermostat berendsen 298.15_K timecon 30.0_fs代表用Berendsen熱浴控溫在298.15 K(此例筆者用的ORCA 4.2.1只能用這個熱浴,據悉從ORCA 5.0開始可以用更好的velocity rescale熱浴),時間常數為30 fs,通常這個時間常數是合適的。
? dump position stride 1 format xyz filename "pos.xyz"代表每一步都往當前目錄下的pos.xyz文件里寫入當前的坐標,因此pos.xyz是多幀xyz格式的軌跡文件。此格式的介紹見《談談記錄化學體系結構的xyz文件》(http://www.shanxitv.org/477)。
? dump force和dump velocity開頭的行都被注釋掉了,這兩行用于把模擬過程中的原子受力和原子速度分別保存到相應xyz文件里。dump gbw開頭的行也被注釋掉了,它可以在MD過程中每隔指定步數保存一次gbw文件,之后可以通過寫腳本調用Multiwfn對它們進行批量分析,從而考察動力學過程中電子結構變化,得到豐富的有化學意義的信息(如動力學中的電荷轉移情況、成鍵變化情況、電子定域性變化等等,在北京科音高級量子化學培訓班里筆者會給出很多這種例子)
? constraint add center 0..22代表將當前整個體系(0號到22號原子)的質心約束在初始位置。之所以這么做,是因為盡管ORCA在產生初速度時已經去掉了整體平移的速度分量,但實際模擬過程中由于數值問題,仍然往往會看到有明顯的整體漂移的現象,因而有礙觀察(在VMD里觀看軌跡時還得做一下align才能消掉)。因此直接把質心位置約束住就沒有這個問題了。
? run 2000代表總共跑2000步,當前步長是1 fs,即最多跑2 ps。當前這個模擬的目的是觀察到質子轉移,跑多長時間合適并不好預估,所以可以一次先跑2000步看看,若不夠到時候再續跑。值得一提的是,至少對于筆者現在用的ORCA 4.2.1,我發現如果一次跑的步數很多,達到2000步左右之后,之后每一步的耗時會有逐漸的上升趨勢,原因不明。因此我建議每次跑最多不宜超過3000步,如果需要跑更長的話,最好分多段來跑。
重要提示:如果你用的是ORCA較新版本,應去掉constraint add center 0..22,而把run 2000改為run 2000 CenterCOM,否則可能跑不了。
現在用ORCA運行complex.inp,模擬過程中可以看到每一步的實時情況:
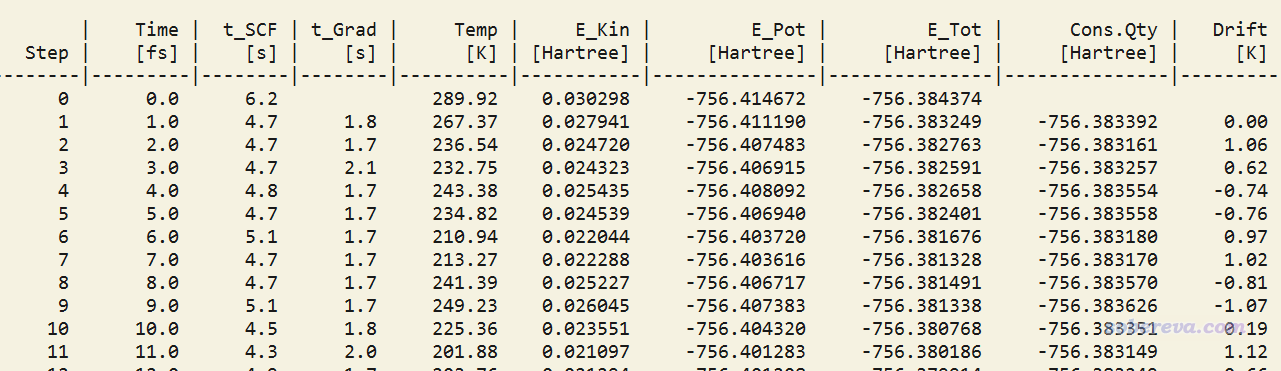
Step是當前的步數,Time是當前的時間,t_SCF和t_Grad分別是算這一步的SCF過程和計算受力的耗時,二者加和就是這一步的總耗時。可見每一步耗時約6~7秒,乘以要跑的步數就可以估計跑完整個任務的耗時。后面還可以看到每一步的溫度、動能、勢能等信息。由于當前體系原子數很少,溫度相對于熱浴的參考溫度波動大是很正常的事。而且由于用了熱浴,所以總能量E_tot也有明顯波動。
VMD是觀看動力學軌跡最好的程序,可以在http://www.ks.uiuc.edu/Research/vmd/免費下載。建議在模擬過程中,隔一陣子就用VMD把pos.xyz載入進去,看看當前的動態行為如何、跑成什么結構了。跑到289步的時候,筆者看了一眼pos.xyz,發現質子不僅已經完全轉移,而且NH4+都已經跑走一定距離了,所以就沒必要繼續跑了,故把ORCA直接殺掉了。
模擬過程中ORCA還產生其它一些文件。complex.md.log是ORCA的MD模塊輸出的信息,相當于整個輸出文件中的子集。complex.mdrestart是用來續跑的文件,每一步都會往里面寫入當前步的時間、坐標、速度、受力等信息。complex-md-ener.csv是把每一步的時間、耗時、溫度、能量等信息以csv格式保存的文件。還有其它一些零碎的文件,通常不是一般用戶關心的,這里就不說了,都可以放心刪掉,留著也沒用。
現在我們來看模擬軌跡。建議大家根據《VMD初始化文件(vmd.rc)我的推薦設置》(http://www.shanxitv.org/545)里說的修改VMD的初始化文件,從而添加自定義命令bt,這樣在播放軌跡的時候對每一幀會自動重新判斷成鍵關系。
啟動VMD,載入pos.xyz(在本文文件包里已提供,共290幀)。然后在文本窗口輸入bt,按回車,使得每幀都更新連接關系。然后再輸入bw,按回車使得背景變為白色。選Graphics - Representation,將Drawing Method改為CPK。然后點擊VMD界面右下角的三角播放動畫,會看到如下結果。如果想在VMD圖形窗口中顯示出幀號或時間,看《使VMD播放軌跡時同步顯示幀號》(http://www.shanxitv.org/13)。
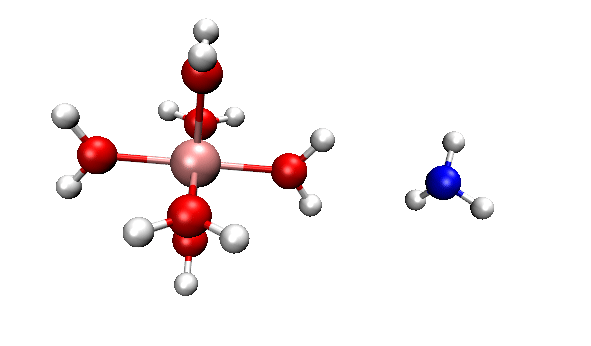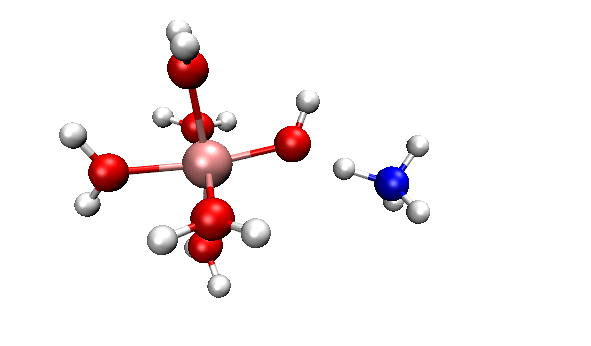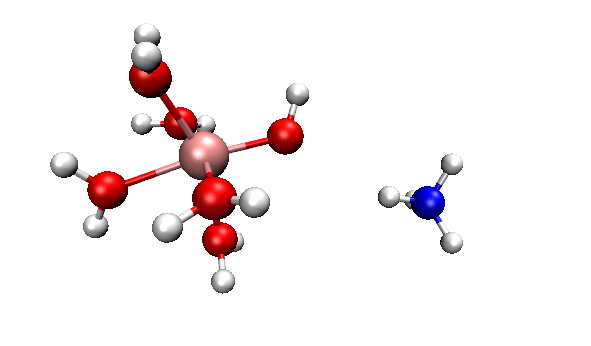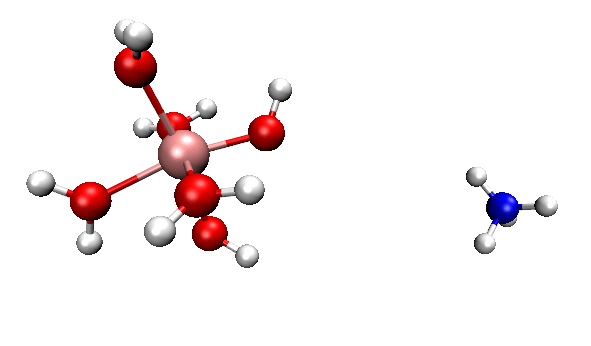
可見,我們跑出來的動力學現象和本文一開始的那個官方動畫幾乎完全一致。都是兩個反應物先接近,然后形成復合狀態時質子在二者之間震蕩,最后NH4+跑掉。
如果想把質子轉移情況通過距離隨時間的變化曲線方式呈獻給讀者,可以在點擊VMD的三維圖形窗口后,按鍵盤上的2鍵(之后按r可以恢復為默認的旋轉模式),然后點擊兩個原子正中心,二者之間就會增加Bond label(默認是以白色字顯示距離,在黑背景下才看得清楚),這里筆者把N和轉移過去的質子之間增加了Bond label。然后進入Graphics - Labels,切換到Bonds,選Graph,點擊Show preview復選框,然后點擊1:H 1:N這項,就會看到距離變化已經顯示在預覽窗口了,如下所示,其中紅色和藍色豎條標記的分別是最小距離和最大距離位置和數值。
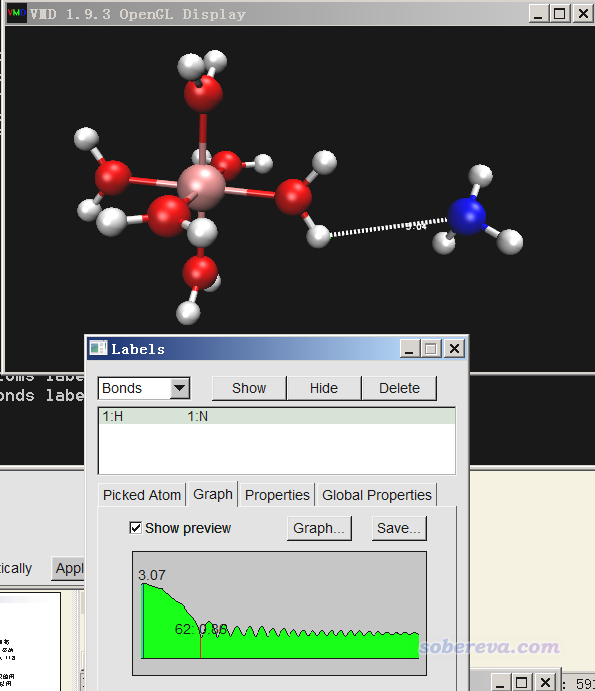
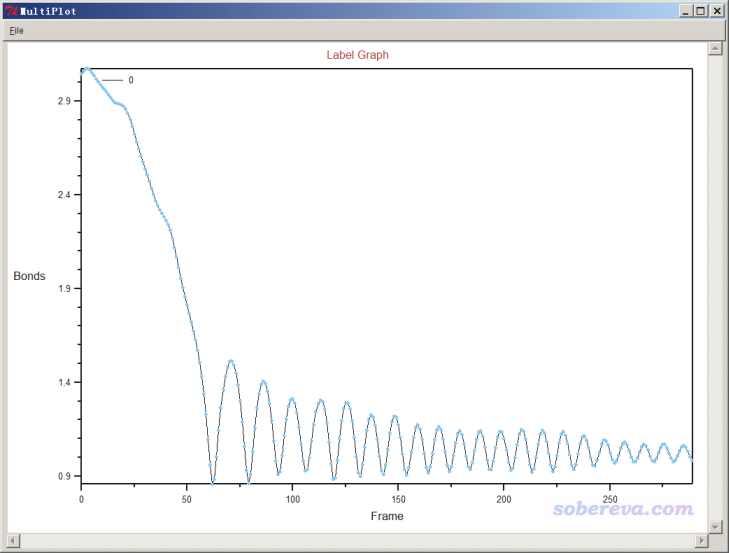
如果點擊Graph按鈕,就會把曲線顯示在大窗口中,如下所示。可見,在大約60幀,也即60 fs左右,N-H鍵就算是基本形成了,之后N-H鍵不斷振動。
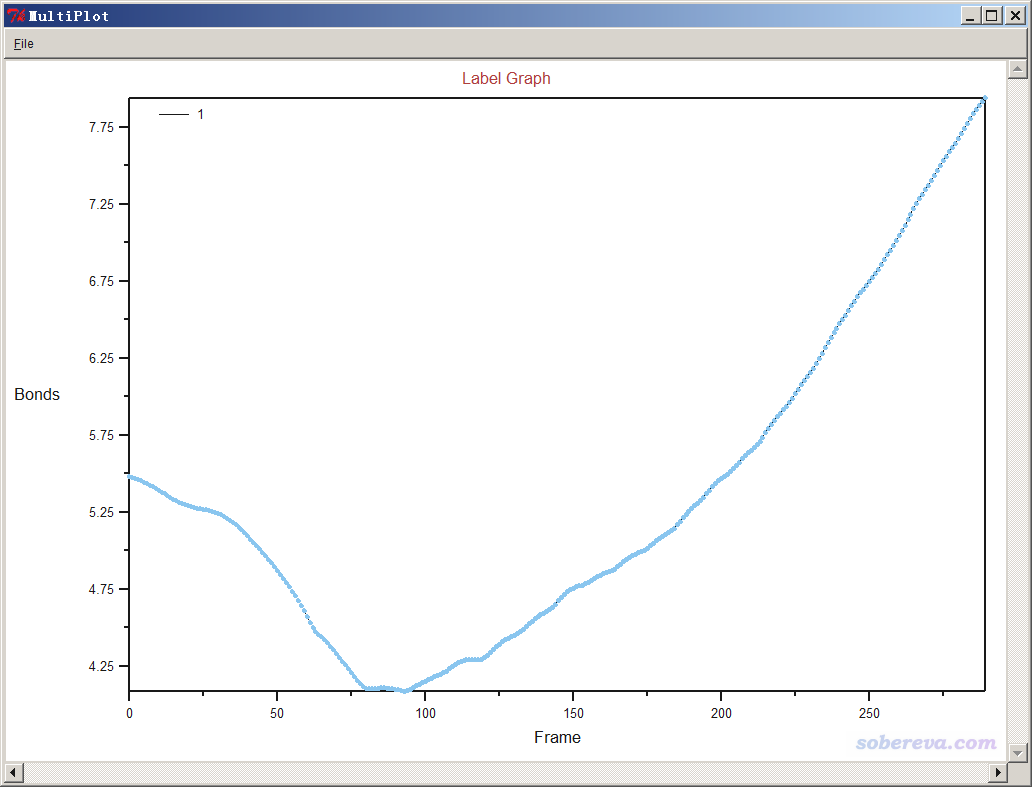
以類似方式,我們可以標記Al與N的距離,隨時間變化如下所示。可見Al與N先接近,質子轉移完畢后,二者就逐漸遠離了。
在Labels窗口里還可以點擊save,把距離變化數據保存到文本文件里,之后可以導入Origin等程序里繪制曲線。
用VMD還可以測量角度、二面角的變化,分別是在圖形窗口里按3然后點三個原子、按4然后點四個原子進行標記,之后在Graphics - Labels里觀看。
3 總結&其它
通過上面的例子,可以看到ORCA做AIMD是相當容易的,只要把Multiwfn支持的含有結構信息的文件(如pdb/gjf/xyz/mol/mol2/fch等等,見Multiwfn手冊2.5節)載入Multiwfn,敲幾下鍵盤產生標準AIMD任務的輸入文件,然后根據實際情況稍微改幾個設定就可以跑了。
以幾十核的一般雙路服務器的運算能力,ORCA里用B97-3c跑幾十原子有機體系的幾十ps的動力學不是特別困難的事。不過,能跑的時間尺度仍遠遠比不上xtb跑半經驗層面DFT的GFN-xTB方法的動力學,xtb跑動力學的粗淺介紹和例子看此文的相應部分:《使用Molclus結合xtb做的動力學模擬對瑞德西韋(Remdesivir)做構象搜索》(http://bbs.keinsci.com/thread-16255-1-1.html)。因此,拿ORCA跑DFT的動力學之前,先拿xtb初步跑跑,找找感覺,大體摸索出自己期望的現象能出現的條件(如溫度、初始結構、反應物相對位置和碰撞方式等),然后再用DFT跑通常是比較好的做法,免得做昂貴的DFT的MD試來試去把時間都耽誤了。
本文只涉及了VMD一丁點皮毛,VMD對于做動力學的人是必須玩得非常溜的。筆者在北京科音分子動力學與GROMACS培訓班(http://www.keinsci.com/workshop/KGMX_content.html)里對VMD有非常深入全面的介紹,包括tcl腳本的編寫。利用VMD的tcl腳本可以對軌跡做千變萬化的分析,有些分析諸如質心距離變化、平面間夾角變化、某幾何變量分布統計、不同結構出現比率等,是必須靠寫腳本才能實現的。
光是分析分析動力學過程的能量、結構變化是很膚淺的。利用Multiwfn,可以對ORCA跑的動力學的全過程的電子結構做深入透徹的分析,從而考察化學鍵、弱相互作用、電荷分布等等在動力學過程中的變化,由此能夠從提供深入的視角,使研究文章信息更豐富、明顯更上檔次。非常建議詳細閱讀《詳談Multiwfn的命令行方式運行和批量運行的方法》(http://www.shanxitv.org/612),里面第4節專門講了怎么做這樣的分析,你會發現特別容易也特別有價值。
如果Multiwfn創建ORCA做動力學輸入文件的功能對你的實際研究產生了幫助,希望在寫文章的時候提及諸如The input file of ab-initio molecular dynamics was prepared with the help of Multiwfn program并引用Multiwfn原文,這是對Multiwfn此功能開發最好的支持。
]]>Displaying atomic forces calculated by Gaussian in VMD
文/Sobereva@北京科音 2020-Sep-13
在量子化學研究中,原子受力往往是非常值得關注的量。Gaussian程序可以通過force關鍵詞計算原子的受力,然后在GaussView中可以根據原子受力大小進行著色,但是沒法繪制出受力矢量。本文介紹如何在免費、靈活、強大的VMD程序中,通過筆者自寫的tcl腳本,將Gaussian計算的原子受力非常直觀地通過箭頭展現出來,這對于一些量子化學問題的研究很有益處。VMD可以在http://www.ks.uiuc.edu/Research/vmd/免費下載,本文使用VMD 1.9.3,Gaussian使用G16 A.03。
在此處下載繪圖腳本和示例文件:http://www.shanxitv.org/attach/568/file.zip。將其中的forcevec.tcl和drawarrow.tcl復制到VMD目錄下。
用文本編輯器打開forcevec.tcl,可看到開頭有幾個設置:
set filename:載入的Gaussian輸出文件路徑
set sclfac:以a.u.為單位的原子受力矢量乘上這個系數就是繪制出的箭頭的長度。應根據實際圖像效果反復嘗試找到最合適的
set rad:箭頭的粗細
set color:箭頭的顏色名
set crit:如果某個原子的受力大小小于所有原子最大受力大小乘以這個系數,則這個原子的受力就不會用箭頭繪制出來,由此可避免在受力相對非常小的原子上出現非常短的、沒什么實際意義的箭頭。默認為0.1
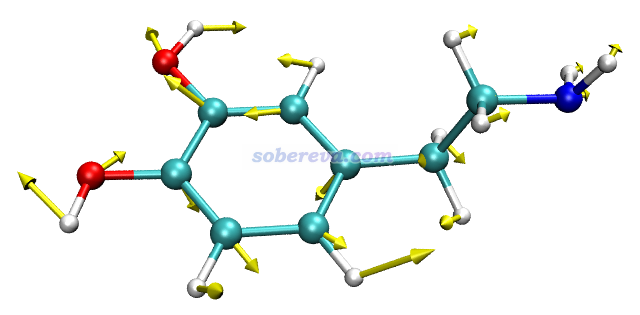
例1:聯苯的基態極小點結構下在S1勢能面上的原子受力
本文文件包里的biphenyl_S1force.out是在優化過的聯苯的基態極小點結構下,用TDDFT算的S1激發態的原子受力的輸出文件,用到的關鍵詞在此文件里已經體現了。
把biphenyl_S1force.out載入GaussView,保存成pdb文件,然后把pdb文件載入到VMD里。然后將biphenyl_S1force.out挪到VMD目錄下,用文本編輯器打開forcevec.tcl,把set filename后面的內容改為biphenyl_S1force.out,把set sclfac后面的內容改為20,然后保存文件。之后在VMD的文本窗口里輸入source forcevec.tcl,腳本就從biphenyl_S1force.out里讀取原子受力,然后繪制出了箭頭。把背景改成白色,在Graphics - Representation里把顯示方式改為Licorice后看到下圖
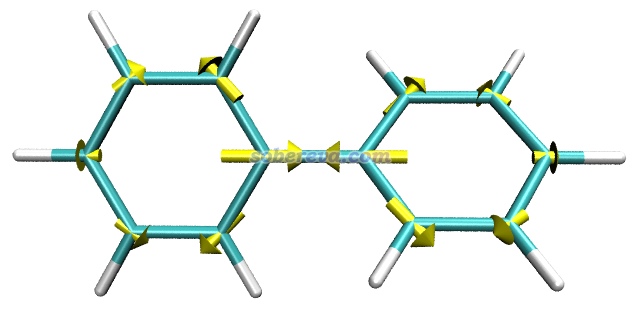
根據原子受力可見,在體系所處的這個Franck-condon點位置,聯苯中央的C-C鍵傾向于縮短。實際上在PBE0/6-311G*級別下,聯苯的基態極小點結構下這個C-C鍵鍵長為1.478埃,而同級別下用TDDFT優化的S1態極小點下這個鍵長為1.413埃,明顯短了很多。另外,從上圖可見其它原子也有顯著受力,這體現出苯環結構將要自發進行調整,確實S1態極小點下苯環的各個鍵長相對于S0態都有顯著變化。
例2:暈苯與富勒烯復合物
筆者之前在PM6-D3下對暈苯與富勒烯形成的復合物進行了優化。這里看一下如果在優化的復合物結構的基礎上,若把富勒烯與暈苯距離稍微拉遠一點,原子受力是什么樣的。本文文件包里complex_force.out就是在人為拉遠的結構下用PM6-D3做force任務的輸出文件,complex_force.pdb是相應的結構文件。將complex_force.pdb載入VMD,把complex_force.out復制到VMD目錄下,將forcevec.tcl里的set filename后面寫上complex_force.out。由于弱相互作用對應的原子間相互作用很弱,所以在set sclfac后面寫上一個比較大的值,這里用1500。在VMD文本窗口里運行source forcevec.tcl,進一步調節下作圖設置后看到下圖
可見,由于富勒烯與暈苯之間的色散吸引作用,再加上當前結構下二者間距比平衡距離更遠,兩個分子彼此間挨得較近的原子上都出現了令二者距離進一步拉近的力。
例3:外電場下的18碳環的受力
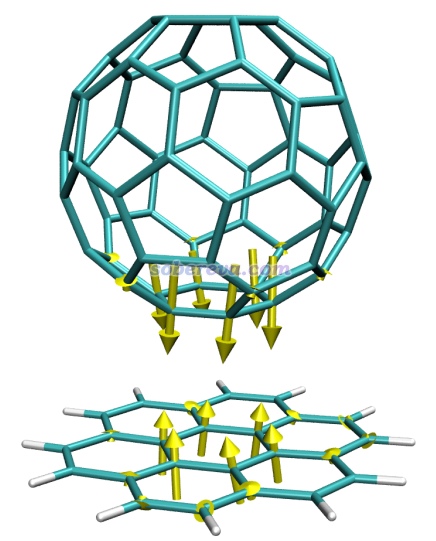
筆者對18碳環及類似體系做過大量的理論研究,匯總見http://www.shanxitv.org/carbon_ring.html。其中,筆者在名為Ultrastrong Regulation Effect of the Electric Field on the All-Carboatomic Ring Cyclo[18]Carbon(https://chemistry-europe.onlinelibrary.wiley.com/doi/10.1002/cphc.202000903)的論文中全面、深入研究了外電場對18碳環的幾何結構、電子結構、電子吸收光譜等特征的影響,此文的介紹見《一篇文章深入揭示外電場對18碳環的超強調控作用》(http://www.shanxitv.org/570)。在此文中通過類似于上文的做法繪制了下面的圖(需要利用專門的tcl腳本)
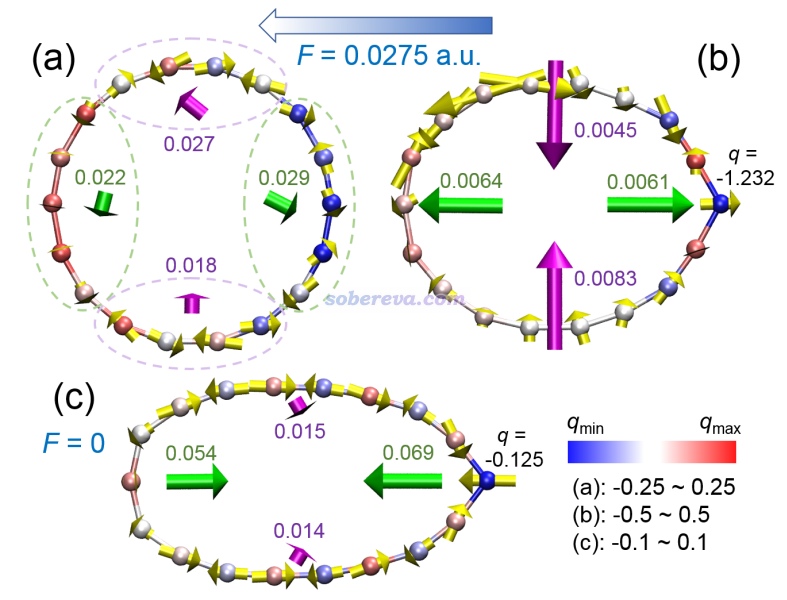
此圖體現的信息的詳細解釋請看上面提到的論文,這里只是簡單提一下。這個圖繪制了各個原子的受力,還用綠色箭頭展現了左、右兩側原子的總體受力,用紫色箭頭展現了上、下兩側原子的總體受力,劃分方式在圖(a)中通過虛線體現了。原子的顏色體現的是ADCH方法算的原子電荷,實現方法在《使用Multiwfn+VMD以原子著色方式表現原子電荷、自旋布居、電荷轉移、簡縮福井函數》(http://www.shanxitv.org/425)中介紹了。圖(a)體現出18碳環在原始結構下,如果剛加上外電場,此刻的受力并不會立刻造成碳環的整體變形,但會明顯造成鍵長發生改變的傾向。圖(b)體現出,在外電場下當碳環的結構已稍微經過弛豫、出現了一些變形后,此刻的原子受力就會明顯傾向于讓18碳環在順著電場方向拉長,而在垂直于電場方向壓扁,從而使碳環變得更加像橢圓。圖(c)是在電場下優化后的結構,但是此刻撤掉了外電場,由綠色箭頭可見此時18碳環有明顯的恢復成原本圓形的傾向,體現出彈性特征。此圖清晰地展現出電場下18碳環這個體系的獨特的力學特性,如果不把受力繪制成箭頭,很難予以這樣深入的探討。
若要繪制一批原子的總受力,可以在source forcevec.tcl之后,把下面的代碼拷到VMD文本窗口里。這部分代碼原本是筆者用來繪制上圖中左側5個原子總受力用的。4 5 6 7 8是被計算總受力的原子序號,總受力矢量及其模也會被輸出到屏幕上。在繪制箭頭時,為了讓位置比較舒服,箭頭中心位置取的是$atmleft 3 9 2 10的幾何中心,即4 5 6 7 8加上相鄰的3 9 2 10四個原子。
set atmleft "4 5 6 7 8"
draw color green
set fxleft 0
set fyleft 0
set fzleft 0
foreach i $atmleft {
set fxleft [expr $fxleft+$fx($i)]
set fyleft [expr $fyleft+$fy($i)]
set fzleft [expr $fzleft+$fz($i)]
}
puts "Left side force: $fxleft $fyleft $fzleft"
set normval [expr sqrt($fxleft**2+$fyleft**2+$fzleft**2)]
puts "Norm: $normval"
drawarrow "serial $atmleft 3 9 2 10" $fxleft $fyleft $fzleft $sclfac 0.2
Method for plotting molecular vibrational vectors calculated by Gaussian in VMD
文/Sobereva@北京科音
First release: 2020-Sep-8 Last update: 2024-Feb-4
Gaussian用戶觀看freq任務產生的振動矢量一般都是通過GaussView看(雖然也有ChemCraft等其它一些程序也可以看)。然而,起碼對于GaussView 6來說,GaussView顯示振動矢量的一個很大不足是箭頭太細,而且頭部不夠粗,導致有時候都看不清楚,放在文章里不夠美觀。另外,GaussView繪制分子結構的作圖選項不夠靈活,而且還收費。VMD是極其流行的化學體系可視化程序,免費、靈活、圖像效果好,本文介紹如何通過筆者寫的VMD作圖腳本非常方便地繪制Gaussian的振動分析任務產生的振動矢量。VMD可以在http://www.ks.uiuc.edu/Research/vmd/免費下載。
在這里下載筆者編寫的繪圖腳本和示例文件:http://www.shanxitv.org/attach/567/file.zip。此腳本至少對于目前撰文時的VMD正式版中最新的1.9.3、Gaussian 09和16是完全適用的。
這里以繪制多巴胺的振動矢量為例進行演示。把文件包里的dopamine.out放到VMD目錄下,這是多巴胺的Gaussian的freq任務的輸出文件。然后我們得把這個.out文件轉化成一個VMD可以認的結構文件的格式,比如可以把此文件載入GaussView,然后另存為.pdb或.mol2文件。也可以下載Multiwfn(http://www.shanxitv.org/multiwfn),啟動Multiwfn后載入此文件,然后選主功能100的子功能2,通過相應選項導出為.pdb或.xyz文件。
把文件包里的drawarrow.tcl和GauNorm.tcl都放到VMD目錄下,然后用文本編輯器打開GauNorm.tcl,把開頭的set filename后面的文件名改為dopamine.out。之后啟動VMD,把多巴胺的結構文件載入VMD,然后在文本窗口輸入source GauNorm.tcl執行此腳本,此時振動矢量信息就被讀入了,與此同時定義了名為norm的繪制振動矢量的命令。之后在VMD的文本窗口輸入比如norm 4,就可以把4號振動模式通過箭頭畫出來。
norm后面還可以接第2個參數,用來設置箭頭長度是正則矢量的幾倍,數值越大箭頭越長,默認是3。norm后面還可以接第3個參數,用來設置箭頭的半徑,默認為0.05。比如norm 5 6 0.07就代表用6倍長度、0.07的半徑繪制第5個振動矢量。默認是用黃色繪制箭頭,如果想用別的顏色,把GauNorm.tcl中的draw color后面的yellow改成其它顏色名,比如cyan。
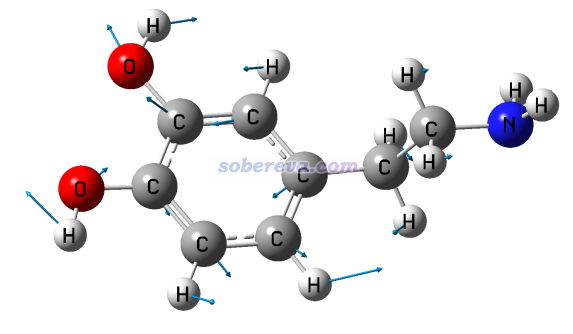
此例輸入norm 17 5,然后令分子以CPK方式顯示(在Graphics - Representation里把Drawing method改為CPK,再把Sphere Scale設為0.6),效果如下,可見非常理想!和GaussView顯示的相對比,可見展現的信息是相同的,而GaussView畫的箭頭相比之下明顯太小氣了。
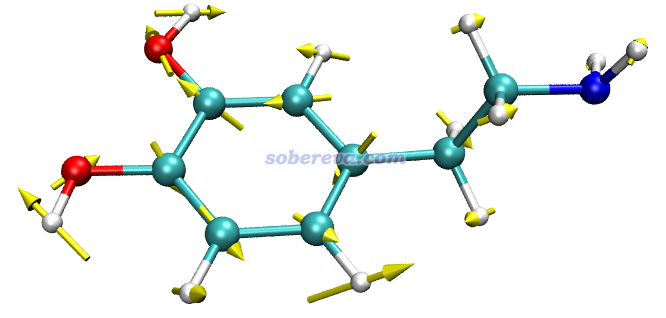
注意GauNorm.tcl開頭還有個set ilinear語句,如果當前體系是線型體系,必須把后面的值改為1。
如果你在Gaussian做freq或opt freq任務中按照《在Gaussian中做限制性優化的方法》(http://www.shanxitv.org/404)中的做法將N個原子的笛卡爾坐標凍結了,運行source GauNorm.tcl之前必須把里面set nfreeze后面的值設為N(默認為0,沒有原子被凍結)。
如果你希望讓箭頭的始端位于各個原子上(和GaussView的風格一致),就把本文文件包里的drawarrow2.tcl放到VMD目錄下,把GauNorm.tcl里的兩處drawarrow都改為drawarrow2并保存。之后再按上文繪圖即可。用norm 17 4命令,效果如下
Calculation of dipole moment of molecular fragments and dipole moment of monomers in complexes using Multiwfn
文/Sobereva@北京科音 2020-Jun-23
1 原理
時不時有人問怎么計算一個分子中某些片段的偶極矩,或者問怎么計算一個分子復合物中各個分子的偶極矩,確實這對于分析局部的極性很有意義,在本文就通過例子對做法進行說明。做這種分析必須使用2020-Jun-22之后更新的Multiwfn程序,可以在http://www.shanxitv.org/multiwfn免費下載,相關基本知識看《Multiwfn FAQ》(http://www.shanxitv.org/452)和《Multiwfn入門tips》(http://www.shanxitv.org/167)。此功能需要使用含有波函數信息的文件作為輸入文件,比如wfn、wfx、mwfn、fch、molden等,產生的方式見《詳談Multiwfn支持的輸入文件類型、產生方法以及相互轉換》(http://www.shanxitv.org/379)。
簡單來說,計算一個體系中由某些原子構成的片段F的偶極矩矢量就是計算
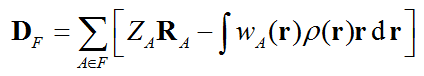
其中R是原子核位置,Z是原子核電荷,ρ是電子密度,w是原子的權重函數。計算片段偶極矩矢量需要用Multiwfn的主功能15(模糊空間分析),目前支持Hirshfeld、Becke、Hirshfeld-I形式的原子權重函數。本文就用比較常用,而且物理意義比較清楚的Hirshfeld權重函數來算。
需要注意的是,如果某個片段的凈電荷不為零,則這個片段的偶極矩是有原點依賴性的,即平移體系會令它發生改變。多數情況下被計算的片段的凈電荷不為0,卻也沒有辦法完全避免原點依賴性,只能是在討論的時候留意這個問題,予以恰當討論。
筆者之前還寫過一篇與本文有一定關系的文章《使用Multiwfn+VMD繪制片段貢獻的躍遷偶極矩矢量》(http://www.shanxitv.org/396),感興趣者推薦看看,這對于考察體系的不同區域對電子激發的振子強度的影響很有益。
2 計算分子片段的偶極矩實例
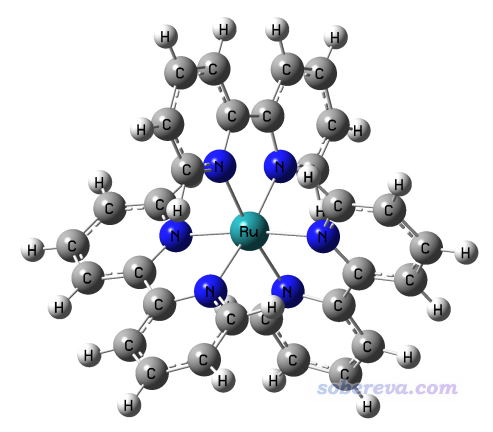
本節計算下面這個單重態[Ru(bpy)3]2+陽離子體系的各個配體的偶極矩,其wfn文件可以在這里下載:http://www.shanxitv.org/attach/558/Ru_bpy_3.zip。此文件的結構使用BP86泛函結合SDD和6-31G*進行優化,波函數用B3LYP結合SDD和6-311G*在IEFPCM模型描述的水環境下產生。此體系分子結構如下:
啟動Multiwfn后載入Ru_bpy_3.wfn,然后依次輸入
15 //模糊空間分析
-1 //選擇原子權重函數
3 //基于內置原子密度的Hirshfeld權重函數
-5 //定義功能1、2計算時考慮的原子范圍
2-21 //體系中一個配體里的原子序號
2 //計算原子和分子多極矩
此時Multiwfn依次計算各個原子的多極矩,最后給出這些原子對應的片段的電子數、偶極矩和不同形式的多極矩:
***** Molecular dipole and multipole moments *****
Total number of electrons: 81.427849
Molecular dipole moment (a.u.): -0.000000 4.348703 0.000000
Molecular dipole moment (Debye): -0.000000 11.053300 0.000000
Magnitude of molecular dipole moment (a.u.&Debye): 4.348703 11.053300
Molecular quadrupole moments (Standard Cartesian form):
XX= -41.258381 XY= -0.000000 XZ= -15.828189
YX= -0.000000 YY= -13.049129 YZ= -0.000000
ZX= -15.828189 ZY= -0.000000 ZZ= -33.042376
Molecular quadrupole moments (Traceless Cartesian form):
XX= -18.212629 XY= -0.000000 XZ= -23.742283
YX= -0.000000 YY= 24.101250 YZ= -0.000000
ZX= -23.742283 ZY= -0.000000 ZZ= -5.888621
Magnitude of the traceless quadrupole moment tensor: 25.129610
Molecular quadrupole moments (Spherical harmonic form):
Q_2,0 = -5.888621 Q_2,-1= -0.000000 Q_2,1= -27.415227
Q_2,-2= -0.000000 Q_2,2 = -24.429930
Magnitude: |Q_2|= 37.189945
Molecular octopole moments (Cartesian form):
XXX= 0.0000 YYY= -455.8273 ZZZ= -0.0000 XYY= -0.0000 XXY= -217.0765
XXZ= 0.0000 XZZ= 0.0000 YZZ= -176.7460 YYZ= 0.0000 XYZ= -85.0737
Molecular octopole moments (Spherical harmonic form):
Q_3,0 = -0.0000 Q_3,-1= -20.8698 Q_3,1 = 0.0000
Q_3,-2= -329.4892 Q_3,2 = -0.0000 Q_3,-3= -154.4790 Q_3,3 = 0.0000
Magnitude: |Q_3|= 364.5030
這些量的具體定義看Multiwfn手冊3.18.3節,這里不細說。我們當前關心的只有
Molecular dipole moment (a.u.): -0.000000 4.348703 0.000000
這一行,即偶極矩矢量為(0.0,4.3487,0.0) a.u.。
然后我們計算其它片段的偶極矩,依次輸入
-5 //重新定義片段
42-61
2 //計算原子和分子多極矩
-5 //重新定義片段
22-41
2 //計算原子和分子多極矩
匯總一下,三個配體的偶極矩分別為:
2-21片段: ( 0.000000 4.348703 0.000000) a.u.
42-61片段:( 3.766463 -2.173911 0.001788) a.u.
22-41片段:(-3.766463 -2.173911 -0.001788) a.u.
下面,我們用VMD程序將偶極矩的箭頭畫出來以便于觀看。VMD可以在http://www.ks.uiuc.edu/Research/vmd/免費下載,本文用的是VMD 1.9.3。wfn格式沒法載入VMD,所以我們先把當前體系轉換為VMD可以載入的xyz格式。在當前Multiwfn窗口中輸入
0 //返回主菜單
100 //其它功能(Part 1)
2 //導出文件
2 //導出為xyz格式
直接按回車用默認路徑,然后Ru_bpy_3.xyz就產生在當前目錄下了。
將Ru_bpy_3.xyz載入VMD,然后將Multiwfn的examples\scripts目錄下的drawarrow.tcl里的全部內容復制到VMD文本窗口里,這樣就定義了一個新的名為drawarrow的命令,我們用這個命令來繪制片段偶極矩。
在VMD的文本窗口里運行以下命令
draw color green
drawarrow "serial 2 to 21" 0.000000 4.348703 0.000000 0.7
draw color red
drawarrow "serial 42 to 61" 3.766463 -2.173911 0.001788 0.7
draw color yellow
drawarrow "serial 22 to 41" -3.766463 -2.173911 -0.001788 0.7
這些命令代表分別用綠、紅、黃色箭頭把三個片段偶極矩繪制出來。雙引號里面是選擇相應片段里的原子的選擇語句,語法詳見《VMD里原子選擇語句的語法和例子》(http://www.shanxitv.org/504)。之后的三個參數是偶極矩的X/Y/Z分量,最后的參數0.7是在繪圖時把偶極矩數值乘上這個系數再繪制,起到控制箭頭長度、令圖像美觀的目的。繪制出來的圖像如下。分子的顯示方式可以在Graphics - Representation界面調節。
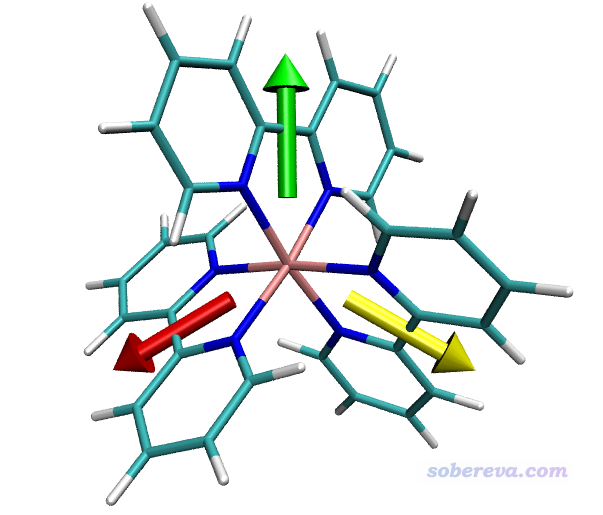
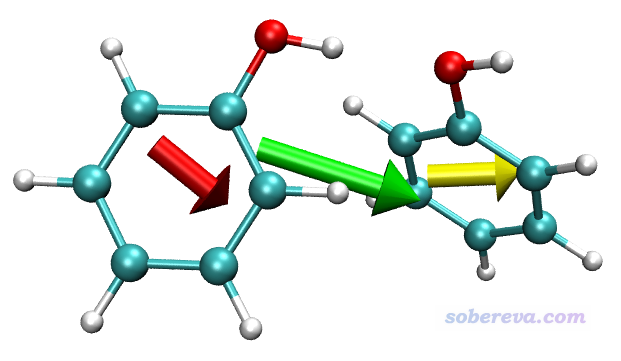
偶極矩是從負電中心指向正電中心。由圖可見每個片段的始端都是兩個氮的方向,這是因為氮帶負電,而且有豐富的孤對電子。顯然當前的結果完全合理。
如果之后想刪掉箭頭,在文本窗口運行draw delete all命令。
值得一提的是,當前這個體系里三個片段的偶極矩是對稱分布的,這是因為中心金屬正好在坐標原點上。由于每個配體的凈電荷不為零,因此如果在做產生波函數的任務之前把體系平移一下,最終得到的三個箭頭就不以Ru為中心對稱了。所以要注意原點位置問題。
有讀者問如果片段里的原子序號是不連續的怎么輸入。假設片段里原子序號是4,9-15,89-100,111,上面例子里的雙引號里就寫serial 4 9 to 15 89 to 100 111,即把-替換為[空格]to[空格],把逗號替換為[空格],即可。
3 計算分子復合物中單體偶極矩實例
下面這個例子我們計算一下苯酚二聚體中每個苯酚單體的偶極矩,并且連同二聚體的總偶極矩一起用VMD畫出來。
啟動Multiwfn,然后輸入
examples\phenoldimer.wfn //自帶的苯酚二聚體波函數文件
15 //模糊空間分析
-1 //選擇原子權重函數
3 //Hirshfeld
2 //計算原子和分子多極矩
由于我們當前還沒定義片段,所以Multiwfn最后給出的下面的信息直接就是二聚體的偶極矩了
Molecular dipole moment (a.u.): 1.227306 -0.128087 0.650833
之后和上一節的例子一樣,對第一個苯酚(1~13號原子)和第二個苯酚(14~26號原子)分別計算片段偶極矩,結果分別為(0.570356 -0.356257 0.284025) a.u.和(0.656950 0.228171 0.366808) a.u.。這兩個苯酚的偶極矩的矢量和與前面輸出的二聚體的偶極矩是相同的。
和前例一樣,把當前結構導出成xyz文件并載入VMD,并且把drawarrow.tcl里的內容拷到VMD的文本窗口里運行,之后輸入以下命令進行繪制:
draw color green
drawarrow all 1.227306 -0.128087 0.650833 2
draw color red
drawarrow "serial 1 to 13" 0.570356 -0.356257 0.284025 2
draw color yellow
drawarrow "serial 14 to 26" 0.656950 0.228171 0.366808 2
這些語句代表二聚體的偶極矩用綠色箭頭繪制,all代表整個體系。兩個苯酚的偶極矩分別用紅色和黃色繪制。由于苯酚的偶極矩較小,為了讓圖中箭頭足夠長,所以命令末尾用了2來讓箭頭長度變成偶極矩大小的兩倍。
得到的圖像如下
可見由于兩個苯酚的偶極矩的方向基本相同,二者的矢量和所對應的二聚體的偶極矩比單體偶極矩大得多。
]]>My recommended settings of VMD initialization file (vmd.rc)
文/Sobereva@北京科音
First release: 2020-Apr-1 Last update: 2023-Apr-4
VMD(http://www.ks.uiuc.edu/Research/vmd/)是用得極為廣泛的化學體系可視化程序,由于其極度靈活,有很多技巧可以使其用起來更方便。
VMD啟動時會先用初始化文件對一些設置進行初始化,即執行里面的各種命令,用戶也可以往里添加額外的命令。對于Windows版來說,這個文件就是VMD目錄下的vmd.rc。對于Linux版來說,這個文件叫.vmdrc,VMD會先在當前目錄下搜索,沒有的話就去找~/.vmdrc,還沒有的話去找$VMDDIR/.vmdrc(這里$VMDDIR環境變量是沒有預先定義的),如果還找不到此文件,就會用默認設置。
注:Linux下的.vmdrc文件默認出現在安裝目錄下,比如以默認路徑安裝會出現為/usr/local/lib/vmd/.vmdrc,但如果不配置VMDDIR環境變量的話這個文件并不會被VMD在啟動時自動載入。Linux下.vmdrc一般都是自行在用戶主目錄下創建。
在此我將我自己的初始化文件里的設置進行分享,其中額外添加的內容如下(放到原有內容后頭即可)。下文的敘述是對撰文時最新版本VMD 1.9.3而言的。
mol default style {Lines 2.0}
display depthcue off
#color Display Background white
#axes location Off
display rendermode GLSL
display distance -8.0
proc bw {} {color Display Background white}
proc bb {} {color Display Background black}
user add key Right {animate next}
user add key Left {animate prev}
user add key Up {animate goto [expr $vmd_frame([molinfo top])+10]}
user add key Down {animate goto [expr $vmd_frame([molinfo top])-10]}
user add key b {mol bondsrecalc all; topo retypebonds}
proc bt {} {
global vmd_frame
trace variable vmd_frame([molinfo top]) w updatebond
}
proc updatebond {args} {
mol bondsrecalc all
topo retypebonds
}
proc bn {} {
global vmd_frame
trace vdelete vmd_frame([molinfo top]) w updatebond
}
proc fog {} {
display depthcue on
display cuemode Linear
display cuestart 1.75
display cueend 2.5
}
下面解釋一下做這些設置有什么好處。
程序默認的顯示方式是Lines,但是線的粗細太細,往往看不清楚,所以用mol default style {Lines 2.0}將默認的顯示方式改為兩倍粗細的Lines。
程序默認開著霧化,即讓距離鏡頭越遠的物體的顏色混入越多的背景色。這會導致在黑色背景下物體的顏色顯得不夠鮮艷,而在白色背景下物體又顯得有點霧蒙蒙,因此用display depthcue off將霧化效果關掉。
#color Display Background white這行是被注釋掉的。如果你想讓VMD啟動后默認就用白背景,就把#去掉。
#axes location Off這行也是被注釋掉的,如果你想讓VMD默認不顯示坐標軸,就把#去掉。
VMD默認用稱作Normal的Rendermode,但此時有些材質的顯示效果很差,甚至Transparent材質根本沒法正確顯示出透明效果。因此通過display rendermode GLSL將默認的Rendermode設為效果好得多的GLSL。
有很多人肯定早已發現畫面邊緣的物體畸變得特別厲害,很難看。通過display distance -8.0語句可以充分避免。但導致一個問題就是原本在窗口左下方的坐標軸看不到了,需要坐標軸的時候可以選Display - Axes - Origin讓坐標軸顯示在窗口中央。
下面這兩行是自定義命令。在VMD的文本窗口里輸入bw(意為background white)就可以令背景立刻變為白的,輸入bb就可以令背景立刻變為黑的,非常方便。
proc bw {} {color Display Background white}
proc bb {} {color Display Background black}
下面的內容是設置用戶自定義快捷鍵。載入軌跡后,在圖形窗口處于被激活的狀態時(激活窗口就是鼠標點擊這個窗口的意思),按左、右鍵就可以分別后退1幀、向前1幀,按上、下鍵就可以分別增加10幀、后退10幀。這使得觀看軌跡方便很多。
user add key Right {animate next}
user add key Left {animate prev}
user add key Up {animate goto [expr $vmd_frame([molinfo top])+10]}
user add key Down {animate goto [expr $vmd_frame([molinfo top])-10]}
關于VMD判斷原子間連接關系的問題我在《談談VMD可視化程序的連接關系的判斷和設置問題》(http://www.shanxitv.org/534)里有非常詳細的說明。為了能很方便地讓VMD對當前幀根據當前結構重新判斷連接關系,增加了下面這句。圖形窗口處于激活狀態時按b鍵(意為bond)就能重新判斷連接關系。
user add key b {mol bondsrecalc all; topo retypebonds}
如果想播放的時候實時自動更新連接關系,而不需要每次都按b鍵,靠以下語句可以實現。也就是定義了一個bt命令,如果在命令行窗口輸入了bt,那么每當當前top體系的幀號發生了變化,就會調用updatebond命令自動來更新連接關系。這樣做的代價就是對較大體系,每播放一幀都要根據距離重新判斷連接關系,播放時會比較卡。
proc bt {} {
global vmd_frame
trace variable vmd_frame([molinfo top]) w updatebond
}
proc updatebond {args} {
mol bondsrecalc all
topo retypebonds
}
下面的內容定義bn命令。如果不想自動更新成鍵方式了,可以輸入bn命令取消掉對幀號的跟蹤即可。
proc bn {} {
global vmd_frame
trace vdelete vmd_frame([molinfo top]) w updatebond
}
霧化效果絕非毫無意義。只要恰當使用,就可以讓近距離的物體完全不受影響,而讓偏遠的原子恰當地霧化,避免擾亂視覺、妨礙清楚地觀看近距離的物體。下面的語句是自定義命令,只要在文本窗口輸入fog,就可以打開霧化并且使用在我來看比較合適的霧化設置。如果覺得實際效果不好,需要進一步調節,可以用Display - Display Settings,修改里面的Cue Start和Cue End。
proc fog {} {
display depthcue on
display cuemode Linear
display cuestart 1.75
display cueend 2.5
}
On the determination and setting of the connection relationship in VMD visualization program
文/Sobereva@北京科音
First release: 2020-Feb-25 Last update: 2023-Dec-29
化學體系可視化程序VMD的用戶越來越多,經常有人在網上問我怎么在VMD里修改鍵連關系,這里就專門說一下這個問題。本文內容對應于撰文時最新的VMD正式版1.9.3,對其它版本可能適用也可能不適用。
當VMD載入pdb、xyz、gro等格式的結構文件時,程序就會自動判斷連接關系。判斷規則和Multiwfn、GaussView等程序類似,都是根據兩個原子間距離以及這兩個原子半徑之和來判斷的,參看此文的相關說明:《談談原子間是否成鍵的判斷問題》(http://www.shanxitv.org/414)。VMD里默認的line方式,以及常用的CPK、Licorice等顯示風格都是根據連接關系顯示的。
具體來說,若原子間距離小于兩個原子半徑之和的0.6倍就會被VMD視為成鍵。原子半徑是VMD內置的,大部分是Bondi范德華半徑。元素的默認半徑沒法在VMD圖形界面里或配置文件里改,但載入結構后可以通過命令行改特定原子的半徑。
如果兩個原子間沒有被判斷為成鍵,但你希望讓它們之間顯示鍵,就選擇Mouse - Add/Remove Bonds,然后點擊這兩個原子的正中心,它們就連上了。如果你點擊兩個已經成鍵的原子,它們之間的鍵就會被去掉。
在你點擊原子時,VMD會自動給你點擊的原子加上標簽并顯示在圖上,如果不想顯示,就按鍵盤上的1鍵,再點擊有標簽的原子,標簽就被隱藏了。如果你想把一批標簽都隱去或者徹底刪掉,就進入Graphics - Labels,按住鼠標左鍵選中一批,然后點Hide按鈕就可以隱藏,點Delete按鈕就可以刪掉。如果想讓批量刪除標簽的操作盡可能省事,對于Windows版VMD,你可以在VMD目錄下的vmd.rc里加入如下內容
proc da {} {
label delete Atoms all
}
啟動VMD后,隨時都可以在文本窗口里輸入自定義命令da來刪除所有原子標簽。
如果有很多同類的鍵,它們的連接關系判斷得都和你要求的不符,一個一個靠鼠標點擊來修改連接關系很麻煩。此時可以在Graphics - Representation界面里把Drawing Method改為DynamicBonds,這代表基于當前結構實時判斷成鍵并顯示,判斷閾值可以用旁邊的Distance Cutoff控件來修改,恰當改成令顯示的連接方式滿足你要求的情況即可。但這種顯示方式有個缺點就是不顯示原子球,而只顯示鍵,而且鍵是空心的,很不好看。因此為了讓原子球也顯示出來,你可以點Add Rep增加一個Representation,把Drawing Method設成VDW,并且把Sphere Scale改小到合適。
有的時候在用DynamicBonds方式顯示時,通過調節Distance Cutoff雖然讓A-B之間鍵連方式滿足要求了,但同時C-D之間的連接方式變得卻與期望的不符了,這種時候就需要靈活變通,多設幾個Representation,每個都恰當用不同的選擇語句選定繪制的區域(參考《VMD里原子選擇語句的語法和例子》http://www.shanxitv.org/504),并用恰當的Drawing Method和參數設定,從而讓顯示出的效果疊加起來正好滿足你的要求。
有的文件格式包含了連接關系信息。比如AMBER、NAMD的拓撲文件都可以被VMD載入,載入的時候就相當于提供了連接關系了,之后再往這個ID里載入結構文件或軌跡時也因此都是按照拓撲文件里的連接關系來顯示。VMD雖然不能載入GROMACS的拓撲文件,但是如果安裝了http://bbs.keinsci.com/thread-37839-1-1.html中提供的tpr插件的話,載入GROMACS的tpr時也會從中讀取正確的連接關系。還有的結構文件格式自己也帶了連接關系段落,典型的就是mol2格式(mol格式也有,但VMD不能載入),相減《談談記錄化學體系結構的mol2文件》(http://www.shanxitv.org/655)。你用文本編輯器打開它的話,會看到@<TRIPOS>BOND字段,這部分記錄的是鍵的形式,諸如5 3 27 2就代表這是第5號鍵,對應3與27原子間的雙鍵。當VMD載入mol2格式文件的時候,就不會自動根據結構來判斷連接關系,而是直接用文件里的連接關系。pdb格式里有個CONECT字段,也是記錄連接關系用的,但是VMD并不會采用這里的連接關系。如果你想讓VMD直接利用這里的連接關系,方法看《使VMD根據pdb文件中的CONECT字段設定原子連接關系》(http://www.shanxitv.org/121)。
在GaussView里可以編輯鍵連關系,而且從GaussView 6開始,可以自定義不同元素之間成鍵的判斷閾值,這在前述的《談談原子間是否成鍵的判斷問題》文中已經說過了。改過之后只點擊Edit - Rebond就可以按照用戶自定義的閾值重新判斷成鍵,因此在批量修改特定類型元素間的成鍵方式的時候比較方便。GaussView可以保存mol2格式的文件,里面的連接關系信息正對應于你在GaussView圖形界面里當前看到的。因此,可以先讓GaussView正確顯示出你期望的連接關系,保存成mol2文件,再用VMD讀取,此時VMD里顯示的連接關系就和GaussView里精確一致了。
注意VMD里沒法以GaussView的風格來顯示雙鍵、三鍵等成鍵形式。比如載入VMD的mol2文件離記錄了某兩個原子間形成的是二重鍵,但在VMD的Line、CPK、Licorice顯示風格顯示時看起來和單鍵一樣。VMD的Bonds顯示風格倒是能區分形式鍵級,但雙鍵和三鍵分別是以兩個、三個套筒形式顯示的,非常難看,沒實際意義。
VMD自身也可以保存mol2文件,因此你在VMD里通過Add/Remove bonds好不容易把連接關系改合適后,不妨通過File - Save coordinate功能保存成mol2文件,以后再次顯示時就免得再修改一遍了。
常有人問我怎么載入動力學軌跡后,雖然第一幀的成鍵方式正確,但播放軌跡時成鍵方式有時連得亂七八糟,甚至跨越盒子。這是因為如前所述,連接關系要么是從拓撲文件里讀取的(比如看Amber軌跡前要載入拓撲文件),要么是載入結構文件時自動判斷的(比如看GROMACS軌跡前通常要載入與模擬體系原子信息對應的gro或pdb文件),或者是根據軌跡文件的第一幀結構判斷的(比如載入molclus、xtb程序產生的多幀xyz文件),之后再載入軌跡或播放軌跡時都不會改變連接關系。由于動力學過程中可能發生分子解離、異構化,或者由于周期邊界條件的原因分子時而完整時而被盒子截斷,因此內存里記錄的連接關系列表可能對于軌跡的某些幀并不適用,極度影響正常觀看。像這種情況,可以用DynamicBonds顯示方式,這樣就會按照當前幀的坐標實時判斷成鍵并顯示(但并不會改寫內存里的連接關系列表);但如果你想用其它顯示方式,就得讓VMD根據當前這一幀的結構重新判斷連接關系并改寫內存里的連接關系列表,做法是在文本窗口輸入mol bondsrecalc all; topo retypebonds。此時連接關系就是滿足當前這一幀的情況了,而對其它幀可能就不適合了。為了方便,你可以定義重新判斷連接關系的快捷鍵,甚至可以讓VMD自動對每一幀實時判斷連接關系,做法見《VMD初始化文件(vmd.rc)我的推薦設置》(http://www.shanxitv.org/545)。
如果你想把當前內存里的連接關系列表以文本方式顯示出來,就在文本窗口輸入[atomselect top all] getbonds,輸出的信息比如
{1 2 3 4 7} 0 0 0 {5 6 7 0} 4 4 {4 0}
這是個列表,里面共有8個成員(有的成員自身就是個列表),對應體系一共8個原子的成鍵情況,比如第0號原子與1、2、3、4、7成鍵,第2號原子與第0號原子成鍵,第3號原子也與第0號原子成鍵...注意這體現的是連接關系,而非鍵級。
連接關系也可以從自行提供的列表里讀取。比如運行
[atomselect top all] setbonds {{1 2 3 4 7} 0 0 0 {5 6 7 0} 4 4 {4 0}}
就代表把當前體系的連接關系改成{1 2 3 4 7} 0 0 0 {5 6 7 0} 4 4 {4 0}所定義的。
使用Multiwfn和VMD繪制平均局部離子化能(ALIE)著色的分子表面圖(含視頻演示)
Using Multiwfn and VMD to plot map of molecular surface colored by averaged local ionization energy (ALIE)
文/Sobereva@北京科音
First release: 2019-Sep-17 Last update: 2020-Oct-10
平均局部離子化能(ALIE)是一個對于考察分子親電反應位點、活性非常有用的量,在《靜電勢與平均局部離子化能綜述合集》(http://bbs.keinsci.com/thread-219-1-1.html)我給出了很多綜述、介紹性文章以及筆者寫過的相關博文,不熟悉ALIE者強烈建議閱讀。Multiwfn是考察ALIE最方便、靈活、強大的程序。通常分析ALIE時都是考察它在分子表面的分布。之前筆者有個文章《使用Multiwfn+VMD快速地繪制靜電勢著色的分子范德華表面圖和分子間穿透圖》(http://www.shanxitv.org/443)介紹了如何將Multiwfn與VMD程序聯用非常方便、快速地繪制高質量的靜電勢著色的分子范德華表面圖。由于后來又有不少人問我怎么類似地也繪制ALIE著色的分子表面圖,因此這里筆者就專門說一下。本文對應于Windows環境,若想在Linux下實現同樣繪制效果,請讀者靈活變通。
本文的過程配有全程演示視頻,強烈建議一看:https://www.bilibili.com/video/av68116168/
首先大家去http://www.shanxitv.org/multiwfn下載Multiwfn程序,必須是2019-Sep-17及之后更新的版本,之前的版本沒有下面提到的腳本。如果還沒裝VMD,去http://www.ks.uiuc.edu/Research/vmd/下載。本文用的是VMD 1.9.3。
之后進行以下操作:
·將examples\scripts里的ALIE.vmd復制到VMD目錄下
·將examples\scripts里的ALIE_isoext.bat和ALIE_isoext.txt復制到Multiwfn.exe所在目錄下
·編輯ALIE_isoext.bat批處理文件,將里面的文件路徑改成實際要考察的,把VMD路徑也改成實際的。輸入文件可以用.wfn、.fch、.molden等
·雙擊ALIE_isoext.bat,Multiwfn將被自動調用進行計算,算完后會在VMD目錄下產生avglocion.cub、density.cub、surfanalysis.pdb
·啟動VMD,在命令行窗口輸入source ALIE.vmd,就可以得到想要的圖像了
下圖是繪制出來的菲的ALIE著色的分子表面圖像。
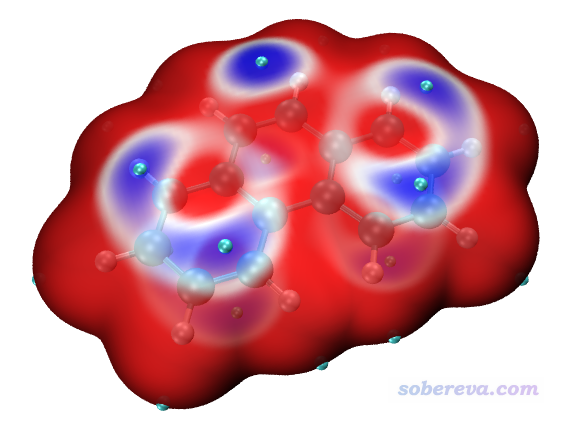
圖中的表面對應的是電子密度為0.0005 a.u.的等值面。之所以不是常用的0.001 a.u.等值面(通常被用來作為分子范德華表面的定義),是因為此時ALIE著色效果不好,不容易通過顏色區分不同區域ALIE分布差異。上圖中青色圓球是分子表面上ALIE的極小點,體現電子被束縛得極弱的位置,也因此體現出容易發生親電反應的位點。ALIE是按照藍-白-紅的色彩過渡方式著色的,因此上圖中藍色區域都是ALIE比較小的區域,電子比較容易參與反應。
為了效果更好,可以用Tachyon渲染器渲染,在視頻里已經演示了。
若你研究的體系不容易通過色彩區分ALIE在不同區域的相對大小,或者整個分子表面顏色全都是紅色或藍色,說明當前用的ALIE的色彩刻度范圍不合適,可以在VMD命令行窗口輸入諸如mol scaleminmax 0 1 0.31 0.37,這里0.31和0.37分別是色彩刻度下限和上限(亦可以在VMD的圖形界面里修改,做法是進入Graphics - Representations,在窗口上方選擇當前是Isosurface的那項,點擊Trajectory標簽頁,在文本框里輸入色彩刻度的下限和上限之后按回車)。色彩刻度范圍怎么設合適和具體體系有關,可以反復試試。如果心里沒譜的話,可以先把下限和上限分別設成當前分子表面上的ALIE實際的最小值和最大值,以讓色彩變化能完整表現分子表面上的ALIE分布。這兩個值在Multiwfn計算過程的輸出信息里就可以看到(將ALIE_isoext.bat第一行末尾加上[空格]> out.txt,腳本調用Multiwfn期間輸出的信息就會導出到當前目錄下的out.txt,其中搜Minimal value:和Maximal value:),然后手動將這兩個值除以27.2114換算為以Hartree為單位的值,作為色彩刻度下限和上限即可。
對于平面體系,如果想準確考察ALIE極值點與原子或者鍵的相對位置,最好改成正交視角,即選擇Display - Orthographic,否則會由于近大遠小的緣故可能導致誤判位置。以正交視角顯示時的菲分子的圖像如下,位于藍色區域的原子或鍵容易參與親電反應。
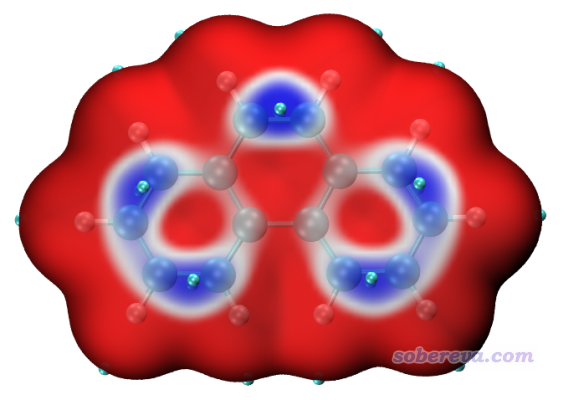
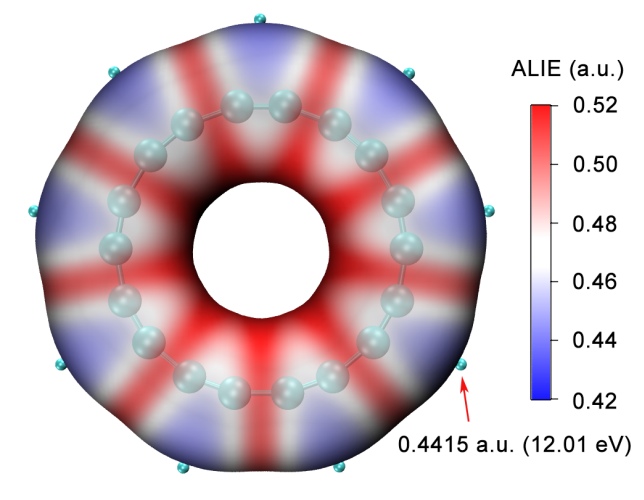
值得一提的是,筆者在《一篇最全面、系統的研究新穎獨特的18碳環的理論文章》(http://www.shanxitv.org/524)介紹的論文中使用本文的方法考察了電子結構非常特殊的18碳環體系,得到的圖像很漂亮(如下所示),體現出的信息又非常有價值,很建議讀者看看此文中關于反應性的相關討論。
如果想像上圖一樣把色彩刻度軸畫出來,按照《使用Multiwfn+VMD快速地繪制靜電勢著色的分子范德華表面圖和分子間穿透圖(含視頻演示)》(http://www.shanxitv.org/443)里面介紹的做法就可以實現。如果你想查詢分子表面ALIE極值點具體的值,也效仿此文里面說的做法。
]]>