談談記錄化學體系結構的mol2文件
談談記錄化學體系結構的mol2文件
文/Sobereva@北京科音 2022-Dec-31
1 mol2文件簡介
mol2文件是計算化學領域非常常用的記錄分子結構的格式,被很多程序所支持和利用。例如VMD、GaussView、Chem3D、OpenBabel、AmberTools里的Antechamber等程序都可以導出mol2文件,筆者開發的Multiwfn(http://www.shanxitv.org/multiwfn)可以基于mol2文件計算EEM原子電荷,筆者開發的Sobtop(http://www.shanxitv.org/soft/Sobtop)可以基于mol2文件產生GROMACS的拓撲文件,等等。
相對于非常常見的在《談談記錄化學體系結構的xyz文件》(http://www.shanxitv.org/477)中介紹的xyz格式,mol2格式關鍵優點在于可以記錄成鍵信息,即誰與誰成什么形式的鍵,這對于判斷原子所處的化學環境非常重要,比如Sobtop需要有這樣的成鍵信息才能自動指認GAFF原子類型,Multiwfn計算EEM原子電荷時需要有這樣的信息才能判斷各個原子要用的EEM參數。另外mol2文件還可以記錄其它諸多信息(距離約束、可旋轉的鍵、原子類型和電荷等等),對于分子模擬、QSAR、化學信息學等一些方面有特殊的意義。
mol2格式略微復雜,不同程序產生的mol2文件有所出入,有的程序產生的mol2文件還不規矩,導致經常有人由于用的mol2文件有問題而無法用Sobtop和Multiwfn等程序正常處理,甚至導致程序崩潰。我遂覺得有必要寫一篇文章介紹一下mol2格式,便于讀者了解怎么讀取mol2文件的信息、判斷自己手頭的mol2文件是否規范,以及拿到不標準的mol2文件時怎么修改。
mol2文件是文本格式,包含大量的字段,每個字段各有各的用處和定義規范。mol2文件的詳細說明可以下載此文檔查看:http://www.shanxitv.org/attach/655/Tripos_Mol2_File_Format.pdf。這些字段并不是全都需要出現的,常見的字段只有幾個而已,每個字段涉及的信息中往往也只有其中少部分會經常涉及到,將在下文進行介紹,若想更全面詳細了解mol2格式可閱讀上述文檔,共54頁。
本文提到的程序的版本是VMD 1.9.3、Multiwfn 3.8(dev, 2022-Dec-18)、Sobtop 1.0(dev3.1)、GaussView 6.0.16、OpenBabel 3.1.1。其它版本可能與本文相同也可能不同。
2 mol2文件的例子和解讀
OpenBabel程序產生的mol2格式相對來說是屬于比較規矩的,這里結合OpenBabel程序產生的苯甲醛的mol2文件的內容進行講解。我先用一個可視化程序畫了一個苯甲醛的結構,保存為了benzaldehyde.pdb文件,然后用obabel benzaldehyde.pdb -O benzaldehyde.mol2命令用OpenBabel把pdb格式轉成了mol2格式的benzaldehyde.mol2文件,其內容如下,GaussView載入此文件后顯示的結構圖也給出了以便于對照
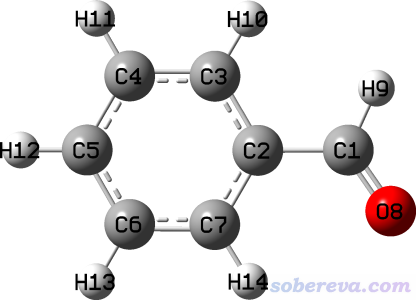
@<TRIPOS>MOLECULE
benzaldehyde.pdb
14 14 0 0 0
SMALL
GASTEIGER
@<TRIPOS>ATOM
1 C 1.9920 0.4700 -0.0000 C.2 0 UNK0 0.1502
2 C 0.5340 0.2150 -0.0000 C.ar 0 UNK0 0.0142
3 C -0.3610 1.2920 -0.0000 C.ar 0 UNK0 -0.0515
4 C -1.7360 1.0600 0.0000 C.ar 0 UNK0 -0.0611
5 C -2.2160 -0.2520 0.0000 C.ar 0 UNK0 -0.0617
6 C -1.3250 -1.3310 -0.0000 C.ar 0 UNK0 -0.0611
7 C 0.0460 -1.1010 -0.0000 C.ar 0 UNK0 -0.0515
8 O 2.8450 -0.3960 0.0000 O.2 0 UNK0 -0.2957
9 H 2.2730 1.5470 0.0000 H 0 UNK0 0.1081
10 H 0.0250 2.3090 -0.0000 H 0 UNK0 0.0624
11 H -2.4300 1.8950 0.0000 H 0 UNK0 0.0618
12 H -3.2860 -0.4350 0.0000 H 0 UNK0 0.0618
13 H -1.7060 -2.3480 -0.0000 H 0 UNK0 0.0618
14 H 0.7610 -1.9170 -0.0000 H 0 UNK0 0.0624
@<TRIPOS>BOND
1 1 2 1
2 1 8 2
3 1 9 1
4 2 3 ar
5 2 7 ar
6 3 4 ar
7 3 10 1
8 4 5 ar
9 4 11 1
10 5 6 ar
11 5 12 1
12 6 7 ar
13 6 13 1
14 7 14 1
首先要知道mol2文件里以#作為第一列的是注釋行,空行也被完全無視。mol2文件是自由格式,因此空格數目完全隨意。
第一列以@開頭的叫做字段,從上面的benzaldehyde.mol2可見,當前文件有@<TRIPOS>MOLECULE、@<TRIPOS>ATOM、@<TRIPOS>BOND三個字段。一般來說這三個字段都是必須出現的,一起提供了描述一個分子最起碼的信息。
2.1 MOLECULE字段
@<TRIPOS>MOLECULE字段記錄了體系的基本信息,包括:
第1行:體系的名字。可見OpenBabel把轉換出mol2文件的源文件的名字benzaldehyde.pdb當做了當前體系的名字
第2行:五個數字分別是體系中的原子數、化學鍵數、子結構數、特征數、set數。對于單純記錄體系結構信息,只要提供前兩者就夠了,后三個可以省略。所謂的子結構是指體系中的一個部分,比如每個分子、每個殘基、蛋白質的每條鏈等等都可以在@<TRIPOS>SUBSTRUCTURE字段里定義為一個子結構。所謂的set是指基于體系中的一些原子/鍵/子結構根據特定規則和需要定義的集合,可以在@<TRIPOS>SET里具體定義。
第3行:體系的類型。可以為SMALL(小分子)、BIOPOLYMER、PROTEIN、NUCLEIC_ACID、SACCHARIDE
第4行:原子電荷。如果mol2文件沒記錄原子電荷信息這里就為NO_CHARGES。而在產生當前benzaldehyde.mol2文件的時候OpenBabel自動計算了Gasteiger電荷,因此此處寫的是GASTEIGER。還可以為MULLIKEN_CHARGES(Mulliken電荷)、MMFF94_CHARGES(MMFF94力場定義的電荷)等等,不同種類電荷都有固定名字。如果記錄的原子電荷是比如Multiwfn算的ADCH、RESP、1.2*CM5等電荷,在mol2格式規范中沒有對應的名字,則這里應當寫USER_CHARGES。
2.2 ATOM字段
@<TRIPOS>ATOM字段每一行定義一個原子的信息,每一列記錄的信息為:
(1)原子序號(整數)
(2)原子名(字符串)
(3)X坐標(埃)
(4)Y坐標(埃)
(5)Z坐標(埃)
(6)原子類型(atom type。字符串)
(7)原子所屬的子結構序號(整數),可省略
(8)原子所述的子結構名字(字符串),可省略
(9)原子電荷(浮點數),可省略
原子名部分可以為比如C2、H4等等,完全隨意。記錄生物分子結構時通常用IUPAC定義的各種殘基中的原子名。
原子類型部分可以記錄做分子模擬用的力場中此原子實際對應的原子類型。mol2格式自己也有一套原子類型定義,見前述的Tripos_Mol2_File_Format.pdf文檔的末尾,比如sp3雜化的碳的原子類型是C.3,C.ar代表芳香碳,Any代表任意,Hal泛指鹵素,Cl代表氯,Ca代表鈣,H代表氫,H.spc特指SPC水模型的氫,LP代表孤對電子(lone pair),Du代表虛原子(dummy),等等。
一定要特別注意,mol2格式雖然定義了一大堆字段,但(居然)沒有一個地方是專門用來記錄元素的,這在我來看是mol2格式的嚴重不足!!!mol2記錄的原子名和原子類型信息可以與元素名相同也可以不同,不同程序產生的mol2文件的情況各有不同。例如如此例可見,OpenBabel產生的這個mol2文件里原子名恰等于元素名,原子類型是根據mol2格式自己定義的原子類型指認的。而在GaussView產生的mol2文件中,原子名是給元素名后面加了數字(因此不會有重名的原子),而原子類型恰等于元素名。由于情況混亂,所以一個程序在讀取mol2文件的時候并沒有嚴格的辦法能準確判斷元素,只能靠猜。Multiwfn和Sobtop在讀取mol2文件時是根據原子類型的字符串判斷元素的:如果字符串中沒有.,就直接將之當做元素名來判斷元素;如果有.,比如是C.3,就把.前面的內容當做元素名判斷元素。因此,讀者應該知道Multiwfn和Sobtop沒判斷對元素時該怎么辦了,最簡單的做法就是手動修改mol2文件以讓@<TRIPOS>ATOM字段每一行的第6列對應元素名。
由于子結構信息在原子電荷前面,因此即便你不想定義原子所屬的子結構信息而只想定義原子電荷,也必須隨便寫上子結構序號和子結構名字來占位,比如此例用0 UNK0來占位。
2.3 BOND字段
@<TRIPOS>BOND字段每一行定義一個鍵的信息,其每一列記錄的信息為:
(1)鍵的序號(整數)
(2)第1個原子的序號
(3)第2個原子的序號
(4)鍵的類型
鍵的類型有以下這些
? 1 = 單鍵
? 2 = 雙鍵
? 3 = 三鍵
? am = 酰胺的N-C鍵(這種鍵有一定pi共軛作用,這是為什么mol2格式里特意用am來與單鍵區分)
? ar = 芳香環(aromatic)上的鍵,以下簡稱芳香鍵
? du = 虛鍵
? un = 未知/無法判斷
? nc = 不相連(倆原子不成鍵就沒必要在BOND字段出現,但可以靠nc強調某兩個原子間就是沒成鍵)
絕大多數程序產生的mol2文件里沒有du、un、nc。
有的程序產生的鍵的類型名不規矩。比如GaussView對于芳香環上的鍵(具體來說,是圖形窗口里看到一個實線+一個虛線的那種鍵)用的類型是Ar,但按照mol2規范應當是ar,因此這會導致一些程序無法識別(而Sobtop在讀取時已經考慮到了GaussView的這個bug,因此用戶不用自己做替換)。GaussView還自行給mol2格式做了擴展,把圖形窗口里看到是一個虛鍵的那種鍵記錄為Wk代表Weak,而這可能導致很多程序無法正常識別和載入。
不同程序對成鍵的指認也往往有很大不同。比如甲酰胺,Avogadro產生的它的mol2里把N-C鍵記錄為am,嚴格符合mol2格式的要求。而GaussView則無法將之記錄為am,而是可能記錄為單鍵也可能記錄為Ar(取決于當前圖形窗口里顯示的成鍵方式)。另外,我之前在《談談原子間是否成鍵的判斷問題》(http://www.shanxitv.org/414)中說過GaussView是根據原子半徑和原子間距離判斷成鍵形式的,導致很有可能判斷出的成鍵方式很不“經典”,甚至很違背化學常識,比如可能顯示一個碳原子連著一個雙鍵和兩個單+虛鍵。而如果把結構保存成pdb然后再用OpenBabel轉成mol2格式,則成鍵方式就很經典了,因為OpenBabel能夠自動讓成鍵方式滿足經典Lewis式且同時識別芳香區域。
VMD程序里如果載入了xyz、gro、pdb等不含成鍵關系信息的文件(雖然pdb有CONECT字段,但VMD不利用),保存出的mol2文件將沒有BOND字段,明顯是不符合規范的。在同時載入拓撲文件以提供拓撲信息后,保存出的mol2文件才是有BOND字段的(與此同時也有了原子類型、原子電荷、原子所屬的殘基號和殘基名信息)。VMD如果載入的是mol2文件,也可以從中獲取成鍵信息,使得保存出的mol2文件里也有BOND部分。但是VMD并不能記錄芳香鍵,而且它自己也沒有像OpenBabel那樣根據幾何結構和元素就能判斷出芳香區域的能力,因此即便載入的是本例的benzaldehyde.mol2,保存出的mol2里苯環上的鍵也都會簡單地記錄成單鍵。
還值得順帶一提的是有個叫mol的格式,介紹見https://en.wikipedia.org/wiki/Chemical_table_file。不要將它和mol2混淆,二者格式截然不同。mol也能像mol2一樣記錄鍵的存在性及其類型。GaussView產生的mol文件中會把芳香鍵用4來記錄,這正是mol標準格式中的芳香鍵的類型序號,而OpenBabel在產生mol文件時則會把芳香環描述成單雙鍵交替的形式。
3 關于記錄晶胞信息
mol2文件通常用來記錄孤立體系,但實際上此格式也定義了記錄晶胞信息的字段@<TRIPOS>CRYSIN,在其下一行寫晶胞的a、b、c三個邊長(埃)以及alpha、beta、gamma夾角(度),每個值之間以逗號分隔。例如:
@<TRIPOS>CRYSIN
3.785,3.785,9.514,90,90,90
Sobtop和Multiwfn在載入mol2文件時都會試圖載入晶胞信息以考慮周期性。GaussView有很好用的對周期性體系建模的功能,但即便在圖形窗口里能看到晶胞邊框,代表當前是周期性體系,其保存出的mol2文件里也沒有@<TRIPOS>CRYSIN字段,因此需要自行補充。在VMD 1.9.3里,即便當前有晶胞信息,保存的mol2文件里也沒有以上字段。VMD 1.9.3載入mol2時也不會從以上字段中載入晶胞信息。