構建被顯式溶劑層包裹的分子的簡單方法
0 前言
溶劑模型分兩大類,顯式溶劑模型、隱式溶劑模型。在量子化學研究中,一般為了省事、方便以及為了考慮溶劑平均效應而用隱式溶劑模型,但是有時候溶劑效應沒法靠連續介質的考慮方式描述,非得用顯式溶劑模型不可。有些情況只需要很少的顯式溶劑即可,比如水催化的反應,只需要把參與反應的水分子納入體系即可。但研究許多問題的時候,顯式溶劑放在哪、怎么放,哪些位置的顯式溶劑會起到關鍵作用并不明確(比如考慮顯式溶劑對UV-Vis光譜影響的問題),那么為了保險就得給整個分子加上一個溶劑層。通常顯式溶劑和隱式溶劑是一起使用的,后者描述顯式溶劑層外部的體相溶劑效應,這稱為雜化溶劑模型。
加溶劑層比較嚴謹、可靠的做法是對體系跑分子動力學,然后從中提取一些幀(可以均勻間隔采樣,也可以做周期性退火,取對應0K的幀),然后把靠近溶質的溶劑分子連同溶質提取出來,再用量化方法優化。但這樣做對于不懂MD的人門檻略高,有時候處理的體系還不好生成拓撲文件、弄到適合的力場參數。
本文目的是介紹比較簡單的給分子加上溶劑層的做法,不需要跑動力學。雖然沒有MD嚴格,但對于搞量化的人來說多數情況也夠用了,本文的方法也比較普適。本文使用的程序為Gaussian16 A.03、Packmol 16.344、VMD 1.9.3、MOPAC2016。本文以構建靛青(indigo)分子被顯式水包圍的體系作為例子。
本文涉及的所有文件可在此處下載:http://www.shanxitv.org/attach/406.7z。
Packmol的下載和使用方法見《分子動力學初始結構構建程序Packmol的使用》http://www.shanxitv.org/473,VMD可在http://www.ks.uiuc.edu/Research/vmd/免費下載。VMD和Packmol的詳細使用會在北京科音的“分子動力學與GROMACS培訓班”里深入講解,本文中只是用到哪說到哪。
1 用Packmol產生溶劑分子球完全包裹溶質的體系
Packmol是常用的構建動力學模擬體系的程序,一般在Linux下運行。下載后解壓,運行make即可編譯,然后把Packmol加入到PATH環境變量中后即可使用。將靛青分子和水分子的pdb文件準備好,分別叫indigo.pdb和H2O.pdb。寫一個叫做mix.inp的Packmol輸入文件,內容如下
tolerance 2.0
output ball.pdb
filetype pdb
structure indigo.pdb
number 1
center
fixed 0. 0. 0. 0. 0. 0.
end structure
structure H2O.pdb
number 350
inside sphere 0. 0. 0. 10.
end structure
這代表將靛青置于體系中央,然后在距離靛青中央10埃的球形空間內填充350個水,而且不能與靛青有不合理接觸。運行packmol < mix.inp,很快在當前目錄下得到ball.pdb。用VMD查看ball.pdb,如下所示
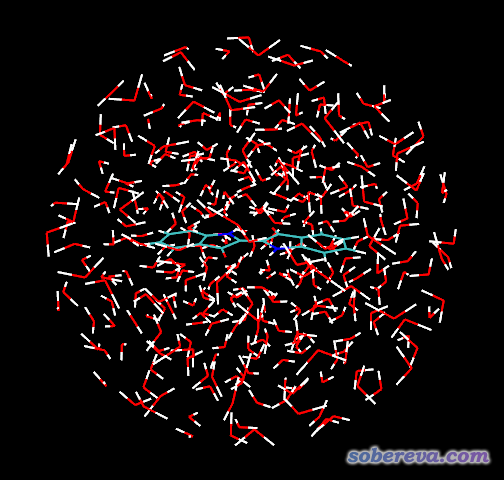
由圖可見,靛青分子完全浸在了水球里,沒有裸露出來的地方。對于其它溶質分子,溶劑球的半徑和填充的溶劑數目應當酌情而定,溶劑球不能太小從而導致溶質裸露;溶劑球也不宜太大,否則會令下一步的耗時過高。填充的溶劑數目不能太少,否則溶劑非常稀疏,之后一優化體系可能嚴重坍塌、出現溶質裸露情況;填充的溶劑數目也不能太大,否則Packmol會反復迭代總也收斂不了。可以反復多試一試。
2 粗略優化溶劑球+溶質體系
Packmol構建體系時并不考慮分子之間的相互作用,只是讓分子以沒有不良接觸的方式簡單堆積起來。因此,上面構建的溶劑球+溶質體系和實際情況相距甚遠。為了快速地讓結構更合理一些,這里用Gaussian在普適型力場UFF下做個簡單的優化。用gview載入上一節得到的ball.pdb,保存Gaussian輸入文件。由于UFF力場終究很粗糙,讓它直接優化整體的話可能會把溶質結構弄糟,因此輸入文件里把靛青分子設為凍結狀態(如果不知道怎么弄,看《在Gaussian中做限制性優化的方法》http://www.shanxitv.org/404)。最終輸入文件是ball.gjf,內容如下#p UFF=qeq opt geom=connectivity
Built with Packmol
0 1
C -1 -0.35400000 5.30600000 0.00000000
C -1 1.04000000 5.15500000 0.00000000
C -1 1.65100000 3.90400000 0.00000000
...略。坐標后面是連接關系。
UFF后面的=qeq代表自動用QEq方式計算每個原子的原子電荷,否則原子將沒有原子電荷,原子間靜電相互作用就表現不出來,連氫鍵基本特征也不可能正確描述(因為常規強度的氫鍵的主要本質就是靜電相互作用)。QEq電荷在《原子電荷計算方法的對比》(http://www.whxb.pku.edu.cn/CN/abstract/abstract27818.shtml)中有介紹。
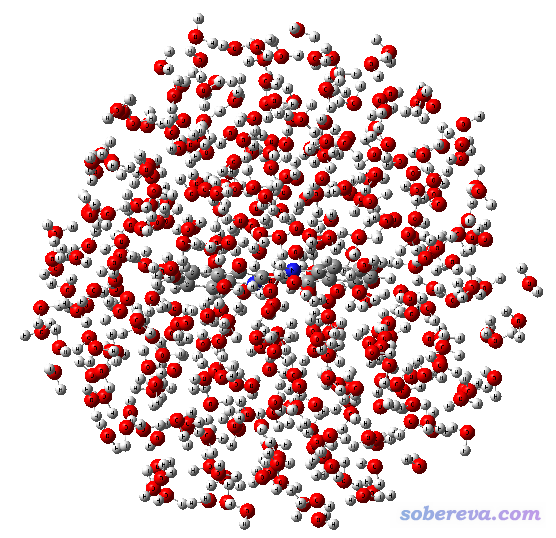
優化的輸出文件是ball.out,圖像如下所示,可見溶劑已不是簡單的球形分布了,而是由于考慮了分子間相互作用,已經呈現出一定結構特征了。不過這個結構還是很粗糙。
3 用VMD挖帶著溶劑層的溶質體系
把ball.out用gview保存為UFF_opted.pdb。將此文件載入VMD。載入后,VMD會自動判斷原子間連接關系,每一批相連的原子構成一個片段,有唯一的fragment號,號碼從0開始。當前體系結構信息中先記錄的是一個靛青,然后是350個水,因此靛青在VMD里叫fragment 0。我們先看看怎么選取原子合適。進入VMD界面里的Graphics-Representation,在Selected Atoms里輸入same fragment as within 5 of fragment 0,這代表選中靛青分子,而且只要水的任意一個原子距離靛青原子小于5埃,則整個水也被選中。此時看到的圖像如下:
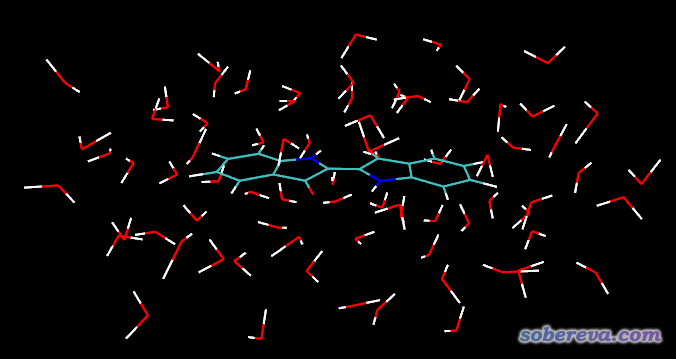
可見,當前的選區設置很符合我們的需要,溶劑層厚度合適。厚度不能太大,否則包含的溶劑原子太多,之后做量化耗時高;厚度也不能太小,要不然溶質分子就露出來了。
在VMD Main窗口中選中當前體系,選File-Save Coordinate,Selected atoms框里還是輸入same fragment as within 5 of fragment 0,File type選xyz,點Save,然后文件名設為solvated.xyz并保存。
4 用MOPAC2016優化溶質+溶劑層體系
上一步得到的溶質+溶劑層模型還是比較粗糙,定性合理都沒法保證,用于量化計算前肯定是要做幾何優化的。通常我們都用DFT來做,但是當前體系對DFT來說已經非常大了,有258個原子呢!比較合適的做法是,先用MOPAC在半經驗級別下對這個分子團簇體系做個優化。MOPAC做半經驗非常快(遠比Gaussian快),優化幾百原子體系花不了多少時間。MOPAC優化完了,體系就定性合理了,然后我們可以再用gview把根據化學直覺認為對自己研究的問題用處不大的溶劑分子再刪掉一些,使得總原子數降低到DFT優化可以接受的程度,再用DFT優化。
我們這里在MOPAC2016中用PM6-D3H4來做幾何優化,它對分子間弱相互作用可以給出基本合理的描述。對PM6-D3H4和分子間相互作用計算不了解的人強烈建議看《大體系弱相互作用計算的解決之道》(http://www.shanxitv.org/214)和亂談DFT-D(http://www.shanxitv.org/83)。不知道MOPAC怎么安裝的看《MOPAC2012安裝方法》(http://www.shanxitv.org/262)。
我們把solvated.xyz改寫成MOPAC輸入文件solvated.mop,內容如下
PM6-D3H4 EF PRNT=2 EPS=78.3 precise
test
All coordinates are Cartesian
C 0.071000 0 -3.083000 0 -4.437000 0
C 0.384000 0 -1.826000 0 -4.974000 0
C 0.490000 0 -0.686000 0 -4.182000 0
C 0.272000 0 -0.832000 0 -2.814000 0
...略
O -2.733000 2.351000 -1.012000
H -3.247000 2.606000 -1.824000
H -2.196000 1.578000 -1.334000
O 1.434000 -3.488000 -11.108000
H 1.574000 -4.345000 -11.596000
H 0.754000 -3.740000 -10.430000
...略
這里寫了EF PRNT=2,目的是使得優化過程可以通過筆者寫的mopac2xyz程序(http://www.shanxitv.org/212)轉化為VMD可以觀看的軌跡,便于了解優化過程以及提取最終優化后的結構。precise關鍵詞是讓計算精度更高一些。EPS=78.3代表通過COSMO隱式溶劑模型表現外部水環境(介電常數為78.3)。靛青的坐標后面都寫了0,代表在優化過程中固定靛青結構不動。對當前例子是否固定溶質其實無所謂,但半經驗方法終究沒有DFT穩健、普適,對于某些特殊的溶質分子結構描述得并不好,如果初始溶質結構是DFT優化過的,但MOPAC優化時不固定其結構,那可能會把分子結構優化得比較糟糕。
這個MOPAC任務在筆者Intel四核機子上一刻鐘就算完了,輸出文件是solvated.out。對于目前的MOPAC2016程序,筆者發現用32bit版本算此任務一開始會報錯,而64bit版無問題。從輸出文件靠末尾部分可以看到提示THE GEOMETRY MAY NOT BE COMPLETELY OPTIMIZED,并且提示加LET DDMIN=0.0可以優化得更充分。確實,用LET DDMIN=0.0的話,優化還可以進一步進行,結構還會進一步發生一些改變,但此處沒有用這個關鍵詞,是因為當前半經驗級別下優化只是個粗略優化,其勢能面就不是很準確,收斂到準確極小點也沒太大必要,畢竟之后還要用DFT再優化。另外,用LET DDMIN=0.0的話對于這種分子團簇體系往往很難達到收斂限(因為容易震蕩)。
用mopac2xyz程序把solvated.out轉化成.xyz軌跡文件,最后一幀,即優化后的結構,如下所示。圖中把水之間的氫鍵在比較松的閾值下(O-H...O距離3.5埃,鍵角>140度)也顯示了出來,可見在溶質分子周圍水分子形成了應有的網狀結構。
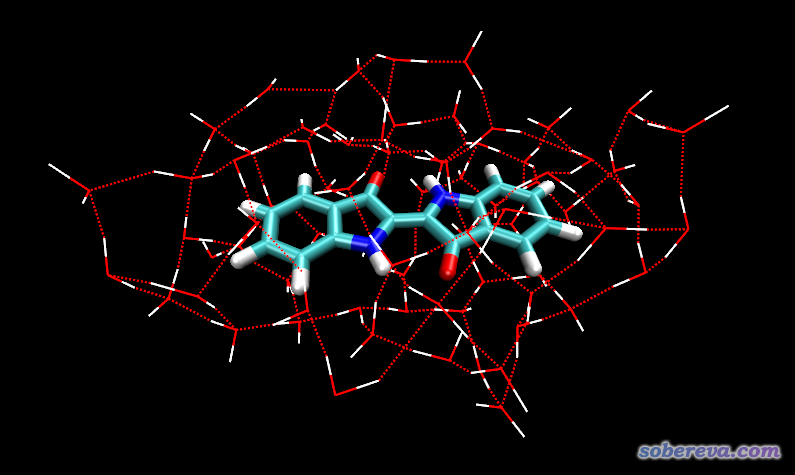
例子到此結束。之后就可以適當修改這個團簇結構,再用DFT優化了。多余的水可以刪,而如果發現溶質有的地方明顯暴露到外頭,也可以在相應位置手動再加點水再優化。
有人會覺得,這樣優化,可能得不到團簇結構能量最小點。確實有這個問題,不過對于比較小的溶劑,特別是水這樣的,這個問題不顯著。本身優化過程步長不是無限小的,優化時是可以翻越勢能面上很淺的、微不足道的勢阱的(比如從MOPAC優化軌跡上就能看到溶劑層分子位置、朝向變化還是很大的),而且最終的結構還是經過UFF->半經驗->DFT三段式優化,雖然可能達不到團簇最小點,但得到的團簇結構起碼也是能量非常低的結構,用它來做量化計算討論問題是合理的(何況也沒有必然要求必須用能量最低的團簇結構,而只要能把溶劑-溶質的顯式作用表達出來就夠了)。但對于尺寸比較大的溶劑,若為了可靠、令溶劑層特征能夠有代表性、反映實際情況,建議還是用MD+量化優化來產生溶劑層包裹的溶質結構。
還有人會想,按照本文的過程,貌似得到的溶劑層排布只有一種,如何才能像MD那樣能夠得到不同的溶劑層排布方式的結構?實際上,在packmol輸入文件中如果寫了seed -1,則Packmol每次運行相同文件得到的結構都是不同的,因為它是利用隨機數的,因此Packmol跑N次,最終就可以得到N種溶劑層排布。