談談怎么判斷分子動力學模擬是否達到了平衡
談談怎么判斷分子動力學模擬是否達到了平衡
文/Sobereva@北京科音 2021-Dec-24
0 前言
時常有人在思想家公社QQ群里或計算化學公社論壇(http://bbs.keinsci.com)里問關于怎么判斷分子動力學模擬中體系是否達到平衡的問題,問是不是看溫度、勢能變化等等信息判斷。然而往往提問的人都不說清楚他的體系是什么特征,導致這根本沒法回答。實際上,對于不同類型的體系,判斷方式有極大的差異。有一點尤其要搞明白:模擬的體系有各種各樣的屬性,達到完全平衡的話,所有這些屬性都不會再有顯著的整體變化(瞬時的波動、漲落不屬于這種變化)。光拿那些較容易達到平穩的屬性來衡量體系是否已經平衡是沒有意義的。衡量是否已達到平衡要用最能反映當前體系從非平衡變化到平衡過程中有關鍵性變化的那些量,或者用最難達到平衡的那些量。本文就用一些有代表性的體系來進行說明,例子都是北京科音分子動力學與GROMACS培訓班(http://www.keinsci.com/workshop/KGMX_content.html)里我詳細講過怎么模擬的體系,有的圖我就直接貼培訓班里的ppt了,分析命令和分析腳本在培訓班里也都講過。
1 小分子液體
水、乙醇、尿素等小分子液態體系的模擬相對來說很容易達到平衡。即便初始結構設置得比較隨意,比如就是用packmol(http://www.shanxitv.org/473)甚至GROMACS自帶的粗糙的gmx insert-molecules來建模,由于小分子擴散運動很容易,通常頂多經過幾百ps的模擬就能達到平衡。對這類體系衡量是否達到平衡用密度(或盒子體積)、壓力、溫度、勢能、動能之類的衡量整體狀態的常見量就可以。對于GROMACS用戶來說,這些量直接就可以通過gmx energy命令從edr文件中提取。
例如,筆者對512個乙醇構成的盒子在常溫常壓下進行100 ps的模擬,在0~30 ps期間參考溫度從0 K線性升到298.15 K。這個模擬對應平衡相(equilibrium phase)階段,目的是讓體系從非平衡達到充分平衡狀態,如果發現這個模擬階段中途就已經達到平衡了,那么就可以延續最后一步的狀態跑較長時間的產生相(production phase)模擬來獲得液體的實際性質。此100 ps平衡相模擬過程中密度變化如下所示
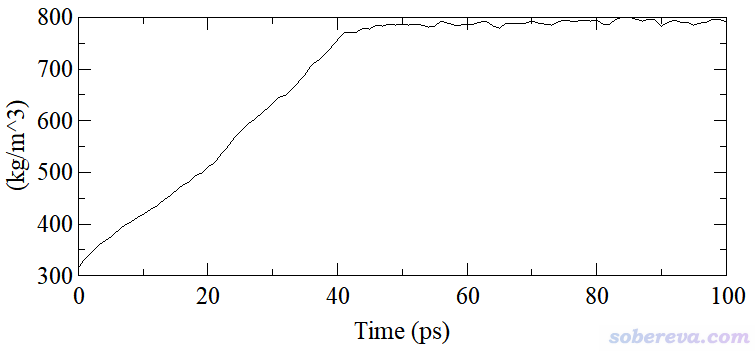
由上圖可見,從密度的變化角度來說,從大約50 ps開始就可以認為已達到平衡了,因為之后僅有瞬時的波動而不再有整體的起落。當前模擬用的初始結構是gmx insert-molecules創建的,為了確保512個分子都能成功插進去,盒子設得比較大,因此初始結構非常稀疏、密度嚴重偏低,這是為什么上圖中模擬前期密度迅速升高。
還可以計算漂移(drift)來進一步從數值上輔助說明上圖已達到平衡。這里所謂的漂移是對屬性隨時間的變化做一個線性擬合,然后求直線的末尾時刻與初始時刻的差值。顯然,如果被考察的屬性如果隨模擬進行沒有整體升高或整體下降,則擬合的直線斜率將為0,drift也就為0。但現實中,就算體系已經平衡了,由于統計誤差,drift也不可能恰好為0,對于密度等屬性來說,只要drift值的數量級遠遠小于統計區段的平均屬性值就行。
計算drift值可以自己在比如Origin等程序里擬合,而對于GROMACS用戶,在使用gmx energy命令從edr文件提取數據后在屏幕上直接就顯示了。對于上圖,如果想對從圖上看已平衡的50~100 ps區間計算drift值,就輸入gmx energy -f eq.edr -b 50 -e 100,屏幕上顯示的信息中Tot-Drift下面的值對于此例是8.7,只有程序輸出的這段區間平均密度789.3的百分之一,因此密度在50 ps后的整體變化可以忽略不計。
對上例再繪制一下溫度的變化,如下所示。可見溫度達到平衡比密度更快,在按照退火設置從0~30 ps期間從0 K上升到300 K后,溫度就不再有明顯變化了。
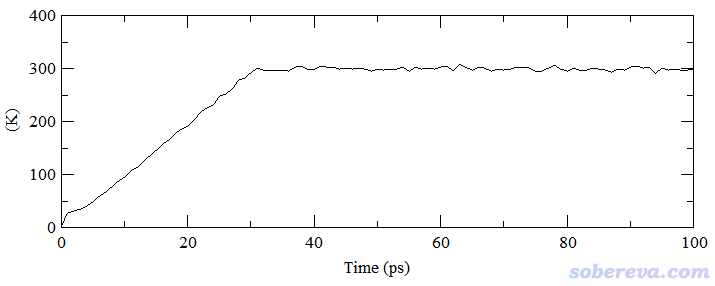
溫度正比于體系內粒子的平均動能的,由于溫度平衡了,動能自然也就平衡了,所以由于前面已經考察過了溫度,故動能就沒必要再檢驗了。通常還會從總能量或勢能的角度再考察一下是否達到平衡。下圖是模擬期間的勢能變化,可見從50 ps開始,勢能也已經平穩了。
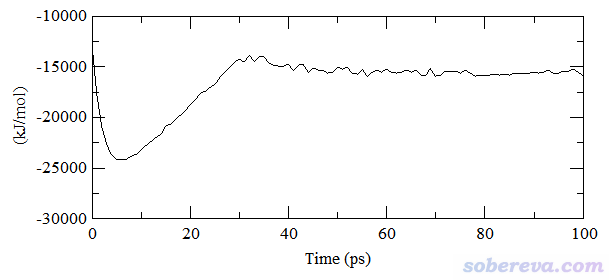
由于上面溫度、密度、勢能這幾個關鍵指標模擬到大約50 ps時都已經基本達到了平衡,所以當前跑的100 ps的平衡相模擬已經夠長了,之后就可以放心地做產生相模擬了。如果有哪一個指標尚未平衡,都不能說體系達到了平衡。
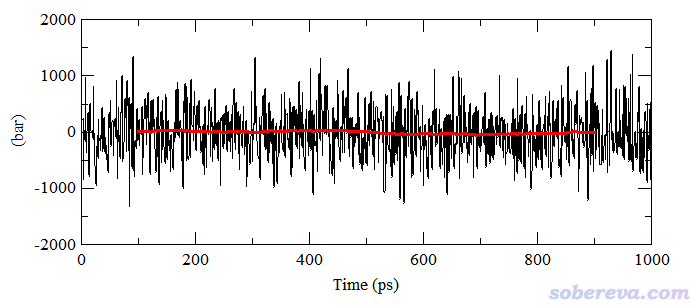
再順帶提及一個初學者經常問的關于壓力隨時間變化的問題。這里我用一個已經達到平衡的含有884個分子的水盒子來說明,此模擬跑了1 ns且記錄了1000次數據。模擬過程壓力隨時間變化如下所示
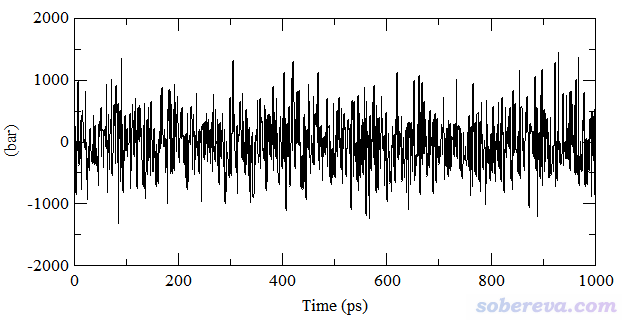
有些人一看這大幅度的壓力波動就慌了,納悶:怎么明明設的參考壓力是1 bar,壓力波動范圍卻達到-1000~1000 bar的程度,而且波動劇烈,這也太夸張了吧?有的初學者還因此以為模擬有問題,或者體系沒有達到平衡。實際上,體系越大,壓力波動會傾向于越小,且壓力的波動隨著粒子數的增多以平方根為比例而減小。對于只有884個水的盒子,這樣的壓力波動是極為正常的!另外,由于液體的可壓縮系數非常小,壓力的很大變化只會導致盒子體積的很輕微變化,所以在模擬過程中盒子尺寸的變化微乎其微。要檢驗模擬過程中控壓是否合理,要關心的是平均壓力而非瞬時壓力。上圖對應的平均壓力為1.8 bar,偏離參考壓力雖然達到80%,但相對于壓力的大范圍波動,這種程度的偏離完全可以忽略不計(哪怕差10 bar都沒關系),也完全不需要顧慮模擬出的性質會由于這個偏差而不準確,畢竟對盒子尺寸影響甚微。另外,上例的壓力drift值算出來為1.98 bar,雖說其數量級和壓力平均值已經差不多大,但由于壓力的劇烈波動造成drift值的統計誤差本來就容易較大,這種程度的drift可以完全忽略不計。
壓力的波動程度相對于其它屬性總是顯得很劇烈,因此難以直接從壓力曲線上觀察壓力的總體變化情況。不過,可以繪制局部平均的壓力曲線來進行考察,也就是對相鄰一定數目的幀的壓力取平均。在Origin、Grace等很多程序中都支持對被載入的數據做這種局部平均化的處理。例如在Grace中,選擇Data - Transformations - Running averages,若將length of average設為200,就代表第i個點的數據取前后相鄰100個點數據的平均值。對上圖的數據做這種處理后,產生的局部平均壓力曲線對應下圖的紅色曲線,可見非常平直,說明在模擬過程中壓力未曾有過明顯的整體的上升或下降。
2 生物大分子
對于蛋白質、核酸這樣的生物大分子,我們關心的對象不是整個體系,而是生物大分子部分,所以檢驗平衡自然是要看生物大分子的狀態是否達到了平衡。最常用的衡量方法是繪制RMSD曲線。RMSD有非質權和質權兩種,分別如下所示,其中粗體r是笛卡爾坐標,ref代表參考結構,A循環所有原子,M是分子總質量。
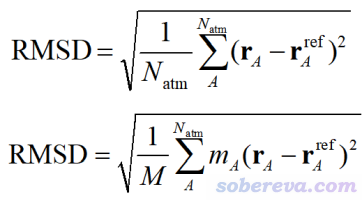
RMSD衡量了當前結構與參考結構的整體差異。考察生物分子是否在模擬中結構達到了平衡狀態,通常以軌跡的第一幀作為參考結構計算各幀的生物分子的RMSD并繪制成曲線,如果從某個時間點開始RMSD曲線基本不再有顯著的整體變化了,就說明結構已經平衡了。通常被計算的是蛋白質或核酸的不包含側鏈的骨架部分原子的RMSD(并且大多不考慮氫原子),也有文章計算的是所有alpha碳原子(這是衡量殘基骨架位置的最關鍵原子)。在計算RMSD之前必須先通過最小二乘法將各幀結構相對于參考結構進行最大程度疊合,從而消除體系的整體運動而令RMSD只體現生物分子內部結構的變化,這稱為align或者least squares fit。GROMACS的gmx rms命令和VMD自帶的Extensions - RMSD Trajectory Tool都可以計算RMSD曲線,分別默認是質權和非質權形式。
在無數生物分子模擬的文章里都給出了RMSD曲線,以證明模擬的時間足夠長。例如下面的圖來自筆者的Biochemistry, 48, 7986 (2009),總共跑了10 ns,這是一個十分典型的蛋白質模擬的RMSD曲線,計算的是alpha碳原子的RMSD,初始結構是晶體結構。
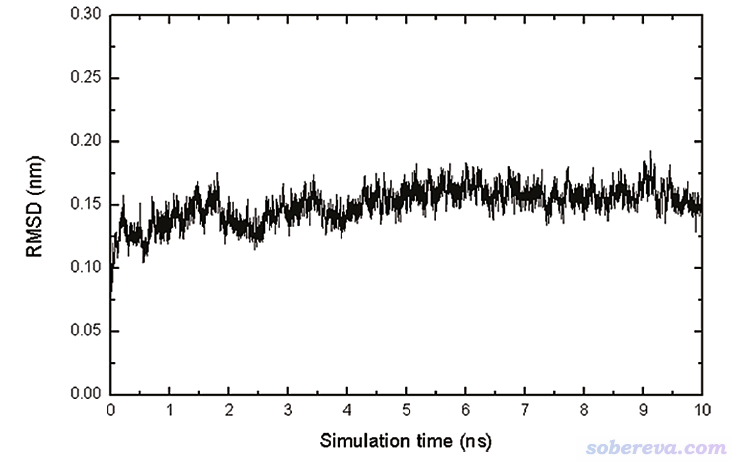
由上圖可見,第一幀是參考幀,其RMSD精確為0。在模擬開始后RMSD迅速增大,一方面是蛋白質的熱運動必然使其結構在一定程度上偏離參考結構,另一方面是體系從非平衡向平衡狀態演變。從上圖來看,蛋白質的骨架在模擬中很快就達到了平衡狀態,也就跑了幾百ps后RMSD就達到了0.13 nm左右,之后就沒有顯著的變化,后續模擬過程中蛋白質都相當于在最穩定構象對應的相空間的勢阱里游歷。
需要注意的是,蛋白質骨架的RMSD曲線并不總能反映出蛋白結構特征變化的全貌。一方面,RMSD的計算涉及到一大堆原子的平均,若一個很大的蛋白某個局部出現一定程度的顯著的、不可忽視的結構變化,在RMSD計算過程中它會被很大程度淹沒掉。另一方面,骨架的RMSD不能反映側鏈上出現的一些關鍵變化。例如上面這篇文章發現,在423 ps的時候磷酸化酪氨酸574殘基側鏈出現了顯著的翻轉,這使得被研究的酪氨酸激酶活性位點打開,這在分子生物學角度是一個很重要的現象,而從alpha碳的RMSD曲線上則完全體現不出來這一點。文中圖4(A)繪制了這個殘基側鏈二面角隨時間的變化,這才讓這關鍵信息被展現出來。
如果蛋白質的RMSD曲線在已經跑的模擬時間內還沒有穩定在一個值附近且持續了較長時間(有零點零幾nm的上下波動是可以接受的),而你又需要蛋白質已達到平衡的結構,通常就需要延長模擬時間續跑,直到RMSD基本平穩。例如下圖的RMSD仍有明顯的整體升高趨勢,況且才跑了1 ns,跑1 ns經常還達不到平衡,所以應當繼續再跑跑。
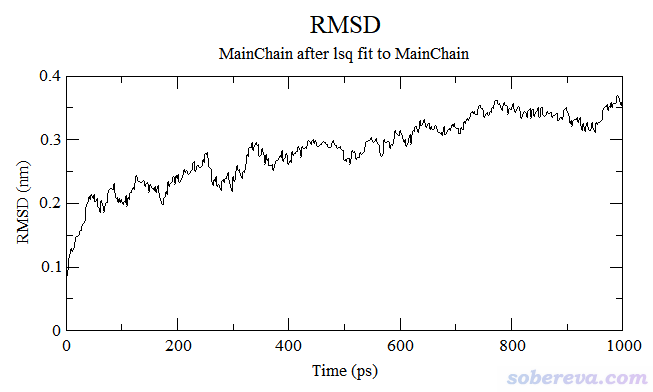
有些人試圖用衡量小分子液體的方式衡量生物分子模擬體系是否達到穩定,即還用溫度、密度、勢能等參數來衡量,這明顯不行。因為這些量的收斂速度遠快于蛋白質的RMSD的收斂,在這些指標已平穩的時候蛋白質部分通常還明顯沒達到平衡。雖然蛋白質整體結構的進一步變化必然會對勢能等因素有多多少少的影響,但在勢能曲線上基本體現不出來。通常模擬蛋白質都是在顯式水模型下模擬的,勢能絕大部分都是水所貢獻的,顯然蛋白質結構的改變對總勢能的影響通常會被淹沒;而且就算在比如GB隱式溶劑模型下模擬(GB模型簡介看《談談分子模擬中的隱式溶劑模型與GB模型》http://www.shanxitv.org/42),蛋白某些局部結構的改變往往在總的勢能曲線上也反映得很不明顯,除非是構象真的有肉眼都能清楚看出的劇烈改變。舉個例子,上面圖中蛋白質骨架的RMSD雖然還沒平衡,但整個蛋白質+水體系的勢能曲線圖已經完全平衡了,如下所示(此模擬之前已經做了限制性動力學,即給蛋白質骨架加了限制勢,水已經弛豫過了一定時間,所以看上去一開始勢能就已經平衡了)。
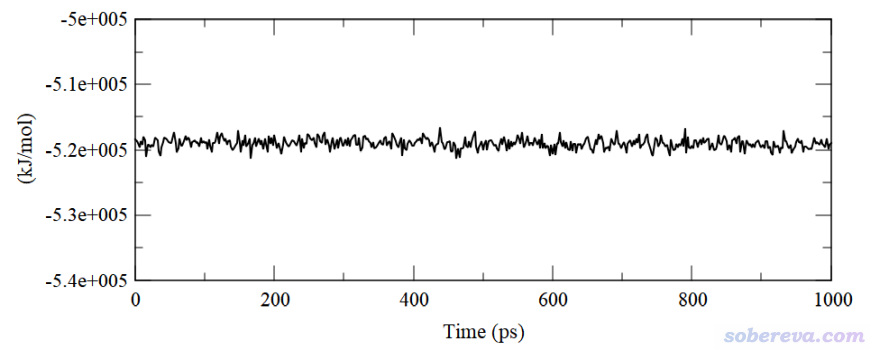
常有人問怎么他的RMSD曲線中途出現了突變。下面兩張圖是計算化學公社論壇上我答疑時看到的,可見中途RMSD都出現過劇烈的變化,完全超越了平衡狀態下的正常波動的程度。這種突變必須給予明確的解釋,要不然審稿人看見肯定會問你是怎么回事。始終要記住,RMSD曲線是軌跡變化的定量反映,想解釋抽象的RMSD曲線的突然變化是什么導致的,當然要去用VMD程序看軌跡動畫才能充分弄清楚原因。絕對不可能RMSD曲線出現厲害的突變時在軌跡動畫上卻看不出任何端倪。如果你的眼力不好,建議先對結構進行疊合,然后在VMD里把突躍前和突躍后的兩幀疊加顯示出來。比如第8200幀相對于第8100幀出現了RMSD的巨大升高,想弄清楚結構什么地方發生了怎樣的巨大變化,在VMD的Graphics - Representation里選Trajectory標簽頁,在Draw Multiple Frames里輸入8100:100:8200,并且把Coloring Method設為Trajectory - Timestep,這樣這兩幀就會用不同顏色一起顯示出來便于比較在哪里結構有明顯變化。
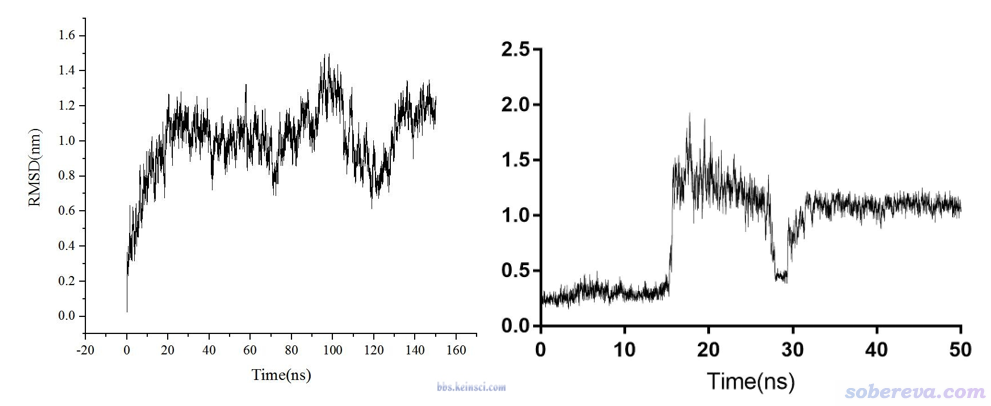
還老有人問他們的RMSD曲線怎么跑成了下面這樣,即出現了RMSD瞬間的上上下下的改變。
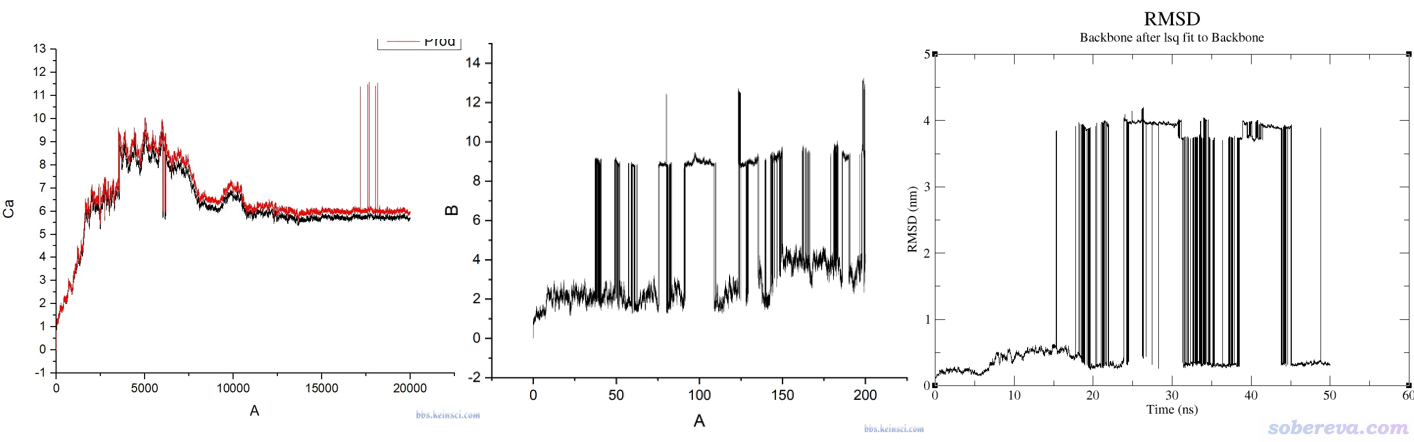
上圖這種情況只要在VMD里觀看軌跡動畫自然能明白原因。就算不看軌跡也100%能斷定肯定是因為周期性方式記錄軌跡,而被計算RMSD的對象在模擬過程中越過了盒子所致。當某一幀突然體系被弄到了盒子另一頭,RMSD自然會出現瞬間的大幅變化。解決這種RMSD曲線不連續性的方法很簡單,消除被計算RMSD的對象的整體運動,并保持此對象的完整性就完了。比如在GROMACS里,可以用gmx trjconv結合-pbc mol使得轉換出的新軌跡中的分子保持完整,計算RMSD前再經過疊合來消除蛋白質的整體運動,其RMSD曲線就不可能突變。還有一種情況是被計算RMSD的對象是由多個分子組成的,比如有多條鏈的蛋白、蛋白+配體復合物、含有兩條鏈的核酸,這種情況可能模擬中途只有其中一部分跑出盒子而另一部分還在盒子內,光是用-pbc mol保持單分子完整的話此對象的不同部分還是分家的,對這種情況需要用gmx trjconv結合-pbc cluster讓指定的組在軌跡中一直保持完整,組的定義就對應于你要計算的RMSD的對象。順帶一提,對生物分子的模擬,我一般建議讓程序在模擬過程中就一直對生物分子消除平動轉動,對于GROMACS跑蛋白質來說就是設comm-grps = protein和comm-mode = angular,這樣就避免了生物分子中途跑出盒子的問題。
不同的生物分子體系的RMSD達到平穩時RMSD的數值會有一定不同。柔性越大的體系,平衡時RMSD越大。比如DNA體系骨架柔性比一般較剛性的蛋白質要大,這會體現在達到平衡時RMSD相對較大上。
通過RMSD考察是否平衡不是必須得用初始幀做參考結構。J. Chem. Phys., 109, 10115 (1998)認為以初始結構當參考幀來觀察RMSD會高估達到平衡的時間,因此會把一部分其實已經達到平衡的軌跡誤認為還沒平衡,導致浪費了一些軌跡。作者的觀點是相對于初始結構,模擬開始后RMSD的增加由兩部分組成:(1)非平衡態到達平衡態 (2)平衡態時在相空間采樣。實際上(1)滿足了的時候本質上生物分子就已經平衡了,這比起以初始結構為參考幀計算的RMSD曲線達到平穩時更早。為了判斷什么時候就已經滿足了(1),作者建議以已經平衡了的結構作為參考幀來計算RMSD。下圖是文中的圖,作者取500、600、700、800、900 ps分別作為參考幀繪制了RMSD曲線,并假定這些時刻體系都已經達到了平衡。由圖可見,每條曲線在參考幀附近都是迅速上升,這體現出已平衡的結構在相空間采樣期間由于熱運動自然而然地相對于參考結構發生的結構改變;而在圖的前200 ps,這幾個曲線都明顯上升,因此體現出前200 ps是確確實實結構沒有達到平衡、有整體性變化的階段。
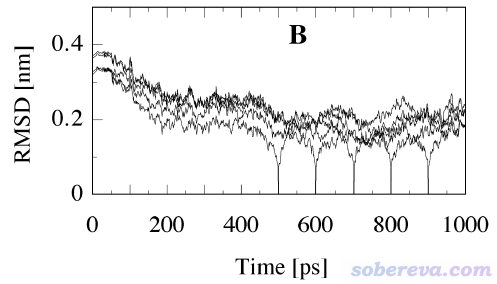
上面這篇文章的做法只是一家之言,不是什么流行的做法,大家僅供參考,許多實際情況也沒有這么理想化。另外,上面這篇文章的做法并不能判斷當前的模擬是否已經跑成了平衡狀態,而只適合你已知體系已經平衡了,然后以它為參考結構,再去判斷軌跡從哪里開始就已經達到了平衡并因此能夠用于正式分析。那篇JCP文章發表時間較早,當時計算條件較差,所以對軌跡比較珍惜,能少浪費一點是一點;而如今GPU加速的計算條件下,普通的蛋白質+顯式水一天就能輕松跑100ns以上,也就沒必要那么較真了,因此此文章的方法也沒有太大實用價值了。不過值得一提的是,如果你是從一個比較離譜的結構開始跑的動力學,那么上文的做法還是比較有價值的。比如模擬一個未折疊的小肽的折疊過程,如果以初始未折疊的散亂結構作為參考幀來算RMSD曲線,那明顯不太適合,最后已平衡的折疊后的結構的RMSD曲線肯定非常大。如果你從已經達到平衡的折疊好的結構當參考幀來算RMSD曲線,就可以比較確切地判斷什么時候體系已經平衡了、已經形成了比較穩定的折疊結構,而且根據RMSD曲線從一開始變化的過程上(必然是整體逐漸降低)還可以考察小肽的構象是怎么一點點趨近于最終平衡結構的。
提醒一下,用結構的RMSD是否平穩來衡量是否達到平衡切勿不分場合地亂用。比如有人做分子團簇的模擬,也試圖用RMSD判斷穩定了沒有,這明顯不行。分子團簇的動力學過程中,團簇的構型不斷變化,分子不斷運動、位置不斷改變,因此RMSD完全亂掉了,什么也說明不了。這一點根據RMSD的定義結合基本邏輯自然就能明白。對磷脂膜的模擬也是,不可能用所有磷脂的RMSD來衡量是否平衡,因為模擬過程中磷脂分子會不斷側向擴散,RMSD不可能最終平穩到基本不變。
3 互溶與分離過程
對于小分子間互溶或分離過程,要判斷是否達到平衡,應當取最能直接反映這種過程特征變化的量。
例如,下面的例子是一開始處于兩相的水和乙醇的體系,在模擬過程中逐漸互溶的過程。在這個過程中什么特征變化特別明顯?顯然可以想到氫鍵的變化。一開始水和乙醇只在界面處彼此間能形成氫鍵,數目相對較少;而模擬到互溶后,水和乙醇充分混到一起,彼此間顯然能形成大量氫鍵。因此,考察水和乙醇之間的氫鍵數目的變化就能準確地考察什么時候互溶過程已充分完成、體系已達到平衡。用GROMACS的gmx hbond命令,或者VMD自帶的Extensions - Analyais - Hydrogen Bonds插件就可以做這種統計。從下面的圖中可見,模擬一開始氫鍵數目迅速增加,說明互溶不斷加劇,到了200 ps時氫鍵數目基本就穩定了,因此可以認為200 ps的時候這個體系已經達到了平衡。
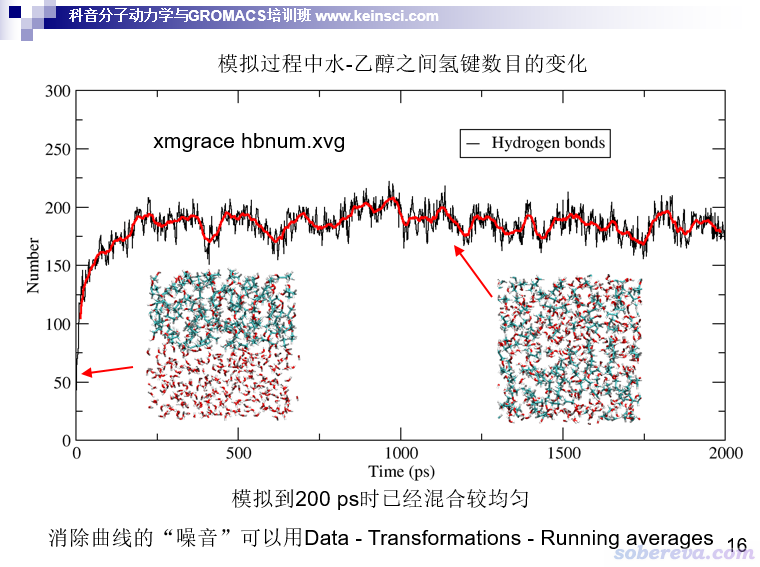
當然,對上面的例子統計氫鍵數目變化不是唯一考察是否達到平衡的做法,還可以自己寫VMD tcl腳本考察乙醇附近水的數目的變化、計算乙醇或者水的溶劑可及表面積(SASA)等等,因為這些量都是對互溶過程很敏感的量,互溶過程中它們都會發生很大變化。此外,還可以監控水和乙醇間非鍵相互作用能,對于GROMACS用戶可以將這兩部分都設為能量組,動力學跑完后就可以從edr文件中提取這兩類分子間非鍵相互作用能隨時間的變化,體系平衡時這個量肯定也已經平穩了。
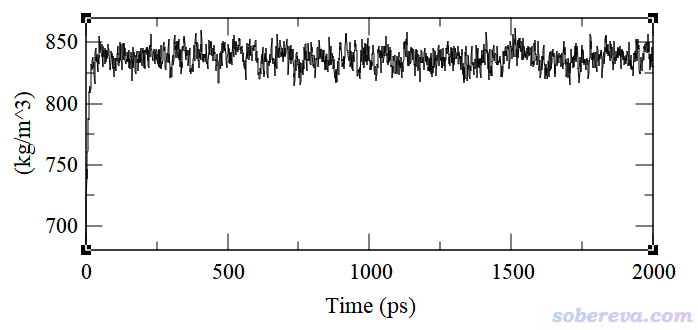
對上面這個體系雖然也可以去考察勢能、密度等屬性的變化,但是憑這些難以判斷什么時候體系真正達到了平衡,因為這些量太容易達到平穩。下圖是水和乙醇互溶過程中體系密度的變化,可見在大約50 ps的時候曲線就已經平穩了,然而如前所見,此時水和乙醇根本還沒完全互溶。筆者也考察了此模擬過程勢能的變化,也是大約50 ps就已經平穩了。所以,像溫度、勢能、密度那些很容易達到平穩的量,只是體系達到平衡的必要而非充分條件,不能只拿這些量說事,除非是本文第1節說的均勻的小分子液體之類的簡單體系。
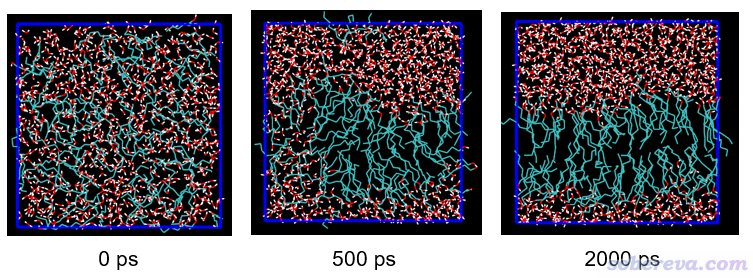
再看另一個例子,模擬的是水-正辛醇自發相分離現象,這在一定程度上是互溶的逆過程。如下圖所示,一開始把正辛醇和水均勻混合在一起,進行一段時間模擬后正辛醇和水相互分離開,并且正辛醇形成類似磷脂雙層膜的結構,親水的羥基頭部朝水,而疏水的烴鏈尾巴朝內。
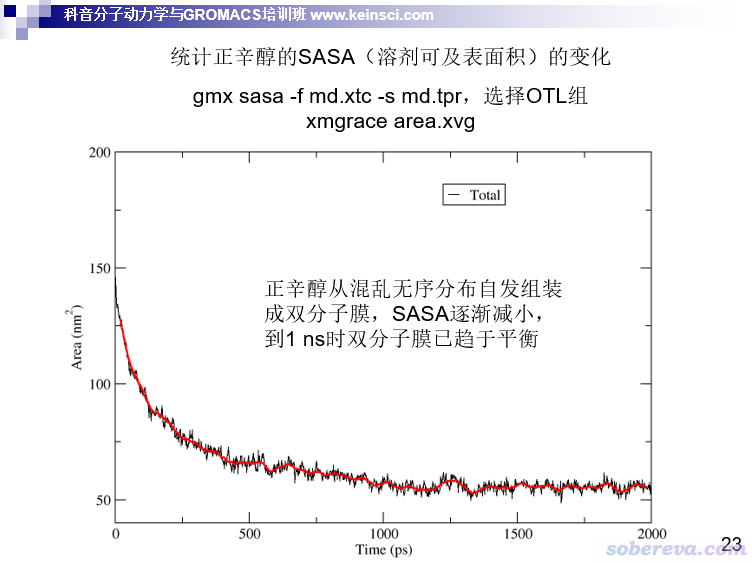
怎么衡量這個過程是否達到了平衡?很容易就想到用SASA來衡量。一開始正辛醇散亂分布,和溶劑水接觸面積必然很大,體現在SASA很大上;而當正辛醇組裝成為膜之后,就只有頭部才能挨著水,自然SASA就比一開始小得多了。什么時候SASA曲線平穩了,就可以說明雙層膜已經組裝好并且結構已經平穩了、體系達到平衡了。如下圖所示,隨著正辛醇與水的分相,SASA不斷下降,在大約1 ns時就不再下降了,因此可以說1 ns的時候體系已經達到了平衡。
如果想為了穩妥,可以再檢驗一下密度和勢能隨時間的變化,筆者發現從勢能變化的角度上看在1 ns的時候也已達到了平衡,而單從密度角度來看則800多ps就已經平衡了。若從溫度角度看,則模擬剛一開始就已經平衡了,所以檢驗溫度是否達到平衡沒多大意義。
再來看一個萃取過程的模擬。一開始把水、甲苯按體積比1:1對應的分子數加入到體系中混勻,并隨機加入少量的乙醚,看看水和甲苯分相后,乙醚是否會被甲苯萃取,即只出現在甲苯一相。
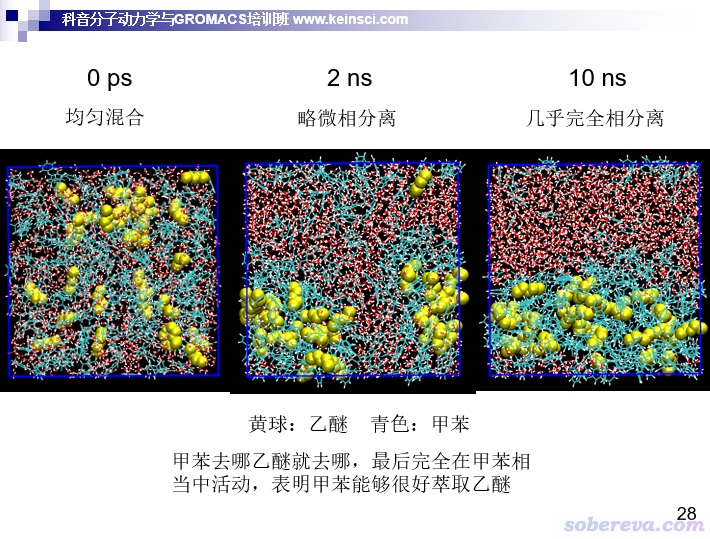
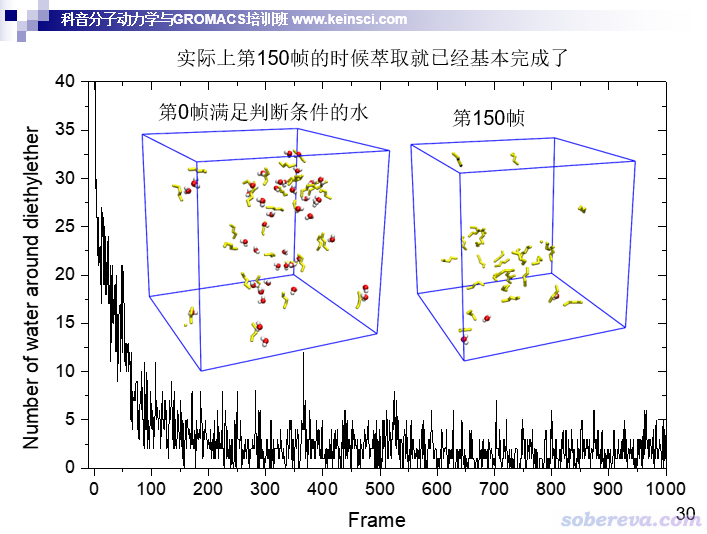
對于上述過程,若達到了平衡,必須水和甲苯兩相已經穩定形成了,并且乙醚的分布也已經穩定了。水和甲苯的分離,可以按照前面說的用兩部分各自的SASA的變化來衡量,而乙醚的分布怎么衡量其穩定與否?方法很多,比如可以考察乙醚和水、甲苯的作用情況或接觸情況。由于乙醚和水之間可以形成氫鍵,故可以觀察它們之間的氫鍵數目隨時間的變化。還可以自己寫個VMD tcl腳本,統計一下乙醚周圍一定距離(比如3.5埃)內水分子的數目,如下所示。下圖還用相同的判據把乙醚(黃色)周圍的水在VMD中也顯示了出來,便于和變化曲線對照以更好地了解實際情況。可見在第150幀對應的時刻,可以認為甲苯已經把乙醚萃取完成了,乙醚幾乎都已經在甲苯相了,因此至少從萃取程度的角度來說體系已經平衡了。
也可以用上面的做法考察乙醚附近的甲苯數、甲苯附近的水數,體系完全平衡時肯定這些量也都平衡了。大家寫文章證明體系已經平衡時可以把這些量隨時間的變化都畫到一張圖上,顯得依據更充分。
4 氣體的蒸發
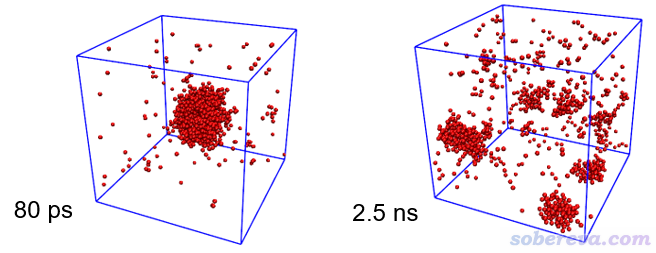
這個例子是在一個10*10*10 nm的盒子中心放一個含有884個水構成的水球,經過常溫下模擬達到平衡后,把參考溫度設到450 K繼續跑5 ns,使水球蒸發,進而變成極高壓的水蒸氣狀態。整個模擬都用NVT系綜。模擬過程中的兩幀如下
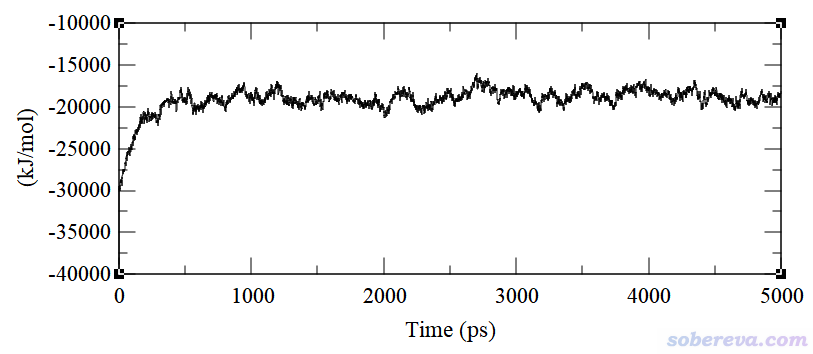
對于這個過程,想判斷模擬到什么時候體系已達到平衡,可以先看看勢能的變化,雖然這不能作為唯一判據,但至少這個量必須能達到平穩才算平衡了(無論對什么體系皆是如此)。模擬過程的勢能變化如下,可見大約500 ps時勢能就已平穩。
勢能只是能量角度上的一個標準,但凡有可能,最好還是再結合一個結構方面的標準來進一步判斷平衡,明顯會更有說服力。考慮到水球蒸發后會形成一大堆孤立的水分子或小水簇,顯然統計一下簇的狀況就可以從結構角度對平衡情況進行考察。在GROMACS程序中自帶了gmx clustsize命令可以做這種分析。其中會給出簇的數目隨時間的變化,如下所示。可見一開始簇的數目極少,因為初始結構主要就是一個在盒子中央的大水球,而隨著高溫下的模擬進行,大水球蒸發并形成一堆水分子或小水簇,因此簇的數目迅速增長,到達約500 ps時不再顯著變化。因此,結合勢能和簇的數目的變化,我們可以下結論這個體系在500 ps時已達到平衡。
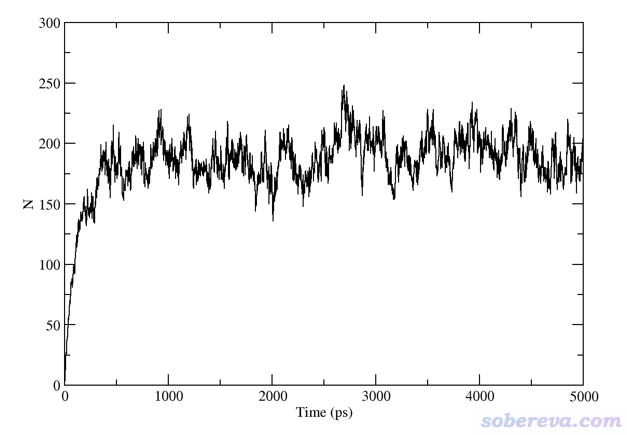
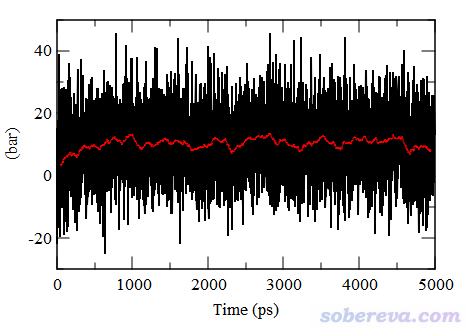
對此體系考察溫度變化依然沒什么用,此體系才模擬了幾ps,溫度就已經平穩在期望的450 K左右了。而考察壓力變化倒還能說明一些問題。如下所示,模擬剛開始壓力很低,畢竟大水球周圍全是真空區。隨著模擬進行、水球蒸發,壓力有明顯的上升趨勢。到了大約500 ps時,壓力就差不多達到了之后模擬過程中壓力的平均值了,這也體現了體系此時達到了平衡。
5 磷脂膜
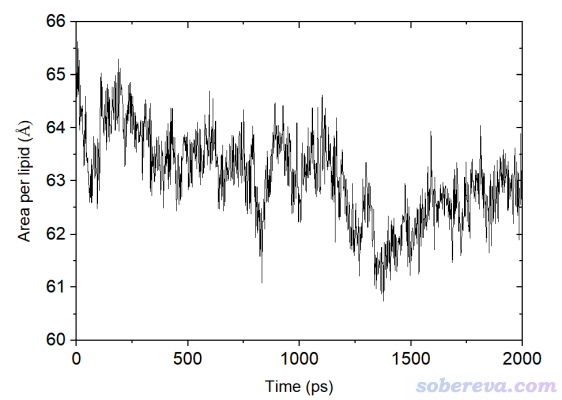
對磷脂膜這類體系模擬,也可以考察密度、勢能等常規屬性的變化,但光是這些易平穩的屬性達到平穩還不足矣判斷磷脂膜已平衡。磷脂膜有個關鍵參數是磷脂頭部平均面積(area per lipid)。假設磷脂膜平行于XY平面,將模擬的每一幀盒子的X和Y尺寸提取出來,相乘,然后再求時間平均,再除以每一層磷脂數,即可得到這個值。膜體系達到平衡的話,至少平均磷脂頭部面積得達到平穩。平衡后的這個參數值和實驗值相比偏差如何也是衡量磷脂力場質量好壞的關鍵指標。下圖是筆者模擬的DPPC磷脂雙層膜的磷脂頭部平均面積隨時間的變化圖,在標況下跑了2 ns,用半各項同性控壓。從這個圖來看,模擬至少在1600 ps之前還沒達到平衡。從1600 ps開始,磷脂頭部平均面積看上去大致平穩了,但是光靠剩下的400 ps模擬的情況還不夠下結論。為了保險起見,應當至少再模擬1000 ps觀察一下趨勢,如果此期間曲線還是平穩的,那么就可以說磷脂膜達到平衡了。
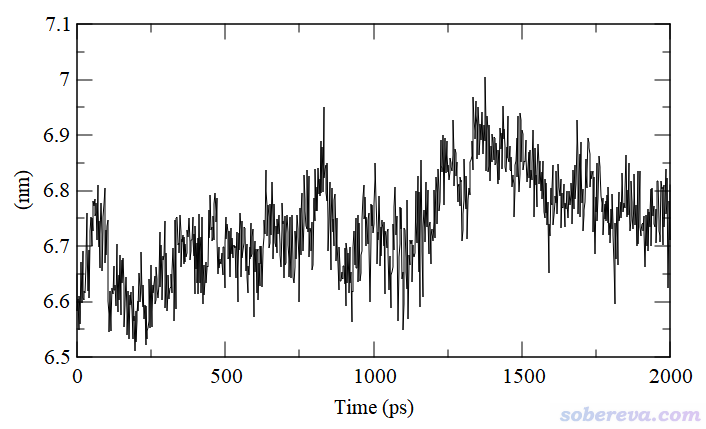
另外,還可以看一下垂直于磷脂方向(Z方向)盒子的尺寸隨時間的變化,如下所示。可見這個方向盒子尺寸變化和磷脂頭部平均面積有顯著的負相關性,也即Z方向尺寸越大、XY面積越小。從目前已經跑的2 ns來看,判斷體系已經達到了平衡還言之過早,還應當再跑跑。
還可以同時從磷脂膜厚度隨時間的變化角度檢驗體系是否達到平衡。可以自己寫個VMD tcl腳本,分別計算磷脂膜上層和下層的所有磷原子的平均Z坐標并求差,看看以這種方式衡量的厚度隨模擬進行什么時候達到穩定。
還有其它一些和膜有關的參數也可以用來檢驗平衡。比如膜的可壓縮度,計算公式如下,尖括號代表對一段軌跡做時間平均,S是瞬時的膜面積。可以取軌跡的不同區間來計算,比如0~2、1~3、2~4 ns...,看看這個量是否隨模擬進行已經趨于平穩。算這個需要自己寫腳本。
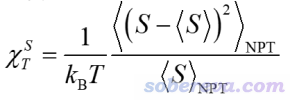
上面這個體系的勢能和密度曲線達到平穩的速度遠快于磷脂頭部平均面積,筆者發現才跑到400 ps左右勢能和密度就都已經算得上達到平穩了,再次體現不要光拿這兩個量說事。
注意,磷脂膜的平衡遠遠遠遠比第1節說的小分子液體那類體系慢得多得多得多,磷脂的側向擴散運動是相當慢的。上面的模擬筆者是用一個他人已經平衡好的磷脂膜為初始結構跑的。如果你是用比如genmixmem程序(見《生成混合組分的磷脂雙層膜結構文件的工具genmixmem》http://www.shanxitv.org/245)從頭搭建一個磷脂膜結構,可能得花十幾甚至幾十ns才能達到平衡。如果是多種磷脂組成的混合膜,有時甚至可能得跑到上百100 ns才能達到絕對充分平衡。
6 柔性小分子
要注意,并不是所有體系都可以像前面幾節的例子一樣達到一般意義上的平衡。有些體系在模擬過程中會不斷翻越勢壘,在不同構象之間反復切換,這種情況下不可能指望結構的RMSD曲線能收斂到一個特定值附近并一直保持基本不變,也不可能指望勢能能平穩在一個值附近始終穩定。比如一些小肽,沒有普通蛋白質那樣單一的穩定構象,就是會在不同構象之間變來變去,這可以通過繪制自由能面圖(見《淺談PCA與g_covar+g_anaeig+ddtdp+sigmaplot做自由能面圖的方法》http://www.shanxitv.org/73)之類的方式討論。很多高度柔性的小分子也是如此,有大量低能量的構象,且這些構象間的勢壘也比較低。筆者之前寫過一篇文章《使用Molclus結合xtb做的動力學模擬對瑞德西韋(Remdesivir)做構象搜索》(http://bbs.keinsci.com/thread-16255-1-1.html),當時在比較高的400 K下(為了加速采樣)對瑞德西韋分子跑了100 ps動力學,50 fs保存一幀,RMSD圖如下所示,共2000幀,每隔500幀我繪制了一個結構附在圖上。可見,此分子的構象變化始終沒有消停,RMSD曲線反復明顯波動。
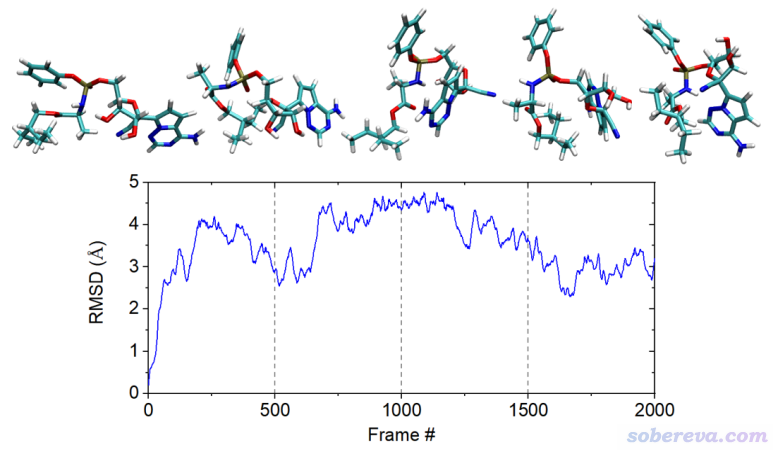
當然了,在常溫下,由于分子的熱運動比400 K時弱得多,瑞德西韋的構象變化肯定不會這么頻繁、劇烈,但跑的時間足夠長的話,對這樣的柔性分子還是會看到中途有顯著的構象變化并導致RMSD曲線出現突變。所以對于柔性體系,不要指望能達到平衡,除非溫度低到體系總是會保持一種構象。或者說,在一般溫度及更高溫度下,對于高度柔性的分子,構象反復變化的狀態本身就是這個體系對當前情況而言的平衡狀態。
7 總結
本文以一系列模擬體系作為例子,示例了如何判斷分子動力學模擬有沒有達到平衡。可見判斷是否平衡沒有唯一方法,問別人怎么判斷平衡時如果不說清楚被模擬的體系,誰也不可能給出確切答案。顯然本文的例子不可能面面俱到,讀者應充分理解被模擬的體系的特點、搞清楚判斷平衡的各種指標的定義和意義,結合具體問題選擇最適合的方式進行判斷,務必要在本文的例子的基礎上靈活變通、舉一反三。對于一些比較特別的模擬體系,還可能需要讀者自己設計一些最能反映體系平衡情況的專一性的幾何判斷標準,由于其特殊性往往沒有現成的計算程序,這時就需要自己寫腳本來實現計算。