使用Multiwfn繪制分子和固體表面的距離投影圖
使用Multiwfn繪制分子和固體表面的距離投影圖
文/Sobereva@北京科音 2021-Feb-28
1 前言
不止一次在思想家公社QQ群和計算化學公社論壇上有人問我下面的這種圖是怎么繪制的
實際上上面這種圖就是定義一個平面,把體系的范德華表面上各個位置距離此平面的垂直距離的大小通過不同顏色展現而已。可能有人覺得直接看范德華表面圖不就完了,何必作這種圖,實際上這種圖是有一定獨特價值的,可以把不同原子所在位置的深淺以直觀且明確的方式顯示,便于某些情況的討論,比如原子被包埋情況、周圍的原子產生的位阻情況等。下面筆者管這種圖叫做“表面距離投影圖”(surface distance projection map),我覺得這么稱呼比較形象、貼切。
筆者在Multiwfn中已經實現了繪制類似上面這種圖的功能。這個功能設置靈活,作圖效果理想,而且支持不同方式定義體系表面。下文先簡要介紹此功能的特征和用法,然后將通過一個[Ru(bpy)3]2+配合物的例子和一個Cu(111)表面的例子演示怎么繪制表面距離投影圖。
讀者請使用2021-Feb-28及以后更新的Multiwfn。Multiwfn可以在官網http://www.shanxitv.org/multiwfn免費下載。不了解此程序的話請閱讀《Multiwfn FAQ》(http://www.shanxitv.org/452)和《Multiwfn入門tips》(http://www.shanxitv.org/167)。
2 Multiwfn中的表面距離投影圖的繪制功能的使用
表面距離投影圖的繪制功能是Multiwfn的主功能300的子功能8。此功能支持三種體系表面的定義:
(1)準分子(promolecular)電子密度的等值面。這種電子密度是將各個原子自由狀態下的電子密度簡單疊加得到的近似的電子密度,沒有考慮原子間相互作用導致的電子轉移和極化
(2)基于電子波函數計算的電子密度的等值面
(3)通過原子球疊加得到的表面。原子球半徑是Bondi范德華半徑乘以用戶指定的系數
耗時關系是(2)>(1)>(3)。通常建議用(1),因為耗時低,表面處處光滑,還不需要提供波函數信息。(3)雖然耗時最低,但其表面在原子球交界處有明顯縫隙。原理上(2)最理想,但耗時相對較高,而且需要做量子化學計算才能得到波函數。使用(1)或(2)時電子密度等值面取什么數值在很大程度上是隨意的,可以根據實際需要并反復嘗試。等值面數值取得越小,表面就越大。
對于以(1)或(3)方式定義表面的情況,需要用的輸入文件只需要含有原子坐標即可,比如pdb/mol/mol2/xyz/gjf等等等等。對于用(2)的情況,輸入文件必須包含波函數信息,比如wfn/wfx/mwfn/fch/molden等格式。Multiwfn支持的這些格式說明以及產生方式詳見《詳談Multiwfn支持的輸入文件類型、產生方法以及相互轉換》(http://www.shanxitv.org/379)。
此功能還支持周期性體系,考慮周期性的話輸入文件必須包含晶胞信息。Multiwfn支持的包含坐標信息又包含晶胞信息的文件在《使用Multiwfn非常便利地創建CP2K程序的輸入文件》(http://www.shanxitv.org/587)第2節列舉了,包含波函數信息又包含晶胞信息的文件在《使用Multiwfn結合CP2K通過NCI和IGM方法圖形化考察固體和表面的弱相互作用》(http://www.shanxitv.org/588)的2.1節說明了。
表面距離投影圖在Multiwfn中的計算原理示意如下:
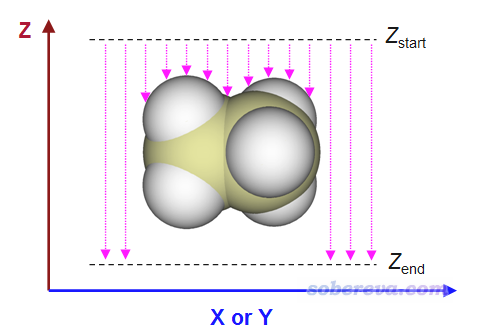
用戶需要定義X和Y范圍,這決定了作出來的圖涵蓋什么區域。還需定義Z_start和Z_end,對每個(x,y)坐標,程序會從Z_start往Z_end逐漸掃描,直到達到體系表面內或者達到Z_end。上圖中粉色箭頭長度就是表面距離投影圖里每個(x,y)位置的數值的負值。根據計算原理可知,給Multiwfn用的輸入文件中分子的朝向必須合適,這樣得到的距離投影圖才能說明你想說明的問題,比如原子在你感興趣的方向的暴露情況。
程序在掃描的時候是有固定步長的,默認為0.05埃,這是精度與耗時的較好權衡。步長越小得到的Z位置精度越好,但耗時也越高。對于基于前述(1)、(2)方式定義體系表面的情況,程序在判斷最終Z位置時還會自動做個線性插值,使得得到的Z坐標精度能提升一個數量級。
程序默認的X、Y范圍是根據體系的邊界原子位置自動往四周延展一些來確定的。Z_start默認為Z坐標最大的那個原子的坐標,用戶也可以改得稍微再比它大一點。默認的Z_end是Z坐標最小的那個原子的坐標。X、Y方向的格點數越多,圖像越精細,但耗時越高。默認這兩個方向都是300個點,這就已經很大了。
程序默認繪制的是平面填色圖+等值線圖。色彩刻度下限和上限分別默認是Z_end-Z_start和0,等值線默認是25條,數值均勻分布在Z_end-Z_start和0之間。等值線的數目可以在計算前由界面里的相應選項修改,等值線具體定義也可以在計算后的后處理菜單里的等值線設定界面里自行修改。
3 實例1:繪制[Ru(bpy)3]2+陽離子配合物的表面距離投影圖
此例對[Ru(bpy)3]2+陽離子配合物繪制表面距離投影圖,Multiwfn自帶的examples\excit\Ru(bpy3)2+.gjf文件里包含了其結構。將此文件載入Multiwfn,進入主功能0可以看到下圖
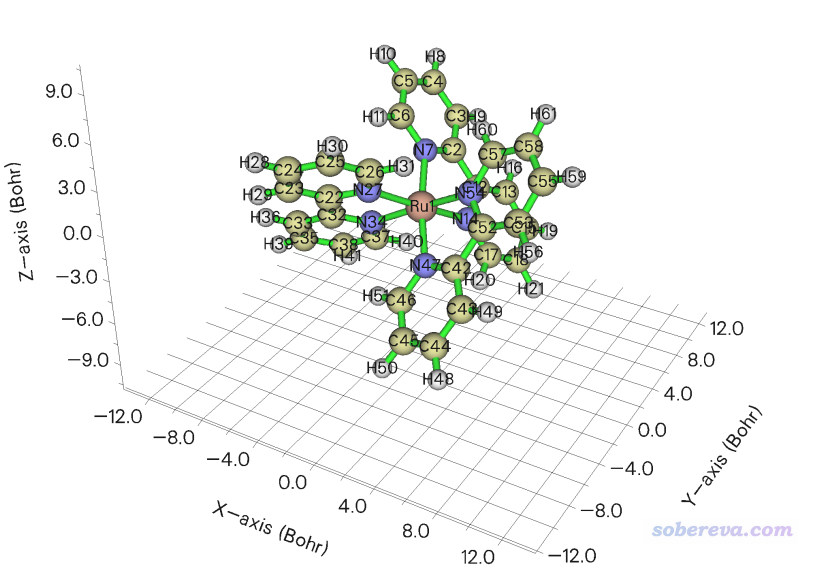
可見此體系中Ru被周圍三個聯吡啶配體所包圍。表面距離投影圖可以直觀地反映過渡金屬配合物中中心金屬被包埋的程度,這和它是否容易與外界分子接觸而發生反應有密切關系。當前我們就使用上圖的分子朝向來繪圖,如果想繪制其它朝向的話就在GaussView里按住alt鍵拖動分子旋轉其朝向然后重新保存gjf文件。
點擊主功能0的圖形窗口右上角的RETURN按鈕返回,然后輸入
300 //其它功能(Part 3)
8 //繪制距離投影圖
0 //基于默認設置直接繪圖
此時程序就開始計算了,在默認設置下是以準分子密度0.05 a.u.等值面作為體系的表面。算完后會進入一個后處理菜單,里面提供了非常豐富的選項用于調節作圖設置,請大家自行嘗試,這里就不一一累述了。這里就直接選0把圖像顯示在屏幕上,你會看到下圖(用于發文章的話記得應當用選項1保存圖像文件,比直接在屏幕上顯示的線條更平滑)
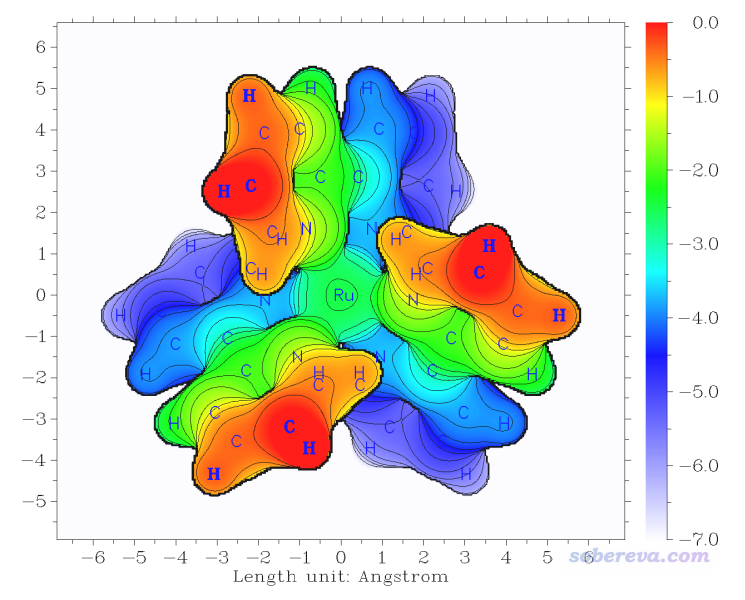
可見在默認設置下作圖效果就已經非常好了,令人眼前一亮。Z位置不同的區域通過顏色很鮮艷地區分開了,等值線使得圖像更有層次感。圖中每個點的數值為Z'-Z_start,其中Z'是這個(x,y)位置處從Z_start向Z_end掃描過程中滿足判斷條件時的Z坐標。圖中純紅色的地方數值為0,是因為剛開始掃描時,第一步在Z = Z_start的位置處就已經滿足了判斷條件(即電子密度>0.05 a.u.,處在體系表面內)。更直觀地說,如果你把Z_start位置想象成當前屏幕的位置,圖中越藍的地方距離屏幕越遠,而純紅的地方表示相應位置的表面在屏幕里側。
下面我們再看改用原子球疊加定義分子表面時候的距離投影圖是什么樣子。輸入如下命令
-1 //返回之前的界面
1 //設置對體系表面的定義
3 //通過范德華球疊加定義表面
1 //系數設為1,即直接使用Bondi范德華半徑作為原子球半徑
0 //開始計算
0 //在屏幕上顯示圖像
此時看到的圖像如下所示
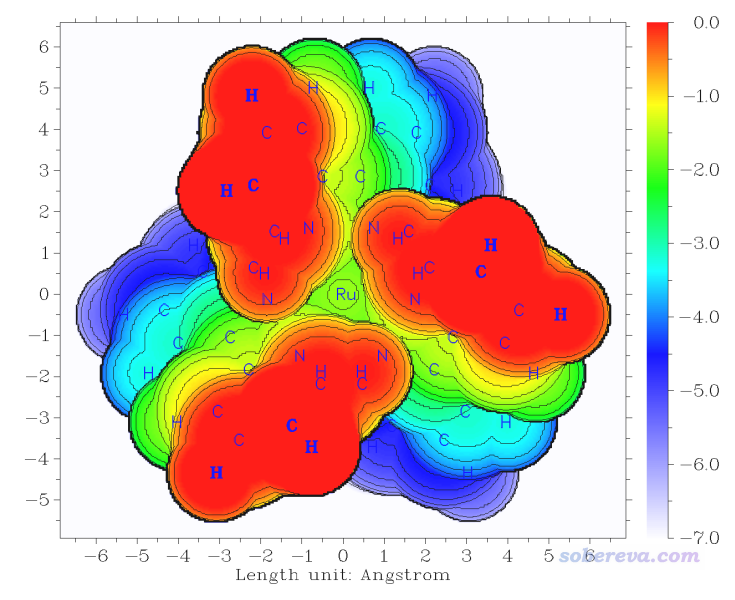
當前的圖對應范德華表面的距離投影,可以很直觀地看出Ru原子被顯著地包埋于三個配體之間,因此外部物質很難有機會接觸到Ru。通過這種圖,可以通過配體的位阻效應討論相似配合物發生金屬-有機反應的難易程度、催化能力高低等問題。大家還可以根據圖中的坐標刻度近似測量配體間距離以對位阻效應予以一些定量討論。
上面基于原子球疊加定義的體系表面圖在原子縫隙處不太平滑,而且對這種表面的定義Multiwfn沒法做插值提升Z位置判斷精度,所以線條稍有鋸齒感(雖然也可以減小掃描步長來減輕,但會提升耗時)。
下面我們還是基于準分子密度做距離投影圖,但使用比之前的0.05 a.u.小一個數量級的0.005 a.u.作為等值面數值。輸入下面的命令
-1 //返回之前的界面
1 //設置對體系表面的定義
1 //準分子密度
0.005 //等值面數值
0 //開始計算
0 //在屏幕上顯示圖像
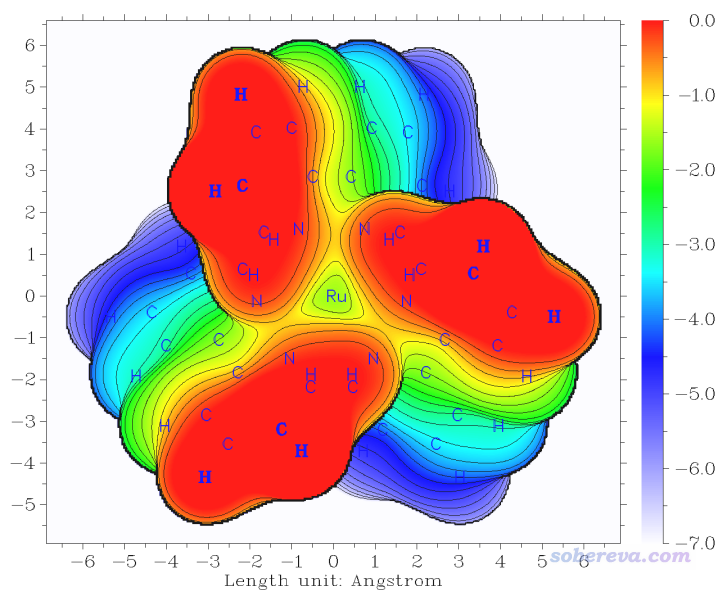
此時看到的圖像如下所示。這個圖和基于范德華球疊加時的圖的基本特征極為相似,但是線條明顯非常平滑了。我個人比較推薦以這種方式定義范德華表面來討論位阻問題(雖然Bader用0.001 a.u.等值面作為氣相下的范德華表面,但此時的等值面范圍有點太大了,Ru完全被配體糊住了,顯得太夸張了)
4 實例2:繪制Cu(111)晶面的表面距離投影圖
下圖舉一個周期性體系的例子,對Cu(111)晶面繪制表面距離投影圖。首先用GaussView基于Cu晶胞的cif文件切一個(111)晶面,厚度為三層,并且再平移復制一下,最終成為下面的樣子,俯視圖和側視圖都給了
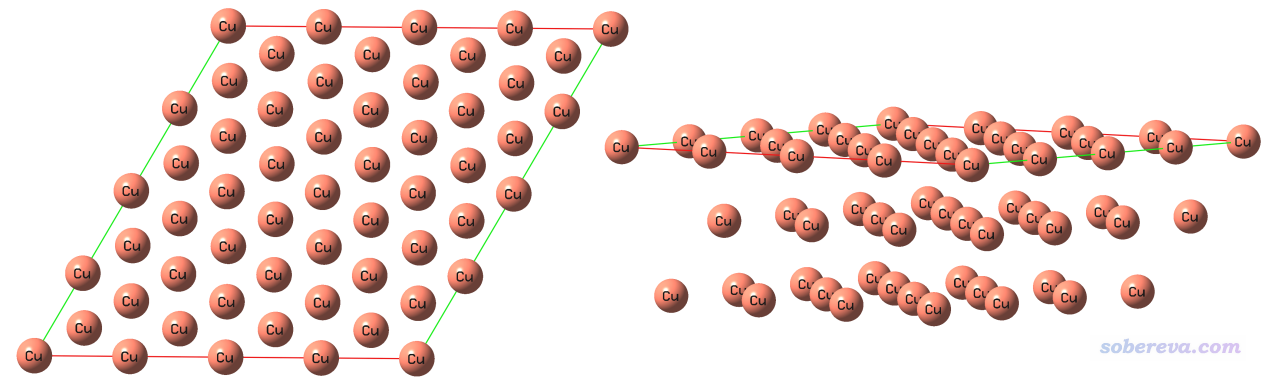
將此結構保存為Cu111_slab.gjf,此文件可以在此下載:http://www.shanxitv.org/attach/589/Cu111_slab.gjf。注意此文件里有兩個Tv(translation vector),這是兩個平移矢量,體現這個體系是個二維周期性體系。Multiwfn載入這樣含有周期性信息的文件后,在計算繪制表面距離投影圖所需的數據的時候就會相應地考慮周期性。
啟動Multiwfn,然后輸入
Cu111_slab.gjf
300 //其它功能(Part 3)
8 //繪制距離投影圖
7 //設置Z_start位置。默認情況正好是在最上層的Cu的原子核位置,這樣體現不出Cu表面的形貌
2 //令Z_start為2埃,相當于讓掃描的起點處在Cu表面上方2埃處,這樣可以把最上層Cu表面的形態展現出來
0 //開始計算。此例用的是默認的準分子密度為0.05 a.u.的等值面作為體系表面
算完之后選0繪圖,看到的圖像如下。圖中顯示Cu標簽的原子是原本晶胞范圍內的原子。
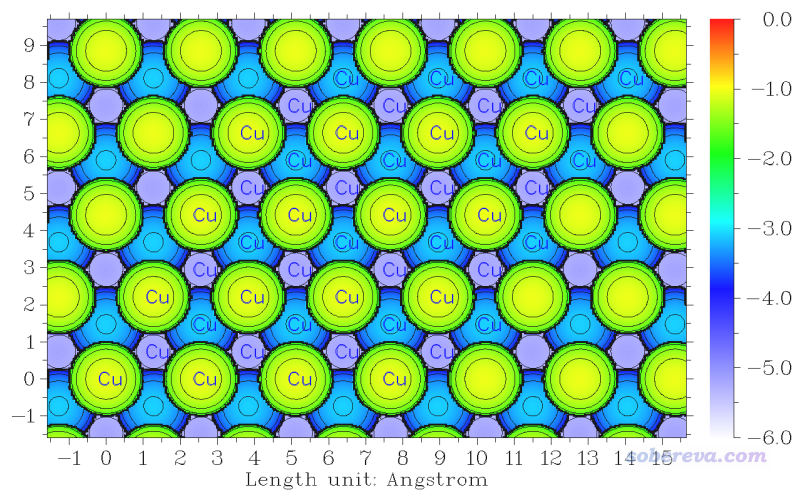
下面根據個人喜好可以調節作圖設置,比如關閉圖像后輸入
9 //修改色彩變化方式
4 //光譜
8 //修改色彩刻度
-6,-0.5
3 //切換是否顯示原子標簽
7 //修改XYZ標簽間隔
2,2,0.5
重新選0作圖,效果如下
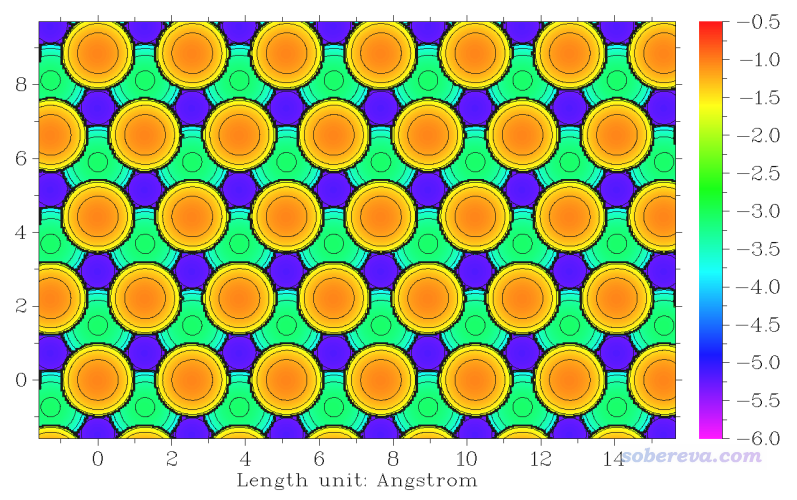
可見表面原子位置的相對深淺可以通過顏色很清晰地區分,在討論固體表面特征的時候可以利用這種圖。
順帶一提,如下圖所示,這種晶面上有不同的特征位點,這在上面我們繪制的表面距離投影圖中都可以非常清楚地看到,不同位點的深淺差異體現得十分清楚。
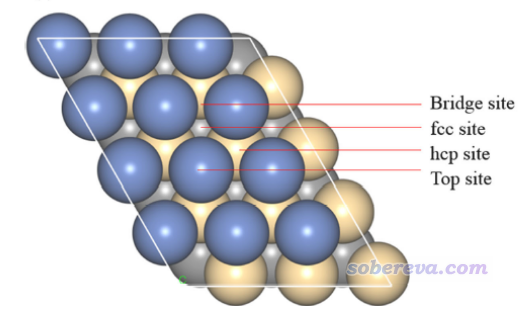
5 文獻中的例子
唐本忠等人的Sci. Adv., 7, eabj2504 (2021)一文中使用了本文介紹的Multiwfn的功能,繪制了一系列類似分子的表面投影圖,將體系結構特征差異很清楚地展現了出來。
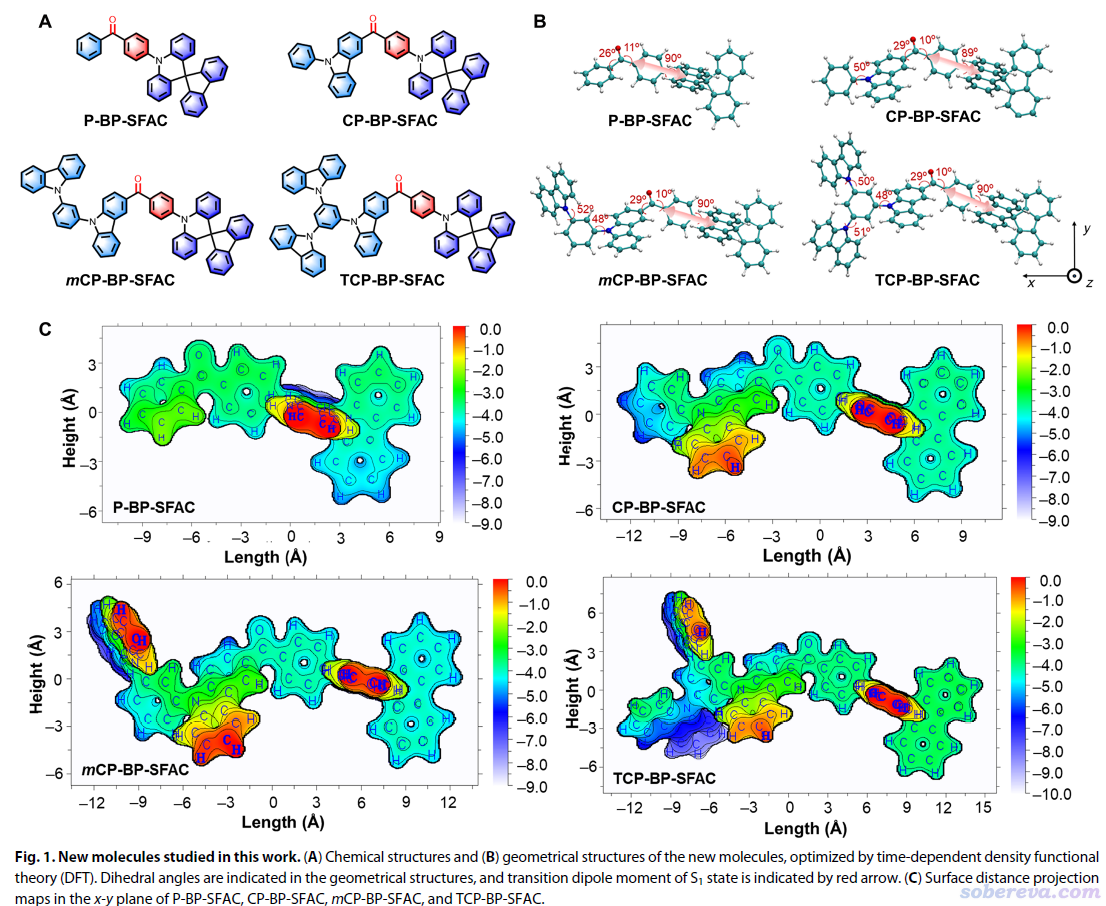
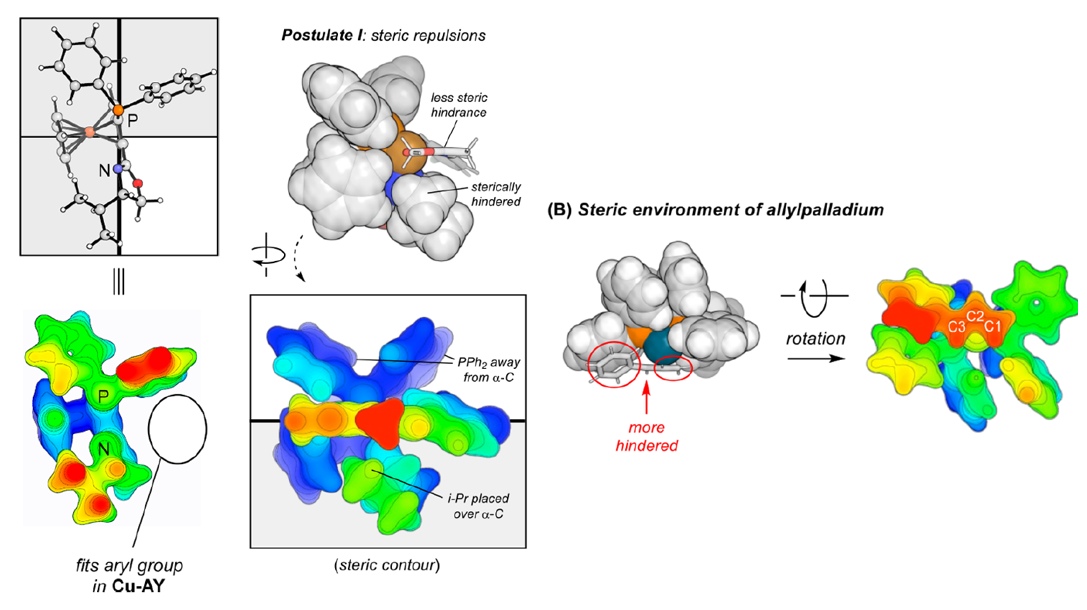
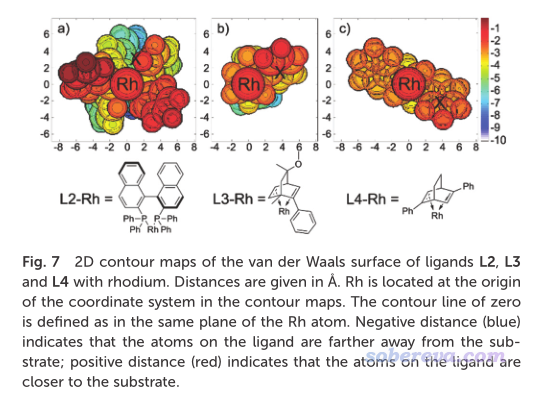
Houk等人的JACS (2022) https://doi.org/10.1021/jacs.1c12664一文中使用了本文介紹的Multiwfn的功能,考察了位阻效應。文中相關的圖匯總放在下面了。