使用Multiwfn計算過渡金屬的d-band center(d帶中心)
使用Multiwfn計算過渡金屬的d-band center(d帶中心)
文/Sobereva@北京科音
First release: 2021-Jan-14 Last update: 2022-Jun-2
1 前言
Multiwfn具有靈活強大的繪制態密度(DOS)曲線的功能,見《使用Multiwfn繪制態密度(DOS)圖考察電子結構》(http://sobereva.com/482),沒看過此文的話務必先仔細看一下,否則無法理解后文的敘述和例子。d-band center與PDOS有密切的關系,在本文中將說明怎么用Multiwfn算d-band center位置。此功能目前只適合孤立體系,如過渡金屬團簇。
Multiwfn可以在http://sobereva.com/multiwfn免費下載,相關知識見《Multiwfn入門tips》(http://sobereva.com/167)和《Multiwfn FAQ》(http://sobereva.com/452)。注意必須使用2021-Jan-14及以后更新的Multiwfn,否則沒有本文提到的功能。
雖然本文第4節以孤立的團簇作為例子演示計算,但本文的方法也同樣可以用于周期性體系。按照《詳談使用CP2K產生給Multiwfn用的molden格式的波函數文件》(http://www.shanxitv.org/651)的做法用CP2K程序對周期性體系產生molden文件,就同樣可以按http://www.shanxitv.org/482的做法繪制d原子軌道的PDOS,也因此可以同樣以下文的方式得到d帶中心。
2 d-band center簡介
Hammer和N?rskov提出的d-band center模型被廣泛并成功用于解釋和預測不同過渡金屬表面的催化活性,一些介紹參見、PNAS, 108, 937 (2011)、Sci. Rep., 6, 35916 (2016)。簡單來說,過渡金屬表面與小分子發生化學吸附時,過渡金屬的d帶與小分子的軌道會混合,產生成鍵態和反鍵態,如下所示,黑色和灰色分別是占據和非占據狀態,εF是Fermi能級,其以下的態都是占據的。成鍵態總是被占據的,反鍵態被部分占據。過渡金屬的d帶相對于Fermi能級越高,反鍵態能量相對于Fermi能級也就越高、被占據的程度就越低、成鍵作用被削弱得就越少,因而過渡金屬和小分子的化學結合作用就越強。
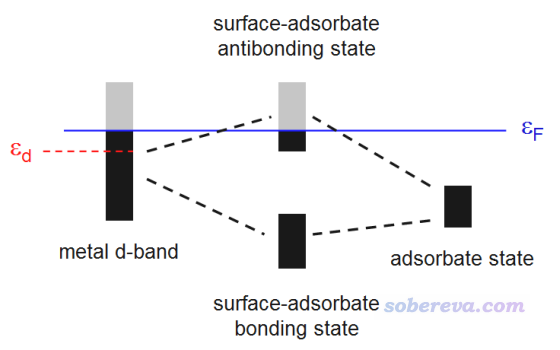
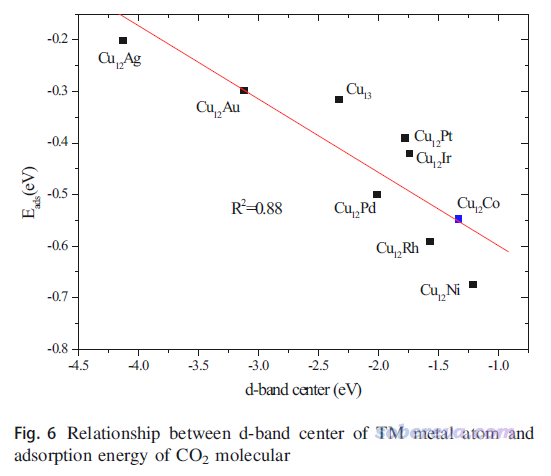
d-band center是指過渡金屬表面體系的d態對應的PDOS的中心位置與Fermi能級的差值,即上圖的εd,它可以視為衡量過渡金屬表面對小分子化學吸附能力的簡單的描述符。d-band center不僅常被搞第一性原理的研究表面催化的人拿來說事,做量子化學研究過渡金屬團簇的很多人也拿這個試圖解釋計算的不同過渡金屬團簇對小分子吸附能的差異,比如J. Clust. Sci., 29, 867 (2018)這篇文章就考察了Cu12TM (TM=Cu, Co, Rh, Ir, Ni, Pd, Pt, Ag, Au),確實發現這些團簇的d-band center越低,團簇與CO2的結合就整體越強,如下所示。雖然相關性不算特別好,但還是能說明一定問題的
3 在Multiwfn中計算d-band center的方法
Multiwfn的主功能10在繪制PDOS時,對每個用戶定義的片段(記為F),在文本窗口都會輸出按照下式計算的中心位置,其中low和high分別是當前繪圖時用的橫坐標的下限和上限
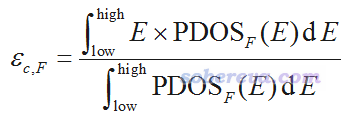
所以,想計算d-band center的話,你只需要將片段定義為你想要考察的那些過渡金屬的D基函數,并且讓作圖范圍框住d帶出現的能量范圍,然后讀取屏幕上輸出的中心位置,再手動減去費米能級值即可。對于孤立體系來說,費米能級沒有確切的定義,但習慣上可以直接取屏幕上顯示的HOMO能級值,這點在文中應當交代一下。由于Multiwfn計算PDOS中心位置的功能很靈活,所以不僅可以用來計算d-band center,還可以試圖用來計算別的類型band的center。
4 在Multiwfn中算d-band center一例:Cu13團簇
此例試圖重現前述的J. Clust. Sci., 29, 867 (2018)一文中的Cu13的d-band center值。那篇文章用的是PBE/lanl2DZ算的此團簇,我用Gaussian在這個級別下也優化了一下,得到的fchk文件可以在這里下載:http://sobereva.com/attach/582/Cu13.zip。
啟動Multiwfn,然后輸入
Cu13.fchk
10 //繪制DOS
-1 //定義片段
1 //定義第1個片段
cond //以條件方式選擇哪些基函數被加入
a //對原子序號范圍不設限制
a //對基函數序號范圍不設限制
D //基函數必須是D型
q //保存片段
q //返回DOS繪制界面
8 //把單位從默認的a.u.切換為eV
2 //設置橫坐標范圍
-13,0,2 //下限、上限、作圖的標簽間隔分別設-13、0、3
0 //作圖
此時看到下圖,紅線是Cu13的d-band對應的PDOS。這和J. Clust. Sci.那篇文章里給的PDOS圖的形狀相差較大,主要是因為Multiwfn默認用的FWHM(半高全寬)比那篇文章里大得多。FWHM的設置在原理上不影響給出的d-band center值,故不用顧慮這點。另外,此圖橫坐標的數值和那篇文章里的圖相差較大,那是因為那篇文章里對軌道能級進行了平移讓HOMO能級恰好為0,我們不用管這個。
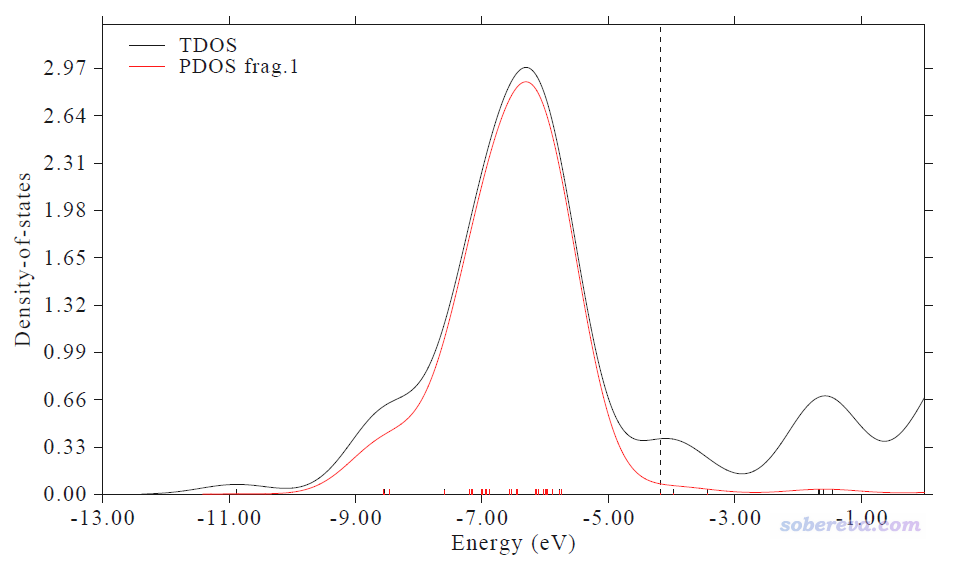
在文本窗口可看到下面的輸出
Center of TDOS: -5.732445 eV
Center of PDOS 1: -6.510656 eV
Note: The vertical dash line corresponds to HOMO level at -4.16932 eV
我們用HOMO能量當Fermi能級的話,此體系的d-band center就是-6.510656-(-4.16932)=-2.34 eV,J. Clust. Sci.那篇文章表3里給的值是-2.33 eV,可見相符極好。
作圖的橫坐標范圍需要注意,這對給出的Center值有影響。從上面顯示的PDOS圖來看,我們此例用的-13.0 ~ 0.0 eV能量區間是適當的,充分擴住了d-band的出現范圍。如果把橫坐標范圍設寬再作圖,看到的是下圖的情況
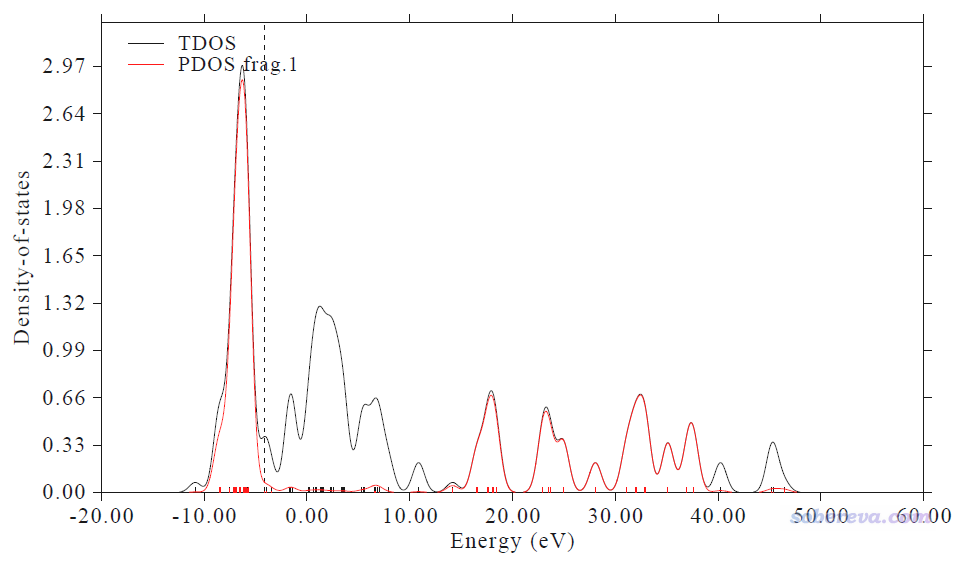
可見作圖下限取得更低一些,比如到-20也完全沒關系,反正在-13 eV往下都沒有d-band出現了,因此設得更低不影響給出的center值。一般作圖的能量上限取0就行了,千萬不能把上限取得特別高,因為如上圖所示,在>10 eV的高能區域d態PDOS又變得非常大,這是沒有化學意義的非價層的態,純粹是因為lanl2DZ是個擴展基(每個d軌道用兩個D型基函數描述)所帶來的現象。用上圖的-20~60 eV的范圍給出的PDOS中心位置是10.373 eV,明顯是沒意義的。
要注意對非限制性方法計算開殼層體系,由于自旋極化,alpha和beta自旋的d-band center的位置是不同的。對這類體系,Multiwfn默認是對alpha軌道繪制PDOS并計算其center。如果要考察beta的,必須在DOS繪制界面里選6 Choose orbital spin再選beta。如果alpha和beta軌道都考慮,就選Both。考慮的是什么自旋的應當在寫文章的時候說清楚。